


Abstract
Nicotine can enhance working memory and attention. Activation of both α7 and β2* nicotinic acetylcholine receptors (nAChRs) in the prefrontal cortex (PFC) has been implicated in these processes. The ability of presynaptic nAChRs to modulate neurotransmitter release, notably glutamate release, is postulated to contribute to nicotine's effects. We have examined the cellular mechanisms underlying α7 and β2* nAChR-mediated [3H]d-aspartate release from the PFC in vitro. Using the α7 and β2* nAChR-selective agonists (R)-N-(1-azabicyclo[2.2.2]-oct-3-yl)(5-(2-pyridyl)thiophene-2-carboxamide) (compound A) and 5-iodo-3-(2(S)-azetidinylmethoxy)pyridine (5-iodo-A-85380), respectively, in conjunction with inhibitors of voltage-operated Ca2+ channels (VOCCs) and intracellular Ca2+ stores, we show that [3H]d-aspartate release evoked by activation of β2* nAChRs occurs via VOCCs. In contrast, α7 nAChR-evoked release was unaffected by VOCC blockers but was abolished by modulators of Ca2+ stores, including ryanodine. The α7 nAChR ligand α-bungarotoxin and ryanodine receptors were colocalized to a subpopulation of PFC synaptosomes. Compound A-evoked [3H]d-aspartate release was also blocked by mitogen-activated protein kinase kinase 1 inhibitors, implicating extracellular signal-regulated kinase (ERK)1/2 in α7 nAChR-evoked exocytosis. Western blotting confirmed that compound A, but not 5-iodo-A-85380, application increased ERK2 phosphorylation in PFC synaptosomes, and this was dependent on ryanodine-sensitive stores. Compound A also promoted synapsin-1 phosphorylation at ERK1/2-dependent sites, in a ryanodine-sensitive manner. Thus, β2* and α7 nAChR subtypes in the PFC mediate [3H]d-aspartate release via distinct mechanisms as a result of their differential coupling to VOCCs and Ca2+-induced Ca2+ release (CICR), respectively. The ability of α7 nAChRs to promote the phosphorylation of presynaptic ERK2 and synapsin-1, downstream of CICR, provides a potential mechanism for presynaptic facilitation in the PFC.
In the mammalian central nervous system, nicotinic acetylcholine receptors (nAChRs) are implicated in cognitive and attentional processes (Mansvelder et al., 2006), making them therapeutic targets for neuropsychiatric disorders, including Alzheimer's disease and schizophrenia. nAChRs constitute a family of pentameric ligand-gated cation channels that are widely distributed in the brain (Gotti et al., 2006); both α7 and non-α7 subtypes of nAChR in the prefrontal cortex (PFC) have been implicated in effects of nicotine on attentional processes and working memory (Hahn et al., 2003; Mansvelder et al., 2006; Chan et al., 2007; Young et al., 2007). Recently, nicotine was shown to increase the frequency and amplitude of spontaneous excitatory postsynaptic currents in pyramidal neurons in mouse PFC, consistent with the facilitation of glutamate release (Couey et al., 2007). In several other brain regions, including hippocampus and ventral tegmental area, presynaptic α7 nAChRs on glutamatergic afferents can contribute to long-term changes in synaptic function (McKay et al., 2007).
α7 nAChRs are distinguished by their high relative permeability to Ca2+ (Dajas-Bailador and Wonnacott, 2004). The ability of nicotine to enhance excitatory postsynaptic currents and to increase intracellular Ca2+ in mossy fiber terminals in the presence of Cd2+, a nonselective blocker of voltage-operated Ca2+ channels (VOCCs), encouraged the view that Ca2+ entry via the α7 nAChR channel is sufficient to support glutamate release (Gray et al., 1996). However, Ca2+ entry can further augment intracellular Ca2+ concentration by Ca2+-induced Ca2+ release (CICR) from internal Ca2+ stores, via Ca2+ release channels: notably, ryanodine receptors, and also inositol 1,4,5-trisphosphate receptors (Verkhratsky, 2005; Bardo et al., 2006). Indeed, increases in Ca2+ in response to α7 nAChR activation are amplified by CICR in diverse preparations (Dajas-Bailador and Wonnacott, 2004), including hippocampal mossy fiber terminals (Sharma et al., 2008) and neuronal cell lines (Dickinson et al., 2007).
The modulation of spontaneous and evoked synaptic transmission by ryanodine is compatible with CICR from presynaptic ryanodine receptors driving changes in synaptic efficacy (Emptage et al., 2001; Collin et al., 2005). One candidate for translating CICR into more sustained changes in presynaptic function is the extracellular signal-regulated kinase/mitogen-activated protein kinase (ERK/MAPK) pathway (Kushner et al., 2005). ERK1/2-mediated phosphorylation of synapsin-1 can influence glutamate release (Jovanovic et al., 2000) and has been linked to presynaptic plasticity in hippocampus-dependent learning (Kushner et al., 2005).
We recently reported that nicotine can elicit [3H]d-aspartate release from rat frontal cortex synaptosomes (Rousseau et al., 2005). Here, we have exploited nAChR subtype-selective agonists to elucidate the presynaptic cellular mechanisms that couple nAChR activation to excitatory amino acid release. We show that in PFC synaptosomes α7 and β2* nAChR operate via distinct pathways that would enable these nAChR subtypes to make distinct contributions to the modulation of synaptic transmission.
Materials and Methods
Materials. [3H]d-aspartic acid (10-25 Ci/mmol) was purchased from PerkinElmer (Zaventem, Belgium). 125I-α-bungarotoxin (125I-αBgt 249 Ci/mmol) was purchased from GE Healthcare (Little Chalfont, Buckinghamshire, UK). (R)-N-(1-azabicyclo[2.2.2]oct-3-yl)(5-(2-pyridyl)thiophene-2-carboxamide) (compound A; Fig. 1A, left inset) was synthesized at GlaxoSmithKline (Harlow, Essex, UK). dl-threo-β-Benzoyloxyaspartic acid (TBOA), ω-conotoxin GVIA, ω-conotoxin MVIIC, U0126, PD 98059, and 5-iodo-A-85380 dihydrochloride (5-I-A-85380; Fig. 1A, right inset) were obtained from Tocris Cookson Inc. (Bristol, UK). αBgt and αBgt-Alexa Fluor 488 were purchased from Invitrogen (PoortGebaow, The Netherlands). Xestospongin-C, anti-ryanodine receptor (anti-mouse), and anti-synaptophysin (anti-rabbit) antibodies were purchased from Calbiochem (Beeston, Nottingham, UK). Another anti-ryanodine receptor pan-specific antibody was a generous gift from Prof. Anthony Lai (University of Wales, Cardiff, UK). Phospho-ERK1/2 (anti-rabbit) antibody was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), and total-ERK1/2 (anti-rabbit) antibody was purchased from Calbiochem (EMD Biosciences, San Diego, CA). Phospho-synapsin-1 (sites 4 and 5, anti-rabbit) and total synapsin (anti-rabbit) antibodies were generous gifts from Dr. Angus Nairn (Yale University, New Haven, CT). Dihydro-β-erythroidine (DHβE), cadmium chloride, verapamil, ryanodine, and mecamylamine were purchased from Sigma Chemical (Poole, Dorset, UK). Optiphase Safe was purchased from Wallac Scintillation Products, Fisher Chemicals (Loughborough, UK). All other chemicals were of analytical grade and obtained from Sigma Chemical. Sprague-Dawley rats were obtained from the University of Bath (Bath, UK) animal house breeding colony.
125I-αBgt Saturation and Competition Binding Assay. P2 membranes were prepared from rat PFC, hippocampus, or whole brain, and binding of 125I-αBgt was determined as described by Whiteaker et al. (2000). In brief, membranes (75 μg) were incubated in 100 μl of phosphate buffer (40 mM K2HPO4, 10 mM KH2PO4, 1 mM EDTA, 0.1 mM phenylmethylsulfonyl fluoride, 0.01% NaN3, and 0.1% BSA, pH 7.4) with 125I-αBgt (0.2-5 nM) for saturation binding. For competition binding assays, PFC membranes (75 μg) were incubated with 1 nM 125I-αBgt and serial dilutions of compound A (1 fM-100 μM) or 5-I-A-85380 (1 pM-10 mM). Each assay was performed in triplicate, and nonspecific binding was determined in parallel in the presence of 1 mM nicotine. Samples were incubated for 4 h at 37°C, followed by 1 h at 4°C and filtration through a double layer of GFA filters (presoaked overnight in 4% nonfat milk powder, top filter; 0.5% polyethylenimine, bottom filter; Gelman Instrument Co., Ann Arbor, MI) using a Brandel cell harvester (Semat, St. Albans, Hertfordshire, UK). Filters were washed three times with ice-cold phosphate-buffered saline (PBS; 10 mM phosphate buffer containing 0.9% NaCl, pH 7.4) and counted for radioactivity in Optiphase Safe in a Packard 1600 Tri-Carb scintillation counter (counting efficiency, 65%; PerkinElmer).
Data were fitted to the Hill equation to determine Kd and Bmax for saturation binding experiments or IC50 for competition assays. Values for Ki (inhibition binding constant) were derived, assuming a Kd value of 0.5 nM for 125I-αBgt binding to rat PFC membranes (Table 1).
Saturation binding of 125I-αBgt to rat brain P2 membranes
Preparation and Superfusion of PFC Synaptosomes. For each experiment, two male Sprague-Dawley rats (250-300 g) were killed by cervical dislocation, and the brains were rapidly removed. PFC tissue (approximately 80 mg/brain) was dissected by cutting approximately 3 mm posterior to the tip, after removing the olfactory lobes. PFC was homogenized 10% (w/v) in ice-cold 0.32 M sucrose and 5 mM HEPES, pH 7.4. P2 synaptosomes were isolated by differential centrifugation as described previously (Rousseau et al., 2005). In brief, the homogenate was centrifuged for 10 min at 1000g, and the resultant supernatant fraction, S1, was removed and centrifuged for 20 min at 20,000g. The P2 pellet containing synaptosomes was washed in Krebs-bicarbonate buffer (KB; 118 mM NaCl, 2.4 mM KCl, 2.4 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, 25 mM Na2HCO3, and 10 mM glucose, oxygenated with 95% O2/5% CO2 for 1 h and adjusted to pH 7.4). Finally, the P2 pellet was resuspended in 1.5 ml of KB.
Synaptosomes were loaded with [3H]d-aspartate (final concentration, 0.2 μM) and superfused as described previously (Rousseau et al., 2005). Approximately 300 μg of protein was loaded into open superfusion chambers of a modified Brandel superfusion apparatus (model SF-12; Semat) (Soliakov and Wonnacott, 1996) and superfused with KB at 0.8 ml/min. After a 24-min washout period, 2-min fractions of perfusate were collected. After a further 6 min, a 90-s pulse of stimulus (nAChR agonist or KCl in KB, or KB alone) was delivered, followed by superfusion for a further 22 min with KB. When present, antagonists and other drugs were included in the perfusing buffer for 10 min before agonist application (except for αBgt, which was applied throughout the 30-min wash period). All inhibitors were present during the stimulation and remained throughout the rest of the experiment. To determine the Ca2+ dependence of [3H]d-aspartate release, synaptosomes were perfused throughout the experiment with Ca2+-free KB containing 2 mM EGTA, replacing CaCl2 with MgCl2 (final concentration of Mg2+, 3.6 mM).
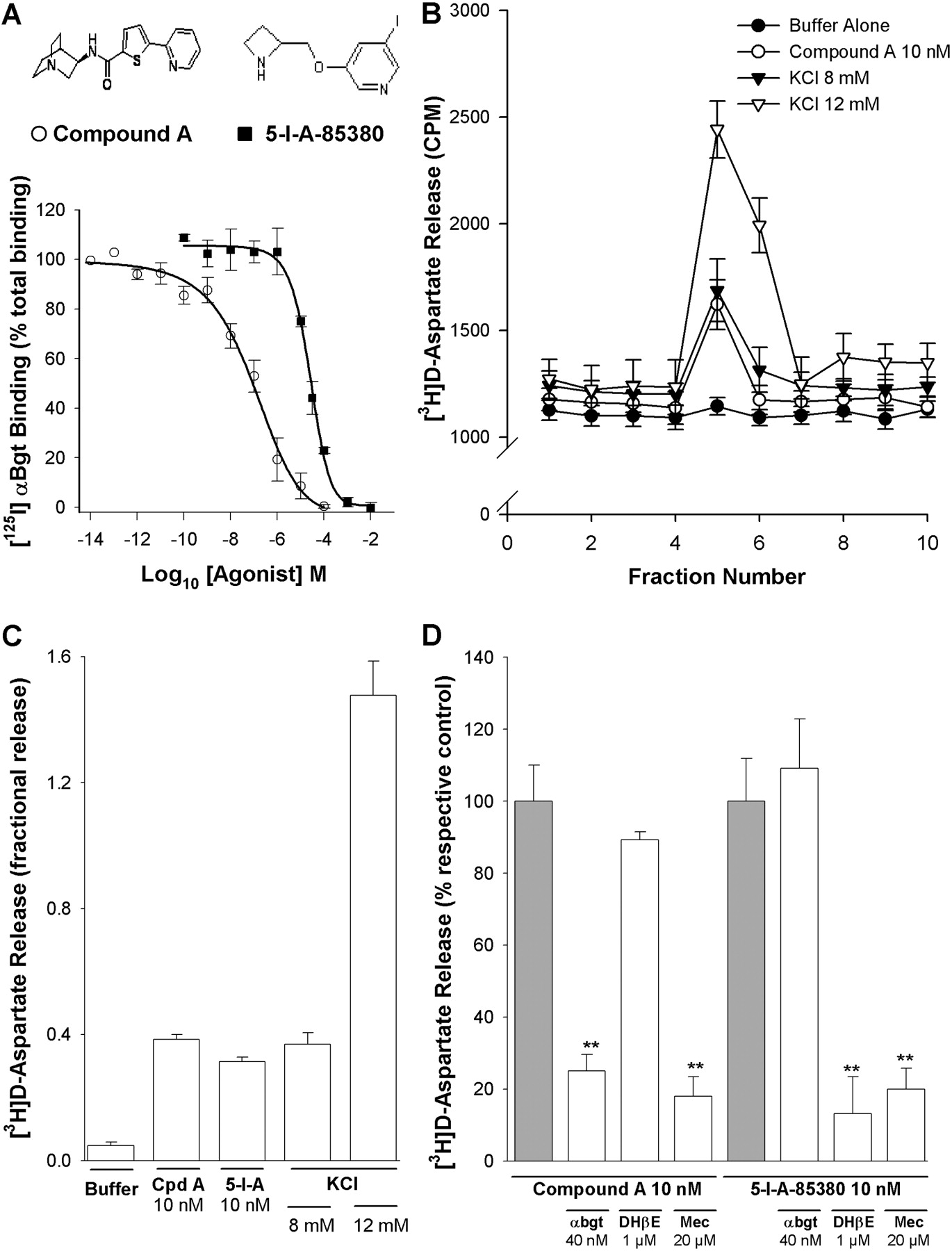
α7- and non-α7 nAChRs mediate [3H]d-aspartate release from PFC synaptosomes. A, inhibition by compound A (open circles; inset left) and 5-I-A-85380 (closed squares; inset right) of 125I-αBgt binding to PFC P2 membranes. P2 membranes were incubated with 1 nM 125I-αBgt and serial dilutions of compound A or 5-I-A-85380 as described under Materials and Methods. Data points are expressed as percentage of specific binding in the absence of test drug and were fitted to the Hill equation: Ki = 32.9 ± 1.4 nM and 10.7 ± 1.1 μM for compound A and 5-I-A-85380, respectively; mean ± S.E.M. of at least three experiments, each conducted in triplicate. B to D, rat PFC synaptosomes were loaded with [3H]d-aspartate, superfused with KB, and stimulated for 90 s with KCl or agonist as described under Materials and Methods. B, representative profiles of [3H]d-aspartate release in response to stimulation with 12 mM KCl (open triangles), 8 mM KCl (closed triangles), 10 nM compound A (open circles), or KB (closed circles). Values (CPM tritium released) are from a single experiment and correspond to the mean ± S.E.M. of three replicate superfusion chambers per condition. C, fractional release of [3H]d-aspartate from rat PFC synaptosomes, evoked by 10 nM compound A (Cpd A), 10 nM 5-I-A-85380 (5-I-A), and KCl. Bars represent the mean ± S.E.M. of at least four independent experiments. D, pharmacological characterization of nAChR agonist-evoked [3H]d-aspartate release from rat PFC synaptosomes. Synaptosomes were stimulated with 10 nM compound A or 10 nM 5-I-A-85380 in the presence or absence of mecamylamine (Mec; 20 μM), DHβE(1 μM), or αBgt (40 nM). Where present, antagonist was introduced into the perfusing buffer 10 min (Mec and DHβE) or 30 min (αBgt) before agonist application and remained throughout the experiment. Responses were calculated as fractional release and expressed as a percentage of the respective agonist control; bars represent the mean ± S.E.M. of at least four independent experiments. **, p < 0.01, significantly different from control.
Radioactivity in each of the collected perfusate fractions and that remaining in the synaptosomes at the end of the experiment was determined by addition of 4 ml of Optiphase Safe and counting in a Packard 1600 Tri-Carb scintillation counter (counting efficiency, 43%). Total radioactivity present in synaptosomes at the time of agonist stimulation was calculated as the sum of subsequently released [3H]d-aspartate plus radioactivity remaining in the synaptosomes. Data were analyzed by fitting a double exponential decay equation to the baseline data (Soliakov and Wonnacott, 1996) using the Sigma Plot (version 2.0) program for Windows: y = ae-bx + ce-dx, where a, b, c, and d are the curve parameters and x is the fraction number.
Evoked [3H]d-aspartate release was calculated as a percentage of the total radioactivity present in the synaptosomes at the point of stimulation (fractional release). The [3H]d-aspartate release in response to a pulse of KB (2.4 mM KCl; buffer-evoked release) was subtracted from agonist- or KCl-evoked release. The mean value for buffer-evoked release was 0.065 ± 0.005 (n = 30). None of the inhibitors or modulators used significantly increased [3H]d-aspartate release above buffer-evoked release. Fractional release in the presence of inhibitor was expressed as a percentage of the control response in the absence of inhibitor, included in all experiments. Data are expressed as the mean ± S.E.M. from at least four independent experiments, each carried out in triplicate. Statistical significance was determined using one-way analysis of variance with post hoc Dunnett's test; p values less than 0.05 were taken to be statistically significant.
Immunocytochemistry of Synaptosomes. PFC P2 synaptosomes were prepared from two male Sprague-Dawley rats (250-300 g) as described above. Synaptosomes were resuspended in 2 ml of KB and incubated with 200 nM Alexa Fluor 488-tagged αBgt at 37°C for 4 h in the presence or absence of 1 mM nicotine. The synaptosomes were washed in PBS (10 mM phosphate buffer containing 150 mM NaCl and 2 mM KCl, pH 7.4) by centrifugation and layered onto poly-l-lysine-coated slides and incubated for 45 min at room temperature to facilitate adhesion. Slides were rinsed and fixed in PBS containing 4% formaldehyde for 45 min. Slides were washed in PBS to remove fixative and incubated in PBS containing 10% normal goat serum, 0.5% BSA, and 0.05% Triton X-100 for 1 h at room temperature. Primary antibodies, rabbit anti-synaptophysin (1/500 dilution) and mouse anti-ryanodine receptor (1/100 dilution) were added in PBS containing 1% normal goat serum, 0.5% BSA, and 0.05% Triton X-100. Slides were incubated overnight at 4°C in a humidified chamber. After three 10-min washes with PBS, slides were incubated with secondary antibody solution (anti-rabbit IgG and anti-mouse IgG, both 1:750 in 1% normal serum, 0.5% BSA, and 0.05% Triton X-100 in PBS) for 2 h at room temperature in the dark. Slides were washed in PBS to remove excess secondary antibodies before mounting with VECTASHIELD (Vector Laboratories, Burlingame, CA) and viewing under an Axiovert 100 M microscope combined with an LSM 510 confocal system (Carl Zeiss GmbH, Jena, Germany). In the presence of nicotine, labeling by αBgt was abolished. Omission of primary antibodies also abolished labeling of the respective markers.

Ca2+ dependence of evoked [3H]d-aspartate release. Rat PFC synaptosomes were loaded with [3H]d-aspartate, superfused with KB, and stimulated for 90 s with 10 nM compound (A and D), 10 nM 5-I-A-85380 (B and E), or 8 mM KCl (C and F) in the presence or absence of Ca2+-free KB, Cd2+ (50 μM), or TBOA (10 μM) (A-C) or ω-conotoxin GVIA (GVIA; 1 μM), ω-conotoxin MVIIC (MVIIC; 1 μM), or verapamil (Ver; 10 μM) (D-F). Inhibitors were introduced into the perfusing buffer 10 min before stimulation and remained throughout the experiment. Fractional release is expressed as a percentage of control responses to 10 nM compound A, 10 nM 5-I-A-85380, or 8 mM KCl in the absence of inhibitors. Bars represent the mean ± S.E.M. of at least four independent experiments. *, p < 0.05; **, p < 0.01, significantly different from control.
Western Blotting of Synaptosomes. PFC P2 synaptosomes were prepared from two male Sprague-Dawley rats (250-300 g) as described above. Synaptosomes were resuspended in 2 ml of KB, and 90-μl aliquots were equilibrated at 37°C for 30 min before stimulation with agonist or KCl. When present, antagonists and other inhibitors were added 10 min before stimulation (except for αBgt, which was applied throughout the 30-min equilibration period). nAChR agonist or KCl (10 μl) was added for 90 s (unless otherwise stated), and then the response was stopped by addition of 2× lysis buffer (final concentrations: 50 mM Tris, 50 mM glycine, 2% SDS, 4% β-mercaptoethanol, 4 M urea, and 1.5 mM sodium orthovanadate) and heating at 95°C for 5 min, as described in Pereira et al. (2002).
Equivalent amounts from each synaptosome sample (ERK1/2 detection: 10 μg of protein; phospho-synapsin-1 detection: 5 μg of protein; total synapsin-1 detection: 0.5 μg of protein determined using the bicinchoninic acid protein assay kit; Pierce Chemical, Cramlington, Northumberland, UK) were resolved using 10% (ERK1/2 detection) or 7.5% (synapsin detection) SDS-polyacrylamide gel electrophoresis and blotted onto Hybond-ECL nitrocellulose (GE Healthcare) membranes. Western blotting was performed with phosphospecific [anti-phospho-p44/42 ERK1/2, 1:1000 dilution; or anti-phosphosynapsin-1 (sites 4 and 5); 1:2000 dilution] or phosphorylation state-independent (anti-p44/42 ERK1/2, 1:2000 dilution; or anti-synapsin, 1:30,000 dilution) primary antibodies. Following extensive washing, the membrane was incubated with horseradish peroxidase-conjugated anti-rabbit IgG (diluted 1:3000) for 2 h at room temperature. In all cases, loading controls were carried out using the phosphorylation state-independent anti-p44/42 ERK1/2 antibody. Electrochemiluminescence Western blotting reagents (ECL-PLUS; GE Healthcare) were used for the detection of corresponding bands and visualized using Epi Chemi II Darkroom (UVP, Inc., Upland, CA).
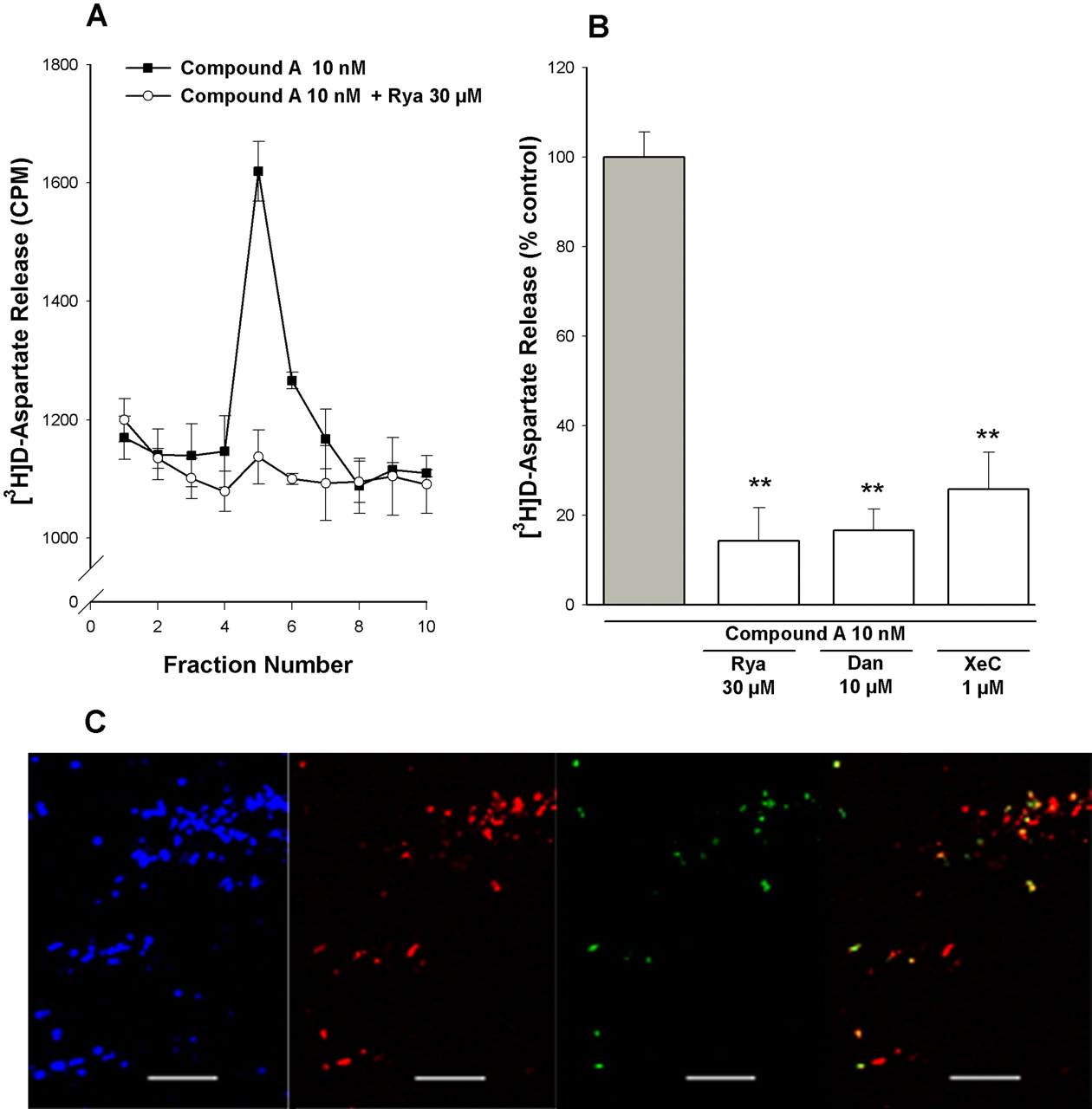
Contribution of CICR to α7 nAChR-mediated [3H]d-aspartate release. A and B, rat PFC synaptosomes were loaded with [3H]d-aspartate, superfused with KB, and stimulated for 90 s with, 10 nM compound A, in the presence or absence of ryanodine (Rya; 30 μM), dantrolene (Dan; 10 μM), and xestospongin-C (XeC; 1 μM). Antagonists were introduced into the perfusing buffer 10 min before applying the 90-s stimulus and remained throughout the experiment. A, representative profiles of [3H]d-aspartate release in response to stimulation by 10 nM compound A, in the absence (closed squares) and presence (open circles) of 30 μM Rya. Values (cpm tritium release) are from a single experiment and correspond to the mean ± S.E.M. of three replicate superfusion chambers per condition. B, fractional release of [3H]d-aspartate was expressed as a percentage of the compound A, 5-I-A-85380, or KCl control response determined in parallel in the absence of inhibitors. Bars represent the mean ± S.E.M. of at least four independent experiments. **, p < 0.01, significantly different from control. C, confocal imaging of synaptosomes tripled labeled with anti-synaptophysin (blue), anti-ryanodine (red), and αBgt-Alexa Fluor 488 (green). Right, overlay of anti-ryanodine and αBgt. Scale bar, 20 μm. A representative image using a pan-acting anti-ryanodine antibody is shown. αBgt-Alexa Fluor 488 labeling was abolished in the presence of 1 mM nicotine (data not shown).
Bands generated were quantified by densitometry using Scion Image (Scion Corporation, Frederick, MD). Results are expressed as a percentage of buffer-treated or drug-stimulated samples, carried out in parallel in each experiment. None of the antagonists or inhibitors used significantly altered the control response in the presence of buffer only. Data represent the mean ± S.E.M. of at least four independent experiments, and statistical significance was determined using one-way analysis of variance with post hoc Dunnett's test, as stated in the figure legends.
Results
α7 and Non-α7 nAChRs Mediate [3H]d-Aspartate Release from PFC Synaptosomes. Isolated nerve terminals (synaptosomes) facilitate direct measurement of transmitter release for characterizing the contribution of presynaptic receptors. Previous reports have provided evidence for nAChR-mediated [3H]d-aspartate release from cortical synaptosomes (Rousseau et al., 2005; Patti et al., 2006). In the present study, we have used synaptosomes isolated from anterior PFC, a functionally and anatomically more homogeneous cortical area that is relevant for the local nicotinic mechanisms that contribute to working memory and attention (Mansvelder et al., 2006). [3H]d-Aspartate mimics the behavior of endogenous glutamate and is used as a convenient surrogate in release studies (Patti et al., 2006).
We have used a novel α7 nAChR-selective agonist, compound A (Cilia et al., 2005; Dickinson et al., 2007; Fig. 1A, left inset), to explore the contribution of α7 nAChRs to [3H]d-aspartate release from PFC synaptosomes. Compound A displaced 125I-αBgt binding to PFC membranes, with a Ki value of 32.9 ± 1.4 nM (Fig. 1A). Compared with whole brain, the PFC expresses a relatively high density of α7 nAChRs, indicated by the number of 125I-αBgt binding sites, although the density is significantly lower than in hippocampus (Table 1). The apparent Kd value for 125I-αBgt binding (0.5 nM) is comparable in all three regions analyzed (Table 1).
Compound A (10 nM) elicited [3H]d-aspartate release from superfused PFC synaptosomes (Fig. 1, B and C). This concentration of compound A was maximally effective, consistent with our previous functional study (Dickinson et al., 2007) and was used for all subsequent experiments. KCl-evoked [3H]d-aspartate release was monitored in parallel; the release evoked by 10 nM compound A corresponded in magnitude to the response to 8 mM KCl, whereas 12 mM KCl produced a substantially larger response (Fig. 1, B and C). The lower concentration of KCl was therefore selected for comparative experiments. Compound A-evoked [3H]d-aspartate release was blocked by the nonselective nAChR antagonist mecamylamine (20 μM) and αBgt (40 nM; 81.9 ± 5.2 and 75.0 ± 4.5% inhibition, respectively; Fig. 1D), confirming the involvement of α7 nAChRs. In contrast, the β2*-selective nAChR antagonist DHβE(1 μM) had no significant effect on compound A-evoked [3H]d-aspartate release (89.3 ± 2.2% of control).
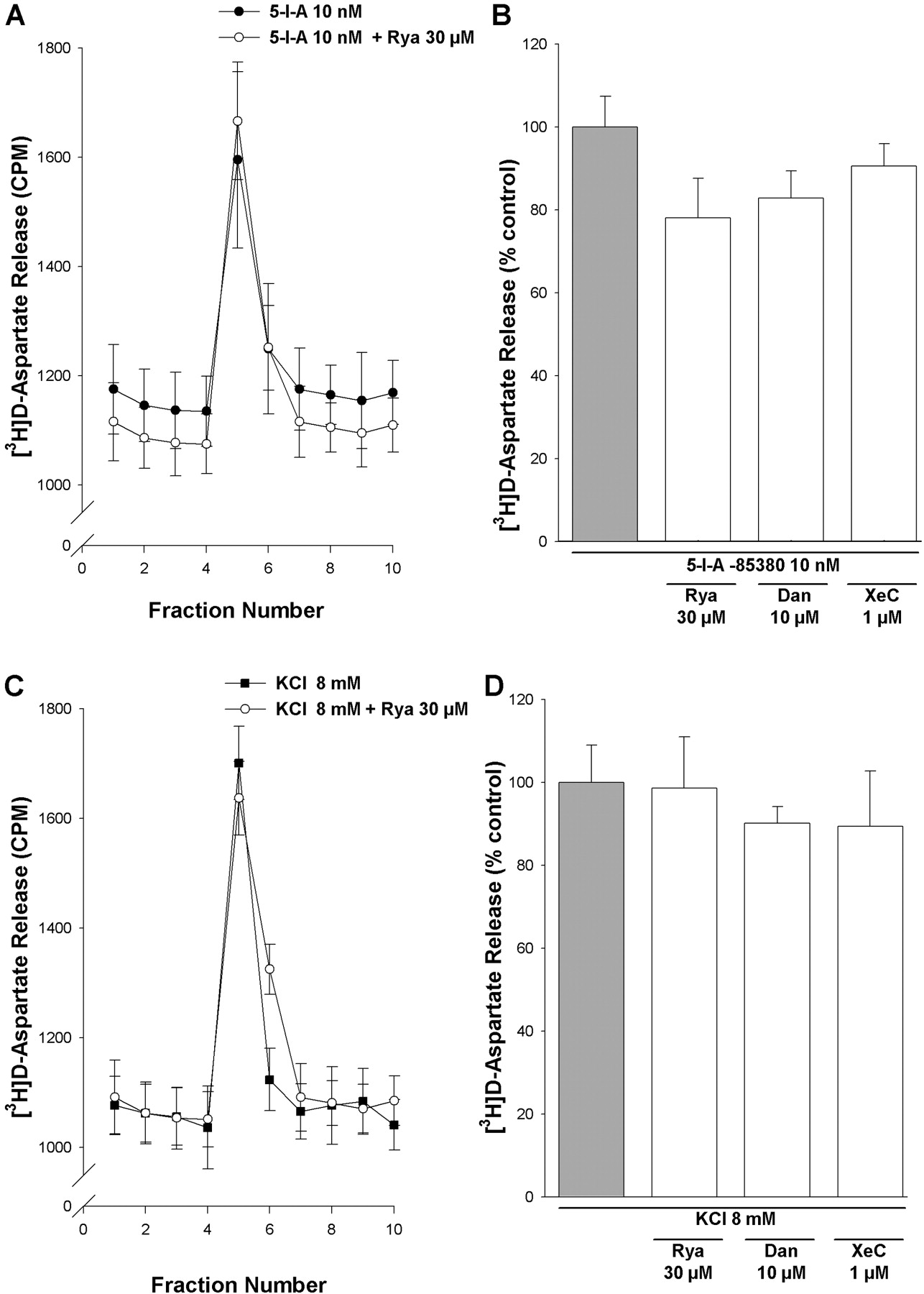
Contribution of CICR to non-α7 nAChR-mediated [3H]d-aspartate release. Rat PFC synaptosomes were loaded with [3H]d-aspartate, superfused with KB, and stimulated for 90 s with 10 nM 5-I-A-85380 (A and B) or 8 mM KCl (C and D), in the presence or absence of Rya (30 μM), dantrolene (Dan; 10 μM), and xestospongin-C (XeC; 1 μM). Antagonists were introduced into the perfusing buffer 10 min before applying the 90-s stimulus and remained throughout the experiment. A and C, representative profiles of [3H]d-aspartate release in response to stimulation by 10 nM 5-I-A-85380 (A) or 8 mM KCl (C) in the absence (▪) and presence (○) of 30 μM Rya. Values (cpm tritium release) are from a single experiment and correspond to the mean ± S.E.M. of three replicate superfusion chambers per condition. B and D, fractional release of [3H]d-aspartate was expressed as a percentage of the 5-I-A-85380 or KCl control response determined in parallel in the absence of inhibitors. Bars represent the mean ± S.E.M. of at least four independent experiments. **, p < 0.01, significantly different from control.
β2* nAChRs have also been implicated in excitatory amino acid release from frontal cortex (Gioanni et al., 1999; Rousseau et al., 2005). 5-Iodo-A-85380 is a potent β2*-selective nAChR agonist (Mukhin et al., 2000; Mogg et al., 2004; Fig. 1A, right inset) that was approximately 300 times less potent than compound A in displacing 125I-αBgt binding to PFC membranes (Ki value of 10.7 ± 1.1 μM; Fig. 1A). A low concentration of 5-I-A-85380 (10 nM) that should not interact with α7 nAChRs elicited [3H]d-aspartate release that was insensitive to αBgt but blocked by mecamylamine and DHβE (80.0 ± 5.9 and 82.3 ± 10.2% inhibition, respectively; Fig. 1, C and D). Therefore, both α7 and β2* nAChR can modulate [3H]d-aspartate release from PFC synaptosomes, in agreement with a previous study of frontal cortex terminals (Rousseau et al., 2005).

Effect of MEK inhibitors on evoked [3H]d-aspartate release. Rat PFC synaptosomes were loaded with [3H]d-aspartate, superfused with KB, and stimulated for 90 s with 10 nM compound (A), 10 nM 5-I-A-85380 (B), or 8 mM KCl (C) in the presence or absence of PD 98059 (PD; 10 μM) or U0126 (U0; 10 μM). The MEK inhibitors were introduced into the perfusing buffer 10 min before applying the 90-s stimulus and remained throughout the experiment. Fractional release of [3H]d-aspartate was expressed as a percentage of the compound A, 5-I-A-85380, or KCl control response determined in parallel in the absence of inhibitors. Bars represent the mean ± S.E.M.; n ≥ 3 (A), n = 5 (B), and n ≥4 (C). **, p < 0.01, significantly different from control.
Compound A-Evoked [3H]d-Aspartate Release Requires Ca2+Entry through the α7 nAChR and CICR. The ability of compound A, 5-I-A-85380, and 8 mM KCl to evoke [3H]d-aspartate release was highly Ca2+-dependent (Fig. 2, A-C). In contrast, the excitatory amino acid transporter inhibitor TBOA (10 μM) was without significant effect (Fig. 2, A-C), consistent with Ca2+-dependent exocytotic release of [3H]d-aspartate rather than release by transporter reversal. In the presence of the nonspecific VOCC inhibitor Cd2+ (50 μM), compound A-evoked [3H]d-aspartate release was diminished by 20.8 ± 2.5% (Fig. 2A), but this was not significantly different from control. However, this concentration of Cd2+ produced a marked inhibition of both 5-I-A-85380- and KCl-evoked release (71.5 ± 14.0 and 73.4 ± 8.8% inhibition, respectively; Fig. 2, B and C).
Selective VOCC inhibitors ω-conotoxin GVIA (N-type; 1 μM,), ω-conotoxin MVIIC (P-/Q-, N-type; 1 μM), and verapamil (L-type; 10 μM), at concentrations previously shown to fully block specific VOCCs (Herrero et al., 1999), failed to diminish compound A-evoked [3H]d-aspartate release (Fig. 2D). However, 5-I-A-85380-evoked [3H]d-aspartate release was sensitive to ω-conotoxin GVIA and ω-conotoxin MVIIC (67.0 ± 7.7 and 46.7 ± 7.4% inhibition, respectively; Fig. 2E). KCl-evoked [3H]d-aspartate release was also partially inhibited in the presence of ω-conotoxin GVIA and ω-conotoxin MVIIC (60.1 ± 4.5 and 71.8 ± 10.9% inhibition, respectively; Fig. 2F). Verapamil was without effect on 5-I-A-85380- or KCl-evoked [3H]d-aspartate release (Fig. 2, E and F), consistent with a lack of L-type VOCC involvement.
The insensitivity of compound A-evoked [3H]d-aspartate release to VOCC inhibitors and 50 μMCd2+ suggests that the predominant route of Ca2+ entry into PFC synaptosomes in response to compound A stimulation is via the α7 nAChR channel itself. Ca2+ influx through α7 nAChR is coupled to CICR in various preparations (Dajas-Bailador and Wonnacott, 2004; Sharma et al., 2008), and CICR has been linked to synaptic plasticity (Emptage et al., 2001). To explore this possible relationship in PFC synaptosomes, we examined the effects of ryanodine (30 μM) and dantrolene (10 μM), which both inhibit Ca2+ release from ryanodine-sensitive endoplasmic reticulum stores, and xestospongin-C (10 μM), a membrane-permeant, noncompetitive inhibitor of inositol 1,4,5-trisphosphate receptors. Xestospongin-C also blocks the endoplasmic reticulum Ca2+ ATPase, resulting in the emptying of stores and inhibition of CICR (Verkratsky, 2005). [3H]d-aspartate release evoked by compound A was markedly reduced in the presence of each of these inhibitors (Fig. 3, A and B). Immunolabeling of PFC synaptosomes revealed a subpopulation of synaptophysin-positive structures labeled with Alexa Fluor 488-tagged αBgt (Fig. 3C). These α7 nAChR-positive structures corresponded to a subpopulation of ryanodine receptor-positive synaptosomes (Fig. 3C), consistent with the presence of α7 nAChRs on nerve terminals that contain ryanodine-sensitive Ca2+ stores.

Phosphorylation of ERK1/2 in response to nAChR agonists. A, rat PFC synaptosomes were incubated with 10 nM compound A or 10 nM 5-I-A-85380 for 90 s before Western blot analysis. Top, p-ERK2 (molecular mass, 42 kDa) levels. Bottom, total ERK1/2 (molecular mass, 44 and 42 kDa, respectively) levels. B and C, time course of stimulation of synaptosomes with 10 nM compound A (B) or 5-I-A-85380 (C) and subsequent Western blot analysis to detect p-ERK2 levels. Representative blots are shown. ERK2 phosphorylation was quantified by densitometry as described under Materials and Methods and is expressed as a percentage of the basal levels of ERK2 phosphorylation determined in parallel. Bars represent the mean ± S.E.M. of at least four independent experiments. Total ERK1/2 levels were unaltered in any of the conditions. *, p < 0.05; **, p < 0.01, significantly different from basal.
These data support the hypothesis that compound A-evoked [3H]d-aspartate release is dependent upon CICR triggered by Ca2+ influx through the α7 nAChR. In contrast, [3H]d-aspartate release evoked by 10 nM 5-I-A-85380 or 8 mM KCl was insensitive to ryanodine, dantrolene, and xestospongin-C (Fig. 4A-D). Therefore, α7 and β2* nAChR differentially mediate [3H]d-aspartate release through distinct Ca2+-dependent pathways.
α7 nAChR-Mediated Release of [3H]d-Aspartate Is Dependent on ERK1/2 Activation. Stimulation of α7 nAChRs results in activation (phosphorylation) of ERK1/2 in vivo and in vitro (Dajas-Bailador and Wonnacott, 2004). A link between presynaptic ERK1/2 activation and glutamate release has also been reported previously (Jovanovic et al., 2000; Pereira et al., 2002). To explore the possible involvement of presynaptic ERK1/2 activation in nAChR-evoked [3H]d-aspartate release from PFC synaptosomes, we used two structurally distinct inhibitors of the upstream kinase MEK1, PD 98059 and U0126.
In the presence of 10 μM PD 98059 or 10 μM U0126, compound A-evoked [3H]d-aspartate release was substantially reduced (81.3 ± 19.8 and 79.2 ± 15.4% inhibition, respectively; Fig. 5A). Responses to 10 nM 5-I-A-85380 and 8 mM KCl were much less affected (Fig. 5, B and C). PD 98059 and U0126 decreased 10 nM 5-I-A-85380-evoked [3H]d-aspartate release by 32.3 ± 21.5 and 51.5 ± 18.4% and 8 mM KCl-evoked release by 34.1 ± 13.3 and 51.5 ± 7.1%, respectively. The latter rather variable responses may reflect nonspecific actions of these inhibitors at other targets, notably, VOCCs (Pereira et al., 2002).
Confirmation that stimulation of α7 (but not β2*) nAChRs results in the activation of ERK1/2 in PFC synaptosomes was provided by Western blot analysis. Total ERK1/2 protein was unchanged under any of the conditions examined. Compound A (10 nM) significantly increased ERK2 phosphorylation (molecular mass, 42 kDa; Fig. 6A; 150 ± 5% of buffer control), but ERK1 was unaltered (molecular mass, 44 kDa). In contrast 5-I-A-85380 did not significantly alter the phosphorylation of ERK1 or 2 (Fig. 6A). The lack of effect of 5-I-A-85380 (10 nM) was confirmed over the time course of 0 to 10 min (Fig. 6B). Compound A increased ERK2 phosphorylation in a time-dependent manner, with maximum increases seen at 1.5 and 3 min (Fig. 6C), compatible with the 1.5-min application of compound A in release experiments. This response was mediated by α7 nAChRs because compound A-evoked increases in ERK2 phosphorylation were abolished in the presence of mecamylamine or αBgt, but not DHβE (Fig. 7A). Both of the MEK1 inhibitors also blocked the compound A-mediated increases in ERK2 phosphorylation (Fig. 7B), consistent with the involvement of the ERK/MAPK pathway.

Pharmacological specificity of compound A-evoked increases in phospho-ERK2. Rat PFC synaptosomes were stimulated with a 90-s application of 10 nM compound A in the presence or absence of DHβE(1 μM), Mec (20 μM), or αBgt (40 nM) (A); or PD (10 μM), U0126 (U0; 10 μM), or Rya (30 μM) (B). When present, antagonists were introduced 10 min (30 min αBgt) before agonist application and remained during stimulation. Representative blots of p-ERK2 (molecular mass, 42 kDa) are shown. ERK2 phosphorylation was quantified by densitometry as described under Materials and Methods and is expressed as a percentage of the basal levels of ERK phosphorylation determined in parallel. Total ERK1/2 (loading control) was unaltered in any of the conditions (data not shown). Bars represent the mean ± S.E.M. of at least four independent experiments. *, p < 0.05; **, p < 0.01, significantly different from agonist alone.
α7 nAChR-mediated activation of ERK1/2 is Ca2+-dependent (Dineley et al., 2001; Dajas-Bailador et al., 2002). To determine whether CICR is required, compound A-evoked phosphorylation of ERK2 was determined in the presence of ryanodine, under conditions that block compound A-evoked [3H]d-aspartate release (Fig. 3, A and B). There was a significant attenuation of ERK2 phosphorylation in the presence of ryanodine (56.4 ± 6.2% inhibition; Fig. 7B), consistent with CICR mediating a substantial proportion of this response to compound A.
α7, but Not β2*, nAChR Stimulation Causes Synapsin-1 Phosphorylation via ERK1/2 Activation. Synapsin-1, a presynaptic protein that tethers vesicles to F-actin when in its nonphosphorylated state, has been proposed as a target for presynaptic plasticity and hippocampus-dependent learning (Kushner et al., 2005). ERK1/2 phosphorylates synapsin-1 at Ser62, Ser67, and Ser551 (sites 4, 5, and 6, respectively), allowing vesicles to move to the presynaptic membrane (Matsubara et al., 1996). Here, we have used a site 4 and 5 phospho-specific synapsin-1 antibody (Jovanovic et al., 1996) to investigate the ability of nAChRs to activate this protein.
Under conditions that evoke [3H]d-aspartate release, compound A significantly increased synapsin-1 phosphorylation (Fig. 8A; 63.7 ± 10.9% increase over buffer control), whereas 5-I-A-8530 application did not alter synapsin-1 phosphorylation (Fig. 8A). The inhibition of compound A-evoked increases in synapsin-1 phosphorylation by αBgt or mecamylamine, but not DHβE, (Fig. 8B), confirms that this response was mediated by α7 nAChRs. To verify that phosphorylation was occurring via the ERK1/2 pathway, stimulation of PFC synaptosomes by compound A was carried out in the presence of PD 98059 (10 μM). PD 98059 abolished compound A-evoked increases in synapsin-1 phosphorylation to basal level (Fig. 8C). Ryanodine (30 μM) also abolished this response to compound A, consistent with a requirement for CICR (Fig. 8C).
Discussion
The key finding of this study is that presynaptic α7 and β2* nAChRs modulate excitatory amino acid release through different cellular mechanisms. Presynaptic α7 nAChRs are predominantly found on nerve terminals that are immunopositive for ryanodine receptors, and stimulation of α7 nAChRs leads to CICR. We have identified novel presynaptic targets of α7 nAChR activation, downstream of CICR, namely, ERK1/2 and synapsin-1 phosphorylation. In contrast, stimulation of β2* nAChRs recruits VOCCs.
A link between activation of α7 nAChRs and CICR has been reported for diverse cellular systems using acetylcholine or nicotine and α7 nAChR-selective antagonists in conjunction with blockers or modulators of intracellular Ca2+ stores (Dajas-Bailador and Wonnacott, 2004). Here, we used the subtype-selective agonists compound A and 5-I-A-85380 to independently target α7 and β2* nAChRs on glutamatergic synaptosomes. This approach demonstrated unequivocally that α7 and β2* nAChRs are functionally coupled to distinct downstream targets: internal Ca2+ stores and VOCCs, respectively. We have observed the same dichotomy in PC12 cells, in which compound A evoked increases in intracellular Ca2+ that were independent of VOCC inhibitors but blocked by ryanodine, whereas 5-I-A-85380 elicited Ca2+ increases that were attenuated by verapamil but insensitive to ryanodine (Dickinson et al., 2007). Taken together, these various reports are consistent with the hypothesis that in diverse cell types and in different subcellular domains, highly Ca2+-permeable α7 nAChRs are preferentially localized to directly elicit CICR from intracellular stores.

Phosphorylation of synapsin-1 at sites 4 and 5 in response to compound A or 5-I-A-85380 application. Before Western blot analysis, rat PFC synaptosomes were incubated for 90 s with 10 nM compound A or 10 nM 5-I-A-85380 [top, p-synapsin levels (molecular mass, 90 kDa); bottom, total synapsin levels (molecular mass, 85 and 90 kDa)] (A); 10 nM compound A in the presence or absence of DHβE(1 μM), Mec (20 μM), or αBgt (40 nM) (B); and 10 nM compound A in the presence or absence of PD (10 μM) or Rya (30 μM) (C). When present, antagonists were introduced 10 min (30 min for αBgt) before agonist application and remained during stimulation. Representative blots are shown. Synapsin-1 phosphorylation was quantified by densitometry as described under Materials and Methods and is expressed as a percentage of the basal levels of synapsin-1 phosphorylation determined in parallel. Total ERK1/2 (loading control) was unaltered in any of the conditions (data not shown). Bars represent the mean ± S.E.M. of at least five independent experiments. *, p < 0.05; **, p < 0.01, significantly different from buffer (A) and agonist alone (B and C).
In contrast, β2* nAChRs promote depolarization-activated Ca2+ influx via VOCCs, analogous to KCl depolarization. 5-I-A-85380 preferentially activates α4β2- or α6β2-containing nAChRs (Mukhin et al., 2000; Mogg et al., 2004). In PFC synaptosomes, α4β2* nAChRs are likely to mediate the effects of nanomolar concentrations of 5-I-A-85380 on [3H]d-aspartate release. The decrease in [3H]nicotine binding in rat PFC after lesions of the mediodorsal thalamic nucleus (Gioanni et al., 1999), together with evidence from mice lacking the β2 subunit (Lambe et al., 2003), support the presence of α4β2* nAChRs on thalamocortical afferents to the PFC. The α6 subunit can be discounted because its expression is largely restricted to catecholaminergic neurons (Gotti et al., 2006). The activation of N- and P/Q-type VOCCs by presynaptic β2* nAChR is consistent with previous studies of striatal dopamine release (Soliakov and Wonnacott, 1996; Kulak et al., 2001).
In PC12 cells, activation of α3β2* nAChRs by higher concentrations of 5-I-A-85380 resulted in increases in intracellular Ca2+ that were blocked by L-type VOCC inhibitors (Dickinson et al., 2007), whereas β2* (but not α7) nAChRs on somatodendritic regions of dopaminergic neurons in the substantia nigra elicited tetrodotoxin-sensitive Ca2+ responses that were mediated by T-type VOCCs (Tsuneki et al., 2000). Thus, different non-α7 nAChR subtypes in various cellular settings can alter intracellular Ca2+ by local depolarization and activation of distinct VOCCs, probably reflecting the types of VOCC expressed locally, but they do not directly promote CICR. Activation of VOCCs can in turn elicit CICR in certain neurons, by either functional or physical coupling to ryanodine receptors (Collin et al., 2005). In nigral neurons, nicotine-induced, VOCC-dependent increases in intracellular Ca2+ may be further amplified by Ca2+ release from intracellular stores (Tsuneki et al., 2000), but this is not the case in PFC synaptosomes (this study) or PC12 cells (Dickinson et al., 2007).
The coupling of α7 and β2* nAChRs to distinct cellular signaling pathways in PFC synaptosomes raises the interesting question of whether these nAChR subtypes are segregated to separate populations of glutamatergic terminals. We have been unable to address this question because of the lack of reliable markers for β2* nAChRs. However, the complimentary laminar distributions of [3H]nicotine and 125I-αBgt binding in rat cortex are compatible with this view (Clarke et al., 1985). Moreover, as mentioned above, α4β2 nAChRs residing on thalamocortical afferents facilitate glutamate release in the PFC (Gioanni et al., 1999; Lambe et al., 2003), whereas α7 mRNA is sparse in the thalamus and α7 nAChRs are more likely to be associated with other glutamatergic inputs to the PFC.
The PFC is an important locus for working memory in the rodent (Mansvelder et al., 2006) and a site of action for the attention-enhancing effects of nicotine (Hahn et al., 2003). Both β2-containing and α7 nAChRs have been implicated in different aspects of sustained attention and working or reference memory tasks (Hoyle et al., 2006; Chan et al., 2007; Young et al., 2007), and both subtypes have been shown to increase glutamate or excitatory amino acid release in rodent PFC (Gioanni et al., 1999; Lambe et al., 2003; Rousseau et al., 2005; Couey et al., 2007). Plastic changes at glutamatergic synapses in the PFC have been found to occur during working memory tasks (Laroche et al., 2000), and it is plausible that nicotine and other agonists exert their influence on attentional paradigms by contributing to synaptic plasticity. Presynaptic α7 nAChRs in other brain regions can enhance synaptic efficacy at glutamatergic synapses, facilitating long-term potentiation (or, in some circumstances, long-term depression; McKay et al., 2007). Therefore, the modulation of excitatory amino release by α7 nAChRs via CICR provides one mechanism that might underpin the behavioral effects of nicotine.
Releasable Ca2+ stores contribute to the modulation of spontaneous and evoked synaptic transmission (Emptage et al., 2001; Bardo et al., 2006), and it has been proposed that presynaptic ryanodine receptors are specifically responsible for driving changes in synaptic efficacy (Collin et al., 2005). Therefore, the ability of presynaptic α7 nAChRs to promote CICR from these stores is a first step in coupling nicotinic stimulation to presynaptic plasticity. Indeed, CICR was recently reported to contribute to nicotine-induced long-term changes in synaptic strength at CA3-CA1 synapses in rat hippocampus (Le Magueresse and Cherubini, 2007). An analogous mechanism, namely, coupling Ca2+ influx via a ligandgated ion channel to CICR, has been proposed to support neurotransmitter release controlled by presynaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (Chávez et al., 2006) and kainate receptors (Lauri et al., 2003; Collin et al., 2005); in the latter case, this contributed to the kainate receptor-dependent induction of long-term potentiation at mossy fiber synapses.
Sustained or enhanced transmitter release in response to increased intracellular Ca2+ generated by CICR will require the mobilization of vesicles from a reserve or depot pool. Synapsin-1 has a well established role in regulating the availability of synaptic vesicles for release, that involves tethering reserve vesicles to the cytoskeleton, a process regulated by phosphorylation (Jovanovic et al., 1996; Matsubara et al., 1996). Several kinases phosphorylate synapsin-1, including ERK/MAPK. ERK1/2-dependent phosphorylation of sites 4, 5, and 6 reduces actin binding and mediates neurotrophin-evoked enhancement of glutamate release from cortical synaptosomes (Matsubara et al., 1996; Jovanovic et al., 2000). Moreover, ERK1/2-mediated phosphorylation of synapsin-1 has been proposed as a mechanism of presynaptic plasticity in hippocampus-dependent learning (Kushner et al., 2005). Because α7 nAChRs can activate ERK1/2 in various systems (Dineley et al., 2001; Dajas-Bailador et al., 2002; Dajas-Bailador and Wonnacott, 2004), and CICR has also been linked to ERK1/2 activation (Kemmerling et al., 2007), ERK1/2 was a plausible candidate for mediating α7 nAChR-induced changes in excitatory amino acid release. The selective phosphorylation of ERK2 in response to compound A is consistent with the predominant role of this isoform in memory and learning (Satoh et al., 2007). The parallel changes in phosphorylation of ERK2, synapsin-1, and [3H]d-aspartate release, combined with the ability of ryanodine and MEK1 inhibitors to block all three measures, support this as a downstream pathway selectively activated by presynaptic α7 nAChRs.
In this study, we have identified some key intermediate steps linking presynaptic α7 nAChR activation to enhanced excitatory amino acid release in PFC nerve terminals. The involvement of CICR that is coupled to presynaptic ERK2 activation and synapsin-1 phosphorylation provides a cellular mechanism for presynaptic facilitation in response to α7 nAChR activation. The occurrence of non-α7 nAChRs (likely to be α4β2* nAChRs) on glutamatergic terminals, and their facilitation of [3H]d-aspartate release by a distinct Ca2+-dependent mechanism that recruits VOCCs, offer the opportunity for multiple nicotinic signals and complex cholinergic modulation.
Acknowledgments
We are grateful to Dr. Angus Nairn for providing the phosphosynapsin-1 antibody.
Footnotes
-
This work was supported by Biological and Biotechnological Research Council (BBSRC) grant BBS/B/15600 (to S.W.) and a Ph.D. studentship from the BBSRC in conjunction with GlaxoSmithKline (to J.A.D.).
-
ABBREVIATIONS: nAChR, nicotinic acetylcholine receptor; PFC, prefrontal cortex; VOCC, voltage-operated calcium channel; CICR, calcium-induced calcium release; ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinase; αBgt, α-bungarotoxin; compound A, (R)-N-(1-azabicyclo[2.2.2]oct-3-yl)(5-(2-pyridyl)thiophene-2-carboxamide); TBOA, dl-threo-β-benzoyloxyaspartic acid; U0126, 1,4-diamino-2,3-dicyano-1,4-bis(methylthio)butadiene; PD 98059 (PD), 2′-amino-3′-methoxyflavone; 5-iodo-A-85380, 5-iodo-3-(2(S)-azetidinylmethoxy)-pyridine; DHβE, dihydro-β-erythroidine; BSA, bovine serum albumin; PBS, phosphate-buffered saline; KB, Krebs-bicarbonate buffer; p-, phospho-; MEK, mitogen-activated protein kinase kinase; Rya, ryanodine; Mec, mecamylamine.
- Received February 25, 2008.
- Accepted April 28, 2008.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}