


Abstract
Kv1.3 potassium channels maintain the membrane potential of effector memory (TEM) T cells that are important mediators of multiple sclerosis, type 1 diabetes mellitus, and rheumatoid arthritis. The polypeptide ShK-170 (ShK-L5), containing an N-terminal phosphotyrosine extension of the Stichodactyla helianthus ShK toxin, is a potent and selective blocker of these channels. However, a stability study of ShK-170 showed minor pH-related hydrolysis and oxidation byproducts that were exacerbated by increasing temperatures. We therefore engineered a series of analogs to minimize the formation of these byproducts. The analog with the greatest stability, ShK-192, contains a nonhydrolyzable phosphotyrosine surrogate, a methionine isostere, and a C-terminal amide. ShK-192 shows the same overall fold as ShK, and there is no evidence of any interaction between the N-terminal adduct and the rest of the peptide. The docking configuration of ShK-192 in Kv1.3 shows the N-terminal para-phosphonophenylalanine group lying at the junction of two channel monomers to form a salt bridge with Lys411 of the channel. ShK-192 blocks Kv1.3 with an IC50 of 140 pM and exhibits greater than 100-fold selectivity over closely related channels. After a single subcutaneous injection of 100 μg/kg, ∼100 to 200 pM concentrations of active peptide is detectable in the blood of Lewis rats 24, 48, and 72 h after the injection. ShK-192 effectively inhibits the proliferation of TEM cells and suppresses delayed type hypersensitivity when administered at 10 or 100 μg/kg by subcutaneous injection once daily. ShK-192 has potential as a therapeutic for autoimmune diseases mediated by TEM cells.
Toxins are a valuable source of tools for understanding a variety of physiological processes. Some have been optimized into drugs for treatment of human diseases and conditions; for example, ziconotide (Prialt), a peptide derived from cone snail venom, is approved for the treatment of chronic pain (Wallace, 2006). Sea anemones possess peptide toxins that block potassium channels (Chandy et al., 2001). One of the most potent inhibitors of these potassium channel toxins is ShK, a 35-residue polypeptide isolated from the sea anemone Stichodactyla helianthus (Castañeda et al., 1995). It blocks the voltage-gated Kv1.3 channels in T lymphocytes with an IC50 of ∼11 pM (Kalman et al., 1998). The three-dimensional structure of ShK and its key Kv1.3 channel-binding residues have been elucidated. The peptide is stabilized by three disulfide bridges and consists of two short α-helices comprising residues 14 to 19 and 21 to 24 (Tudor et al., 1996). The N-terminal eight residues of ShK adopt an extended conformation, followed by a pair of interlocking turns that resemble a310 helix, whereas its C-terminal Cys35 residue forms a nearly head-to-tail cyclic structure through a disulfide bond with Cys3 (Pohl et al., 1995). ShK interacts with all four subunits in the Kv1.3 channel tetramer, Lys22 occluding the channel pore like a cork in a wine bottle (Pennington et al., 1996a,b; Kalman et al., 1998; Rauer et al., 1999).
Kv1.3 is widely regarded as a therapeutic target for preferential suppression of effector memory (TEM) T cells that mediate autoimmune diseases such as multiple sclerosis (MS), type 1 diabetes mellitus, rheumatoid arthritis (RA), and psoriasis (Ellis and Krueger, 2001; Markovic-Plese et al., 2001; Viglietta et al., 2002; Schönland et al., 2003; Wulff et al., 2003; Kivisäkk et al., 2004; Rus et al., 2005; Beeton et al., 2006; Krakauer et al., 2006; Haegele et al., 2007). When activated, TEM cells change into CCR7- TEM-effector cells with up-regulated Kv1.3 expression (Kv1.3high). Naive and central memory (TCM) T cells, when activated, change into CCR7+ effector cells with up-regulated expression of calcium-activated KCa3.1 channels (KCa3.1high) (Wulff et al., 2003). In CCR7- effector cells, opening of Kv1.3 channels causes membrane hyperpolarization that promotes Ca2+ influx through calcium-release-activated calcium channels during the activation process. KCa3.1 channels play the equivalent functional role in CCR7+ effector cells. Consequently, Kv1.3 blockers suppress cytokine production and proliferation of CCR7- effector cells while sparing CCR7+ effector T cells (Wulff et al., 2003). This difference in potassium channel physiology between CCR7- effectors that contribute to the pathogenesis of autoimmune diseases and CCR7+ effectors that protect against infections and cancers (Yue et al., 2004; Klebanoff et al., 2005; Bengsch et al., 2007) raises the possibility of using specific Kv1.3 blockers to ameliorate autoimmune diseases while avoiding side effects associated with broad immunosuppression.
Over the last decade, we have generated 380 ShK analogs to improve Kv1.3-specificity, and in 2005, we successfully synthesized a 37-amino acid peptide designated ShK-L5 (ShK-170) with picomolar affinity for Kv1.3 (IC50, 69 pM), 100-fold selectivity for Kv1.3 over Kv1.1 and greater than 200-fold selectivity over other closely related channels (Beeton et al., 2005). ShK-170 contains an l-phosphotyrosine attached via an AEEA (“mini-PEG”) hydrophilic linker to Arg1 of ShK. The peptide selectively inhibited the proliferation of CCR7- TEM-effector cells. When administered by daily subcutaneous injection (100 μg/kg), ShK-170 effectively treated At-EAE induced by the adoptive transfer of myelinspecific CCR7- TEM-effector cells and suppressed the DTH response mediated by skin-homing TEM-effectors (Beeton et al., 2005). Furthermore, it did not cause cardiac toxicity in rats, as assessed by continuous EKG monitoring, and did not alter clinical chemistry and hematological parameters after 2 weeks' therapy at the therapeutic dose.
Improved delivery systems now available have allowed peptides to be considered much more favorably as lead drug candidates. These delivery systems offer the patient an alternative to daily injections of peptide; by using biodegradable polymers that slowly release the entrapped drug, it is possible to maintain a stable circulating drug concentration. Polymers such as PLGA lactate would be ideal for a ShK analog that was stable at acidic pH values. Determining stability at different pH and temperatures will be critical in designing the optimal formulation of ShK, and minimizing oxidative processes will be important in the future handling and formulation of oxygen sensitive peptides such as ShK.
In this study, we have investigated the stability of ShK-170 at different temperatures and pH values. Analysis of the by-products from ShK-170 led us to design additional stabilizing elements to minimize degradation. A novel analog with improved stability and selectivity, ShK-192, was developed by making three changes to ShK-170: replacing the N-terminal phosphotyrosine with the nonhydrolyzable phosphate mimetic para-phosphonophenylalanine (Ppa), Met21 with the isosteric homolog norleucine (Nle) to remove methionine oxidation, and the C-terminal carboxyl with an amide to minimize digestion by carboxypeptidases. ShK-192 blocks Kv1.3 with an IC50 of 140 pM, exhibits excellent specificity over closely related channels, and is an effective immunosuppressant.
Materials and Methods
Peptide Synthesis. Fmoc-amino acids (Bachem AG, Bubendorf, Switzerland) included Arg(Pmc), Asp(OtBu), Cys(Trt), Gln(Trt), His-(Trt), Lys(Boc), Ser(tBu), Thr(tBu), and TyrPO3Bzl. Stepwise assembly was carried out on an Applied Biosystems 431A peptide synthesizer at the 0.25-mmol scale using Fmoc-Ramage-amide-resin. Residues 34 through 22 were single-coupled. At this point, half of the resin was removed to effect better mixing as well as to facilitate substitution for Nle at position 21. The remainder of the native peptide sequence was double-coupled to the remaining resin aliquot. All couplings were mediated by diisopropyl carbodiimide in the presence of 2 eq of 1-hydroxybenzotriazole. After synthesis of the 35-residue peptide, the hydrophilic linker was coupled onto the N-terminal Arg residue. The resin was then divided into three portions for coupling of pTyr, Pmp, and Ppa, respectively. The same synthetic protocol was followed for the Nle-substituted analogs starting from the resin aliquot removed at Lys22. After final removal of the Fmoc group, the peptide resin (2.42 g) was cleaved from the resin and simultaneously deprotected using reagent K (King et al., 1990) for 2 h at room temperature. After cleavage, the peptide was filtered to remove the spent resin beads and precipitated with ice-cold diethyl ether. The peptide was collected on a fine filter funnel, washed with ice-cold ether, and finally extracted with 20% AcOH in H2O. The peptide extract was subsequently diluted into2LofH2O and, after pH adjustment to 8.0 with NH4OH, was allowed to air oxidize at room temperature for 36 h. After oxidation of the disulfide bonds, the peptide solution was acidified to pH 2.5 and pumped onto a Dynamax C18 column (5.0 × 30 cm; Rainin Instruments, Woburn, MA). The sample was eluted with a linear gradient from 5 to 30% acetonitrile into H2O containing 0.1% TFA. The resulting fractions were analyzed using two analytical RP-HPLC systems: TFA and triethylammonium phosphate. Pure fractions were pooled and lyophilized.
Analysis. Synthetic peptide samples were hydrolyzed in 6 N HCl at 110°C for 22 h in vacuo. Amino acid analysis was performed on an amino acid analyzer (126AA System Gold; Beckman Coulter, Fullerton, CA). Matrix-assisted laser desorption ionization/time-of-flight mass spectroscopic analysis was performed on a Kratos Kompact mass spectrometer using α-cyano-4-hydroxycinnamic acid as a matrix.
Synthesis of Boc-Ppa(OtBu)2-OH. Synthesis of Boc-Ppa(OtBu)2-OH was performed as reported recently (Chauhan et al., 2007). In brief, Boc-Phe(4-I)-OEt (8.38 g, 20 mM) and tetrakis(triphenyl)phosphinepalladium (0) (1.155 g, 1 mM) were dissolved in 400 ml of anhydrous acetonitrile. Di-t-butyl phosphite (11.64 g, 60 mM) and triethylamine (6.06 g, 60 mM) were then added, and the reaction mixture was heated at 70 + 4°C for 40 h. The solvent was evaporated and the syrupy residue was purified by silica gel column chromatography using 5% methanol in dichloromethane as the solvent mixture. Fractions containing the product were pooled and evaporated to afford 6.1 g (62.8%) of the ethyl ester as a light yellow solid with a mass of m/e 507.9 (M + Na)+.
The ester (6.0 g, 12.37 mM) was dissolved in a mixture of 150 ml of methanol and 25 ml of water. After cooling the mixture in an ice bath, 1 N aqueous sodium hydroxide (15 mM) was added dropwise. After 4 h at ambient temperature, methanol was evaporated. The product was extracted with ethyl acetate after neutralizing the mixture with 0.5 N aqueous HCl. The crude product was purified by silica gel column chromatography using 5% methanol in dichloromethane as a solvent system to afford 3.8 g (67.2%) pure product as a white solid. Electrospray ionization-mass spectrometry: m/e 479.8 (M + Na) + α[D]24 = -15.89° (c 0.5, DMF). 1H NMR (CDCl3): δ 7.76 to 7.54 (m, 2H), 7.37 to 7.24 (m, 2H), 5.32-5.15 (d, 1H), 4.69 to 4.55 (m, 1H), 3.34 to 3.17 (m, 2H), 1.54 to 1.28 (S, 27H).
Measuring Stability. Peptide samples were dissolved in 50 mM sodium phosphate containing 1 mM EDTA. The pH of the solution was adjusted with either NaOH or H3PO4. No special precautions were used in buffer preparation to exclude oxygen from the solution. Samples were incubated at ambient temperature (22°C), physiological temperature (40°C), or stress temperature (60°C) for the appropriate time point. Stability was accessed by RP-HPLC using a C18 column with an aqueous gradient of acetonitrile into water each containing 0.1% TFA. A linear gradient from 10 to 40% acetonitrile over 30 min at a flow rate of 1 ml/min was used for all analytical results.
Protease Digestion. The peptide was dissolved in 50 mM sodium phosphate, pH 6.5. The peptide was incubated at 37°C over the time course of the experiment at a ratio of 1:50 enzyme [sequencing grade trypsin and chymotrypsin (Sigma) to substrate (w/w)]. Analysis was carried out by RP-HPLC according to the buffer and gradient conditions described above.
NMR Spectroscopy. Synthetic ShK-192 (6 mg) was dissolved in 600 μl H2O containing 10% 2H2O and the pH was adjusted to 5.0. Two-dimensional homonuclear total correlation (TOCSY) spectra with a spin-lock time of 70 ms, nuclear Overhauser enhancement (NOESY) spectra with mixing times of 50, 150, and 250 ms, and double-quantum-filtered correlation spectra were acquired at 600 MHz on a Bruker DRX-600 spectrometer. Spectra were acquired at 20°C unless otherwise stated and referenced to dioxane (3.75 ppm). TOCSY and NOESY spectra were also collected at 10°C. The water resonance was suppressed using the WATERGATE pulse sequence. Amide exchange rates were monitored by dissolving lyophilized material in 2H2O containing 10 mM sodium acetate buffer at pH 4.5 then recording a series of 1D spectra, followed by 70-ms TOCSY, 50-ms NOESY, and exclusive correlation spectra at 5°C. In addition, 1H-13C and 1H-15N heteronuclear single-quantum correlation spectra for the assignment of 13C and 15N chemical shifts were collected at 20°C on a spectrometer (Avance 500; Bruker, Newark, DE) equipped with a Triple Resonance Inverse (TXI) Cryoprobe (Avance 500; Bruker BioSpin GmbH, Rheinsetten, Germany). Diffusion measurements were performed at 5 and 20°C using a pulsed field gradient longitudinal eddy-current delay pulse sequence. Spectra were processed using TOPSPIN (Version 1.3; Bruker Biospin) and analyzed using XEASY (Version 1.3.13) (Bartels et al., 1995).
Structural Constraints.3JHNHA coupling constants were measured from double-quantum-filtered correlation spectroscopy spectra at 600 MHz and then converted to dihedral restraints as follows: 3JHNHα > 8 Hz, φ = -120 ± 40°; 3JHNHα < 6 Hz, φ = -60 ± 30°. The final dihedral bond angle constraints are listed in Supplemental Table S4, and details have been deposited along with distance constraints in BioMagResBank (Ulrich et al., 2007) as entry 15983. The half-cystines for ShK-192 have been mapped previously (Pohl et al., 1995) and were included as structural restraints in preliminary calculations. Hydrogen bonds (Supplemental Table S3) were identified in MOLMOL using a maximum C-N distance of 2.4 Å and a maximum deviation of 35° from linearity.
Structure Calculations. Intensities of NOE cross-peaks were measured in XEASY and calibrated using the CALIBA macro included with the CYANA software package (version 1.0.6) (Herrmann et al., 2002). NOEs providing no restraint or representing fixed distances were removed. The constraint list resulting from the CALIBA macro of CYANA was used in XPLOR-NIH to calculate a family of 200 structures using the simulated annealing script. The 50 lowest energy structures were then subjected to energy minimization in water; during this process, a box of water with a periodic boundary of 18.856 Å was built around the peptide structure, and the ensemble was energy-minimized based on NOE and dihedral restraints and the geometry of the bonds, angles, and improper dihedral angles. From this set of structures, a final family of 20 lowest energy structures was chosen for analysis using PROCHECK-NMR (Laskowski et al., 1996) and MOLMOL (Koradi et al., 1996). In all cases, the final structures had no experimental distance violations greater than 0.2 Å or dihedral angle violations greater than 5°. The structures have been deposited in the Protein Data Bank with ID 2K9E. Structural figures were prepared using the programs MOLMOL (Koradi et al., 1996) and PyMOL (http://www.pymol.org).
Modeling and Docking. The NMR-derived structure of ShK-192 was subjected to molecular dynamics (MD) simulation using the GROMACS (v3.3.1) package of programs (Lindhal et al., 2001). All simulations consisted of an initial minimization of water molecules followed by 100 ps of MD with the peptide fixed. After positional restraints MD, the restraints on the peptide were removed and MD continued for a further 10 ns. The computational methods applied here are identical to those used previously to model ShK and its d-allo homolog (Beeton et al., 2008). Structures along the MD trajectory were collected at 1-ns intervals.
Comparative models of the transmembrane region (only) of the murine Kv1.3 channel were constructed using the X-ray structure of the K+ channel from Streptomyces lividans (KcsA, Protein Data Bank accession code 1BL8) as a template. The MODELLER (6v2) program (Fiser and Sali, 2003) was used to create nine models based on the sequence alignment described previously (Beeton et al., 2008).
Complexes of ShK-192 with mKv1.3 were modeled using the ZDOCK program (Chen et al., 2003). Each of the structures of ShK-192 collected during the MD simulation was docked with one of the nine models of the channel; thus, we considered all 90 possible combinations of toxin with channel. The top 2000 scoring predictions from each combination were then refined using the RDOCK program (Li et al., 2003), in which the binding interface was refined using molecular mechanics minimization. The docking predictions from all 180,000 complexes were ranked according to the RDOCK scoring function. The top 20 ranked predictions were then subjected to additional MD refinement using the CHARMM package (MacKerell et al., 1998). Initial heating and equilibration at 600 K was followed by constant temperature MD (100 ps) and subsequent quenched MD and minimization—all backbone atoms of the channel were harmonically restrained to their initial positions during these MD simulations. The interaction energy between ligand and receptor was evaluated from the sum of van der Waals, electrostatic, and hydrogen-bond energies.
Buried surface areas were calculated from the difference in surface areas of channel and toxin from the complex. Surface areas were calculated using the NACCESS program (Hubbard and Thornton, 1993).
Ion Channels. Cells stably expressing mKv1.1, mKv1.3, and hKv1.5 have been described previously (Grissmer et al., 1994). Cell lines stably expressing other mammalian ion channels were gifts from several sources: hKv1.4 in LTK cells from Michael Tamkun (University of Colorado, Boulder, CO); Kv11.1 (HERG) in human embryonic kidney 293 cells from Craig January (University of Wisconsin, Madison, W0049; and human embryonic kidney 293 cells expressing hKCa3.1 from Khaled Houamed (University of Chicago, Chicago, IL). hKv1.6 and rKv3.2 (both in pcDNA3) were obtained from Protinac GmbH (Hamburg, Germany) and transiently transfected into COS-7 cells with Fugene-6 (Roche, Indianapolis, IN) according to the manufacturer's protocol.
Electrophysiological Analysis. Experiments were conducted in the whole-cell configuration of the patch-clamp technique. KV currents were elicited by 200-ms depolarizing pulses from a holding potential of -80 to 40 mV as described previously (Beeton et al., 2001b). Each channel blocker was tested at multiple concentrations. The measured reduction in peak current at 40 mV for each concentration was used to generate a dose-response curve with Origin software (OriginLab Corp, Northampton, MA) as described previously (Beeton et al., 2001b). For Kv11.1 channels, current block was measured both at 20 and -50 mV (tail current) (Zhou et al., 1998). For KCa3.1 channels, the change in slope conductance by the ShK analogs was measured (Wulff et al., 2003).

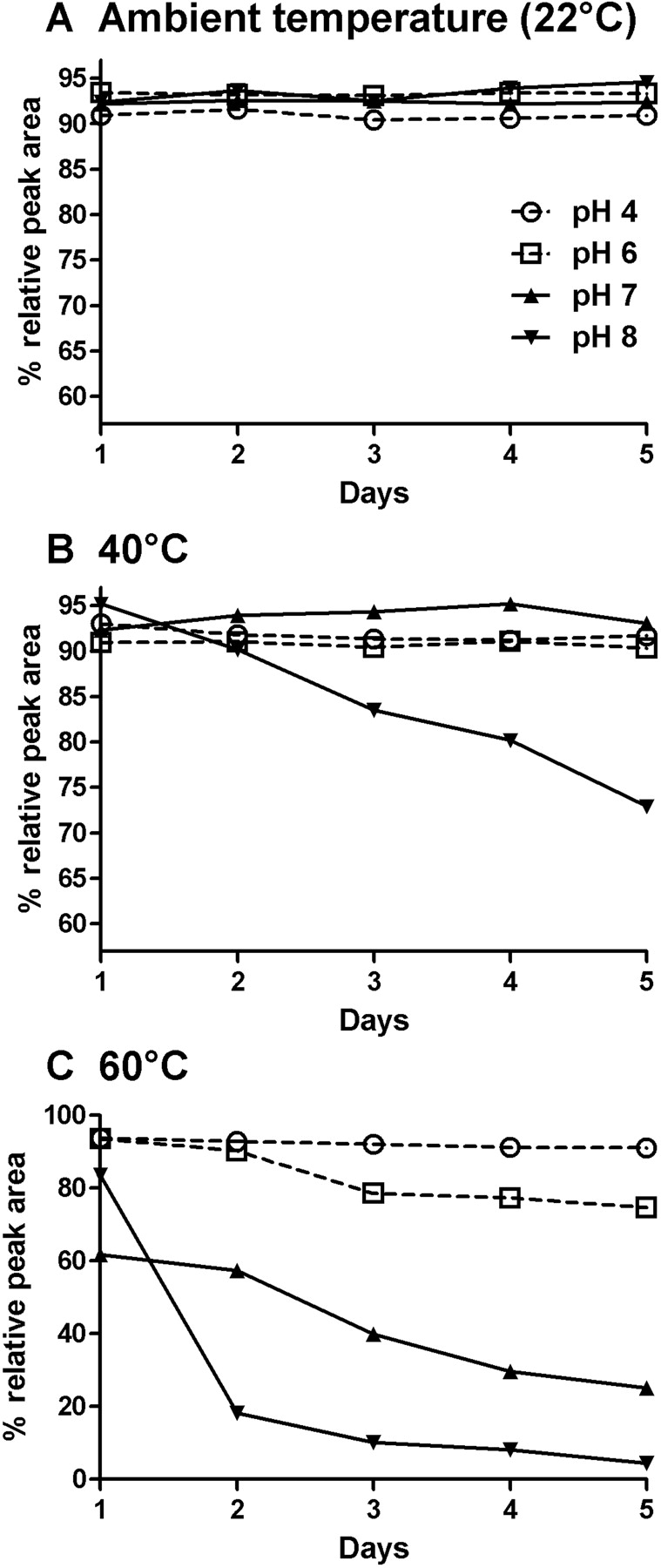
ShK-170 Stability profile over 5 days at pH 4 (○), 6 (□), 7 (▴), and 8 (▾) at temperatures of 22°C (A), 40°C (B), and 60°C (C).
Proliferation Assays on Human Synovial Fluid T Lymphocytes. T cells were isolated by negative selection (EasySep cocktail; StemCell Technologies, Seattle, WA) from the synovial fluid of patients with rheumatoid arthritis under a protocol approved by the University of California, Irvine Institutional Review Board. The majority of these cells (>90%) are CCR7-Kv1.3high TEM cells (Beeton et al., 2006). [3H]Thymidine incorporation assays were conducted as described previously (Wulff et al., 2003).
Animals. Female inbred DA and Lewis rats 7 to 9 weeks old were purchased from Harlan-Sprague-Dawley (Indianapolis, IN) and housed under clean conditions with irradiated rodent chow and acidified water ad libitum. All experiments were in accordance with National Institutes of Health guidelines and approved by the University of California, Irvine and the Baylor College of Medicine Institutional Animal Care and Use Committees.
Circulating Half-Life Determination. Known amounts of ShK-192 were added to normal DA rat serum, and the blocking activity on Kv1.3 channels was tested by patch-clamp after a 1:10 dilution in bath solution to establish a standard dose-response curve. Serum samples from DA rats were obtained at various times after a single subcutaneous injection of 100 μg/kg ShK-192 and were tested for Kv1.3 blocking activity by patch-clamp after a 1/10 dilution in bath solution. The levels of ShK-192 were determined from the standard curve as described previously (Beeton et al., 2001b).
Prevention of Active Delayed Type Hypersensitivity. Active DTH was induced in Lewis rats by immunization with an emulsion of ovalbumin in complete Freund's adjuvant (Difco, Detroit, MI). Seven days later, the rats received an injection of ovalbumin dissolved in saline in the pinna of one ear and saline in the other ear. Rats then received subcutaneous injections of ShK-186 (100 μg/kg/day), ShK-192 (1, 10, or 100 μg/kg/day), or vehicle (PBS + 2% Lewis rat serum, pH 6.0; 0.5 ml/rat). Ear swelling was measured 24 and 48 h later using a spring-loaded micrometer (Mitutoyo, Spokane, WA). Statistical analysis was carried out with the Mann-Whitney U-test (Prism; GraphPad Software, La Jolla, CA).
Results
Stability Studies on ShK-170. Stabilization is an essential step in formulating a peptide therapeutic because many slow-release formulations (e.g., polymers such as PLGA lactate) require that the peptide be stable at acidic pH and high temperature during the formulation process. To gain insight into the solution stability of ShK-170, samples were incubated at three different temperatures and a pH range from 4 to 8. The temperatures selected were 22°C (ambient temperature), 40°C (near the physiological temperature for mammals), and 60°C (stressed). ShK-170 at ambient temperature showed no evidence for degradation at any pH value tested (Fig. 1A). The main peptide peak maintained the same over-all peak area (within normal experimental error), with no evidence of new side peaks, during the course of the 5-day experiment. The 40°C incubation study showed very little change in peak area at pH values of 7 and 8, indicating that the peptide is stable near physiological pH values (Fig. 1B). At acidic pH values of 4 and 6, the peptide exhibited some minor instability, with an 8% decrease in peak area (Fig. 1B). A later eluting new peak was observed to form in a time-dependent manner. LC-MS analysis of one of the samples (pH 4) determined this new peak to be the dephosphorylated species (Fig. 2A). The elution time for this new peak was identical in each of the other acidic pH studies. Thus, the principal degradation product observed was the acid hydrolysis product of the pTyr residue, releasing the phosphate moiety. Although ShK-170 contains a potentially oxidation-sensitive Met residue at position 21, no evidence of Met(O) was observed at ambient temperature at any of the pH values tested. At 40°C and 60°C, however, Met(O) formation was detected as a small peak eluting slightly before the major peak (Fig. 2B). The formation of Met(O) was time-dependent and accounted only for a change in peak area of less than 3%.

RP-HPLC profiles for selected ShK-170 stability samples. HPLC conditions were as follows: C18-Vydac 0.46 × 250 mm, 300 Å, 4 μm. Gradient conditions were 10 to 40% B in 30 min at a flow rate of 1 ml/min where A = 0.1% TFA in H2O and B = 0.1% TFA in MeCN. A, ShK-170 sample from 40°C and pH 4 at day 5. B, ShK-170 sample from 60°C and pH 7 at day 5. C, ShK-170 sample from 60°C and pH 8 at day 5.
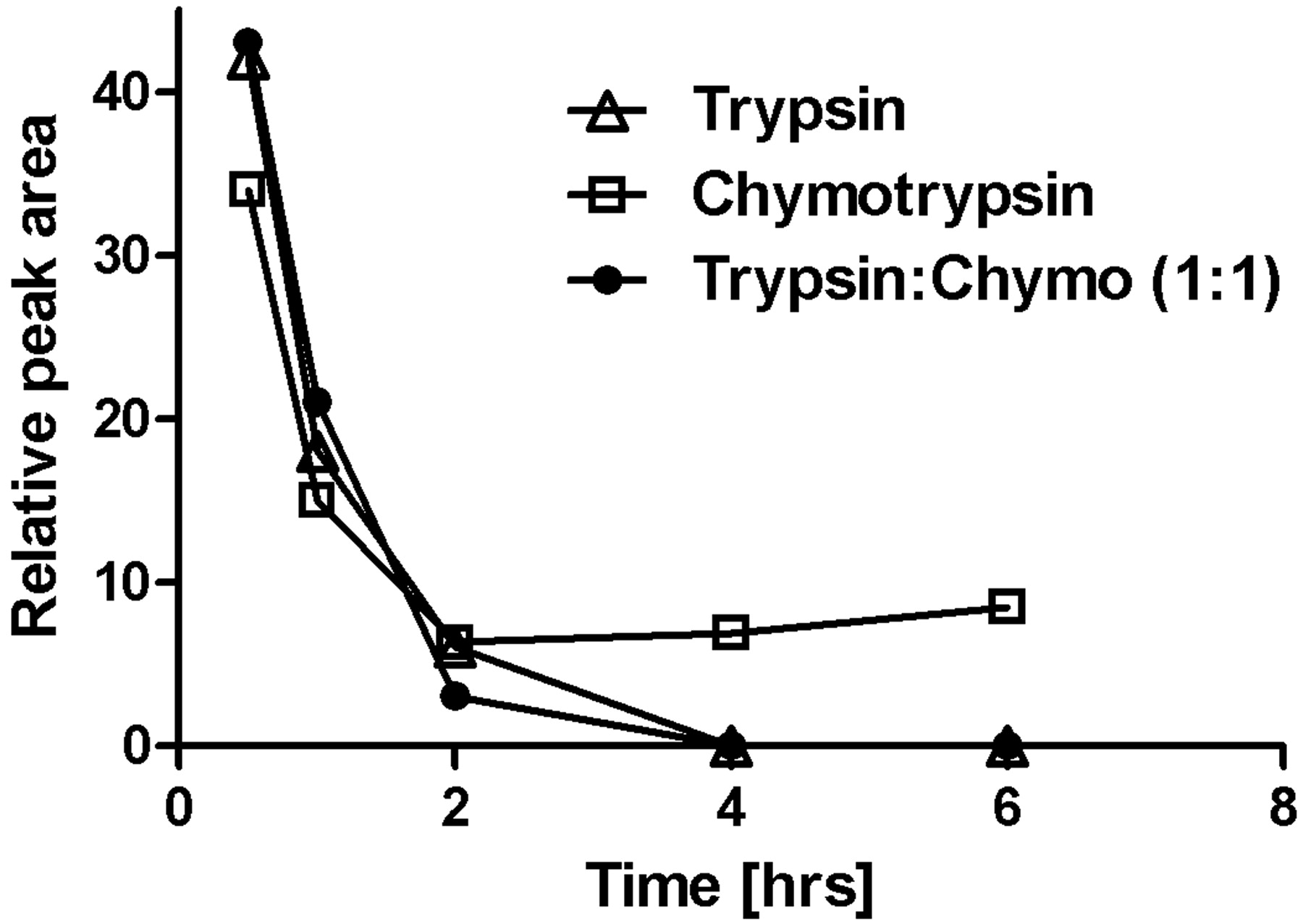
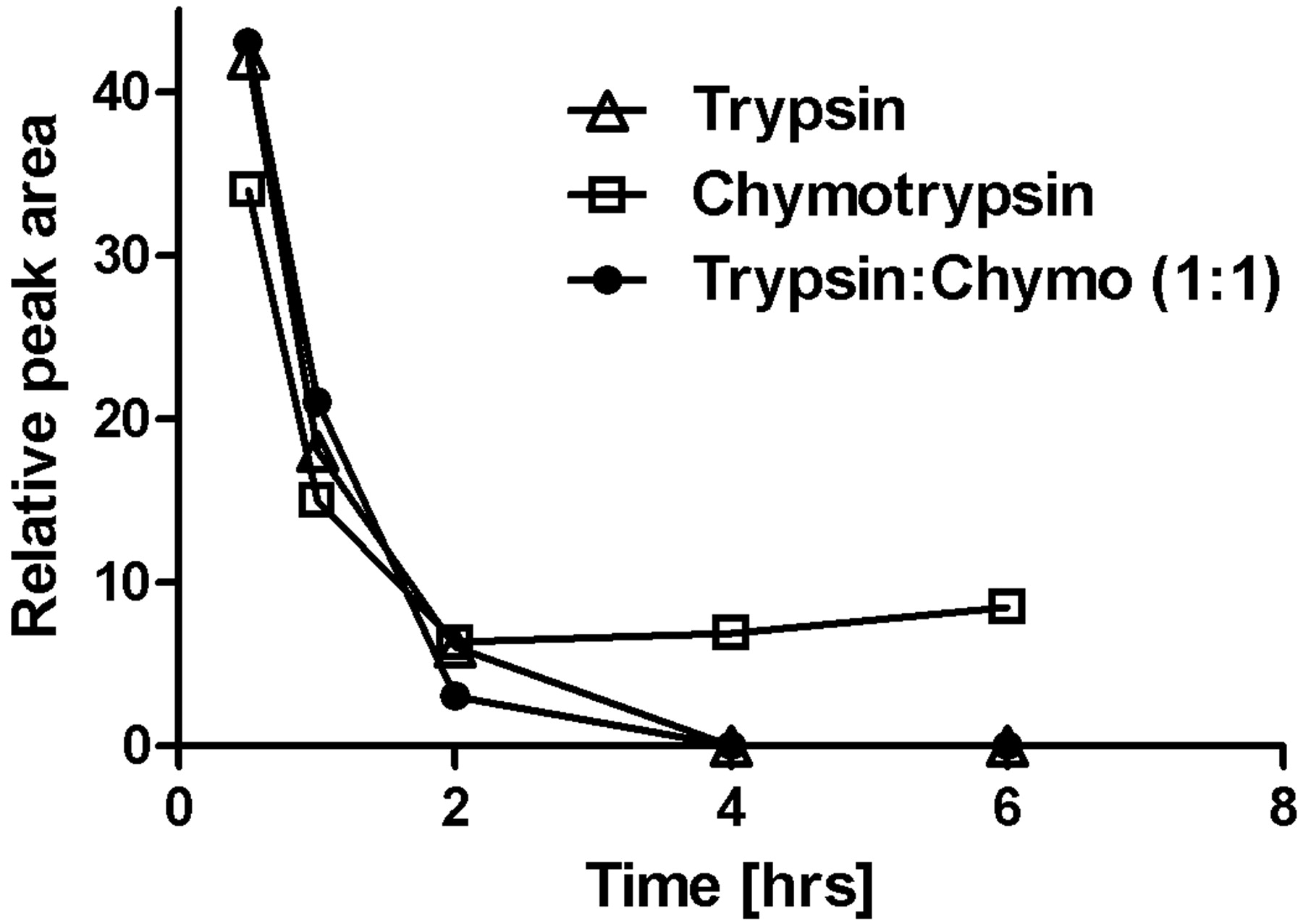
Stability of ShK-170 upon treatment with proteolytic enzymes trypsin (▵), chymotrypsin (□), and a combination of both enzymes (•) at 37°C. Plot was derived from the reduction in peak area determined by RP-HPLC using the same gradient parameters mentioned earlier.
The 60°C study was carried out to gain insight into possible formulation parameters. The stressed conditions clearly had a major effect on the stability of the peptide over time (Fig. 1C). At pH 7, ShK-170 maintained the greatest stability, with a 23% decrease in peak area (Fig. 1C). The dephosphorylated species and the Met(O) product were found to be the principal new species at this pH (Fig. 2B). At acidic pH values, the peptide exhibited accelerated loss of the phosphate moiety to the largest extent (Fig. 1C). The Met(O) byproduct was also present but only in minor amounts. This instability precludes ShK-170 from being formulated in polymers such as PLGA-lactate, which require that the peptide be stable at acidic pH and high temperature. The most deleterious pH for ShK-170 at 60°C was pH 8. LC-MS analysis showed that the phosphotyrosine residue was rapidly hydrolyzed to tyrosine at pH 8. Evidence for disulfide bond rear-rangement was also observed in the appearance of the HPLC profile. The major peak had a time-dependent disappearance with the formation of a later eluting large hump on the HPLC chromatogram (Fig. 2C). Our experience with preparing peptides containing multiple disulfide bonds suggests that this large hump represents misfolded and multimeric forms of the peptide. Thus, basic pH at high temperature must be avoided with ShK-170.
To test the protease susceptibility of ShK-170, we examined the effects of trypsin, chymotrypsin, and a combination of both (Fig. 3). Because ShK-170 has multiple cleavage sites for both of these enzymes, proteolysis was expected. In all three experiments, ShK-170 degraded rapidly to a series of disulfide-stabilized fragments, consistent with cleavage of the product at the Lys and Arg (trypsin) or Phe andTyr (chymotrypsin). The results confirm that this peptide would not survive oral administration without proper protection because of the enzymes and acidic pH in the gut. Such protection might be achieved by encapsulation of the peptide for delivery.
Synthesis of New Analogs to Improve Stability While Retaining Potency and Selectivity. Guided by the results of our stability studies on ShK-170, we generated four new analogs to improve the stability of ShK-170. Amidation of peptides is known to decrease susceptibility of the C terminus to carboxypeptidase-mediated cleavage (Matsueda and Steward, 1981) and could therefore improve the half-life of the peptide. Because ShK-170 has a C-terminal Cys, cleavage of this residue would certainly disrupt the tertiary structure and lead to loss of biological activity. We previously reported that the 3-35 disulfide linkage was the least important for biological activity, but replacement of Cys with Abu isosteres results in a 100-fold drop in activity (Pennington et al., 1999).

A, structures of phospho-tyrosine used in ShK-170, ShK-186, and ShK-188 (left), Pmp used in ShK-191 (middle), and Ppa used in ShK-192 (right). B, table summarizing the structural differences and effects of the different analogs on Kv1.1 and Kv1.3. Current block was measured at 40 mV (Beeton et al., 2001b). C, effects of ShK-192 on Kv1.3 (top) and Kv1.1 (bottom). D, dose-response curve of ShK-192 on Kv1.3 (closed symbols) and Kv1.1 (open symbols). Error bars represent S.E.M.
As the first step to improve stability, the C terminus of the ShK-170 was amidated to prevent carboxypeptidase degradation (Fig. 4). The resulting peptide, ShK-186, was equipotent (IC50 71 pM) to ShK-170 and showed the same selectivity for Kv1.3 over Kv1.1 (Fig. 4B). It suppressed cytokine production and proliferation of CCR7- effector T cells without affecting the function of CCR7+ T cells (Beeton et al., 2006). Furthermore, it treated pristane-induced arthritis, a rat model of RA, and, upon repeated administration for a month, induced no discernible change in blood counts, blood chemistry, or histopathologic examination results of organs tested (Beeton et al., 2006). ShK-186 exhibits the same stability as ShK-170 at acidic pH and 40°C (near the physiological temperature for mammals), but it is less stable at alkaline pH values (Fig. 5). The main breakdown product is dephosphorylated ShK-186 (not shown), which is similar to ShK(L4), a peptide that blocks Kv1.3 (IC50 48 pM) with roughly 3-fold selectivity over Kv1.1 (IC50 159 pM) (Beeton et al., 2005).
To eliminate oxidation of the single methionine in ShK-186, we replaced Met21 (the amino acid numbering of native ShK was used) with the nonprotein amino acid Nle (Anfinson and Corley, 1969). Nle has the same hydrophobic character as Met but contains a carbon atom in place of the oxidation-sensitive sulfur atom in Met. To reduce susceptibility to acid-catalyzed hydrolysis of the phosphate from the N-terminal pTyr residue, we replaced phosphotyrosine with the non-hydrolyzable phosphate mimetics (Fig. 4A) Pmp or Ppa (Nomizu et al., 1994). ShK-170, ShK-186, and the new analogs (ShK-188, ShK-191 and ShK-192) are shown in Fig. 4B. All three new analogs contain a C-terminal amide and additional stability elements; ShK-188 contains Nle21 in place of Met21, ShK-191 contains Pmp instead of pTyr, and ShK-192 contains Nle21 and Ppa.

ShK-186 stability profile over 5 days at pH 4 (○), 6 (□), 7 (▴), and 8 (▾) at a temperature of 40°C.
Figure 4C shows the effect of ShK-192 on Kv1.3 and Kv1.1 currents elicited with 200-ms depolarizing pulses to 40 mV from a holding potential of -80 mV. The peptide blocked these channels reversibly in a dose-dependent manner with a Hill coefficient of 1. All three new analogs were less potent on Kv1.3 than either ShK-170 or ShK-186. Furthermore, ShK-188 and ShK-191 demonstrated less selectivity for Kv1.3 over Kv1.1 than ShK-170 or ShK-186 (Table 4B). In contrast, ShK-192 showed ∼160-fold selectivity for Kv1.3 over Kv1.1. The difference in selectivity of the Ppa-containing ShK-192 and the Pmp-containing ShK-191 is probably caused by the greater rigidity of the aryl phosphonate in ShK-192, in contrast to the higher flexibility of the phosphonic acid in ShK-191 as a consequence of its linkage to the methylene carbon.
Because ShK-192 retains picomolar potency, shows increased selectivity for Kv1.3 over Kv1.1, and contains all three stabilizing elements, we assessed the specificity of ShK-192 on a panel of related channels. Like ShK-170, ShK-192 showed >700-fold selectivity over Kv1.4, Kv1.5, Kv11.1/HERG, and KCa3.1 (Table 1). However, ShK-192 was less selective than ShK-170 for Kv1.3 over Kv1.6 and Kv3.2 (Table 1).
Selectivity of ShK-170 and ShK-192 determined by patch-clamp
Data are shown as mean ± S.E.M. Current block was measured at 40 mV for Kv1.x and Kv3.2 currents (Beeton et al., 2001b) and at both 20 mV and −50 mV for KV11.1 currents (Zhou et al., 1998). For KCa3.1 currents, we measured the change in slope conductance at −80 mV (Wulff et al., 2003).
Stability Studies on ShK-192. At ambient temperature, ShK-192 was stable over 5 days at all of the pH conditions tested (Fig. 6A). At 40°C, it was stable at acidic and neutral pH values, but there was some minor decomposition at pH 8.0 (Fig. 6B, 12% decrease). ShK-192 was stable at 60°C and pH 4.0 (Fig. 6C). ShK-192 would therefore be suitable for use in slow-release formulations (e.g., PLGA lactate polymers). Slight degradation was observed at 60°C and pH 6.0, but the peptide degraded rapidly at neutral and basic pH, being nearly completely denatured by day 2 at pH 8.0 (Fig. 6C).
NMR Spectroscopy. ShK-192 gave good quality NMR spectra at pH 5 and 20°C. Two-dimensional 1H-15N hetero-nuclear single-quantum correlation (Supplemental Fig. S1) and homonuclear NOESY (Supplemental Fig. S2) spectra at 20°C were used to resolve peak overlap in the amide region. No spectral inhomogeneities were observed under these experimental conditions, indicating the presence of a single conformation in solution. The fingerprint regions of TOCSY and NOESY spectra are shown in Fig. S2. Chemical shift assignments are presented in Supplemental Table S1, and have been deposited in the BioMagResBank with accession number 15983 (Ulrich et al., 2007).
The trans orientation of the peptide bond preceding Pro8 was established by the intense Hα-Hδ NOEs between residues 7 and 8. Self-diffusion coefficients for ShK-192 were (0.14 ± 0.03) × 10-10 m2/s and (0.21 ± 0.06) × 10-10 m2/s at 10° and 20°C, respectively. The temperature dependences of amide proton chemical shifts were determined to assess possible hydrogen bonding (Supplemental Table S3). Among those that could be experimentally measured, the backbone amide temperature coefficients for Arg11, Lys18, and Ser26 were smaller than 3 parts per billion/K in magnitude, and those for Arg24, Cys28 and Gly33 were around 3 to 4 parts per billion/K, indicating that these amides were partially protected from solvent. Amide exchange experiments conducted at pH 4.2 and 5°C indicated that amide protons of Ile4, Thr6, Ile7, Arg11, Thr13, Cys17, Arg24, Arg29, Lys30, Cys32, and Gly33 exchanged relatively slowly compared with the rest, whereas Asp5, Lys9, Ser10, Cys12, Lys18, Ser20, Tyr23, Leu25, Ser26, Cys28, Thr31, Thr34, and Cys35 were in intermediate exchange (Supplemental Table S3).
Deviations of backbone NH, Hα and Hβ chemical shifts for ShK-192 from random coil values are compared with those of ShK in Supplemental Fig. S3. The largest differences were observed at the N and C termini and at Met21, which is substituted by Nle in ShK-192. Differences at the N and C termini are a consequence of modification with the Ppa adduct and C-terminal amidation, respectively. Overall, the predominantly similar pattern of chemical shift deviations confirms that ShK and ShK-192 adopt similar backbone conformations, the dominant secondary structure being helical.
Solution Structure. A summary of experimental constraints and structural statistics for ShK-192 is given in Supplemental Table S2. More than 94% of residues have φ/ψ values in the generously allowed regions of the Ramachandran plot, Gly34 being the only residue with a positive φ angle. The angular order parameters for φ and ψ angles in the final ensemble of 20 structures were both >0.8 over residues 3-34 (Supplemental Fig. S4), indicating that the backbone is well defined over most of the molecule. The mean pairwise root-mean-square deviation over well defined backbone heavy atoms was 0.81 Å (Table S2). The structure of ShK-192 is characterized by two short α-helices encompassing residues 17 to 20 and 22 to 25 (Supplemental Fig. S5). The presence of medium-range NOEs [dαN(i,i+3) and dαN(i,i+4)] (Supplemental Fig. S6), low coupling constants (<6 Hz), and slowly exchanging backbone amide protons in these regions supported the presence of these helices. No long-range NOEs were observed between the N-terminal Ppa adduct and other ShK-192 residues, and its location is poorly defined across the family of structures (Supplemental Fig. S5), indicating that the adduct does not interact with the rest of the molecule.

ShK-192 stability profile over 5 days at pH 4 (○), 6 (□), 7 (▴), and 8 (▾) at temperatures of 22°C (A), 40°C (B), and 60°C (C).

Solution structure of ShK-192 (left) and ShK (right). The backbone is shown in gray with secondary structure elements in light purple, the disulfide brides are in orange, Lys22 in blue, and Nle21 or Met21 in dark purple.
Comparison of the closest-to-average structure of ShK with that of ShK-192 confirms their structural similarity (Fig. 7 and Supplemental Fig. S7). The pairwise root-mean-square deviation over the backbone heavy atoms between the closest-to-average structures of ShK and ShK-192 was 1.79 Å over well defined residues. ShK-192 has a similar secondary structure to ShK, with both having an extended conformation up to residue 8, followed by a pair of interlocking turns that defines a 310-helix, and two short helices. In ShK-192 the interlocking turns constitute a short stretch of 310-helix centered on residues 10 to 12 (with 11-8 and 12-9 hydrogen bonds in all 20 structures). In ShK, the 310-helix is centered on residues 9 and 10, even though it possesses an identical hydrogen-bonding pattern within this region. The ShK analog ShK-Dap22 (where Lys22 was mutated to the non-natural amino acid diaminopropionic acid) also exhibited slight differences within this 310-helix region (Supplemental Fig. S7).
The first helix in ShK-192 (residues 17-20) was shorter than the corresponding helix in ShK and ShK-Dap22 (residues 14-19), even though most interactions between the helix N-capping residues Thr13 and Gln16 were conserved (Supplemental Table S5). However, the backbone hydrogen bond 16-13, constituting part of the ST-motif previously identified in ShK was found in only 7 of the final 20 ShK-192 structures (Supplemental Table S3). The second helix of ShK-192 (residues 22-25) was similar in length to that of ShK and ShK-Dap22 (residues 21-24) but was shifted toward the C terminus by one residue (Supplemental Fig. S7). Most residues within this region (i.e., Nle21, Lys22, and Tyr23) had higher temperature coefficients, and Leu25 had a faster backbone exchange rate compared with ShK, suggesting that this region may be less well defined in ShK-192; these differences presumably result from the replacement of Met21 in ShK with Nle in ShK-192. The C-terminal regions of ShK-192 and ShK-Dap22 were similar to one another but slightly different from ShK (Fig. S7).
Docking of ShK-192 with Kv1.3. A model of the complex between ShK-192 and Kv1.3 was constructed following the procedure applied previously to construct models of ShK and d-allo-Shk with Kv1.3 (Beeton et al., 2008). The model with the largest interaction energy predicted from the docking is shown in Fig. 8. It shows many similarities to the model of ShK docked to the channel; most significantly, Lys22 is located in the ion-selectivity filter, blocking passage of K+ ions through the channel. The ammonium group of the Lys side-chain forms hydrogen bonds with the backbone carbonyl oxygen atoms of Gly399, and Tyr27 packs alongside Lys22, in the space created by the two Gly residues of the GYG ion-selectivity filter from neighboring channel monomers. The loss of solvent-accessible surface area upon complex formation of ShK-192 is 2773 Å2 in this model. This is significantly larger than the 1858 Å2 observed in the model of ShK binding to Kv1.3.

Docking of ShK-192 with the Kv1.3 channel. A, surface representation of ShK-192 capping the extracellular face of the channel (in ribbon representation). Lys22 of ShK-192 inserts into the ion-selectivity filter, preventing passage of K+ ions. The view is in the plane of the membrane. B, ShK-192 (green tube) docked onto the channel (ribbon representation in gray). The view is down the 4-fold symmetry (ion-conduction) axis of the channel—90° rotation of the view in A about the horizontal axis. The Ppa moiety extends to the periphery of the channel (atoms are color-coded as follows: cyan, carbon; blue, nitrogen; red, oxygen; gold, phosphorous; white, hydrogen). The region highlighted is expanded in C. C, interactions of the Ppa moiety with the channel are indicated.
The N-terminal Ppa group of ShK-192 lies at the junction of two channel monomers (Fig. 8B), extending to the periphery of the channel. The negatively charged phosphono moiety forms a salt bridge with Lys411 of the channel, whereas the N-terminal ammonium interacts with the side-chain carboxylate of Asp376, the side-chain hydroxyl of Thr407, and the backbone carbonyl oxygen of Val406 (Fig. 8C). The miniPEG linker between the Ppa group and the (formerly) N-terminal Arg make no contact with the channel; the side-chain guanine of this Arg mediates an otherwise repulsive interaction between the phophono moiety and the carboxylate of Asp386 of the channel.
Pharmacokinetic Analysis of ShK-192 in Rats. A patch-clamp bioassay was used to determine circulating levels of functionally active (free) ShK-192 in rats after a single subcutaneous injection of 100 μg/kg. To standardize the assay, known amounts of ShK-192 were added to DA rat serum, and these samples were tested on Kv1.3 channels. These spiked serum samples blocked Kv1.3 currents in a dose-dependent fashion that was similar to that measured in the absence of serum. Levels of ShK-192 in treated animals were determined by comparison with the standard curve. Serum levels of functionally active ShK-192 reached 3.5 nM within 30 min (Fig. 9A). The disappearance of the free ShK-192 from the serum could be fitted by a single exponential decay. The circulating half-life was estimated to be ∼30 min. However, ∼200 pM concentration of functionally active peptide was detected in the blood 24 and 48 h after the injection, and approximately 100 pM was detectable at 72 h (Fig. 9A).
Effect of ShK-192 on TEM Cell Proliferation and Active DTH. ShK-192 suppressed antigen-triggered proliferation of TEM cells in a dose-dependent manner (Fig. 9B). We evaluated ShK-192 for immunosuppressive activity in vivo in a rat model of DTH. Lewis rats were immunized with an emulsion of ovalbumin in complete Freund's adjuvant and were challenged 7 days later with ovalbumin in one ear and saline in the other ear. Rats then received once-daily subcutaneous injections of saline (controls) or ShK-192 (1, 10, or 100 μg/kg/day) or ShK-186 (100 μg/kg/day) in the scruff of the neck. Ear thickness was measured as an indication of tissue swelling in DTH. All control rats developed ear swelling after ovalbumin challenge, whereas the DTH reaction was significantly milder in ShK-186- and ShK-192-treated animals. ShK-192 suppressed DTH in a dose-dependent manner, being effective even at 1 μg/kg/day (Fig. 9C).
Discussion
Autoimmune diseases affect millions of people globally. Although disease-modifying immunotherapies have improved the management of autoimmune diseases, these treatments cause broad-range immune suppression. Autoimmune disease patients may be spared the adverse effects of generalized immunosuppression by selective suppression of TEM-effector cells that mediate tissue destruction in diverse autoimmune diseases. The voltage-gated Kv1.3 channel is an attractive target for preferential suppression of TEM-effector cells. Specific blockers of the channel inhibit cytokine production and proliferation of TEM-effector cells, while naive and TCM cells are spared the suppression because they use the calcium-activated KCa3.1 channel in place of Kv1.3 to regulate membrane potential and calcium signaling (Wulff et al., 2003; Beeton et al., 2006). ShK-186 immobilizes TEM-effectors and prevents their activation at the site of DTH, whereas the homing of naive and TCM cells to lymph nodes and their motility within lymph nodes is unaffected by the peptide (Matheu et al., 2008). Kv1.3 blockers ameliorate disease in rat models of DTH, At-EAE, chronic-relapsing EAE, pristane-induced arthritis, experimental autoimmune diabetes mellitus and contact dermatitis, and they have excellent safety profiles in both rats and monkeys (Beeton et al., 2001a,b, 2005, 2006; Wulff et al., 2003; Azam et al., 2007; Pereira et al., 2007; Matheu et al., 2008).

A, circulating half-life of ShK-192. A single dose of 100 μg/kg of ShK-192 was injected subcutaneously to DA rats. Blood was drawn at the indicated times and serum was tested by patch-clamp to determine levels of ShK-192. B, dose-dependent inhibition by ShK-186 (open symbols) and ShK-192 (closed symbols) of [3H]thymidine incorporation by TEM lymphocytes isolated from the synovial fluid of patients with rheumatoid arthritis stimulated with an anti-CD3 antibody. C, an active DTH reaction was elicited against ovalbumin in rats (n = 8-10 per group). These rats were treated with saline or 100 μg/kg ShK-186 or 1, 10, or 100 μg/kg ShK-192, after which ear swelling was measured. Statistical analysis was carried out using the Mann-Whitney U test. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
ShK, a 35-residue peptide toxin from the Caribbean sea anemone Stichodactyla helianthus, is among the most potent inhibitors of Kv1.3 (Kd = 11 pM) (Kalman et al., 1998). We have determined the three-dimensional structure of ShK and elucidated the key Kv1.3 channel-binding residues. An analog, ShK-170, with improved specificity for Kv1.3 was developed by attaching a phosphotyrosine residue via an 11-atom hydrophilic linker to the N terminus of ShK (Beeton et al., 2005). The peptide had picomolar affinity for Kv1.3 (IC50, 69 pM), selectively inhibited TEM-effector cells, effectively treated At-EAE and DTH, did not cause cardiac toxicity assessed by continuous EKG monitoring and did not alter clinical chemistry or hematological parameters after 2 weeks of therapy.
Stabilization of peptides is an essential step in developing a peptide therapeutic. Several factors contribute to peptide instability, including proteolytic degradation by endo- and exo-peptidases, hydrolysis of sensitive peptide bonds (Asp-Gly, Asp-Pro, Asp-Ser), oxidation of methionine or tryptophan, hydrolysis of side chain groups, and reduction or scrambling of disulfide bonds. The lead compound ShK-170 was subjected to a series of experiments to gain insight into its stability at different pH values and temperatures over a 5-day time course. At acidic pH values, the major degradation product was generated by hydrolysis of the phosphate moiety from the N-terminal pTyr residue. This residue was critical for the enhanced selectivity observed for this peptide, and therefore we needed to develop an analog in which this by-product could be eliminated. A minor species also observed in these stability studies was associated with oxidation of Met21. The Met(O) creates a dipole and chirality issues at the oxidized sulfur atom, and, from a manufacturing perspective, any potential source of instability is undesirable, so replacement of Met with a stable hydrophobic isostere was considered necessary.
Taking into account all the data from the stability studies on ShK-170 and ShK-186, we engineered three stability elements into the molecule—C-terminal amidation, nonoxidable Nle21, and nonhydrolyzable Ppa at the N terminus—to generate ShK-192, which retained picomolar affinity for the channel (IC50, 140 pM), and was 160-fold selective over the neuronal Kv1.1 channel. The unnatural amino acid Nle has the same hydrophobic character as Met but is insensitive to oxidation as its sulfur atom is replaced by a carbon atom. To reduce susceptibility to acid hydrolysis of the phosphate from the phospho-Tyr side chain at the N terminus, the phosphotyrosine was replaced with the nonhydrolyzable phosphate mimetics Ppa or Pmp (Nomizu et al., 1994). Pmp has been used to make peptides that are stable to phosphatase enzymes involved in signal transduction pathways (Domchek et al., 1992). In these studies, the Pmp residue was found to be quite stable to hydrolysis of the phosphonate moiety, but the peptides did not have the same potency as phosphotyrosine-containing analogs (Domchek et al., 1992). It was postulated that the lower potency resulted from the higher pKa2 of the Pmp phosphonate as well as loss of hydrogen bonding interactions. The other phosphate mimetic we used, Ppa, is also not susceptible to hydrolysis of the anionic phosphonate group by phosphatases and has a more acidic pKa than the Pmp. The aryl phosphonate pKa is more similar to that of naturally occurring phosphate in phosphotyrosine (Liu et al., 2002).
ShK-192 was remarkably stable at acid pH values at temperatures ranging from 22°C to 60°C, which supports the feasibility of generating slow-release formulations of the peptide under these conditions. At pH 8.0 and 40°C, the peptide showed 12% decomposition on day-5, most likely due to disulfide rearrangement (the pKa of Cys is 8.5). This degradation was pronounced at 60°C at pH values of 7.0 and 8.0, when nearly all the peptide was denatured by day 2. The development of the large hump in the chromatogram indicated disulfide scrambling, as observed for ShK-170. LC-MS analysis through the entire hump detected the same mass species. Our experience in interpreting these types of data for many different multidisulfide peptides has shown that these humps consist of poorly resolved disulfide isomeric species (15 potential patterns are possible for a peptide with three disulfide bonds). These data imply that the Ppa residue may stimulate this decomposition by acting as a general base near the Cys3-Cys35 disulfide bond. As no hydrolysis of the phosphonate was observed with ShK-192, we are led to believe that Ppa may have facilitated the breakage of disulfide bonds through intramolecular catalysis or a neighboring group effect.
The solution structure of ShK-192 is very similar to that of ShK and ShK-Dap22, indicating that the modifications introduced to create this analog did not affect the native peptide structure. The N-terminal extension does not associate with other regions of ShK, but in fact samples a large volume of conformational space in solution. Upon binding to Kv1.3, the N-terminal adduct is able to engage in electrostatic and hydrogen bonding interactions with channel residues, which presumably contribute to the enhanced selectivity of ShK-192 for Kv1.3 over other channel subtypes. The importance of these interactions will have to be investigated by complementary mutational studies.
After a single subcutaneous injection of 100 μg/kg injection, ShK-192 disappeared rapidly from the circulation to a steady-state value of ∼200 pM at 24 and 48 h. We interpret these results to indicate that most of ShK-192 is excreted in the urine, whereas a small fraction distributes into a deep compartment from which active peptide is released slowly into the blood stream. This blood concentration is sufficient to block more than 50% of Kv1.3 channels in a T cell (Fig. 4), and this concentration is adequate to suppress the function of TEM cells (Fig. 9B) (Beeton et al., 2005). Consequently, ShK-192 administered at 1 to 100 μg/kg once daily by subcutaneous injection suppressed active DTH in rats induced against ovalbumin.
Because of their selective effect on TEM cells, Kv1.3 blockers are not expected to enhance susceptibility to infections. We reported recently that ShK-186 did not affect the clearance of two medically relevant infectious agents (influenza and chlamydia) in rats at concentrations that suppressed DTH (Matheu et al., 2008). Quinine, an antimalarial therapeutic, blocks Kv1.3 channels at concentrations found in the circulation of patients and does not increase the risk of infections. 4-Aminopyridine, a K+ channel blocker used in MS therapy, suppresses the proliferation of TEM cells at concentrations comparable with those found in the cerebrospinal fluid of treated MS patients, and these patients do not exhibit increased susceptibility to infections (Beeton et al., 2006). However, long-term therapy with Kv1.3 blockers may affect immune responses to chronic infections such as cytomegalovirus, HIV, and Epstein-Barr virus (Champagne et al., 2001; Yue et al., 2004; de Bree et al., 2005) that are effectively mediated by TEM cells. PAP-1, a small-molecule Kv1.3 inhibitor, induced a modest increase in CMV viral titer in monkeys that received the drug daily for 28 days; this subsided when the drug was stopped, but the levels were significantly lower than that required to cause clinical symptoms (Pereira et al., 2007).
In summary, ShK-192, a potent and specific Kv1.3 inhibitor, contains three stability elements: C-terminal amide, nonoxidable Nle23, and nonhydrolyzable Ppa at the N terminus. Its stability at acidic pH values and high temperatures suggests that it might be suitable for slow-release formulation. ShK-192 has the potential to ameliorate autoimmune diseases while sparing patients from the serious side effects resulting from broad-range immunosuppression. Many peptides similar in size to ShK-192 have been successfully taken to market and are now used clinically as therapeutics. These include ziconotide (25 amino acids, 3 disulfide bonds), exenatide (Byetta; 39 amino acids, 0 disulfide bonds), teriparatide (Forteo; 34 amino acids, 0 disulfide bonds), pramlintide (Symlin; 37 amino acids, 1 disulfide bond), calcitonin (Calcimar; 32 amino acids, 1 disulfide bond) and enfuvirtide (Fuzeon; 36 amino acids, 0 disulfide bonds). The large-scale synthesis of enfuvirtide to meet the monthly patient requirement (5400 mg/month/patient) has driven down the cost of raw materials for peptide synthesis. ShK-192 has the potential to ameliorate autoimmune diseases while sparing patients from the serious side effects resulting from broad-range immunosuppression.
Footnotes
-
This work was supported by National Institutes of Health National Institute of Neurological Disorders and Stroke [Grant NS48252]; the American Heart Association [Grant 0665009Y]; and a fellowship from the Australian National Health and Medical Research Council.
-
ABBREVIATIONS: MS, multiple sclerosis; TEM, effector memory T cells; RA, rheumatoid arthritis; TCM, central memory T cells; EAE, experimental autoimmune encephalomyelitis; At-EAE, adoptive transfer experimental autoimmune encephalomyelitis; DTH, delayed type hypersensitivity; PLGA, poly(d,l-lactic-co-glycolic acid); Ppa, para-phosphonophenylalanine; Fmoc, 9-fluorenylmethoxycarbonyl; Pmp, phosphonomethyl phenylalanine; TFA, trifluoroacetic acid; RP-HPLC, reversed-phase high-performance liquid chromatography; TOCSY, total correlation spectroscopy; NOE, nuclear Overhauser enhancement; NOESY, nuclear Overhauser enhancement spectroscopy; MD, molecular dynamics; HERG, human ether-á-go-go-related gene; DA, dark Agouti.
-
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material. - Received October 13, 2008.
- Accepted January 2, 2009.

- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}