


Abstract
Stromal cell-derived factor-1 (SDF-1/CXCL-12) and vascular endothelial growth factor (VEGF), which can be secreted by hypoxic tumors, promote the generation of new blood vessels. These potent angiogenic factors stimulate endothelial cell migration via the activation of Rho GTPases and the phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway. Thus, characterization of guanine nucleotide exchange factors critical in the angiogenic signaling cascades offers the possibility of identifying novel molecular targets. We demonstrated previously that mammalian target of rapamycin, an important effector and regulator of PI3K/AKT, activates phosphatidylinositol 3,4,5-triphosphate-dependent Rac exchanger 1 (P-Rex1), a Rac guanine nucleotide exchange factor identified as a target of Gβγ and PI3K, via direct interactions. In this study, we tested the hypothesis that P-Rex1 is involved in the angiogenic responses elicited by SDF-1 and VEGF. Using a knockdown approach, we demonstrate that P-Rex1 is indeed required for SDF-1 promoted signaling pathway, because there is decreased Rac activation, cell migration, and in vitro angiogenesis in P-Rex1 knockdown cells stimulated with SDF-1. In contrast, P-Rex1 knockdown does not affect responses to VEGF, and signaling to extracellular signal-regulated kinase in response to either angiogenic factor is not sensitive to P-Rex1 knockdown. We also demonstrate that in endothelial cells, VEGF promotes an increase in the expression of endogenous P-Rex1 and the SDF-1 receptor CXCR4, In addition, VEGF-pretreated cells show an increased migratory and angiogenic response to SDF-1, suggesting that VEGF stimulation can complement SDF-1/CXCR4 signaling to induce angiogenesis. We conclude that P-Rex1 is a key element in SDF-1-induced angiogenic responses and a potential target for therapeutic intervention.
New blood vessels are formed from pre-existing capillaries during the development and particular circumstances of postnatal life, such as wound healing. This process, widely known as angiogenesis, sustains the progression of pathological conditions, including cancer and chronic inflammatory diseases. The molecules involved in pathological angiogenesis are potential biomarkers and targets of pharmacological intervention (Carmeliet, 2005). Proof of principle that validates the therapeutic value of antiangiogenic intervention is the anti-VEGF treatment used in patients with metastatic colon cancer (Hurwitz et al., 2004). The inhibition of VEGF-dependent angiogenesis, combined with chemotherapy, is clearly effective in some pathological conditions but is limited in others (Ebos et al., 2009), suggesting that the characterization of alternative molecular targets is essential for developing new therapeutic tools.
Endothelial cell migration is a critical step in VEGF and SDF-1/CXCL-12-dependent angiogenesis. VEGF, through its tyrosine kinase receptors, promotes cell migration, proliferation, and expression of proangiogenic molecules, including the chemokine receptor CXCR4 (Salcedo et al., 2003; Kryczek et al., 2005). Stromal fibroblasts in tumors secrete SDF-1, the ligand of Gi-coupled CXCR4, promoting the formation of new blood capillaries and the mobilization of proangiogenic cells from the bone marrow (Kryczek et al., 2005; Orimo et al., 2005; Ruiz de Almodovar et al., 2006; Liang et al., 2007; Zheng et al., 2007; Chavakis et al., 2008; Seandel et al., 2008). VEGF and SDF-1 promote the activation of Rho GTPases, generating an ordered distribution of cellular protrusions and retractions that orchestrate a polarized phenotype during cell migration (Koh et al., 2008; Vega and Ridley, 2008). Thus, Rho guanine nucleotide exchange factors (RhoGEFs), the proteins that activate Rho GTPases by catalyzing the exchange of GDP to GTP, constitute an obligate molecular component in angiogenesis. RhoGEFs are complex multidomain proteins that integrate the intracellular actions of G protein-coupled receptors and tyrosine kinase receptors among other receptors, to define a precise localization and temporality of Rho GTPase activation (Rossman et al., 2005; Garrett et al., 2007; Koh et al., 2008; Vega and Ridley, 2008). As a consequence, RhoGEFs emerge as potential molecular targets in antiangiogenic therapies. Their potential is further sustained by the existence of more than 60 RhoGEFs, suggesting possible selectivity in the activation of Rho GTPases under physiological and pathological conditions. An interesting example is that LARG, a Gα12/13-sensitive RhoGEF, was found recently to be critical in the genesis of salt-induced hypertension but was irrelevant for the maintenance of normal vascular tone in mouse models (Wirth et al., 2008).
Although the role of SDF-1 in endothelial cell migration and tumor-induced angiogenesis is broadly accepted, the identity of the relevant RhoGEFs remains unknown. The aim of our studies is to identify RhoGEFs critically involved in angiogenic signaling cascades. We hypothesized that P-Rex1, a Rac GEF that in neutrophils is activated in the Gβγ/PI3K signaling pathway, is a critical participant in the angiogenic signaling pathways elicited by SDF-1 and VEGF. The role of RhoGEFs in SDF-1-induced cell migration has mainly been studied in cells of the immune system. In these cells, CXCR4 induces cell migration via the intervention of hematopoietic-specific RhoGEFs, such as Vav1 and dedicator of cytokinesis 2 (Fukui et al., 2001; García-Bernal et al., 2005). VEGF signaling to Rac has been demonstrated to be biphasic, with Vav2 related to the late phase peaking at 30 min (Garrett et al., 2007). The PI3K/AKT/mammalian target of rapamycin signaling pathway is tightly linked to angiogenic events (Chen et al., 2005; Barilli et al., 2008; Chavakis et al., 2008), suggesting that RhoGEFs regulated by this pathway could play a role in angiogenesis. We demonstrated previously that P-Rex1 is an effector of mammalian target of rapamycin, linking this kinase to Rac activation and cell migration (Hernández-Negrete et al., 2007). We now postulate that, among the multiple candidates involved in the signaling to Rac and endothelial cell migration, P-Rex1 could be a critical participant. In neutrophils, P-Rex1 is known to be activated by fMLP receptors, leading to Rac activation, production of reactive oxygen species, and chemotactic migration (Welch et al., 2002; Zhao et al., 2007). In addition, P-Rex1 is implicated in neurite differentiation and migration of cortical neurons (Yoshizawa et al., 2005; Waters et al., 2008) and metastatic prostate cancer cells (Qin et al., 2009). Here, we tested the hypothesis that P-Rex1 is critically involved in the angiogenic responses elicited by SDF-1 and VEGF in human microvascular endothelial cells and assessed the possibility that VEGF promotes the expression of this Rac GEF, further contributing to the migratory and angiogenic response elicited by SDF-1.
Materials and Methods
Constructs and Plasmids.
The short hairpin RNA (shRNA) for P-Rex1 and a control shRNA with a scrambled sequence were obtained by cloning the oligonucleotides CGAAGACACACTGTGCTTCCAGATTCAAGAGATCTGGAAGCACAGTGTGTCTTTTTG and CGAAATTGTATGCGATCGCAGACTTCAAGAGAGTCTGCGATCGCATACAATTTTTTTG, respectively, in the lentiviral transfer vector pSP-108 (generously provided by Jing Yang, University of California, San Diego, CA).
Cell Culture, Lentivirus Production, and Infection.
Human microvascular endothelial cells (HMEC) were maintained in MCDB-131 medium (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum, 10 mM l-glutamine, 10 ng/ml epidermal growth factor, and 1 μg/ml hydrocortisone (Sigma-Aldrich, St. Louis, MO). Human embryonic kidney 293T cells were cultivated in Dulbecco's modified Eagle's Media (Invitrogen) containing 10% FBS. Ten-centimeter plates of human embryonic kidney 293T (70% confluence) were transfected with the lentiviral pSP-108 vector along with lentivirus packaging and envelope plasmids using TransIT-LT1 (Mirus Bio LLC, Madison, WI). Each plate was transfected with 2 μg of pSP-108, 1.8 μg of pCMVΔR8.2, and 0.2 μg of pVSV-G. The day after transfection, media were replaced, and viral supernatants were harvested at 48 and 72 h. Viral supernatants, filtered through 0.45-μm filters, were supplemented with protamine sulfate at 6 μg/ml (Sigma-Aldrich) and incubated with HMEC cells during 4 h; then, media were substituted with fresh media. Cell lines used were stable pools. Cells were selected for 48 h with 0.5 μg/ml puromycin. Pilot studies confirmed that these conditions killed 100% of uninfected HMEC cells.
Western Blotting.
Total cell lysates were resolved on SDS-PAGE gels and transferred to Immobilon membranes (Millipore Corporation, Billerica, MA). The following antibodies were used for the immunoblots: mouse antibodies against Rac1 (BD Transduction Laboratories; BD Biosciences, San Jose, CA) and actin (Santa Cruz Biotechnology, Santa Cruz, CA); and rabbit antibodies against GAPDH, p44/42 mitogen-activated protein kinase (Cell Signaling Technology, Danvers, MA), and ERK-2 (Santa Cruz Biotechnology). Anti-P-Rex1 rat monoclonal antibodies were kindly provided by Mikio Hoshino (National Institute of Neuroscience, Kyoto, Japan) (Yoshizawa et al., 2005). Densitometric quantitation of Western blots was determined with the ImageJ software (http://rsbweb.nih.gov/ij/).
Cell Migration Assays.
Migration assays were done in Boyden chambers as described previously (Hernández-Negrete et al., 2007). In brief, serum-starved P-Rex1 knockdown or control HMEC cells were suspended in serum-free media supplemented with 0.1% bovine serum albumin and placed in the upper wells (50,000 cells/well) of the Boyden chamber. Bottom wells were previously filled with 0.1% bovine serum albumin containing the indicated chemoattractant, either SDF-1 (concentration indicated in the figure legends) or 10% FBS (used as positive control), and covered with a polycarbonate filter (8 μm pore; Neuro Probe, Gaithersburg, MD), previously coated with 10 μg/ml fibronectin (Sigma-Aldrich). Chambers were incubated at 37°C for 6 h, and then the filters were fixed with methanol, stained with crystal violet, and the nonmigrating cells were removed from the upper side of the filter with a cotton swab. Densitometric quantitation of migrating cells was determined with the ImageJ software. For experiments assessing the effect of long-term stimulation with VEGF on migratory responses, serum-starved HMEC cells, incubated with or without VEGF (100 ng/ml) for 12 h, were subjected to chemotaxis assays.
Rac-GTP and Phospho-ERK Assays.
P-Rex1 knockdown or control HMEC cells were used 3 days after infection with the corresponding lentiviral particles. The day before the assay, cells were serum-starved overnight and stimulated with 50 ng/ml SDF-1 as indicated in the figure legends. Cells were rinsed once with phosphate-buffered saline and lysed in 1 ml of ice-cold buffer (50 mM Tris, pH 7.5, 5 mM EDTA, 150 mM NaCl, 1% Triton X-100, and 10 mM MgCl2) containing protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, and 10 μg/ml aprotinin). Cell lysates were cleared, and aliquots were recovered to check total protein expression. GST-PAK CRIB beads, 30 μl/sample, were added and incubated on ice for 45 min in a shaker. Beads were washed three times with 1 ml of lysis buffer. Finally, beads were boiled in 35 μl of Laemmli protein sample buffer under reducing conditions and electrophoresed in 15% SDS-PAGE gels and subjected to Western blotting. Relative -fold increase of Rac activation was quantified by densitometric analysis and adjusted to the effect of VEGF in control cells, which was set to 100%. Total cell lysates (total Rac) and pull-downs (Rac GTP) were resolved on the same SDS-PAGE gel, and Rac GTP and total Rac blots were scanned and normalized to total Rac used as loading control after background subtraction. Relative -fold increase of ERK phosphorylation was also quantified by densitometric analysis. Total cell lysates were resolved on 10% SDS-PAGE gels, and the content of ERK in total cell lysates and phosphorylated ERK was determined by parallel Western blots using equal amounts of the same samples that were electrophoresed in parallel in the same gels. Relative -fold increase of ERK phosphorylation was normalized to total ERK and adjusted to the effect of VEGF in control cells, which was set to 100%. To ensure that all samples were consistent, all of the plates were seeded with an equal number of cells and were at equivalent confluence when lysed.
RT-PCR Analysis.
Control or VEGF-stimulated HMEC cells were treated with TRIzol reagent (Invitrogen) to obtain total RNA. cDNA was prepared by using Superscript RTII kit (Invitrogen). Sense and antisense amplification primers were as follows: P-Rex1: sense, CCCTGGTCAGTGAAGAGAGC; antisense, TCATCTCCAGACCCCATCTC; CXCR4: sense, GGTGGTCTATGTTGGCGTCT; antisense, TGGAGTGTGACAGCTTGGAG; CXCR7: sense, GGCTATGACACGCACTGCTA; antisense, CTCATGCACGTGAGGAAGAA; and TIAM1: sense, TGACCAGCTGATTGCTGAAC; antisense, AGACGAATGCTGCCAACTCT. PCR assays were done using JumpStart RED Taq ReadyMix PCR Reaction Mix kit (Sigma-Aldrich). PCR products were electrophoresed in 2% agarose gels. Pictures were captured in a BioDoc-It imaging system (UVP Inc., Upland, CA). Densitometric quantitation was performed with ImageJ software.
In Vitro Angiogenesis Assay.
P-Rex1 knockdown or control HMEC cells, selected with 0.5 μg/ml puromycin, were serum-starved for 12 h. The following day, cells were plated onto 90 6-well plates that had been preincubated with 20 μl of Matrigel (BD Biosciences) at 37°C for 1 h. Cells (104 cells in 25 μl of MCDB 131 medium) were placed into each well and then stimulated with 50 ng/ml SDF-1, 100 ng/ml VEGF, or 5% FBS and incubated for 9 to 12 h. The wells were photographed in different fields, and the length of tubular networks was determined using Image J software. For experiments assessing the effect of long-term stimulation with VEGF, serum-starved HMEC cells, incubated with or without VEGF (100 ng/ml) for 12 h, were subjected to in vitro angiogenesis assays.
Statistical Analysis.
Analysis of the effect of VEGF on P-Rex1 expression was compared using Student's t test. All other experiments were analyzed using analysis of variance tests followed by Newman-Keuls multiple comparison tests, differences of p < 0.05 were considered significant.
Results
SDF-1 Activates Rac, Endothelial Cell Migration, and in Vitro Angiogenesis.
Activation of Rac and cell migration are critical steps in the angiogenic process promoted by the SDF-1/CXCR4 axis (Kryczek et al., 2005), but the RacGEF that permits this activation in endothelial cells is unknown. In light of evidence that P-Rex1 is activated in response to Gβγ and phosphoinositide 3-kinase (Welch et al., 2002), downstream mediators generated through SDF-1 action, we hypothesized that this GEF could be involved in the activation of Rac and relevant for angiogenesis in response to SDF-1 in endothelial cells. First, we evaluated whether HMEC-1 cells expressed CXCR4 or CXCR7 receptors. CXCR-4 is widely recognized as the main SDF-1 receptor in mammalian cells, whereas CXCR7 has been recently identified as a chemokine receptor for SDF-1 that promotes tumor growth in breast, lung, and prostate cancers (Burns et al., 2006; Miao et al., 2007; Wang et al., 2008). As shown in Fig. 1A, HMEC-1 cells expressed CXCR4 but not CXCR7, whereas MDA-MB-231 cells, a breast cancer cell line, expressed both receptors.
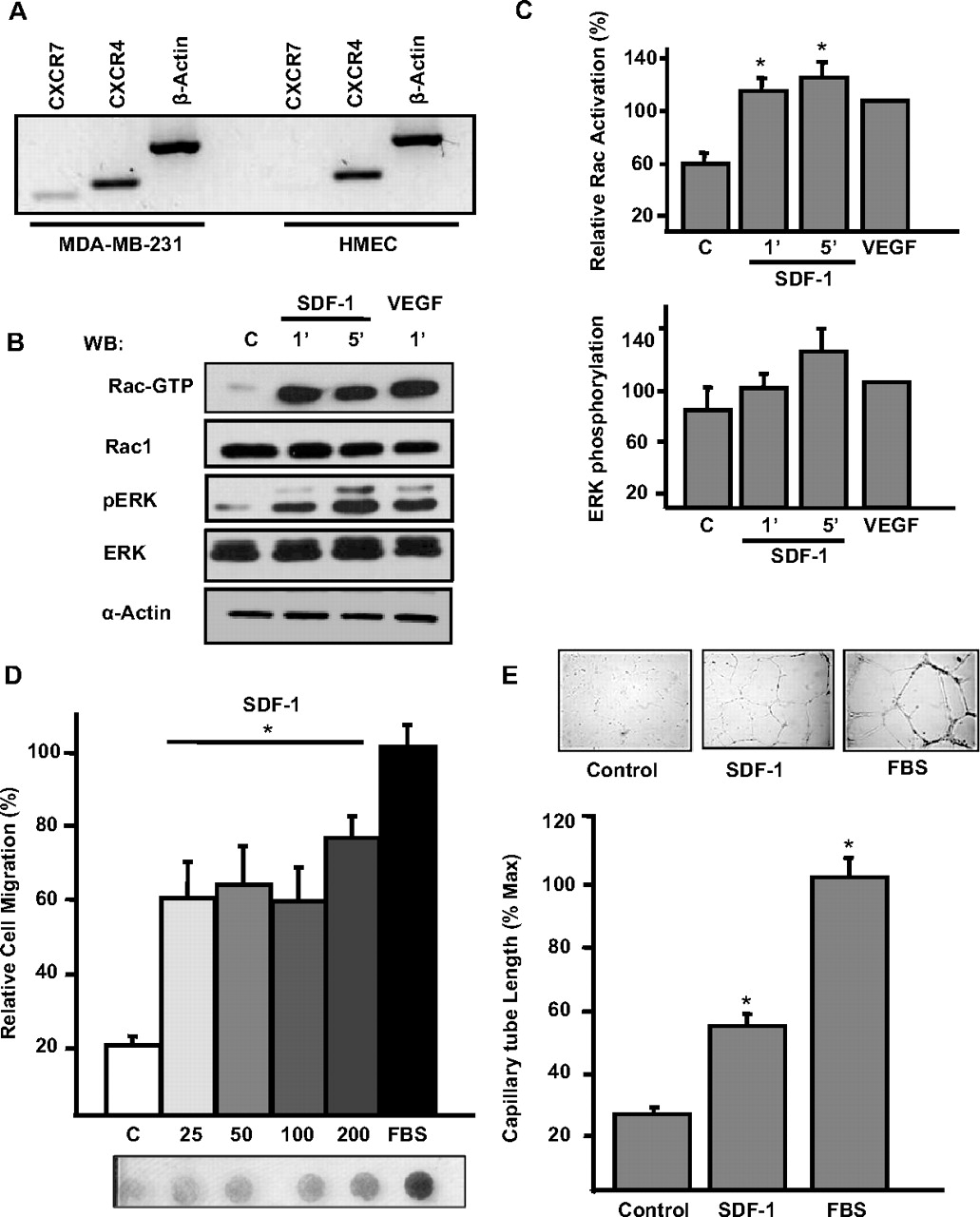
SDF-1 induces Rac1 activation, endothelial cell migration, and in vitro angiogenesis. A, expression of CXCR4 and CXCR7 in HMEC and MDA-MB-231 cells. Extraction of total RNA was performed as described under Materials and Methods, and RT-PCR assays of MDA-MB-231 and HMEC cells were performed. As shown, CXCR4 was the only receptor for SDF1 detected in HMEC cells. B, HMEC cells, starved in serum-free media, were stimulated with 50 ng/ml SDF-1 or 10 ng/ml VEGF for the indicated times. The active form of Rac (Rac-GTP) was isolated using GST-PAK-CRIB beads. Total cell lysates and pull-downs were resolved on SDS-polyacrylamide gels and analyzed by immunoblotting for Rac1, phospho-ERK (pERK), ERK-2, and actin. A representative experiment is shown. C, relative -fold increase of Rac activation and ERK phosphorylation was quantified by densitometric analysis of three independent experiments and adjusted to the effect of VEGF, which was set to 100%. D, serum-starved HMEC cells were subjected to chemotaxis assays in serum-free media containing the indicated concentrations of SDF-1 (from 25 to 200 ng/ml) or 10% FBS used as positive control. Migration assays were done in Boyden chambers for 6 h at 37°C in a 5% CO2 atmosphere. Migrating cells were stained, and the relative cell migration was quantified by densitometry of scanned filters; a representative filter is shown. Graphs represent the average results of three independent experiments, error bars indicate S.E.M. E, serum-starved HMEC cells were subjected to in vitro angiogenesis assays on Matrigel in the presence of 100 ng/ml SDF-1 or 5% FBS at 37°C, 5% CO2 for 9 to 12 h. As a negative control, cells were incubated in the absence of SDF-1 or FBS. Length of cord-like structures was quantified using ImageJ program and normalized to the effect of FBS, which was considered 100%. Bars represent the average results of three independent experiments. Error bars indicate S.E.M.; *, p < 0.05 difference with the corresponding effect in the control group.
Next, we evaluated whether HMEC-1 cells responded to SDF-1 by increasing the levels of active Rac. As shown in Fig. 1, B and C, the amount of GTP-bound Rac increased within 1 min after the addition of SDF-1 and remained elevated for 5 min. In parallel, SDF-1 lead to the phosphorylation of ERK, known to be activated in the signaling cascade downstream of CXCR4 stimulation (Kukreja et al., 2005; Huang et al., 2007) (Fig. 1, B and C). VEGF, used as positive control (Garrett et al., 2007), also induced Rac activation and promoted ERK phosphorylation (Fig. 1, B and C). In addition, HMEC cells were able to migrate and acquired an angiogenic phenotype, in vitro angiogenesis assays, in response to SDF-1 (Fig. 1, D and E).
Role of P-Rex1 in the Signaling of SDF-1 to Rac in Endothelial Cells.
To address the possibility that P-Rex1 is involved in the activation of Rac, cell migration, and in vitro angiogenesis, we first generated a specific shRNA in a lentiviral vector and tested its effect in microvascular endothelial cells. The ability of P-Rex1 shRNA to inhibit the expression of endogenous endothelial P-Rex1 is shown in Fig. 2A. The P-Rex1 shRNA had no effect on the expression of GAPDH used as control in the Western blot and control (scrambled shRNA) was without effect. We then evaluated the participation of P-Rex1 in the activation of Rac. P-Rex1 knockdown and control (scrambled shRNA) HMEC cells were stimulated with SDF-1, and the levels of Rac-GTP were measured (Fig. 2B). The ability of SDF-1 to induce the activation of Rac was decreased by approximately 50% in P-Rex1 knockdown cells (Fig. 2, B and C). In contrast, VEGF activated Rac via a P-Rex1-independent pathway (Fig. 2, B and C). In contrast, the phosphorylation of ERK in response to either SDF-1 or VEGF was not diminished in P-Rex1 knockdown cells (Fig. 2, B and C). These results suggest an important role of P-Rex1 in the signaling cascade leading to Rac activation, in response to SDF-1, in endothelial cells.
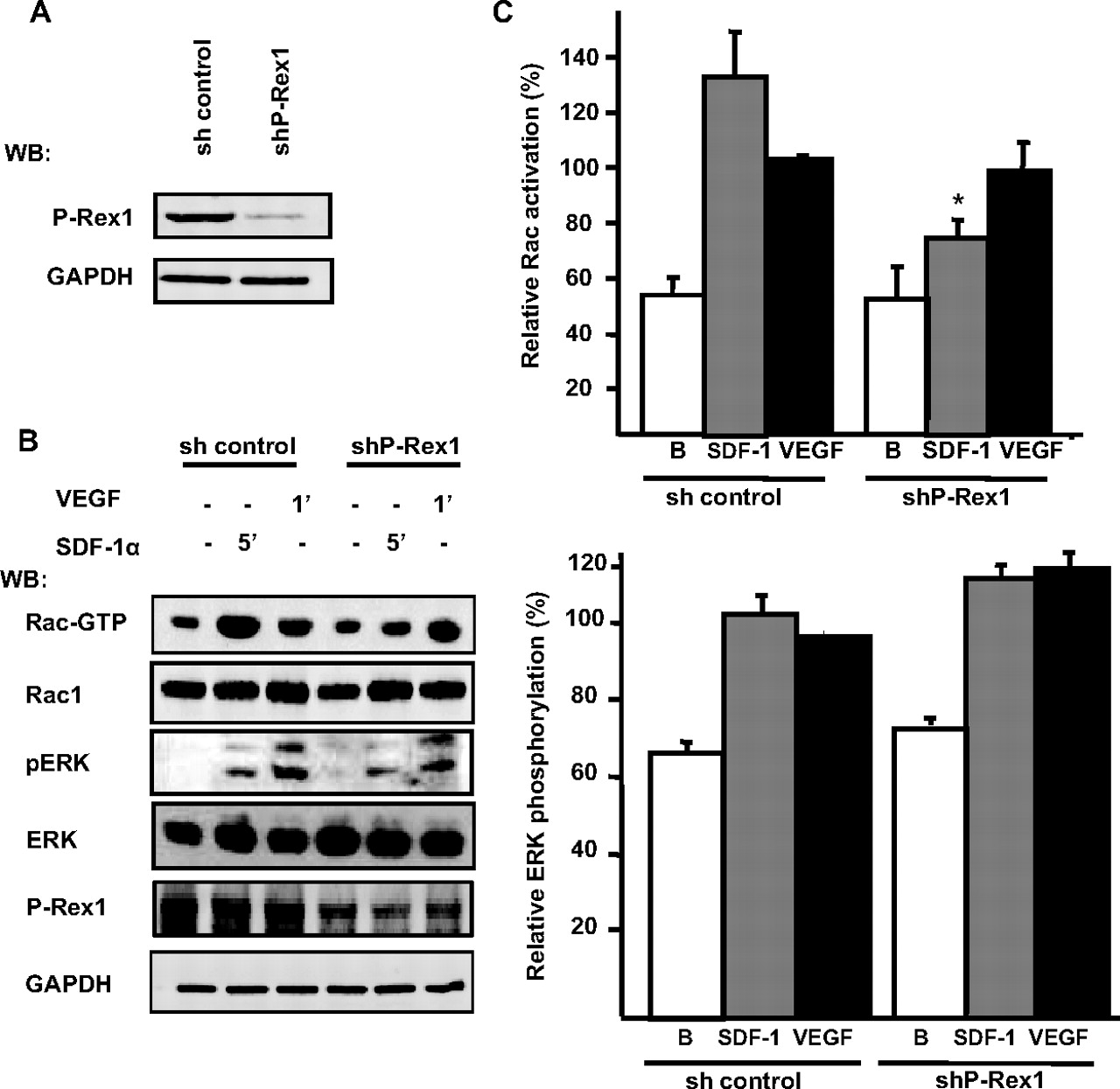
SDF-1 promotes Rac1 activation via P-Rex1 in microvascular endothelial cells. A, P-Rex1 knockdown in endothelial cells. HMEC cells were infected with lentiviral vectors carrying the sequence to produce a P-Rex1-specific shRNA (shPRex-1) or a scrambled sequence for a control shRNA (sh control). Cell lysates were resolved on SDS-PAGE gels and analyzed by immunoblotting with P-Rex1 and GAPDH antibodies. B, P-Rex1 knockdown or control HMEC cells, infected with the corresponding lentiviral vectors, were stimulated with 50 ng/ml SDF-1 or 10 ng/ml VEGF for 5 and 1 min, respectively. The active form of Rac (Rac-GTP) was isolated with GST-PAK-CRIB beads. Total cell lysates and pull-downs were resolved on SDS-PAGE gels and analyzed by immunoblotting for Rac1, phospho-ERK (pERK), ERK-2, and GAPDH. A representative blot detecting the indicated proteins is shown. C, relative -fold increase of Rac activation and ERK phosphorylation was quantified by densitometric analysis of six independent experiments and adjusted to the effect of VEGF in control cells which was set to 100%. Error bars indicate S.E.M.; *, p < 0.05 difference with the corresponding effect in the control group.
SDF-1 Induces Endothelial Cell Migration via P-Rex1.
Migration of endothelial cells is a critical step in the angiogenic process. Because SDF-1 activated Rac in endothelial cells via P-Rex1, we assessed whether P-Rex1 participates in the chemotactic response of endothelial cells stimulated with SDF-1. Cell migration assays were performed on HMEC cells infected with lentiviral vectors carrying either P-Rex1-shRNA or scrambled-shRNA control. As shown, P-Rex1 knockdown cells showed a significant reduction in their migration toward SDF-1, whereas the migration toward FBS was not affected, and the migration in response to VEGF showed a small, nonsignificant reduction (Fig. 3).

SDF-1 induces microvascular endothelial cell migration via P-Rex1. P-Rex1 knockdown or control HMEC cells were subjected to chemotaxis assays in Boyden chambers. Cells were starved for 12 h in serum-free media and stimulated with different concentrations of SDF-1 as indicated (from 50 to 200 ng/ml) or VEGF (100 ng/ml) for 6 h at 37°C, in a 5% CO2 atmosphere. FBS (10%) was used as positive control, basal migration was assessed in media lacking chemoattractants. Migrating cells were stained, and relative cell migration was determined by densitometry of scanned filters. Relative migration was obtained by adjusting to the effect observed in cells migrating toward FBS, which was adjusted to 100%. Three independent experiments were averaged and plotted. Error bars indicate S.E.M., *, p < 0.001 difference with control group. A representative filter of a migration assay is shown.
SDF-1-Dependent Angiogenic Response Requires the Expression of P-Rex1.
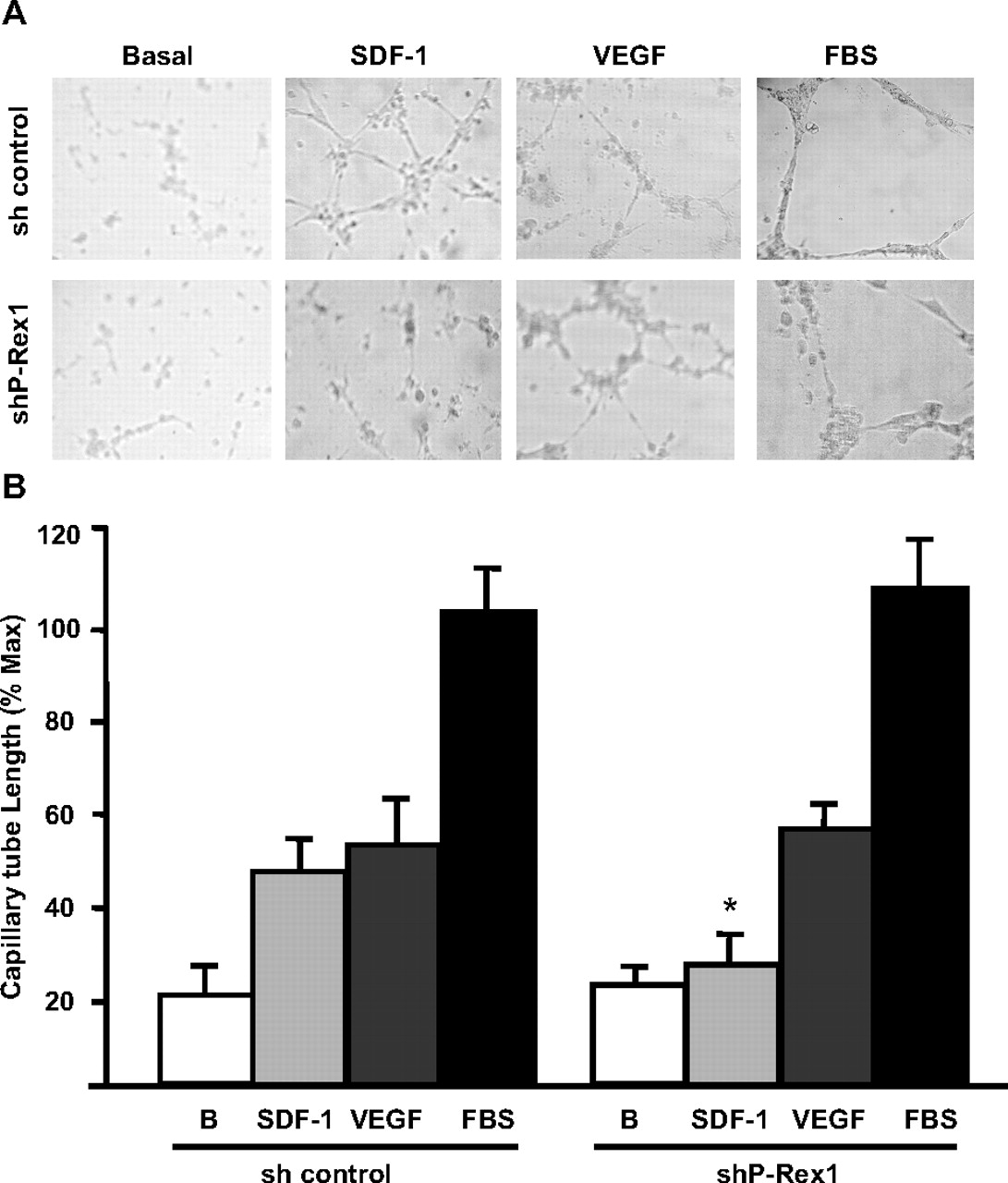
Because SDF-1 activated Rac and induced endothelial cell migration via P-Rex1, we explored the role of this GEF in angiogenesis. These experiments, carried out with cells in Matrigel, demonstrated that P-Rex1 knockdown caused a significant decrease in the assembly of interconnected capillary-reminiscent structures formed in response to SDF-1 (Fig. 4, A and B). In addition, the few cells that acquired a capillary-like phenotype in response to SDF-1 showed a reduced length compared with control cells (Fig. 4, A and B). P-Rex1 knockdown did not affect the ability of endothelial cells to acquire a capillary-like phenotype in response to VEGF or FBS (Fig. 4, A and B), further demonstrating a specific role of P-Rex1 in the CXCR-4 angiogenic pathway. Overall, these results indicate that SDF-1 is able to induce Rac activation, cell migration, and in vitro angiogenesis via a P-Rex1-dependent manner in endothelial cells.

P-Rex1 is an essential element in the angiogenic response elicited by SDF-1. A, P-Rex1 knockdown in endothelial cells caused a marked inhibition of the angiogenic effect elicited by SDF-1 but not by VEGF. Serum-starved P-Rex1 knockdown (shP-Rex1) or control endothelial cells (sh control) were added onto Matrigel and incubated without (negative control) or with 100 ng/ml SDF-1, 100 ng/ml VEGF, or 5% FBS at 37°C in a 5% CO2 atmosphere for 9 to 12 h. Representative photographs show the angiogenic effect of the indicated stimuli assessed as the acquisition of a cord-like interconnected structure of endothelial cells. B, bars represent the quantitative analysis of the angiogenic effect of SDF-1 or VEGF assessed as the capillary tube length normalized to the effect of FBS. Data are shown as mean + S.E.M of six experiments for SDF-1 and FBS and three experiments for VEGF. *, p < 0.001 difference with the corresponding effect in the control group.
Effect of VEGF on P-Rex1 and CXCR4 mRNA Expression in Human Microvascular Endothelial Cells.
The angiogenic properties of VEGF include the activation of signaling cascades that result in the expression of signal transduction proteins and receptors that facilitate the angiogenic response, including CXCR4 (Li et al., 2006; Olsson et al., 2006; Zheng et al., 2007). We postulated that VEGF might induce an increase in the expression of P-Rex1 as well as CXCR4 in HMEC cells. In addition, we determined the effect of VEGF on the expression of TIAM1 (T-cell lymphoma invasion and metastasis-1), a well characterized Rac GEF involved in cell migration, invasion, adhesion, and tumor progression (Habets et al., 1994; Minard et al., 2004). As shown in Fig. 5, endothelial cells constitutively expressed low levels of P-Rex1 mRNA, and the expression of this GEF increased nearly 1.5-fold in cells stimulated for 3 h with VEGF. In agreement with previous reports (Salcedo et al., 1999), VEGF induced a 2-fold increase in CXCR4 mRNA levels (Fig. 5). The expression of TIAM1, was not altered in response to VEGF treatment (Fig. 5). These results demonstrate that P-Rex1 is endogenously expressed in endothelial cells and that its expression is further increased in response to VEGF providing a mechanism that would enable SDF-1-mediated Rac activation and angiogenesis.

VEGF increases the expression of P-Rex1 and CXCR4 in human microvascular endothelial cells. A, total RNA was isolated from control of VEGF-stimulated HMEC cells stimulated with 100 ng/ml VEGF for the indicated times. RT-PCR assays were done to detect the expression of P-Rex1, CXCR4, and TIAM. A representative experiment is shown. B, -fold increase in mRNA expression was determined by densitometric analysis after normalization using β-actin as control. The mean and S.E.M values of seven experiments are shown. *, p < 0.05 difference with respective unstimulated controls.
Effect of VEGF on SDF-1-Induced Endothelial Cell Migration and in Vitro Angiogenesis.
Because VEGF induced an increase on the expression of CXCR4 and P-Rex1 in HMEC cells, we assessed whether long-term incubation with VEGF resulted in an increased migratory and angiogenic response toward SDF-1. As shown in Fig. 6, VEGF pretreatment induced a significant increase in the migratory response of endothelial cells to 5 ng/ml SDF-1, whereas the response to VEGF or serum was not affected. The response to VEGF reached a maximum that was not further increased by costimulation with SDF-1. In the in vitro angiogenesis experiments, there was significant increase on the response to SDF-1 in VEGF-pretreated cells. The response to VEGF or serum was similar in control and VEGF-pretreated cells.
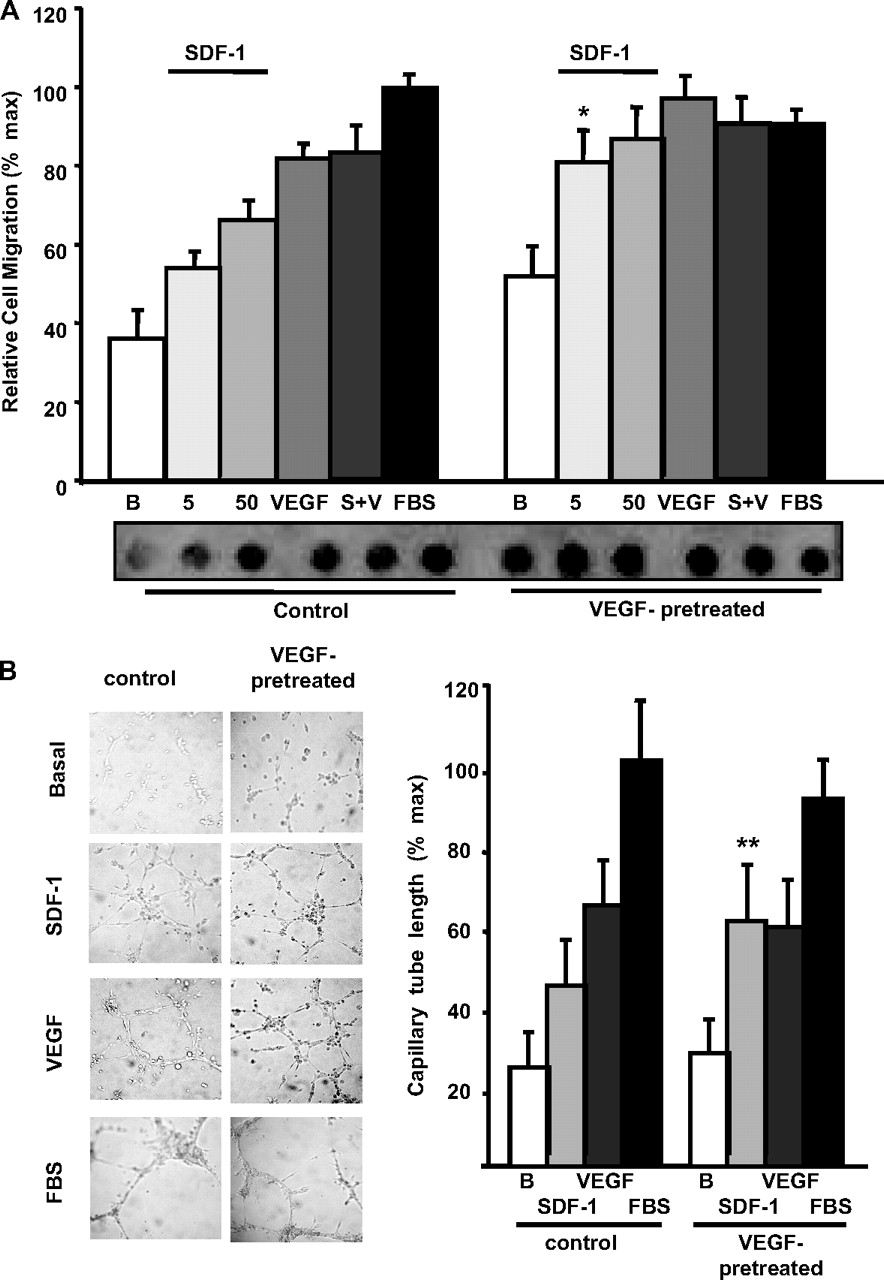
Effect of VEGF-pretreatment on SDF-1-induced migration and angiogenic response in human microvascular endothelial cells. A, VEGF-pretreatment increases SDF-1-induced endothelial cell migration. Serum-starved HMEC cells, incubated with or without VEGF (100 ng/ml) for 12 h, were subjected to chemotaxis assays. Cells were stimulated with different concentrations of SDF-1 (5 or 50 ng/ml), VEGF (100 ng/ml), SDF-1 + VEGF (S+V, 50 and 100 ng/ml, respectively), or 10% FBS for 6 h at 37°C in a 5% CO2 atmosphere. Relative cell migration (percentage of FBS) was determined by comparing control cells with cells preincubated with VEGF. Three independent experiments were averaged and plotted. A representative filter of a migration assay is shown. Graph represents the average results of three independent experiments. Error bars indicate S.E.M., *, p < 0.05 versus the same concentration of ligand for control group (no VEGF pretreatment). B, HMEC control and VEGF-pretreated cells were subjected to in vitro angiogenesis assays on Matrigel in the presence of 100 ng/ml SDF-1, 100 ng/ml VEGF, or 5% FBS at 37°C, 5% CO2 for 9 to 12 h. As a negative control, cells were incubated in serum-free media (basal). Length of cord-like structures was quantified using ImageJ program and normalized to the effect of FBS, which was considered 100%. Graph represents the average results of three independent experiments. Error bars indicate S.E.M., **, p < 0.001 versus the same concentration of ligand for control group (no VEGF pretreatment).
Discussion
In this work, we demonstrate a critical requirement for the Rac guanine exchange factor P-Rex1 in SDF-1-elicited endothelial responses leading to Rac activation, cell migration, and formation of endothelial cord-like structures in an in vitro angiogenesis assay. These results provide evidence for a role of P-Rex1 in the signal transduction pathway elicited by SDF-1 leading to an angiogenic response.
Previous reports have shown that VEGF and basic fibroblast growth factor up-regulate CXCR4 expression (Salcedo et al., 1999, 2003). This response has been interpreted as contributing to a mechanism that prepares endothelial cells to sustain an angiogenic response to SDF-1. Here, we demonstrate that P-Rex1 is endogenously expressed in endothelial cells and that its expression, as is the case for CXCR4, is increased in cells stimulated with VEGF, suggesting that an increase on the expression of this GEF can be linked to an angiogenic response. This possibility is further supported by the findings showing that VEGF-pretreated cells showed an increased migratory and angiogenic response to SDF-1.
Because SDF-1, known to activate Gi-coupled CXCR4 receptors in endothelial cells (O'Hayre et al., 2008), was able to activate Rac, endothelial cell migration, and in vitro angiogenesis, and P-Rex1 is known to respond to Gβγ and phosphoinositide 3-kinase (Welch et al., 2002), we explored the possible involvement of this GEF in the angiogenic response elicited by SDF-1. Our results show a marked decrease in the activation of Rac in response to SDF-1 in P-Rex1 knockdown endothelial cells, indicating participation of this GEF in initiation of Rac-mediated signaling pathways by SDF-1. The specificity of this action is further evidenced by the unaltered ability of these cells to activate Rac in response to VEGF, which is believed to activate Rac through Vav2 and potentially other GEFs (Garrett et al., 2007). As additional evidence for the specificity of the P-Rex-1 knockdown, we demonstrated a lack of effect on other signaling properties of SDF-1 in endothelial cells, as demonstrated by the ability of these cells to promote normal phosphorylation of ERK.
SDF-1-induced endothelial cell chemotaxis is required for the formation of cord-like structures (Liang et al., 2007). Here, we demonstrate that the Rac exchange factor P-Rex1 is also required for endothelial cells to migrate in response to this chemoattractant. In addition, the ability of SDF-1 to elicit an angiogenic response (determined by the formation of interconnected cord-like structures) is significantly decreased in P-Rex1 knockdown cells. The observation that the VEGF- or serum-induced in vitro angiogenic response was not affected in P-Rex1 knockdown cells demonstrates a specific role for P-Rex1 in the CXCR-4 angiogenic pathway. Therefore, we suggest that P-Rex1 is a specific and requisite participant in the signaling pathway by which CXCR4 controls human microvascular endothelial cell migration and morphogenetic changes relevant for angiogenesis. It is interesting that our results showing a role of P-Rex1 in SDF-1-stimulated human microvascular endothelial cell migration support recent findings demonstrating that metastatic prostate cancer cells migrate toward SDF-1 in a P-Rex1-dependent way (Qin et al., 2009) and extend these data to directly demonstrate that P-Rex1 participates in the activation of Rac in response to SDF-1. Our current results indicate that P-Rex1 is up-regulated in response to VEGF, a constituent of the SDF-1-induced Rac signaling pathway and a mediator of the angiogenic response to SDF-1. These findings suggest that under pathological conditions commonly characterized by VEGF-initiated angiogenic responses (Charlesworth and Harris, 2006), increased P-Rex1 and SDF-1 induced Rac activation may be critical links in angiogenesis, tumor dissemination, and tumor progression. Our findings raise the possibility of considering P-Rex1 as a potential target for antiangiogenic therapies in cases in which pathological angiogenesis is due to the action of CXCR4.
Acknowledgments
We thank Jing Yang and Mikio Hoshino, who kindly provided lentiviral vectors and antibodies, respectively. Technical assistance provided by Estanislao Escobar Islas, David Pérez, and Jeffrey Smith is acknowledged.
Footnotes
This work was supported by the University of California Institute for Mexico and the United States, UC-Mexus and the Consejo Nacional de Ciencia y Tecnología [Grants 61127, 79429].
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.109.060400
-
ABBREVIATIONS:
- VEGF
- vascular endothelial growth factor
- SDF-1
- stromal cell-derived factor-1
- GEF
- guanine nucleotide exchange factor
- RhoGEF
- Rho guanine nucleotide exchange factor
- P-Rex1
- phosphatidylinositol 3,4,5-triphosphate-dependent Rac exchanger 1
- ERK
- extracellular signal-regulated kinase
- PCR
- polymerase chain reaction
- PAGE
- polyacrylamide gel electrophoresis
- HMEC
- human microvascular endothelial cells
- GAPDH
- glyceraldehyde 3-phosphate dehydrogenase
- TIAM
- T-lymphoma invasion and metastasis
- VAV
- vav guanine nucleotide exchange factor
- shRNA
- short hairpin RNA
- FBS
- fetal bovine serum
- RT-PCR
- reverse transcriptase-polymerase chain reaction.
- Received September 2, 2009.
- Accepted December 17, 2009.
- Copyright © 2010 The American Society for Pharmacology and Experimental Therapeutics
References
- Barilli et al., 2008.↵
- Burns et al., 2006.↵
- Carmeliet, 2005.↵
- Charlesworth and Harris, 2006.↵
- Chavakis et al., 2008.↵
- Chen et al., 2005.↵
- Ebos et al., 2009.↵
- Fukui et al., 2001.↵
- García-Bernal et al., 2005.↵
- Garrett et al., 2007.↵
- Habets et al., 1994.↵
- Hernández-Negrete et al., 2007.↵
- Huang et al., 2007.↵
- Hurwitz et al., 2004.↵
- Koh et al., 2008.↵
- Kryczek et al., 2005.↵
- Kukreja et al., 2005.↵
- Li et al., 2006.↵
- Liang et al., 2007.↵
- Miao et al., 2007.↵
- Minard et al., 2004.↵
- O'Hayre et al., 2008.↵
- Olsson et al., 2006.↵
- Orimo et al., 2005.↵
- Qin et al., 2009.↵
- Rossman et al., 2005.↵
- Ruiz de Almodovar et al., 2006.↵
- Salcedo et al., 1999.↵
- Salcedo et al., 2003.↵
- Seandel et al., 2008.↵
- Vega and Ridley, 2008.↵
- Wang et al., 2008.↵
- Waters et al., 2008.↵
- Welch et al., 2002.↵
- Wirth et al., 2008.↵
- Yoshizawa et al., 2005.↵
- Zhao et al., 2007.↵
- Zheng et al., 2007.↵





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}