


Abstract
Despite the identification of 2-amino-3-benzoylthiophenes (2A3BTs) as the first example of small-molecule allosteric potentiators of agonist function at a G protein-coupled receptor (GPCR)—the adenosine A1 receptor—their mechanism of action is still not fully understood. We now report the mechanistic basis for the complex behaviors noted for 2A3BTs at A1 receptors. Using a combination of membrane-based and intact-cell radioligand binding, multiple signaling assays, and a native tissue bioassay, we found that the allosteric interaction between 2A3BTs and the agonists 2-chloro-N6-[3H]cyclopentyladenosine or (−)-N6-(2-phenylisopropyl)adenosine (R-PIA) or the antagonist [3H]8-cyclopentyl-1,3-dipropylxanthine is consistent with a ternary complex model involving recognition of a single extracellular allosteric site. However, when allowed access to the intracellular milieu, 2A3BTs have a secondary action as direct G protein inhibitors; this latter property is receptor-independent as it is observed in nontransfected cells and also after stimulation of another GPCR. In addition, we found that 2A3BTs can signal as allosteric agonists in their own right but show bias toward certain pathways relative to the orthosteric agonist, R-PIA. These results indicate that 2A3BTs have a dual mode of action when interacting with the A1 receptor and that they can engender novel functional selectivity in A1 signaling. These mechanisms need to be factored into allosteric ligand structure-activity studies.
Introduction
G protein-coupled receptors (GPCRs) are the largest family of cell surface proteins (Lagerström and Schiöth, 2008) and remain pre-eminent targets for novel drug discovery (Overington et al., 2006). An important paradigm of drug action at GPCRs is the recognition that most of these receptors posses at least one allosteric site that is topographically distinct from the orthosteric site that binds the endogenous agonist (Christopoulos and Kenakin, 2002; Christopoulos, 2002). Upon binding, allosteric ligands modulate the conformation of the GPCR and, therefore, the biological properties of cobound orthosteric ligands, by changing their affinities, their signaling efficacies, or both (May et al., 2007b). More recently, it has been suggested that allosteric modulators can also engender functional selectivity in the signaling of orthosteric ligands; that is, the allosteric ligand may “bias” the stimulus imparted by the orthosteric ligand such that only a subset of the possible repertoire of intracellular signaling cascades linked to the GPCR is activated (Leach et al., 2007). Thus, understanding the mechanisms of action of GPCR allosteric modulators offers potential for the development of novel tools with which to probe receptor function, as well as more selective therapeutic agents (Conn et al., 2009).
The A1 receptor, one of four subtypes of GPCR for the purine nucleoside adenosine, was the first GPCR for which positive allosteric modulators of agonist function were described (Bruns and Fergus, 1990; Bruns et al., 1990). The finding that 2A3BT derivatives, exemplified by compounds such as 2-amino-(4,5,6,7-tetrahydrobenzo[b]thiophen-3-yl) (4-chlorophenyl)methanone (T62), 2-amino-4,5-dimethyl- 3-thienyl-[3-(trifluoromethyl)phenyl]methanone (PD81,723), and 2-amino-(4,5,6,7-tetrahydrobenzo[b]thiophen-3-yl)(3,4-dichlorophenyl)methanone (LUF5484) (Fig. 1), potentiated the actions of adenosine was very promising and opened a new avenue of promoting selective activation of the A1 receptor by its endogenous agonist. Moreover, extracellular adenosine concentrations can quickly rise up to 100-fold over basal levels in response to cellular damage in inflammatory or ischemic tissues (Rudolphi et al., 1992; Latini et al., 1999), providing a rationale for both spatial (tissue-specific) and temporal selectivity of drug action via targeting allosteric sites on adenosine receptors; potentiation of the activity of A1 receptors has been implicated in the treatment of conditions such as ischemia reperfusion injury, paroxysmal superventricular tachycardia, chronic pain, and noninsulin-dependent diabetes mellitus (Gao and Jacobson, 2007; Elzein and Zablocki, 2008).

Structures of 2-amino-3-benzoylthiophene (2A3BT) derivatives used in this study.
Despite the early identification of 2A3BTs as allosteric modulators of A1 receptors, their mechanism of action appears complex and is not fully understood. For instance, when tested against agonist radioligands in equilibrium assays, these modulators typically yield bell-shaped binding curves, characterized by an increase in orthosteric radioligand binding at low modulator concentrations and a decrease in orthosteric binding at high concentrations; similar experiments using antagonist radioligands reveal only inhibition of orthosteric binding by the modulator. This property has been almost invariably interpreted as evidence that 2A3BTs recognize an allosteric site at low concentrations but also bind to the orthosteric site at high concentrations. Within such a scheme, the interaction with agonists presumably reflects a mixed mode of positive cooperativity and competitive inhibition, whereas the interaction with antagonists reflects both negative cooperativity and competition. Accordingly, many structure-activity relationships have been performed to separate the “allosteric component” from the “orthosteric component” of these ligands (van der Klein et al., 1999; Baraldi et al., 2000; Figler et al., 2003; Lütjens et al., 2003; Nikolakopoulos et al., 2006; Aurelio et al., 2008, 2009; Ferguson et al., 2008). Although these studies have yielded ligands with increased allosteric potencies, none has successfully moved away from the 2-aminothiophene scaffold or has been able to completely eradicate the apparently orthosteric/competitive component of their actions. To delineate the basis of this phenomenon, we undertook a detailed study of the binding and functional properties of three 2A3BTs: the “prototypical” allosteric A1 modulator, T62 (Bruns and Fergus, 1990; Childers et al., 2005; Baraldi et al., 2006, 2007) and two novel derivatives, 2-amino-4-(3,5-bis(trifluoromethyl)phenyl)thiophen-3-yl)(phenyl)methanone (VCP520) and tert-butyl 2-amino-3-(4-chlorobenzoyl)-7,8-dihydro-4H-thieno[2,3-d]azepine-6(5H)-carboxylate (VCP333) (Fig. 1) (Aurelio et al., 2009). We show that the divergent effects on agonist and antagonist binding affinities at low concentrations of 2A3BTs can be mechanistically accommodated by interaction with a common allosteric site, without the need to invoke an orthosteric component in the modulator actions; we reveal that these ligands can exhibit novel functional selectivity in their own right as allosteric agonists; we find that the inhibitory effects noted at high concentrations of 2A3BTs actually reflect a receptor-independent inhibition of Gi/o protein activity. This dual mechanism of action of 2A3BTs as allosteric ligands of the A1 receptor has significant implications for structure-activity studies of allosterism at this important GPCR family.
Materials and Methods
Synthesis of 2-Amino-3-benzoylthiophene Derivatives.
T62 and VCP520 were synthesized as described previously (Nakanishi et al., 1973; Aurelio et al., 2009). The synthesis of VCP333 is described in the Supplemental Material.
Materials.
Dulbecco's modified Eagle's medium and hygromycin B were purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum (FBS) was purchased from ThermoTrace (Melbourne, VIC, Australia). 2-Chloro-N6-[3H]cyclopentyladenosine ([3H]CCPA) (42.6 Ci/mmol) and [3H]8-Cyclopentyl-1,3-dipropylxanthine ([3H]DPCPX) (120 Ci/mmol) were purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA), and adenosine deaminase, derived from calf intestine, was purchased from Roche (Basel, Switzerland). [35S]GTPγS (>1000 Ci/mmol) was purchased from both PerkinElmer Life and Analytical Sciences and GE Healthcare (Little Chalfont, Buckinghamshire, UK). The Sure-Fire cellular ERK1/2 assay kits were a generous gift from TGR BioSciences (Adelaide, SA, Australia). AlphaScreen reagents for ERK1/2 and cAMP assays and Ultima gold scintillation cocktail were from PerkinElmer Life and Analytical Sciences. All other reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Cell Culture and Membrane Preparation.
FlpIn Chinese hamster ovary (CHO) cells stably expressing adenosine A1 receptors were generated and cultured as described previously (Stewart et al., 2009). FlpIn-CHO cells stably expressing muscarinic M2 receptors (M2 mAChR FlpIn-CHO; May et al., 2007a) were cultured as described for A1 receptor FlpIn-CHO cells. Membranes of A1 receptor and M2 mAChR were generated as described previously in Stewart et al. (2009) and May et al. (2005), respectively.
Agonist and Antagonist Radioligand Equilibrium Binding Assays.
Radioligand binding assays were performed using two different radioligands. When using [3H]CCPA (2 nM), assays were performed as described previously (Ferguson et al., 2008), except that membrane-based binding assays were performed at 30°C. When using [3H]DPCPX (1 nM), assays were also performed as described previously (May et al., 2005), except that membrane-based binding assays were performed at 30°C. These experiments were performed in the absence of guanine nucleotides in the case of agonist radioligand and antagonist radioligand whole-cell binding or in presence of 100 μM Gpp(NH)p in the case of antagonist radioligand membrane preparation binding. Nonspecific binding was defined using 100 μM R-PIA.
Dissociation Kinetic Binding Assays.
Membrane homogenates (15 μg) were equilibrated with [3H]CCPA (2 nM) in a 1-ml total volume of assay buffer for 90 min at 30°C. R-PIA (10 μM), alone or in the presence of T62, VCP520, or VCP333, was then added at various time points to prevent the reassociation of [3H]CCPA with the receptor. In subsequent experiments designed to investigate the effect of a range of modulator concentrations on [3H]CCPA dissociation rate, a “two-point kinetic” experimental paradigm was used in which the effect of increasing concentrations of allosteric ligand on [3H]CCPA dissociation was determined at 20 and 90 min. This approach is valid to determine [3H]CCPA dissociation rate constants if the full time course of radioligand dissociation is monophasic in both the absence and the presence of modulator (Lazareno and Birdsall, 1995; Kostenis and Mohr, 1996); this was the case in our current study. Incubation was terminated as described previously.
Signaling Assays.
Interaction studies in ERK1/2 phosphorylation assays and [35S]GTPγS binding assays were performed as per Aurelio et al. (Aurelio et al., 2009). For ERK1/2 phosphorylation experiments, 3% (v/v) FBS was used as a positive control, and vehicle controls were also performed; data were normalized to the maximal response elicited by 3% (v/v) FBS at the 6-min time point, unless otherwise specified. For either A1 receptor or M2 mAChR [35S]GTPγS binding assays, identical buffer and condition of incubation were used; data were normalized to the maximal response elicited by 1 μM R-PIA for interaction studies or to the fold over basal when experiments were performed at the M2 mAChR. Calcium mobilization assays and cAMP assays were performed as described previously (Stewart et al., 2009).
Isolated Rat Atria.
Experiments were carried out in accordance with the 1997 Australian National Health and Medical Research Council Code of Practice under a protocol approved by the Institutional Animal Ethics Committee. Sprague-Dawley rats (250–300g), were housed at the Animal Resources Center in cages (North Kent Plastics, Rochester, Kent, UK) with sawdust bedding and maintained on a constant 12-h light/dark cycle at 18–22°C. Animals were given normal tap water and food in the form of ARM cubes (Clark-King and Co., Gladesville, NSW, Australia) ad libitum. Rats were sacrificed by stunning followed by exsanguination, and hearts were rapidly removed and placed in Krebs-Henseleit solution (118 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 25 mM NaHCO3, 11.7 mM glucose, 1.1 mM MgSO4,and 2.5 mM CaCl2). The right atrium was isolated and mounted in an organ bath at 37°C, bubbled with 5% CO2/95% O2, and allowed to contract spontaneously. The rate of atrial contraction was measured using a force-transducer connected to a PowerLab data acquisition system (ADInstruments Pty Ltd., Castle Hill, NSW, Australia). Responses to a low concentration (10 nM; approximately EC10) of R-PIA were determined in the presence of allosteric modulator (0.3 and 3 μM) or vehicle (0.1% DMSO). The modulator or vehicle was added 1 min before R-PIA stimulation, and the magnitude of the resultant decrease in heart rate was determined at the point of peak response, usually 2 to 3 min after addition of the R-PIA.
Data Analysis.
Computerized nonlinear regression was performed using Prism 5.01 or a prerelease version of Prism 6.0 (GraphPad Software, San Diego, CA). Radioligand inhibition binding data were empirically fitted to a one-site inhibition mass action curve to determine inhibitor potency estimates, which were then converted to KI values as appropriate (Cheng and Prusoff, 1973). Radioligand potentiation binding curves were fitted to a simple allosteric ternary complex model to derive estimates of allosteric modulator affinity (KB) and cooperativity (α), the latter parameter being a measure of the strength and direction of the interaction between the orthosteric and allosteric sites (Christopoulos and Kenakin, 2002; May et al., 2007b); values of α > 1 denote positive cooperativity, whereas values of 0 < α < 1 denote negative cooperativity. Dissociation kinetic data were fitted to monoexponential functions to derive observed dissociation rate constants. Where appropriate concentration-response curves were fitted to a three-parameter logistic equation to derive ligand potency estimates. Finally for whole-cell functional ligand combination studies, the interaction between the orthosteric agonist R-PIA and the allosteric ligands T62, VCP520, or VCP333 was fitted to the following two forms of an operational model of allosterism and agonism (Leach et al., 2007; Aurelio et al., 2009) to derive functional estimates of modulator affinity, cooperativity, and efficacy.

 where Em is the maximum attainable system response for the pathway under investigation; [A] and [B] are the concentrations of orthosteric agonist and allosteric modulator/agonist, respectively; KB is the dissociation constant of the allosteric modulator; EC50 is the concentration of orthosteric (full) agonist yielding 50% of the response between minimal and maximal receptor activation in the absence of allosteric ligand; n is a transducer slope factor linking occupancy to response; α and β are the cooperativity factors governing allosteric effects of the modulator on orthosteric agonist binding affinity and signaling efficacy, respectively; and τA and τB are operational measure of the ligands' respective signaling efficacies that incorporate receptor expression levels and efficiency of stimulus-response coupling (Leach et al., 2007; Aurelio et al., 2009). For this analysis, the entire family of curves for a given agonist-modulator combination across three different signaling pathways (ERK1/2 phosphorylation, Ca2+ mobilization, and inhibition of cAMP accumulation) were globally fitted to the model; Ca2+ data were fitted to eq. 1, whereas ERK1/2 and cAMP data were fitted to eq. 2, with the pKB parameter constrained to be shared across the entire family of curves.
where Em is the maximum attainable system response for the pathway under investigation; [A] and [B] are the concentrations of orthosteric agonist and allosteric modulator/agonist, respectively; KB is the dissociation constant of the allosteric modulator; EC50 is the concentration of orthosteric (full) agonist yielding 50% of the response between minimal and maximal receptor activation in the absence of allosteric ligand; n is a transducer slope factor linking occupancy to response; α and β are the cooperativity factors governing allosteric effects of the modulator on orthosteric agonist binding affinity and signaling efficacy, respectively; and τA and τB are operational measure of the ligands' respective signaling efficacies that incorporate receptor expression levels and efficiency of stimulus-response coupling (Leach et al., 2007; Aurelio et al., 2009). For this analysis, the entire family of curves for a given agonist-modulator combination across three different signaling pathways (ERK1/2 phosphorylation, Ca2+ mobilization, and inhibition of cAMP accumulation) were globally fitted to the model; Ca2+ data were fitted to eq. 1, whereas ERK1/2 and cAMP data were fitted to eq. 2, with the pKB parameter constrained to be shared across the entire family of curves.
All affinities, potencies, efficacies, and cooperativity parameters were estimated as logarithms (Christopoulos, 1998). Results are expressed as means ± S.E. unless otherwise stated. Statistical analyses were by Student's t test, or one-way analysis of variance followed by Bonferroni's post test, as appropriate. Values of p < 0.05 were considered statistically significant.
Results
Allosteric Effects of 2A3BTS on Both Orthosteric Agonist and Antagonist Binding Are Quantitatively Consistent with Interaction via a Common Extracellular Allosteric Site on the A1 Receptor.
Mechanistic studies of allosteric modulator effects on orthosteric radiolabeled agonist at A1 receptors have only been performed on broken cell preparations; this likely reflects the fact that 1) commercially available agonists of the A1 receptor display high affinity as radioligands only when interacting with the G protein-coupled state of the receptor, which often cannot be detected in whole cells, and 2) agonists can promote receptor internalization in intact cells, which would confound interpretation of binding results. We thus first investigated the effects of T62 and our two novel derivatives, VCP520 and VCP333, on the equilibrium binding of the radiolabeled agonist [3H]CCPA (2 nM) in FlpIn-CHO A1 receptor membranes (Fig. 2A). As expected, the orthosteric agonist R-PIA completely inhibited [3H]CCPA binding at the A1 receptor, characterized by a Hill coefficient not significantly different from 1 (nH = 0.98 ± 0.04) and preferentially fitted to a one-site competition binding model (pKI = 8.68 ± 0.01 for the high-affinity state; n = 3). In contrast, all three allosteric ligands caused variable degrees of enhancement of the binding of [3H]CCPA, ranging from approximately 10% above control specific binding with VCP333 to approximately 50% with VCP520. However, concentrations higher than 10 μM resulted in a reduction of specific radioligand binding, as expected based on prior studies of 2A3BTs. To quantify the degree of potentiation, therefore, the data points up to the effect of 10 μM of modulator were fitted to an allosteric ternary complex model (Christopoulos and Kenakin, 2002; May et al., 2007b) and the results are shown in Table 1, where it can be seen that all three modulators displayed similar affinities (pKB) for the allosteric site on the unoccupied A1 receptor, but different degrees of cooperativity (α) when the receptor is occupied by [3H]CCPA.

2A3BTs have divergent effects on orthosteric agonist binding. A, effects of the orthosteric agonist R-PIA or the allosteric modulators T62, VCP520, or VCP333 on the binding of [3H]CCPA at A1 receptors expressed in FlpIn-CHO membranes. Data points represent the mean ± S.E. obtained from three experiments conducted in duplicate. Curves drawn through the data points represent the best fit of a competitive (R-PIA) or allosteric ternary complex model (T62, VCP52, VCP333). B, effects of the allosteric modulators on the apparent dissociation rate of [3H]CCPA from A1 receptors expressed in FlpIn-CHO membranes. Data were normalized to the percentage of the control rate constant determined in absence of modulator, and represent the mean ± S.E. obtained from three experiments conducted in triplicate.
A1 receptor binding (pKB) and cooperativity (log α) estimates of the allosteric modulators for interaction with orthosteric radioligands
Data are expressed as the mean ± S.E. of three separate experiments performed in duplicate.
To further validate the results of our analysis, we investigated the effect of the allosteric modulators on the rate of [3H]CCPA dissociation from receptors that had been pre-equilibrated with the radioligand. In theory, the potency (EC50) of an allosteric modulator to affect the rate of dissociation of an orthosteric ligand should equal the ratio of the affinity constant of the modulator for the receptor (KB) divided by the cooperativity factor (α) for the interaction (Lazareno and Birdsall, 1995; Kostenis and Mohr, 1996), provided that the radioligand dissociation was monophasic both in the absence and presence of modulator; the latter property was confirmed in control experiments in which we tested the effects of a single concentration (3 μM) of each modulator for effects on the control [3H]CCPA dissociation rate (koff = 0.042 ± 006; n = 3) at the A1 receptor in a full time course assay (data not shown). We thus constructed complete concentration-response curves for the inhibition of [3H]CCPA dissociation at the A1 receptor with all three ligands (3 nM–10 μM) using a “two-point dissociation kinetic paradigm (Fig. 2B). The estimated potencies (pEC50) for this effect were determined as: T62, 5.94 ± 0.10 (n = 3); VCP520, 6.92 ± 0.11 (n = 3); and VCP333, 5.61 ± 0.18 (n = 3). These values showed excellent concordance with theoretical predictions based on the sum of the pKB and log α estimates from Table 1 (6.13, 6.73, and 5.68, respectively). It is noteworthy, however, that the maximal effect on agonist dissociation observed at the highest concentrations of allosteric modulator trended toward a plateau greater than 0%, which was most evident for VCP520.
We next studied the effects of R-PIA and the three allosteric modulators on the equilibrium binding of two different concentrations of the orthosteric antagonist [3H] DPCPX [1 nM (approximately 1 × KA) and 5 nM (approximately 5 × KA) (Fig. 3A]. In both instances, R-PIA completely inhibited the specific binding of the radioligand. For the experiments performed against 1 and 5 nM [3H] DPCPX, the R-PIA Hill coefficients were 0.99 ± 0.05 and 0.91 ± 0.04, respectively, and the estimated pKI values were 6.33 ± 0.04 and 6.73 ± 0.02, respectively (n = 3). The pKI values were significantly different (p < 0.05) from that determined against [3H]CCPA, suggesting that the interaction between R-PIA and [3H]DPCPX (Fig. 3A) was preferentially reflecting binding to the low-affinity (G protein-uncoupled) state of the A1 receptor. Each of the allosteric modulators also inhibited the binding of [3H]DPCPX, in contrast to their effects on agonist binding. Although the inhibition of 1 nM [3H]DPCPX binding mediated by VCP520 and VCP333 was incomplete, consistent with an allosteric interaction characterized by limited negative cooperativity, the inhibition mediated by T62 appeared complete and may be reconciled with a competitive mechanism. However, when these experiments were repeated against a higher concentration of [3H]DPCPX (5 nM), the allosteric nature of the interaction was readily evident in the behavior of all three ligands, because the inhibition of radioligand binding in each modulator approached a limit over which no further inhibition of specific binding was attainable. Application of an allosteric ternary complex model to the data yielded the parameters shown in Table 1. Of particular importance was the fact that the pKB estimates for each of the modulators were not significantly different (p > 0.05) when determined using [3H]DPCPX as the orthosteric ligand relative to when [3H]CCPA was used as the orthosteric ligand. This indicates that, in each instance, the modulators recognize a common allosteric site on the unoccupied receptor with similar affinity irrespective of whether the interaction is probed using an agonist versus an antagonist orthosteric ligand. However, the nature and magnitude of the cooperativity between the two types of ligands is drastically altered depending on the orthosteric probe (agonist versus antagonist). This phenomenon is a hallmark of GPCR allosteric modulation, and is referred to as “probe dependence” (Leach et al., 2007; Kenakin, 2009).

2A3BTs are negative allosteric modulators of orthosteric antagonist binding. Effects of the orthosteric agonist R-PIA or the allosteric modulators T62, VCP520, or VCP333 on the binding of [3H]DPCPX at A1 receptors expressed in FlpIn-CHO membranes (A) or intact cells (B). Data points represent the mean ± S.E. obtained from three experiments conducted in duplicate. Curves drawn through the data points represent the best fit of a competitive (R-PIA) or allosteric ternary complex model (T63, VCP52, VCP333).
To confirm that the interaction between [3H]DPCPX (1 nM) and the modulators was mediated by an extracellular allosteric site, the experiments were repeated using intact FlpIn-CHO A1 receptor cells; otherwise, assay conditions were identical to those used in the membrane-based assays with respect to buffer, temperature, and incubation time (Fig. 3B). As with the experiments performed in membranes, R-PIA caused a complete inhibition of radioligand binding (pKI = 6.69 ± 0.03; n = 3). In contrast, all three allosteric modulators caused a saturable but incomplete inhibition of [3H]DPCPX-specific binding, unambiguously indicating an allosteric mode of action. Moreover, the pKB and values derived from the whole-cell binding using the ATCM were not significantly different from the values derived from the membrane-based studies (Table 1). A slight discrepancy was noted in the estimates of negative cooperativity for VCP520 and VCP333 in the membrane versus intact cell experiments, but this reached statistical significance only for the case of VCP333, and even then, the overall cooperativity estimates were in the same range (i.e., approximately α = 0.1 versus 0.3). Overall, the results of our binding assays indicate, for the first time, that the potentiation component of orthosteric agonist binding isotherms determined in membranes, as well as the inhibition of antagonist affinity in both membranes and intact cells by 2AB3Ts, can be quantitatively accommodated by interaction with a common extracellular allosteric site on the A1 receptor.
2A3BTs Display Functional Selectivity As Allosteric Agonists of A1 Receptor Function.
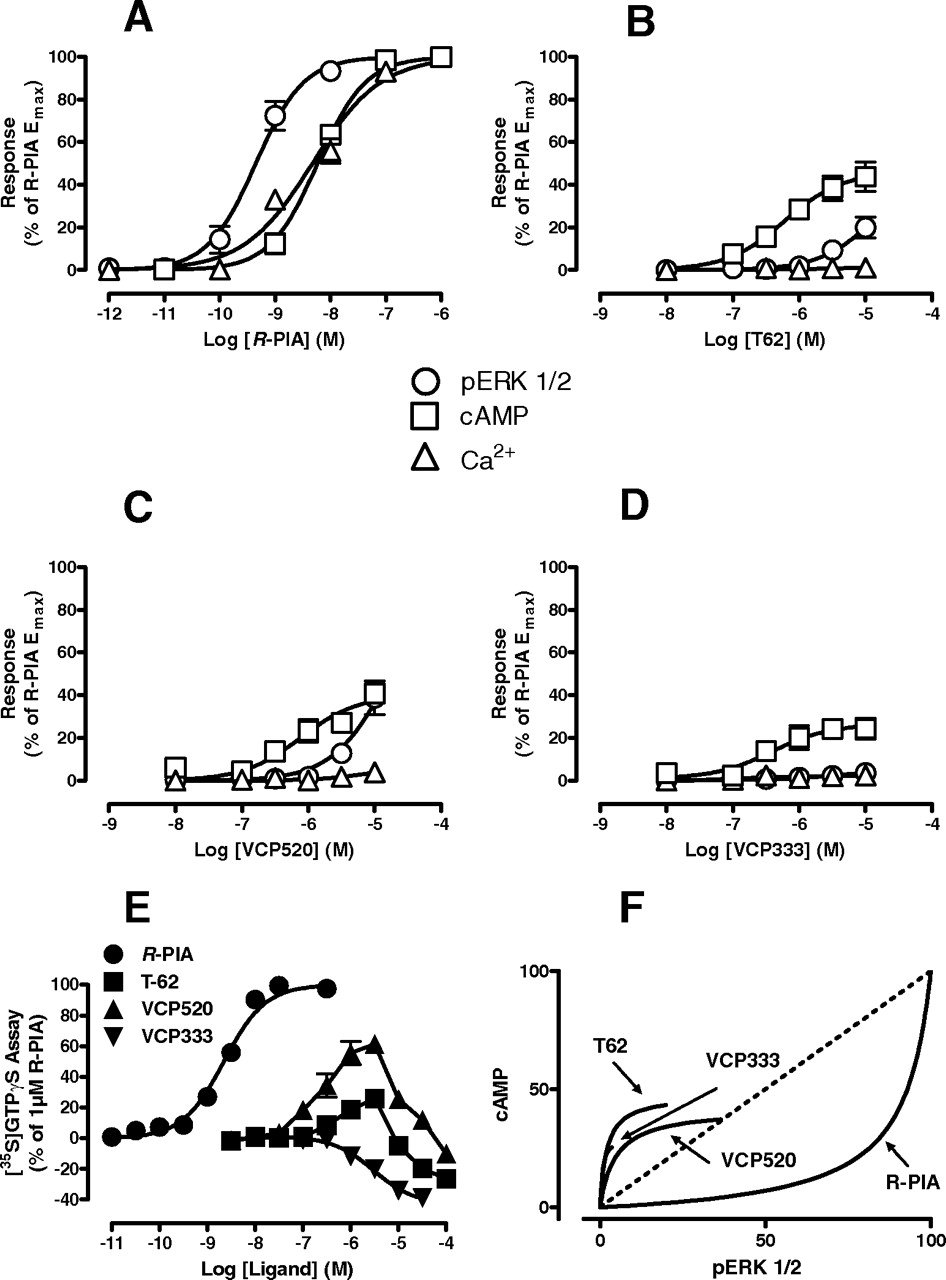
Given that the 2A3BTs selectively potentiated agonist binding to the high-affinity, presumably G protein-coupled, state of the A1 receptor, we investigated whether the compounds on their own can promote receptor activation, and whether the nature of this activation differed relative to the prototypical orthosteric agonist R-PIA. For this purpose, four different assays of A1 receptor activation were used: ERK1/2 phosphorylation, inhibition of forskolin-stimulate cAMP accumulation, mobilization of intracellular Ca2+, and promotion of [35S]GTPγS binding to activated Gαi/o proteins. The results of these studies, in which a number of interesting observations were made, are summarized in Fig. 4. First, except for intracellular Ca2+ mobilization, all allosteric ligands were able to mediate changes in receptor activity in their own right. Second, compared with R-PIA, all three allosteric ligands displayed divergent concentration-response relationships for promoting [35S]GTPγS binding (Fig. 4E), with T62 and VCP520 characterized by both stimulatory and inhibitory effects, whereas VCP333 only showed inhibitory effects on basal G protein activation. Third, when considering the stimulatory components of signaling of the four ligands across the various pathways, it was noted that the rank order of potencies for phosphorylation of ERK1/2 relative to inhibition of cAMP and promotion of [35S]GTPγS binding were reversed for VCP520 and T62 relative to the orthosteric agonist R-PIA; this is more evident in Fig. 4F, where the percentage response for equivalent concentrations of each ligand at the cAMP and ERK1/2 assays were graphed against each other in the form of a “bias plot” (Gregory et al., 2010). This finding is a hallmark of functional selectivity and suggests that the allosteric ligands bias the receptor stimulus in a manner that is different to the orthosteric agonist.

2A3BTs display functional selectivity as allosteric agonists of the A1 receptor. A–D, effects of the indicated agonist on A1-mediated ERK1/2 phosphorylation, inhibition of forskolin-stimulated cAMP accumulation and intracellular calcium mobilization in intact FlpIn-CHO cells. E, effects of the indicated agonist on A1-mediated [35S]GTPγS binding to activated G proteins in FlpIn-CHO membranes. In all instances, data points represent the mean ± S.E. obtained from three to four experiments conducted in duplicate. E, “Bias plot” showing the nonlinear curve fits from panels A to D for cAMP and ERK1/2 responses plotted against each other.
2A3BTs Are Positive Allosteric Modulators of R-PIA in Whole-Cell Functional Assays.
The functional assays suggested differences in the manifestation of compound effects depending on whether receptor function was probed in whole cells versus membranes. Thus, we next focused on the ability of the 2A3BTs to modulate the function of an orthosteric agonist at the A1 receptor in intact FlpIn-CHO cells by performing interaction studies between each modulator and R-PIA in the ERK1/2 phosphorylation, inhibition of cAMP accumulation, and intracellular Ca2+ mobilization assays (Fig. 5). In all instances, each allosteric ligand acted as a potentiator of R-PIA function, although the extent and manifestation of the positive modulation varied with the pathway. For ERK1/2 phosphorylation and cAMP inhibition, where R-PIA was a full agonist, the allosteric effect of the 2A3BTs was manifested as an increase in the potency of R-PIA (Fig. 5, A–F). For intracellular Ca2+ mobilization, the modulation was manifested primarily as an increase in the maximal R-PIA response (Fig. 5, G–I); this finding suggests that R-PIA was not behaving as a full agonist in this assay, and is consistent with the notion that the coupling of the A1 receptor to intracellular Ca2+ mobilization is less efficient than its coupling to the other pathways. By applying an operational model of agonism (eqs. 1 and 2) to the entire set of curves (Fig. 5, A–I), we were able to quantify the allosteric interaction for each modulator at each of the pathways. The results of this analysis are summarized in Table 2, where it can be seen that the functional estimates of modulator affinity were in general agreement with those obtained from the binding assays performed on whole cells (versus [3H]DPCPX, Table 1). It is noteworthy that the magnitude of positive cooperativity, quantified by the composite αβ parameter in our operational model appeared to vary between some pathways. In particular, the potentiation of R-PIA function by VCP520 in the Ca2+ mobilization was significantly greater (one-way analysis of variance, p < 0.05) than that noted for ERK1/2 phosphorylation. This suggests that the allosteric modulators may also engender functional selectivity in the actions of orthosteric ligands acting at the A1 receptor.

Different degrees of positive allosteric modulation of R-PIA function by 2A3BTs in intact cells. Effects of T62 (A, D, G), VCP520 (B, E,H), or VCP333 (C, F, I) on R-PIA-mediated ERK1/2 phosphorylation (A–C), cAMP inhibition (D–F), or intracellular calcium mobilization (G–I). Data points represent the mean ± S.E. obtained from three to five experiments conducted in duplicate. Curves drawn through the data represent the best global fit of an operational model of allosterism to all nine datasets (Table 2).
Allosteric model parameters for the interaction between R-PIA and various allosteric modulators in cell-based assays
pKB is the antilogarithm of the dissociation constant of the allosteric modulator. Log αβ is the logarithm of the composite cooperativity between the allosteric modulator and R-PIA. Log τ is the logarithm of the operational efficacy of the allosteric modulator.
2A3BTs Display Divergent Effects on Allosteric Modulation of G Protein Activation in Broken Cell Preparations.
Functional interaction studies between R-PIA and each of the 2A3BTs were then extended to the [35S]GTPγS binding assay in membranes of A1 receptor FlpIn-CHO cells (Fig. 6). For these experiments, only concentrations of modulator up to 3 μM were used, where it can be seen that both T62 and VCP520 caused an increase in the basal response and the potency of R-PIA (Fig. 6, A and B). Application of our operational model of allosterism (eq. 2) to the data yielded pKB = 6.02 ± 0.09 and Logαβ = 0.46 ± 0.06 (n = 3) for T62, and pKB = 5.96 ± 0.11 and Logαβ = 1.19 ± 0.14 (n = 3) for VCP520. However, VCP333 caused a concentration-dependent decrease in the maximal response to the orthosteric agonist, with no significant effect on agonist potency (R-PIA pEC50 = 8.21 ± 0.06 in the absence of modulator and 8.31 ± 0.19 in the presence of 3 μM VCP333; Fig. 6C). Together with the findings obtained when these compounds were tested alone over a higher concentration range (Fig. 4E), our results suggest that the 2A3BTs actually bind to two different sites, presumably two sites with different structure-activity requirements. Furthermore, given the results of our studies on whole cells, it is likely that the second site of action is not the orthosteric site but rather an intracellular binding site.
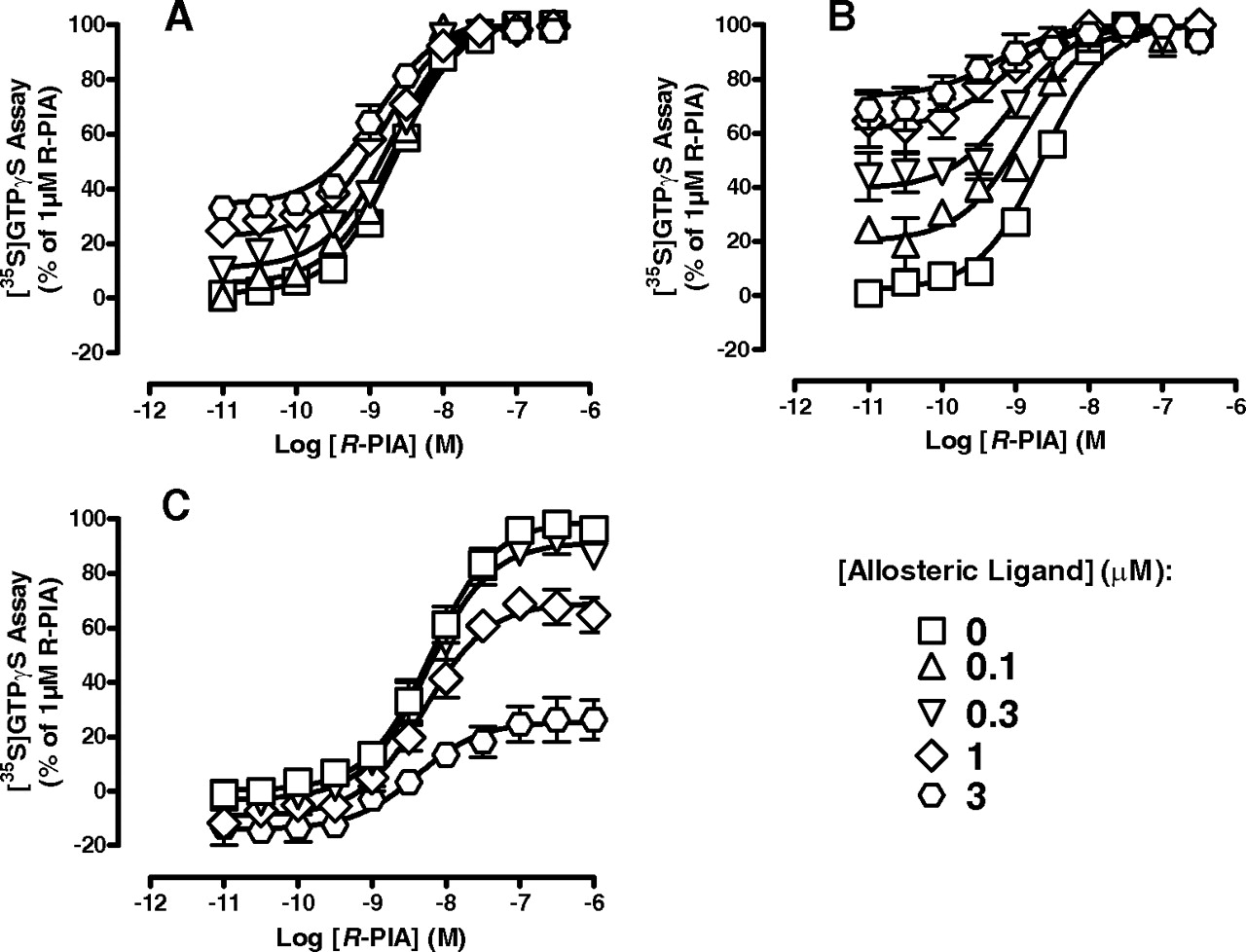
2A3BTs have divergent effects on orthosteric agonist function in a broken cell preparation. Effects of T62 (A), VCP520 (B), and VCP333 (C) on R-PIA-mediated [35S]GTPγS binding to activated G proteins in membranes prepared from FlpIn-CHO cells stably expressing the A1 receptor. Data points represent the mean ± S.E. obtained from three experiments conducted in duplicate and are normalized to 1 μM R-PIA response. Curves drawn through the data in A and B represent the best fit to an operational model of allosterism.
2A3BTs Are Direct G Protein Inhibitors.
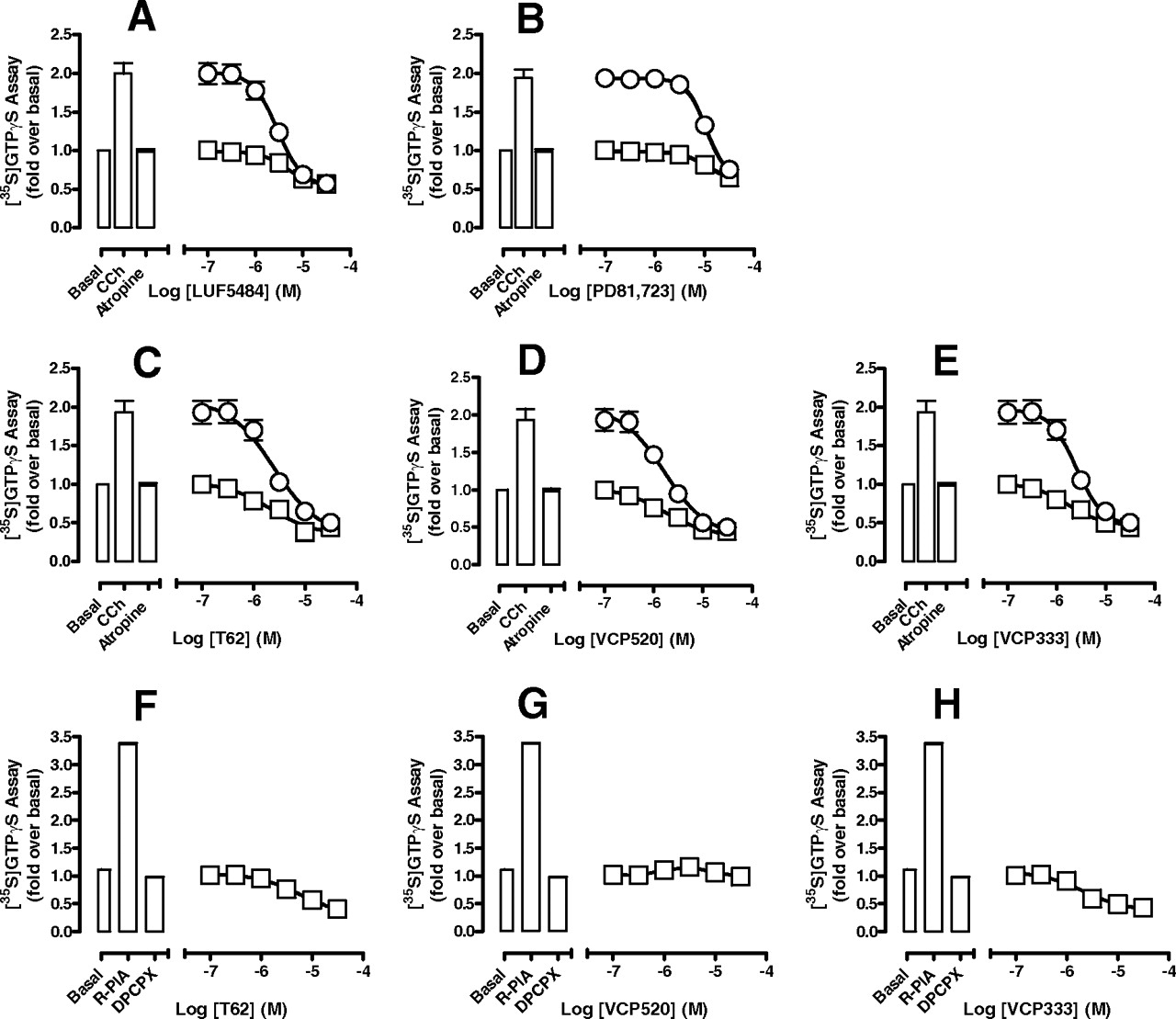
To determine whether the G protein inhibitory effect of the 2A3BTs is specific to the A1 receptor or whether it occurs directly at the level of the G protein, we investigated their ability to inhibit [35S]GTPγS binding promoted by another Gi/o-coupled receptor, the M2 mAChR. Furthermore, we extended our studies to include the effects of two other well characterized 2A3BT allosteric modulators of the A1 receptor, LUF5484 and PD81,723 (Fig. 1), to determine whether this property is common to 2A3BTs as a class. As shown in Fig. 7, A to E, all five allosteric ligands inhibited carbachol (100 μM)-stimulated [35S]GTPγS binding in a concentration-dependent manner and with similar potencies (Table 3). It is noteworthy that the 2A3BTs also reduced [35S]GTPγS binding in the presence of a saturating concentration of the inverse agonist atropine (10 μM), suggesting that their effect is directly on the G protein, rather than the receptor. We also repeated these experiments using T62, VCP520, or VCP333 at the A1 receptor in the presence of a saturating concentration (10 μM) of the antagonist DPCPX. As shown in Fig. 7, F to H, a modest inhibition of activity was noted for T62 and VCP333, but not for VCP520; this finding reflects the fact that, at this receptor, the ligands can also display, via the allosteric site, variable degrees of direct receptor activation, which is most pronounced with VCP520 (Fig. 4E); thus, the observed response profile in the presence of DPCPX will reflect a composite of both stimulatory and inhibitory actions on G protein activity. It is noteworthy that we also found a reduction in basal [35S]GTPγS binding to the 2A3BTs observed in nontransfected FlpIn-CHO cells (Supplemental Fig. 1), confirming that this inhibitory effect is receptor-independent. Although the mechanism underlying the direct inhibitory effect of the 2A3BTs on G protein activity is unknown, we considered whether they promote receptor-G protein uncoupling in a manner akin to that of guanine nucleotides; this could account for their inhibitory effects on A1 receptor agonist binding. To further test this, we monitored the effects of the 2A3BTs on the ability of ACh to compete with the antagonist [3H]NMS at the M2 mAChR in FlpIn-CHO cell membranes. To our surprise, we found that the 2A3BTs had either no effect (VCP333, VCP520) or a modest enhancing effect (T62) on the ability of ACh to compete for [3H]NMS binding; an inhibition of [3H]NMS binding was also noted for VCP333 and T62 (Supplemental Fig. 2). In contrast, the addition of Gpp(NH)p (100 μM) yielded the anticipated result of a reduction in the affinity of ACh (Supplemental Fig. 2). Thus, the results of these experiments confirm that 2A3BTs, as a class, possess at least two modes of action, one via an extracellular allosteric site on the A1 receptor and the other via an intracellular (nonreceptor) site on, or associated with, Gi/o family proteins that is not of the same mechanism as that exhibited by guanine nucleotides.

2A3BTs are inhibitors of Gi/o proteins. Effect of LUF5484 (A), PD81,723 (B), T62 (C), VCP520 (D), or VCP333 (E) on M2 mAChR-mediated [35S]GTPγS binding to activated G proteins in the presence or absence of the orthosteric agonists carbachol 100 μM (○) or atropine (□)10 μM in FlpIn-CHO cells. Data points represent the mean ± S.E. obtained from four experiments conducted in duplicate and normalized to the fold over basal. F–G, effect of the indicated modulator on A1 receptor-mediated [35S]GTPγS binding to activated G proteins in the presence of the orthosteric antagonist DPCPX. Data points represent the mean ± S.E. obtained from three experiments conducted in duplicate and normalized to the fold over basal.
Potency (pEC50) estimates for inhibition of G protein activity by 2A3BTs in [35S]GTPγS binding assay at the M2 mAChR (n = 4) and A1 receptor (n = 3)
2A3BTs Retain Allosteric Enhancing Activity in a Native Tissue Preparation.
Given our findings, an important consideration in the pursuit of 2A3BTs as allosteric modulators of A1 receptor function in a therapeutic setting is the extent to which the secondary, inhibitory, effects of such compounds can potentially compromise their utility as enhancers of adenosine activity in native tissues. To assess this in a physiologically relevant setting, we determined the effects of T62, VCP520, and VCP333 on R-PIA-mediated decrease in heart rate. Figure 8 shows that each allosteric ligand was able to promote a concentration-dependent increase in the effect of an EC10 concentration of agonist. Unfortunately, solubility limitations precluded our ability to test higher concentrations of modulators in the organ bath.

2A3BTs retain allosteric enhancement properties in intact native tissues. Effect of VCP333 (open bars), VCP520 (checked bars), and T62 (filled bars) on R-PIA (10 nM)-mediated decrease in rate of contraction of rat right atrium. Data bars represent the mean ± S.E. obtained from four experiments conducted in duplicate and normalized to the maximum R-PIA response.
Discussion
After nearly 2 decades of research, the prevailing view on the mechanism of action of 2A3BTs at the adenosine A1 receptor invokes interaction with both an allosteric and the orthosteric site. In turn, this has led to many structure-activity studies aimed at removing the apparently competitive properties of 2A3BTs while retaining allosteric properties (see Introduction). Our study now shows that this mechanism is unlikely to be correct and that the pharmacology of 2A3BTs arises from binding to an extracellular allosteric site, which can quantitatively accommodate different interactions with agonists versus antagonists, and, depending on accessibility, another interaction with an intracellular site(s) specifically involved in G protein activity. In addition, we have also shown that 2A3BTs can display functional selectivity, mediated via this allosteric site, in their ability to recruit signaling pathways relative to a prototypical orthosteric agonist.
The detection of allosteric effects at GPCRs requires careful consideration of the signal-to-noise window because of the phenomenon of cooperativity between orthosteric and allosteric sites (May et al., 2007b). On the one hand, weak positive or negative cooperativity can lead to a failure to detect an allosteric interaction. On the other hand, strong negative cooperativity can be difficult to distinguish from competition. As we have now demonstrated, the interaction between 2A3BTs and the orthosteric antagonist [3H]DPCPX can be quantitatively accommodated by a simple ATCM characterized by negative cooperativity between the orthosteric probe and each of the allosteric ligands; for T62 at least, however, the negative cooperativity is high enough, when determined in membrane-based assays using an approximately KA concentration of orthosteric radioligand, as to appear consistent with a competitive effect. Because many published studies of A1 receptor radioligand binding have been performed in membrane preparations, it is perhaps not surprising that negative allosteric interactions have been misclassified as competitive. To our knowledge, only one prior study (Figler et al., 2003) has suggested that the interaction between 2A3BTs and an antagonist ([3H]DCPX) may be allosteric. Application of the ATCM in our study has allowed for the determination of the affinity of each of the modulators for the allosteric site on the A1 receptor, as well as the cooperativity factors governing the interaction with [3H]DPCPX (Table 1). Importantly, the affinity values were not significantly different from those obtained when performing the same interaction in whole cells or when using [3H]CCPA as an agonist probe of the interaction in membranes. These findings suggest that the ATCM is an appropriate mechanistic descriptor of the interaction because, according to the model, the pKB parameter is a measure of the affinity of the modulator for the unoccupied receptor and is thus independent of the nature of the orthosteric probe (Leach et al., 2007). Our findings using the intact cell binding assay also suggest that the 2A3BT allosteric site is located extracellularly.
In contrast, differences were noted in the cooperativity of the interaction depending on the orthosteric ligand that was used. This phenomenon, termed “probe dependence,” is a hallmark of allosterism at GPCRs (Leach et al., 2007; May et al., 2007b; Kenakin, 2009), and indicates that allosteric ligands either sense and/or promote different receptor conformations depending on the orthosteric partner. In our case, the 2A3BTs prefer to bind to the allosteric site of an agonist-occupied receptor. At higher concentrations of modulator, in membrane-based assays, the secondary G protein inhibitory effect that we identified results in a loss of agonist binding, which we observed in the [3H]CCPA experiments (Fig. 2A). This dual mechanism can thus account for the complex behavior of 2A3BTs noted in the past. It is noteworthy that this same mechanism may also account for the inability of high concentrations of 2A3BTs (especially VCP520) to completely prevent [3H]CCPA dissociation (Fig. 2B); the slowing of dissociation promoted by action at an extracellular allosteric site on the A1 receptor may be offset by an increase in dissociation promoted by intracellular actions of the 2A3BT, yielding a net effect of submaximal inhibition of radioligand dissociation.
A consideration of the effects of 2A3BTs arising solely from interaction with an extracellular allosteric site on the A1 receptor leads to a number of interesting conclusions. The first is that these compounds can behave as agonists of the receptor in their own right, as evidenced by whole-cell assays of ERK1/2 phosphorylation and cAMP accumulation; the lack of effect on calcium mobilization most likely reflects the poor coupling of this latter pathway to A1 receptor activation. The finding of direct allosteric agonism with 2A3BTs has been noted previously (Bruns and Fergus, 1990; Bruns et al., 1990; Bhattacharya and Linden, 1995; Musser et al., 1999; Baraldi et al., 2000; Figler et al., 2003; Aurelio et al., 2009), suggesting that their mechanism of allosteric potentiation is likely to involve, at least in part, an increase in the proportion of receptors in an active state and, hence, an increase in receptor-G protein coupling (Bruns and Fergus, 1990; Bhattacharya and Linden, 1995; Hall, 2000). However, a key novel finding in our current study was the reversal in potency orders of the 2A3BTs and R-PIA for signaling to ERK1/2 relative to cAMP inhibition (Fig. 4), which suggests that the active conformation promoted by the allosteric ligands is not the same as that promoted by orthosteric agonists like R-PIA. This finding is a striking example of functional selectivity (Urban et al., 2007) and has significant implications for pathway-selective drug discovery. Importantly, our finding is also the first example of agonist functional selectivity arising solely from interaction with an allosteric site. It is likely that more such examples will be identified in the near future given the current focus on novel small-molecule agonists in GPCR drug discovery programs.
A second important observation on the allosteric properties of the 2A3BTs relates to their ability to act as allosteric potentiators of orthosteric agonist function. Application of our operational model of allosterism and agonism revealed that, in whole cells, the magnitude of the positive modulation of R-PIA function could vary between pathways, especially for VCP520 (Table 2). Based on the conformational hypothesis of 2A3BT action discussed in the preceding paragraph, it may be expected that the degree of positive allosteric enhancement should correlate with the degree of allosteric agonism displayed by the modulators, but this does not seem to be the case. Indeed, VCP520 promoted greater allosteric potentiation at the pathway for which it displayed no signaling efficacy. Therefore, this is an example of allosteric modulator-engendered functional selectivity in the actions of an orthosteric ligand and highlights an additional means by which allosteric ligands can be used to further “fine-tune” orthosteric ligand responses (Leach et al., 2007).
With regard to the intracellular component of 2A3BT action (i.e., the ability to directly inhibit G protein function), it is noteworthy that the potency of the compounds to mediate this effect is similar to their affinity for the allosteric site (in the absence of orthosteric ligand). This has important implications in terms of structure-activity studies. First, our findings in intact CHO cells suggest that the compounds are unlikely to gain appreciable intracellular penetrance. However, this may not always be guaranteed, and thus the ideal aim of future structure-activity work would be to obtain as large a separation as possible between affinity for the allosteric site relative to G protein inhibitory activity. Second, depending on the nature of the assay, the observed response will reflect an interplay between the two properties (allosteric modulation and G protein inhibition); for a compound like VCP333, which is the weakest allosteric potentiator in our series based on the intact cell functional data (Fig. 5), the resultant pharmacology would be in favor of inhibition, which explains why the compound appeared to inhibit [35S]GTPγS turnover at all concentrations tested. Third, we cannot conclude from our data whether the mechanism of G protein inhibition occurs directly at the level of the G protein itself, as opposed to another membrane component; certainly, the discrepant findings between effects on A1 receptor agonist binding (Fig. 2) and M2 mAChR agonist binding (Supplemental Fig. 2) suggest that 2A3BTs do not act as simple receptor-G protein “uncouplers,” unlike guanine nucleotides such as Gpp(NH)p. However, it is nonetheless possible that 2A3BTs may prove to be novel scaffolds for the development of small-molecule G protein inhibitors.
In conclusion, this study has identified the mechanisms underlying the complex mode of action of 2A3BTs as allosteric modulators of A1 receptors and nonspecific G protein inhibitors. Moreover, we have demonstrated how the A1 receptor-specific effects of the compounds against both agonist and antagonist orthosteric ligands can be quantitatively accommodated by a simple ternary complex model. Finally, we have identified novel functional selectivity in the actions of 2A3BTs, both as allosteric agonists and allosteric modulators.
Acknowledgments
We are grateful to Drs. Michael Crouch and Ron Osmond (TGR Biosciences, Thebarton, SA, Australia) for the generous gift of ERK1/2 SureFire kit reagents.
Footnotes
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.This work was funded by the National Health and Medical Research Council (NHMRC) of Australia [Program Grant 519461]; the Australian Research Council [Discovery Grant DP0877497]; and by NHMRC Senior (to A.C.) and Principal (to P.M.S.) Research Fellowships.
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.110.064568.
-
ABBREVIATIONS:
- GPCR
- G protein-coupled receptor
- 2A3BT
- 2-amino-3-benzoylthiophene
- T62
- 2-amino-4,5-dimethyl-3-thienyl-[3-(trifluoromethyl)phenyl]methanone
- PD81,723
- 2-amino-4,5-dimethyl-3-thienyl-[3-(trifluoromethyl)phenyl]methanone
- LUF5484
- 2-amino-(4,5,6,7-tetrahydrobenzo[b]thiophen-3-yl)(3,4-dichlorophenyl)methanone
- VCP520
- 2-amino-4-(3,5-bis(trifluoromethyl)phenyl)thiophen-3-yl)(phenyl)methanon
- VCP333
- tert-butyl 2-amino-3-(4-chlorobenzoyl)-7,8-dihydro-4H-thieno[2,3-d]azepine-6(5H)-carboxylate
- FBS
- fetal bovine serum
- CCPA
- 2-chloro-N6-cyclopentyladenosine
- DPCPX
- 8-cyclopentyl-1,3-dipropylxanthine
- [35S]GTPγS
- guanosine 5′-O-[γ-[35S]thio]triphosphate
- ERK
- extracellular signal-regulated kinase
- CHO
- Chinese hamster ovary
- FBS
- fetal bovine serum
- Gpp(NH)p
- guanosine 5′-(β,γ-imido)triphosphate
- ATCM
- allosteric ternary complex mode
- R-PIA
- (−)-N6-(2-phenylisopropyl)adenosine.

- Received March 8, 2010.
- Accepted June 14, 2010.
- Copyright © 2010 The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}