


Abstract
The VPAC1 receptor belongs to family B of G protein-coupled receptors (GPCR-B) and is activated upon binding of the vasoactive intestinal peptide (VIP). Despite the recent determination of the structure of the N terminus of several members of this receptor family, little is known about the structure of the transmembrane (TM) region and about the molecular mechanisms leading to activation. In the present study, we designed a new structural model of the TM domain and combined it with experimental mutagenesis experiments to investigate the interaction network that governs ligand binding and receptor activation. Our results suggest that this network involves the cluster of residues Arg188 in TM2, Gln380 in TM7, and Asn229 in TM3. This cluster is expected to be altered upon VIP binding, because Arg188 has been shown previously to interact with Asp3 of VIP. Several point mutations at positions 188, 229, and 380 were experimentally characterized and were shown to severely affect VIP binding and/or VIP-mediated cAMP production. Double mutants built from reciprocal residue exchanges exhibit strong cooperative or anticooperative effects, thereby indicating the spatial proximity of residues Arg188, Gln380, and Asn229. Because these residues are highly conserved in the GPCR-B family, they can moreover be expected to have a general role in mediating function.
Introduction
The human VPAC1 receptor is expressed in liver, breast, kidney, prostate, bladder, pancreatic ducts, thyroid gland, lymphoid tissues, and gastrointestinal mucosa and in most of the tumors derived from these tissues. The VPAC1 receptor is a member of family B of G protein-coupled receptors (GPCRs), which have seven transmembrane (TM) helices. This family includes the VPAC2, secretin, PAC1, glucagon, glucagon like-peptide 1 and 2, calcitonin, corticotropin-releasing factor, and parathyroid hormone (PTH) receptors. The physiological ligands of the VPAC1 receptor are vasoactive intestinal peptide (VIP) and pituitary adenylate cyclase activating peptide (Dickson and Finlayson, 2009).
Extensive studies of the largest family of GPCRs, the GPCR-A/rhodopsin family, led to the identification of key steps frequently involved in the early signaling events of this family. These include the disruption of an ionic interaction between the cytoplasmic face of TM3 and TM6 maintaining the receptor preferentially in a ground inactive conformation in absence of agonist (ionic lock) and a “rotamer toggle switch” (modulation of the helix conformation around a proline-kink) in TM6, causing key sequences to be exposed to cytoplasmic binding partners (Ballesteros et al., 2001; Schwartz et al., 2006).
The mechanisms regulating the GPCR-B family signal transduction are less precisely understood, because no X-ray structure of the whole receptor is available, and conserved motifs of the GPCR-A family (E/DRY at TM3, NPXXY at TM7) are absent in the GPCR-B family. Although recent studies have solved the structure of the N terminus of several family B receptors (corticotropin-releasing factor, PTH, phosphatase of activated cells 1, gastric inhibitory polypeptide, glucagon-like peptide-1) and clarified their role in ligand binding (Grace et al., 2007; Parthier et al., 2007; Sun et al., 2007; Pioszak and Xu, 2008; Runge et al., 2008), there is little information on the events that follow ligand binding. Considering the VPAC1 receptor as a paradigm for class B, it actually seems that a large network of interactions must be considered. Indeed, on the basis of mutagenesis studies, it has been proposed that TM1, TM2, TM3, and TM6 and the intracellular loop 3 and the proximal part of the C-terminal intracytoplasmic tail take part in the receptor signal transduction (Gaudin et al., 1998, 1999; Couvineau et al., 2003; Langer and Robberecht, 2007).
In the present study, a network of interactions that stabilize the VPAC1 receptor conformation in the absence of ligand is identified by combining modeling and mutagenesis studies and is proposed to be involved in receptor activation. This network includes an arginine (Arg188) located in TM2, demonstrated previously by complementary-paired mutagenesis to interact with the Asp3 residue of VIP (Solano et al., 2001), an asparagine (Asn229) located in TM3, important for VPAC1 and VPAC2-mediated G protein activation (Nachtergael et al., 2006), and a glutamine (Gln380) conserved among the GPCR-B family members and located in TM7. To our knowledge, this is the first identification of early steps that lead to the receptor activation of a GPCR-B family member upon ligand binding.
Materials and Methods
Comparative Modeling Procedure.
Comparative modeling was carried out by Modeler 9V3 (Martí-Renom et al., 2000) on the basis of alignments between the target and template sequences obtained as described under Results. The modeling was constrained to create an obligate disulfide bond between the residues Cys215 at the extracellular end of TM3 and Cys285 in the extracellular loop 2; this disulfide bridge is indeed known to occur in GPCR-B members. All models were stepwise energy-relaxed in the following way: 1) with all heavy atoms fixed; 2) with backbone atoms fixed; and 3) with Cα atoms fixed. Gromacs 3.3.1 was used for energy calculations (Lindahl et al., 2001).
Quality Assessment of the Structural Models.
To evaluate the quality of the structural models generated from different sequence alignments, we used the membrane score approach (Chugunov et al., 2007a,b), which was developed for the assessment of the packing quality of α-helical TM domains of membrane proteins. In this method, a database-derived scoring function (Smem) is used to quantitatively estimate the fitness of a given amino acid residue for its three-dimensional class of protein-membrane environment. This scoring function was derived from the analysis of a nonredundant set of α-helical membrane protein structures (Chugunov et al., 2007a). The larger Smem the model has, the better it is packed in space. Generally, models with Smem < 0 should be considered to be misfolded. This method has been proven to be useful in discriminating close-to-native structures from large decoy sets built from misleading alignments (Chugunov et al., 2007b). A second quality assessment, performed on the best structural models identified by the membrane score approach, consisted of a detailed analysis of the variability moment vectors. In a first step, all protein sequences homologous to the target are aligned, and the amino acid variability at each position is computed. In a second step, a vector is assigned to each residue in each TM helix of the 3D model of the target. The vector is put in a plane parallel to the surface of the membrane, points out of the helix, and its amplitude is proportional to the variability of the residue among the members of the target family. The resulting vector is computed for each helix and shows thus the most variable side of the helix, which should be exposed to the membrane, because evolution is known to better conserve amino acids that point toward the protein core and are likely to be involved in important interactions. The models that fulfill the previous quality assessments were submitted to a 1-ns molecular dynamics (MD) simulation in vacuum at 500 K, with fixed Cα atoms solely for exploration of side-chain motility (not for model optimization). GROMACS 3.3.1 (Lindahl et al., 2001) was used for that purpose. More sophisticated MD calculations would require an explicitly defined medium (membrane) and advanced setup, but for our purposes, the sampling of the side chain conformations with a fixed backbone is sufficient. During each MD simulation, 1000 frames were memorized, and the Smem values were computed for each of them to cumulate score distributions.
Construction and Expression of VPAC1 Mutant Receptors.
The cell lines expressing wild-type (wt) VPAC1, as well as R188A, R188Q, N229A, and N229Q mutant receptors, have been detailed in previous publications (Solano et al., 2001; Nachtergael et al., 2006). The generation of the other mutated receptors was achieved using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla CA) according to the manufacturer's instructions. Confirmation of the expected mutation was achieved by DNA sequencing on an ABI automated sequencing apparatus using the BigDye Terminator Sequencing Prism Kit from Applied Biosystems (Foster City, CA). After DNA amplification using a Midiprep endotoxin-free kit (Promega, Madison, WI), the complete nucleotide sequence of the receptor coding region was verified by DNA sequencing. DNA (20 μg) was transfected by electroporation in a Chinese hamster ovary cell line expressing aequorin (kindly provided by Vincent Dupriez, Euroscreen SA, Brussels, Belgium) as described by Nachtergael et al., (2006). Selection was carried out in the culture medium (50% Ham's F-12, 50% Dulbecco's modified Eagle's medium, 10% fetal calf serum; 1% 10 mU/ml penicillin, 1% 10 μg/ml streptomycin, 1% 200 mM l-glutamine; PAA Laboratories, Pashing, Austria), supplemented with 600 μg of G418 per milliliter of culture medium. After 10 to 15 days of selection, isolated colonies were transferred to 24-well plates and grown until confluence, trypsinized, and further expanded in 6-well plates, from which cells were scraped and membranes prepared for identification of receptor expressing clones by an adenylate cyclase activity assay in the presence of 1 μM VIP and by binding assay with 125I-VIP (see below).
Membrane Preparation.
Membranes were prepared from scraped cells lysed in 1 mM NaHCO3 followed by immediate freezing in liquid nitrogen. After thawing, the lysate was first centrifuged at 4°C for 10 min at 400g, and the supernatant was further centrifuged at 20,000g for 10 min. The resulting pellet, resuspended in 1 mM NaHCO3, was used immediately as a crude membrane fraction.
Adenylate Cyclase Activation Assay.
Adenylate cyclase activity was determined by the procedure described previously (Salomon et al., 1974). Membrane proteins (3–15 μg) were incubated in a total volume of 60 μl containing 0.5 mM [α32P]ATP, 10 μM GTP, 5 mM MgCl2, 0.5 mM EGTA, 1 mM cAMP, 1 mM theophylline, 10 mM phospho(enol)pyruvate, 30 μg/ml pyruvate kinase, and 30 mM Tris-HCl at a final pH of 7.8. The reaction was initiated by membranes addition and was terminated after 15-min incubation at 37°C by adding 0.5 ml of a 0.5% SDS solution containing 0.5 mM ATP, 0.5 mM cAMP, and 20,000 cpm [3H]cAMP. cAMP was separated from ATP by two successive chromatographies on Dowex 50W-X8 columns and neutral alumina.
Binding Studies.
Binding studies on VPAC1 wt and mutant receptors were performed by using the 125I-VIP. The nonspecific binding was defined as residual binding in the presence of 1 μM unlabeled VIP. Binding was performed for 30 min at 23°C in a total volume of 120 μl containing 20 mM Tris-maleate, 2 mM MgCl2, 0.1 mg/ml bacitracin, and 1% bovine serum albumin, pH 7.4, buffer using 3 to 30 μg of protein per assay. Bound and free radioactivities were separated by filtration through glass-fiber GF/C filters presoaked for 24 h in 0.01% polyethylenimine and rinsed three times with a 20 mM, pH 7.4, sodium phosphate buffer containing 0.8% bovine serum albumin. The binding sites density was estimated by analysis of homologous competition curves assuming that the labeled and unlabeled ligands had the same affinity for the receptors.
Peptide Synthesis.
The peptides used were synthesized in our laboratory as described by Nachtergael et al., (2006). Peptide purity (at least 95%) was assessed by capillary electrophoresis, and conformity was assessed by electrospray MS.
Data Analysis.
All competition curves, dose-response curves, pIC50, and pEC50 values were calculated using nonlinear regression (Prism software; GraphPad Software Inc., San Diego, CA). Statistical analyses were performed with the same software.
Results
Molecular Modeling
Because no experimental VPAC1 structure is available, molecular modeling of its TM domain was performed in view of identifying residues involved in VIP binding and receptor activation, selecting potentially interesting mutations to be studied experimentally and rationalizing the results of these analyses. We took advantage of a preliminary 3D model of the TM domain of VPAC1 (Conner et al., 2005) and designed a new, carefully optimized model.
An important ingredient toward optimal modeling is the production of a correct amino acid alignment between the template and target proteins, given the almost nonsignificant level of sequence identity between the members of the GPCR-B family and the GPCR-A receptors, for which several structures have been solved (Palczewski et al., 2000; Okada et al., 2004; Cherezov et al., 2007; Jaakola et al., 2008; Warne et al., 2008). There is moreover no evidence that the receptors' activation mechanism should be the same in GPCR-A and GPCR-B and involve similar intermediate states. Therefore, there is no clear reason for selecting any particular structural template for modeling GPCR-B proteins. Here we chose the well resolved crystallographic structure of bovine visual rhodopsin (Protein Data Bank code 1U19) (Okada et al., 2004).
Sequence Alignments and TM Model
Because commonly available and automatic sequence alignment methods fail to produce reliable OPSD-VPAC1 alignments because of their very low sequence identity, we turned to an iterative and partly manual procedure of sequence alignment selection. In a first step, we compiled a set of four OPSD-VPAC1 alignments (Aln-1 to Aln-4). Aln-1 was adapted from the approach of Frimurer and Bywater (1999) for the modeling of the glucagon-like peptide-1 receptor based on a comprehensive sequence analysis, a low-resolution structure of frog rhodopsin obtained by electron crystallography, and the so-called “cold-spot” alignment method for sequences with low similarity. Aln-2 was taken from the work of Bissantz et al. (2004), in which they defined a framework for the automated modeling of GPCRs of the three main subfamilies. The latter approach tends to superimpose highly conserved positions, irrespective of their physicochemical nature, rather than residues viewed as similar according to substitution matrices. Aln-3 was built manually by implementing a kind of “cold-spot” approach idea. No gaps were allowed inside the TM domain; they were moved to the middle of loop regions. Aln-4 was produced by mGenThreader (McGuffin et al., 2000) via the BioInfoBank Meta Server (Ginalski et al., 2003). All of these alignments along with final variant are shown in Supplemental Fig. S1.
The four alignments were in agreement for helices TM3, TM6, and TM7, but they differed considerably for the other ones: we obtained four, two, two, and three variants for TM1, TM2, TM4, and TM5, respectively. Considering the sequence as a whole, this corresponds to 48 (4 × 2 × 2 × 3) global alignment variants of the TM region.
Each of the 48 alignments so obtained was used to generate a set of 10 structural models using the comparative modeling approach described under Materials and Methods. The packing quality of the TM helices in these 480 models was assessed using the membrane score Smem (see Materials and Methods). In a second step, the alignment that produced the best structural models, which display the maximum Smem value averaged over the 10 models (< Smem >), was used as starting point for further exploration of the alignment space This involved generating 37 = 2187 alignment variants by shifting each of the 7 TM helices independently by −1, 0, or +1 residue. From each of these new alignments, 10 structural models were built and evaluated on the basis of the membrane score Smem. The best alignment at this stage, referred to as ReAln (see Supplementary Fig. S1), was submitted again to the same helix shifting procedure, leading to 2187 other alignment variants. However, the models produced from these new alignments are not superior to that from ReAln (data not shown). The procedure was therefore stopped, and ReAln was considered to be the optimal alignment as measured by the Smem score.
Given the shortcomings of the empirical membrane score method, it is essential to consider available data on homologous proteins and check whether the model meets the general packing principles for membrane proteins such as hydrophobicity and variability organization. In particular, it is well known that the side of a given secondary structure element (here, an α-helix) that has mutated most during evolution is always exposed to the surrounding medium (here, the membrane). On the contrary, the conserved side, which is likely to play some important structural or functional roles, is buried inside the protein interior (Beuming and Weinstein, 2004). We performed a detailed analysis of the variability moment vectors in the structural model (see Experimental Procedures and Supplementary Fig. S2) and corrected manually the ReAln alignment to fulfill the requirement that the most variable side of the helix should face the membrane. The final alignment, which we refer to as finalAln, is given in Fig. 1 (see also Supplementary Fig. S1).

Alignment used to produce the VPAC1 receptor model (finalAln). Neither the N-terminal nor the C-terminal parts of the template (bovine visual rhodopsin, OPSD) and target (VPAC1) are shown, although loop regions are presented. All gaps were imposed to be approximately in the middle of loops. TM segments (experimentally resolved for rhodopsin and predicted from UniProt data for VPAC1) are highlighted in gray. Residues that were mutated in this work are underlined. The most conserved residues in TM segments of family A and B receptors are in boldface type on OPSD and VPAC1 sequences, respectively. A single residue per helix is given for rhodopsin—the one that is used as an “anchor” point in well known Ballesteros-Weinstein GPCR-A numbering system (Ballesteros and Weinstein, 2005). The conserved residues in TM helices of VPAC1 receptor may serve as a “signature” for automated identification of family B GPCRs from the sequence (Bissantz et al., 2004). Notice that virtually none of such residues coincide in both families. The average calculated degree of sequence identity of TM helices on the alignment is approximately 10%, which is at the lower boundary of the so-called “twilight zone” in comparative modeling (Baker and Sali, 2001). Other alignment variants that were used for construction of the “final” (this one) are shown in Supplementary Fig. S1.
Despite a better variability and hydrophobicity organization, the models based on the final alignment finalAln (Fig. 1) demonstrated seriously impaired Smem values in comparison with the ReAln-based models. This may be the consequence of a known caveat of the membrane score method (i.e., an excessive sensitivity to small conformational changes of the amino acids' side chains). To analyze whether these bad Smem values are indeed due to not accounting for the flexible nature of protein side chains, we performed MD runs with a fixed backbone conformation and computed the Smem values along the MD trajectories, as described under Materials and Methods. The comparison of the resulting Smem distributions (Fig. 2) shows that the final alignment finalAln generally leads to better packed models than the ReAln alignment, even though the starting conformations of the MD simulation present a lower Smem score in the case of finalAln than for ReAln. Note that the four initial alignments (Aln-1 to -4), which came from a single source and not from an iterative semimanual alignment procedure (Fig. S1), exhibit much worse Smem distributions than ReAln and finalAln (Fig. 2). We thus definitely consider finalAln as the optimal alignment and the resulting 3D models as the optimal model structures.

Schematic view of the packing quality of VPAC1 models built from different alignments. The curves represent distributions of the membrane score values (Smem), each for an ensemble of 1000 MD conformers with fixed Cα atoms positions. Thick solid lines correspond to distributions obtained from the ReAln alignment (gray) and the manually corrected version finalAln (black). Arrows of the same colors show Smem values for the starting conformations before MD sampling. Broken lines correspond to models built using the four initial alignments Aln-1 to -4 (see Supplementary Fig. S1). Notice that the finalAln model, initially worse packed than the ReAln model, becomes superior in terms of the <Smem> value of the MD-derived distribution. This suggests that the backbone of the finalAln model permits more advantageous conformations and environments for side chains, and therefore this model should be considered to be better packed.
Analysis of the TM Model
The resulting model of VPAC1 TM domain, depicted in Fig. 3, has native-like variability (Fig. S2) and hydrophobicity organization (data not shown). In addition, all residues that are known to be functionally important are located in an environment that provides a reasonable explanation of their function. The only polar stretch in contact with the membrane is located on the TM4 surface (Supplementary Fig. S3), which has been shown experimentally to correspond to a dimerization site in the case of the secretin receptor (Harikumar et al., 2007). This site may thus be expected to correspond to a dimerization site for the VPAC1 receptor too.
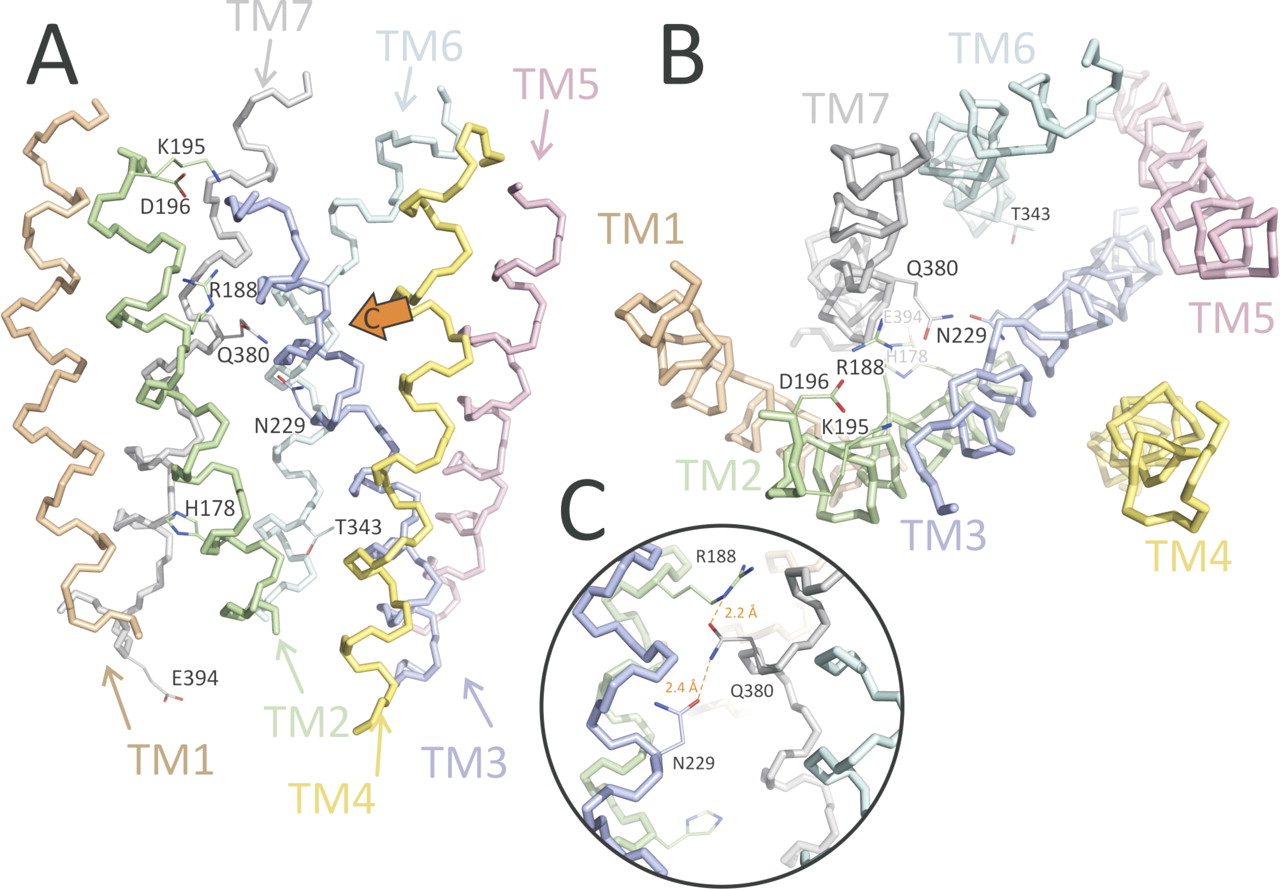
3D model of the TM domain of VPAC1 receptor. Each of the TM α-helices is individually colored and marked. The most important residues that are discussed in the main text are shown including Arg188, Asn229, and Gln380 that were mutated in this study. A, side view (from the membrane). B, top view (from the extracellular space). C, zoomed view of the mutated region (the view direction is shown by the orange arrow in A). Mutated residues form a chain: Arg188 in TM2 to Gln380 in TM7 to Asn229 in TM3. They possibly form hydrogen bonds, as shown with dashed orange lines. Note that the figure illustrates the possibility of forming H-bonds but does not imply that these two interactions should (or may) exist simultaneously.
As seen in Fig. 3, residues that are known to mediate ligand binding—Arg188, Lys195, and Asp196 in TM2 (Solano et al., 2001; Langer and Robberecht, 2007)—form a cavity close to the exterior surface of the membrane, with the charged group of Arg188 at the very bottom of the cavity. An interhelical interaction Arg188 to Gln380 between TM2 and TM7, analogous to the Arg233 to Gln451 interaction shown to be important for PTHR1 receptor (Gardella et al., 1996), is moreover observed in the model. This interaction can partially compensate for the unfavorable presence of the positive charge of Arg188 inside the helix bundle in the absence of ligand. These two residues belong to a chain of polar residues inside the receptor bundle: Arg188 in TM2 to Gln380 in TM7 to Asn229 in TM3 (Fig. 3c). His178 in TM2 and Thr343 in TM6, described as important for the activation and constitutive activity of some family B receptors (Hjorth et al., 1998; Gaudin et al., 1999), are also incorporated in a polar network in the cytoplasmic half of the TM domain of the receptor.
Experimental Analyses
Gln380 Located in TM7 Is Important for VPAC1 Activation.
Mutagenesis and functional studies identified previously an asparagine located in TM3 (Asn229 and Asn216 in VPAC1 and VPAC2 receptor, respectively) that is essential for receptor activation (Nachtergael et al., 2006). Indeed, as reported in Table 1, the N229A and N229Q mutations were shown to reduce the potency to stimulate adenylate cyclase by 10-fold. Furthermore, Arg188 in TM2 was demonstrated previously to establish a salt bridge interaction with Asp3 of VIP, which is essential for VPAC1 activation (Solano et al., 2001). Indeed, the R188A and R188Q mutations drastically impair both VIP binding and adenylate cyclase stimulation (Table 1), whereas the double mutant R188Q in VPAC1 and D3N in VIP is fully functional. These results led us to postulate that both Asn229 and Arg188 residues could be involved in an interaction network between TM helices, stabilizing the active receptor conformation, as often observed in GPCR-A family receptors.
Effect of mutation of Arg188, Asn229, and Gln380 in human VPAC1
pIC50 of binding, pEC50 and Emax of adenylate cyclase activation for VIP on membranes from CHO cells expressing the wild-type VPAC1 or mutated receptor. ΔpEC50 is defined as pEC50(mutant) − pEC50(wt) and ΔpIC50 as pIC50(mutant) − pIC50(wt). The expected values are the changes that would be expected if there was strict additivity. Emax is measured in response to 1 μM VIP, and values represent the mean ± S.E.M. of at least three independent experiments.
To identify other residues likely to take part in the network, we took advantage of the 3D model presented here and searched for amino acids located in vicinity of Asn229 and Arg188. As shown in Fig. 3, the ideal candidate is Gln380 in TM7, which seems to be sandwiched between Asn229 and Arg188. To evaluate the potential role of Gln380, we substituted this residue into alanine, arginine, or asparagine and performed binding competition curves and dose-response curves of adenylate cyclase stimulation. As shown in Table 1 and Fig. 4, the Q380A and Q380N mutants preserve the affinity for VIP, whereas Q380R shows a 20-fold decrease in affinity (ΔpIC50 = −1.35). The three mutants moreover display a decrease in the maximal cAMP stimulation for a comparable receptor density and a decrease in the pEC50 value of adenylate cyclase activation. This effect is stronger for Q380A and Q380R and relatively weak for Q380N. These results indicate that Q380 is important for VPAC1 activation, but probably is not directly involved in VIP binding. Note that the reason why Q380R displays a decreased affinity for VIP is probably related to the proximity of Arg188 and the repulsion between positive charges.

Binding and adenylate cyclase assay of wt and mutated VPAC1. Left, inhibition of 125I-VIP binding to membranes from Chinese hamster ovary cells expressing the wt or mutated VPAC1 receptors by increasing concentrations of VIP. Right, adenylate cyclase stimulation by VIP of the same membranes used at left. The pIC50 and pEC50 and efficacy values, averaged over three experiments, are given in Table 1, along with the standard error.
Effect of Double Mutations of Arg188, Asn229, and Gln380.
To evaluate whether Arg188, Asn229, and Gln380 are functionally interdependent, as proposed by the model, we next introduced in VPAC1 double mutations and compared the results with the effects obtained with the corresponding single mutants. Indeed, functionally independent residues should exhibit additive effects, whereas some synergy, either positive or negative, may appear for functionally interdependent residues. We thus constructed mutant receptors containing double substitutions of the three residues of interest and the double mutation in which both Asn229 and Gln380 are replaced by an alanine. Note that we did not test double mutants containing mutation of Arg188 into alanine because the R188A mutant was so much affected that we were unable to characterize it (Solano et al., 2001). Cell surface expression of all mutants tested was evaluated by fluorescence-activated cell sorting analysis using specific monoclonal anti-VPAC1 antibody to ensure that the effect observed is not due to receptor misfolding or altered cell surface targeting (data not shown).
As shown in Table 1, the capability to stimulate adenylate cyclase activity of the N229A/Q380A mutant is lower than that of the wt and similar to that of the individual single-site mutants. Furthermore, the double substitution N229Q/Q380N only slightly reduces VIP potency and efficacy to activate adenylate cyclase and displayed a pharmacological profile intermediary between the N229Q and the Q380N mutants (Fig. 4). The Q380N mutation thus partly restores the loss of activity caused by the N229Q mutation. These results tend to confirm the model, in particular the proximity of Asn229 and Gln380 and the importance of their interaction for VPAC1 activation. Note that both double mutants present some decreased affinity for VIP, whereas no such effect is observed for the single mutants. This may result from packing defects that indirectly affect the surrounding ligand-binding residues. This is consistent with the larger decrease in affinity caused by N229A/Q380A, compared with N229Q/Q380N.
The double substitution R188N/N229R resulted in a marked synergistic effect on the decrease in binding compared with R188N and N229R mutants. Indeed, we were unable to detect any VIP-specific binding for the R188N/N229R mutant, suggesting that VIP affinity is much more affected than for R188N mutant despite the fact that it was preserved for the N229R mutant. Likewise, VIP potency to stimulate adenylate cyclase was reduced by 100-fold for the R188N/N229R mutant, by 10-fold for R188N mutant, but was preserved for N229R; the VIP efficacy was reduced by 25% for both R188N/N229R and N229R mutants (Table 1 and Fig. 4).
Moreover, R188Q/Q380R showed synergy in reducing the potency of VIP on cAMP production and a less-than-additive effect on binding compared with R188Q and Q380R mutants. Indeed, VIP affinity was reduced by 100-, 20-, and 100-fold for R188Q, Q380R, and R188Q/Q380R mutants, respectively. VIP potency to stimulate adenylate cyclase was reduced by 7- and 6-fold for R188Q and Q380R mutants, respectively, but by 150-fold for the R188Q/Q380R mutant. VIP efficacy was dramatically impaired for the Q380R and R188Q/Q380R mutants but preserved for the R188Q mutant (Table 1 and Fig. 4). Altogether, these data suggest that Arg188, Asn229, and Gln380 are functionally interdependent and important for both VIP affinity and VPAC1 activation.
Discussion
G protein-coupled receptors, also referred to as seven-transmembrane domain receptors, represent the largest family of signal transducers for extracellular stimuli. The determination of the high-resolution structure of members of family A of GPCRs (Palczewski et al., 2000; Okada et al., 2004; Cherezov et al., 2007; Jaakola et al., 2008; Warne et al., 2008) has confirmed that receptor activation is mediated by relative movements among the seven transmembrane helices that are stabilized by different network of interactions. However, because these key residues are not conserved in family B GPCRs, and structural data are only available for the N-terminal extracellular domain, little is known about the precise mechanisms involved in the activation of this family of receptors.
The commonly accepted model for agonist action uses the PTH receptor as template and suggests that the N-terminal domain is the principal binding site for the C-terminal region of the exogenous ligand, whereas binding of residues 1 to 3 of the ligand to the extracellular loops and TM helices are believed to drive the receptor activation and subsequent G protein coupling. After agonist binding, subsequent conformational changes are expected within the TM domain of the receptor. This is illustrated by the fact that a Zn(II) bridge between helices 3 and 6 of the PTH receptor constrains the receptor in a conformation unable to promote PTH-mediated G-protein activation, whereas agonist-induced internalization or phosphorylation was preserved (Vilardaga et al., 2001; Castro et al., 2005).
In the present study, by combining pharmacological and in silico approaches, we have identified a network of interactions between residues located in helices 2, 3, and 7 of the VPAC1 receptor, which are involved in the stabilization of the receptor in absence of agonist and in early steps of receptor activation. We propose that, in the absence of VIP, the Gln380 residue of TM7 interacts with Arg188, located in TM2 and identified previously as essential for recognition of the Asp3 side chain of VIP and subsequent receptor activation. Upon VIP binding, the interaction between Arg188 and Gln380 is broken and a stronger interaction (salt bridge) is established between Arg188 and the Asp3 side chain of VIP. This necessarily has an affect on the network of interactions essential for G protein activation, in which Gln380 and Asn229 are proposed to play an important role. This view is supported by several experimental and modeling results.
First, we showed that the substitution of Gln380 to alanine or asparagine significantly reduced the VIP efficacy to stimulate adenylate activation, similarly to what happens for the Asn229 substitutions (Nachtergael et al., 2006), and that the substitution of both residues to alanine had a less than additive effect. The altered activation observed with these mutants cannot be attributed to the disruption of the binding pocket because the affinities for VIP were not affected for N229A, N229Q, Q380A, and Q380N mutants. As suggested previously for Asn229, it is likely that Q380A and Q380N mutants still bind the G protein but fail to activate it properly. As shown for several GPCRs, it is expected that reciprocal exchange of two residues involved in a direct interaction should restore the activity of the receptor. We found that the N299Q/Q380N substitution partially restored the receptor activity. The N299A/Q380A shows a similar anticooperativity because the loss in activity of the double mutant is only slightly larger than that of each single-site mutation. Note that both double mutants present a loss in binding affinity, which contributes to the loss in activity. As a consequence, the actual anticooperative effect is even stronger than suggested by the comparison of the measured and expected ΔpEC50 values (Table 1). Thus altogether the results suggest that the interaction between Asn229 and Gln380 is important for VPAC1-mediated G protein activation.
The 3D model can be taken to suggest that Gln380 functions as a floating “ferry boat,” switching between Arg188 and Asn229 residues' side chains. This triad lines up in the model almost perfectly (Fig. 3c), so disruption of the Arg188 to Gln380 interaction upon VIP binding probably modifies the Asn229 to Gln380 interaction, hence contributing to signal transduction propagation and activation of G protein. However, the exact mechanism by which this occurs cannot be determined at this stage, because this would require a model of the activated receptor in complex with VIP. In particular the two N-terminal residues of VIP, His1 and Ser2, are likely to affect, directly or indirectly, the interaction network surrounding Asn229 and Gln380.
It is also interesting to note that none of the tested single-site mutations of Asn229 or Gln380 affects the affinity for VIP, except for Q380R. This exception can be explained by the proximity in the mutant of two arginine residues at positions 188 and 380, which will create repulsive interactions modifying the relative position of the two side chains compared with the wt receptor. These results indicate that Asn229 or Gln380 are probably not directly involved in VIP binding.
Reciprocal substitution mutants are in agreement with the importance of Arg188 for the high-affinity binding of VIP. Indeed, no specific VIP binding was detected for the R188N/N229R mutant, and VIP affinity was reduced by 150-fold for R188Q/Q380R. In those mutants, the localization of the arginine could actually be much deeper into the helices, thus preventing interaction with the Asp3 side chain of VIP.
To our knowledge, this is the first study that identified, in a member of family B GPCRs, interactions between residues located in transmembrane helices that are involved in the stabilization of the receptor conformation. Interestingly, Arg172 in the closely related VPAC2 receptor and Arg166 in the secretin receptor (these positions correspond to Arg188 in VPAC1) also interact with the Asp3 side chain of VIP and secretin, respectively (Di Paolo et al., 1998; Langer and Robberecht, 2007). Some of us also demonstrated previously that Asn216 in VPAC2, corresponding to Asn229 in VPAC1, was essential for receptor activation (Nachtergael et al., 2006). Likewise, other studies have pointed out the importance of TM2 and TM7 in G protein activation. Indeed, the mutation of His178 into arginine located at the bottom of TM2 in VPAC1 led to a constitutively activated receptor (Gaudin et al., 1998). Because the mutation of this residue into alanine, aspartic acid, or even lysine did not confer ligand-independent activation, the authors proposed that the replacement by an arginine provokes subtle conformational changes that do not simply remove some stabilizing interactions, as seen in family A GPCRs (Gaudin et al., 1998). On the other hand, it has also been shown that Glu394 located at the junction of TM7 and the C terminus of VPAC1 was important for G protein activation but not for coupling (Couvineau et al., 2003).
In agreement with these data and on the basis of the present results, we propose that, in absence of ligand, interaction between Arg188, Asn229, and Gln380 ties helices 2, 3, and 7 together. Because there is no evidence for increased constitutive activity with any mutants studied, this suggests that the wild-type network of interactions is not the unique determinant for maintaining the receptor in its inactive conformation in the absence of ligand. On the other hand, it is possible that multiple interactions constrain the receptor in an inactive conformation, and just disrupting one doesn't lead to enhanced basal activity. Upon interaction of the N-terminal tail of VIP with the TM domain of VPAC1, which includes the Asp3(VIP) to Arg188(VPAC1) salt bridge, TM2 and probably other helices undergo conformational modulations causing key sequences located in intracellular loops to be exposed and to interact with the G protein. In the meantime, the interaction network involving Asn229 and Gln380 maintains TM7 in a conformation necessary for proper activation of G protein, mediated through interaction with Glu394.
Note that the importance of Arg188, Asn229, Gln380, and Glu394 residues in VPAC1 activity is further supported by their high degree of conservation among all members of GPCR-B family (see Supplemental Fig. S4 in for sequence alignment of TM2, 3, and 7 of GPCR-B family members). These residues may therefore be involved in a binding and activation mechanism that is common to the whole GPCR-B family. However, additional experiments on other family members should be performed to support this view.
Acknowledgments
All experiments, except molecular modeling, were performed in the Laboratory of Biological Chemistry and Nutrition (School of Medicine, Université Libre de Bruxelles) under the direction of Dr. P. Robberecht, whom we thank for constructive comments.
Footnotes
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.This work was supported by the Brussels Region TheraVip project; the Belgian State Science Policy Office through an Interuniversity Attraction Poles Programme (DYSCO); the Belgian Fund for Scientific Research [Grant 3.4553.06]; and the President of Russian Federation [Grant MK-125.2008.4].
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.110.063578.
-
ABBREVIATIONS:
- GPCR
- G protein-coupled receptor
- 3D
- three dimensional
- Aln
- alignment
- MD
- molecular dynamics
- OPSD
- bovine visual rhodopsin
- TM
- transmembrane
- VIP
- vasoactive intestinal peptide
- wt
- wild type
- PTH
- parathyroid hormone.

- Received January 13, 2010.
- Accepted June 23, 2010.
- Copyright © 2010 The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}