


Abstract
In view of the therapeutic importance of dopamine D3 and D2 receptors, there remains considerable interest in novel ligands. Herein, we show that the tetrahydroisoquinoline 1H-indole-2-carboxylic acid {4-[2-(cyano-3,4-dihydro-1H-isoquinolin-2-yl)-ethyl]-cyclohexyl}-amide (SB269,652) behaves as an atypical, allosteric antagonist at D3 and D2 receptors. Accordingly, SB269,652 potently (low nanomolar range) abolished specific binding of [3H]nemanopride and [3H]spiperone to Chinese hamster ovary-transfected D3 receptors when radioligands were used at 0.2 and 0.5 nM, respectively. However, even at high concentrations (5 μM), SB269,652 only submaximally inhibited the specific binding of these radioligands when they were employed at 10-fold higher concentrations. By analogy, although SB269,652 potently blocked D3 receptor-mediated activation of Gαi3 and phosphorylation of extracellular-signal-regulated kinase (ERK)1/2, when concentrations of dopamine were increased by 10-fold, from 1 μM to 10 μM, SB269,652 only submaximally inhibited dopamine-induced stimulation of Gαi3. SB269,652 (up to 10 μM) only weakly and partially (by approximately 20–30%) inhibited radioligand binding to D2 receptors. Likewise, SB269,652 only submaximally suppressed D2 receptor-mediated stimulation of Gαi3 and Gαqi5 (detected with the aequorin assay) and phosphorylation of ERK1/2 and Akt. Furthermore, SB269,652 only partially (35%) inhibited the dopamine-induced recruitment of β-arrestin2 to D2 receptors. Finally, Schild analysis using Gαi3 assays, and studies of radioligand association and dissociation kinetics, supported allosteric actions of SB269,652 at D3 and D2 receptors.
Introduction
The monoamine dopamine modulates motor activity, cognition, mood, endocrine secretion, and a broad range of other physiological functions via actions at two families of G-protein-coupled receptors. D1-class receptors (D1 and D5) are principally coupled to stimulatory Gα-proteins and enhance the production of cAMP, whereas D2-class receptors (D2, D3, and D4 subtypes) are primarily coupled to inhibitory Gα-proteins and suppress the activity of adenylyl cyclase (Zhuang et al., 2000; Ahlgren-Beckendorf and Levant, 2004; Neve et al., 2004). D2 receptors display a high degree of sequence similarity with D3 receptors, and they share a predicted binding site for dopamine and synthetic ligands at the interface of transmembrane helices (Shi and Javitch, 2002). D2 and D3 receptors also show similar patterns of signal transduction, although under certain conditions, the latter couple less broadly and robustly to intracellular messengers such as adenylyl cyclase (Cussac et al., 1999; Ahlgren-Beckendorf and Levant, 2004; Neve et al., 2004; Sokoloff et al., 2006). Substantial efforts have been made to synthesize ligands that competitively interact with orthosteric dopamine-binding sites on D3 and/or D2 receptors, but an alternative route toward clinically useful drugs is offered by allosteric modulation. Allosteric modulators are agents that “remotely” alter the interaction of cognate ligands with their receptors, reflecting conformational changes and alterations in binding and coupling parameters (Christopoulos and Kenakin, 2002; Schetz, 2005; May et al., 2007). The quality of the allosteric effect is said to be positive if the modulator facilitates orthosteric agonist-induced receptor function, and negative where function is diminished. A third possibility is that the binding of an allosteric modulator does not affect the affinity of a ligand at the primary site, an observation termed “neutral” cooperativity (Lazareno and Birdsall, 1995).
Sodium and zinc ions (Schetz et al., 1999), amiloride and its nitrogen-substituted derivatives (Hoare and Strange, 1996), and analogs of the tripeptide proline-leucine-glycine (PLG) (Verma et al., 2005) were suggested to interact allosterically with D2 receptors. More recently, the distinctive in vivo profiles of (3S)-3-[3-(methylsulfonyl)phenyl]-1-propylpiperidine hydrochloride [(−)-OSU6162] and ACR16 (pridopidine) were suggested to involve allosteric actions at D2 receptors, although supporting data are limited (Tamminga and Carlsson, 2002; Rung et al., 2008). In general, allosteric modulators are well tolerated and can fine-tune pharmacological responses to endogenous neurotransmitters and exogenous agents, underpinning interest in their clinical application either alone or as adjunctive treatments (Christopoulos and Kenakin, 2002; May et al., 2007). Accordingly, (−)-OSU6162 displayed antipsychotic-like properties in rats in the absence of extrapyramidal motor effects and with little induction of dyskinesia (Tamminga and Carlsson, 2002; Natesan et al., 2006; Rung et al., 2008), and PLG potentiated the induction of contralateral rotation by l-DOPA in unilateral 6-hydroxydopamine lesioned rats without exacerbating the induction of dyskinesia (Ott et al., 1996). These observations support the notion that, in addition to orthosteric agents, positive and negative allosteric modulators at D2 and/or D3 receptors could be therapeutically useful agents, used either alone or as adjunctive therapy.
The present study characterized a novel tetrahydroisoquinoline derivative 1H-indole-2-carboxylic acid {4-[2-(cyano-3,4-dihydro-1H-isoquinolin-2-yl)-ethyl]-cyclohexyl}-amide (SB269,652), which was found to behave as an allosteric antagonist at dopamine D3 and D2 receptors employing a broad and complementary range of cellular approaches. To date, only a preliminary description of this drug has appeared in abstract form (Taylor et al., 1999).
Materials and Methods
Materials.
[3H]Nemonapride and [3H]spiperone were purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA); dopamine, haloperidol, and sulpiride were obtained from Sigma (St. Louis, MO). Tissue culture media and sera were acquired from Sigma and Invitrogen (Carlsbad, CA). SB269,652 (Fig. 1) was synthesized by G. Lavielle (Paris, France).
Chemical structure of SB269,652 (1H-indole-2-carboxylic acid {4-[2-(cyano-3,4-dihydro-1H-isoquinolin-2-yl)-ethyl]-cyclohexyl}-amide).
Mutant Plasmid Constructs.
The wild-type human D2L receptor (subsequently referred to as D2) inserted in a pcDNA 3.1 eukaryotic expression plasmid was purchased from the Guthrie cDNA resource center. The human dopamine D3 receptor (subsequently referred to as D3) was kindly provided by Dr. Robert Levenson. Human β-arrestin2 was fused to mVenus with a seven-amino acid linker (GAGALAT). Chimeric dopamine D2/D3 receptors were constructed as described in the Supplemental Data.
Cell Cultures and Transfection.
Chinese hamster ovary (CHO)-D3-DHFR− cells (subsequently referred to as CHO-D3) were provided by Servier (Paris, France). CHO-D2 cells were constructed by inserting the pcDNA 3.1-D2 plasmid into wild-type CHO cells and selected in G418. African green monkey kidney COS-7 cells were transiently transfected with the plasmid DNA encoding wild-type or chimeric receptors by the DEAE-dextran chloroquine method (Cullen, 1987). The total amount of DNA used for each transfection was 4 μg. Receptor densities (Bmax) in stably transfected CHO-D3 and CHO-D2 cells were, respectively 5058 ± 241 and 3159 ± 184 fmol/mg protein (calculated with [3H]nemonapride). Bmax values ranged between 1000 to 1200 fmol/mg protein in COS-7 cells transiently transfected with wild-type and chimeric receptors.
Flp-In T-REx 293 cells were stably transfected with human β-arrestin2-mVenus in pIRESpuro3 and individual colonies were selected in 2 μg/ml puromycin. Colonies were screened for mVenus fluorescence, an appropriate clone was transfected with Flag-D2L-Rluc8 in pcDNA5/FRT/TO together with pOG44, and a stable pool was selected in 100 μg/ml hygromycin. This led to constitutive expression of the bioluminescence resonance energy transfer (BRET) acceptor β-arrestin2-mVenus and tetracycline-inducible expression of the donor Flag-D2L-Rluc8.
Membrane Preparations and Binding Assays.
Three days after transfection (COS-7) or one day after plating (CHO) cells were lysed, and membrane preparation and binding assays were performed as described previously (Maggio et al., 2003). Membranes were incubated with [3H]nemonapride (82 Ci/mmol) or [3H]spiperone (100 Ci/mmol) at 30°C for 1 h in a final volume of 1 ml. In competition experiments, unless otherwise specified, the concentration of the radioligands were 50 or 200 pM for [3H]nemonapride in COS-7 and CHO cells, respectively, and 500 pM for [3H]spiperone. Nonspecific binding was determined in the presence of 2 mM dopamine. The total amount of protein in each sample was ∼5 μg for CHO cells and ∼20 μg for COS-7 cells. Dissociation kinetics assays were performed at 30°C by first equilibrating dopamine D2 receptors with 200 pM [3H]nemonapride or 800 pM [3H]spiperone in 20 μl of binding assay buffer (50 mM Tris HCl, pH 7.4, 155 mM NaCl, and 0.01 mg/ml bovine serum albumin). Two milliliters of binding buffer containing 10 μM haloperidol, sulpiride, or SB269,652 were added at time 0, and sequential samples were taken. Association kinetics assays were performed at 30°C with 0.65 nM [3H]nemonapride. When SB269,652 was present, it was allowed to equilibrate for 1 h before radioligand addition.
Antibody-Capture/Scintillation-Proximity Assays Studies of Coupling to Gαi3.
The influence of SB269,652 upon dopamine-induced activation of Gαi3 subunits coupled to CHO-transfected D3 and D2 receptors was determined using a [35S]GTPγS binding assay coupled to scintillation proximity detection in 96-well OptiPlates (PerkinElmer Life and Analytical Sciences) as described previously (Millan et al., 2004a). SB269,652 was preincubated for 30 min before addition of a fixed concentration of dopamine (1 or 10 μM). After incubation, 20 μl of Tergitol-type NP-40 (0.27% final concentration; Roche Diagnostics, Mannheim, Germany) was added, and plates were incubated for 30 min at 22°C. Then, after addition of 10 μl of mouse anti-Gαi3 monoclonal antibody (Enzo Life Sciences, Farmingdale, NY), incubation continued for 1 h. Scintillation proximity assay beads coated with secondary, anti-mouse antibodies (GE Healthcare, Vélizy, France) were added in a volume of 50 μl, and plates were incubated overnight under gentle agitation. They were centrifuged (15 min; 1300g) and radioactivity determined on a TopCount microplate scintillation counter (PerkinElmer Life and Analytical Sciences).
Aequorin Assay.
A functional assay based on luminescence of mitochondrial aequorin after intracellular Ca2+ release was performed as described previously (Blanpain et al., 1999; Han et al., 2009). Flp-In T-REx 293 cells stably expressing aequorin, the human D2S receptor, and a Gαqi5 protein that can signal from Gαi-coupled receptors (Conklin et al., 1993) were seeded in a 15-cm dish and grown in antibiotic-free medium for ∼48 h. Tetracycline (1 μg/ml) was added to the medium for 3 to 24 h to induce receptor expression. Cells were detached and pelleted and then resuspended at a final concentration of 5 × 106 cells/ml in the presence of 5 μM coelenterazine h. After 4-h rotation at room temperature (20°C) in the dark, the cell solution was diluted 10-fold, followed by 1-h incubation under the same conditions. Concentration-response curves were obtained by injecting 50 μl of cell solution into wells of a 96-well microplate containing 50 μl of a 2× concentration of the desired compounds in medium. Luminescence signals from the first 15 s after injection were read by a POLARstar OPTIMA reader (BMG Labtech GmbH, Offenburg, Germany). Total response was defined as the signal resulting from injecting 50 μl of cell solution into 50 μl of assay medium containing 0.1% (v/v) Triton X-100, which raises the Ca2+ concentration directly by membrane permeabilization as described previously (Han et al., 2009).
Phosphorylation of ERK1/2.
CHO cells expressing D2 or D3 receptors were grown in Ham's F12 medium or Dulbecco's modified Eagle's medium, respectively, as described previously (Millan et al., 2008). SB269,652 was preincubated for 30 min at 37°C before stimulation by dopamine for 5 min. For immunoblotting, cell extracts (10 μl) were loaded on 15-well 12% polyacrylamide gels; after migration, proteins were transferred onto nitrocellulose membranes. Activated MAPK was detected with a monoclonal antibody specifically recognizing the phosphorylated pp42mapk (ERK2) and pp44mapk (ERK1) forms on both threonine and tyrosine residues (Cell Signaling Technology Inc., Danvers, MA).
BRET.
D2/arrestin cells were seeded in a 100-mm dish (1.5 × 106 cells); after 48 h, 0.01 μg/ml tetracycline was added to induce Flag-D2L-Rluc8 expression. After an overnight incubation, the cells were harvested, washed, and resuspended in phosphate-buffered saline. One percent of the cells were distributed into each well of a 96-well plate. Cells were treated with sulpiride or SB269,652 at room temperature for 30 min, incubated with 5 μM coelenterazine h for 5 min and incubated for 5 min with 10 μM dopamine or phosphate-buffered saline. The fluorescence (excitation at 510 nm and emission at 540 nm, 1-s recording) and luminescence (no filters, 1-s recording) were quantified (PHERAstar; BMG Labtech). The BRET signal (Barak et al., 1997) was determined by calculating the ratio of the light emitted by Venus (510–540 nm) over that emitted by RLuc8 (485 nm).
Analysis of Data.
Kd and Bmax values of [3H]nemonapride were determined in direct saturation experiments. IC50 values of SB269,652 were calculated in competition curves fitted to one-site binding models using the iterative, nonlinear, least-squares regression analysis of OriginPro 7.5 (OriginLab Corporation) software. When inhibition reached a plateau above the basal level, curves were fitted to the plateau. IC50 values were converted to IC50corr values according to the equation of Cheng and Prusoff (1973): IC50corr = IC50/(1 + [Radioligand]/Kd). In experiments assessing the functional interaction between SB269,652 and dopamine, estimates of the modulator-receptor dissociation constant (K) and co-operativity factor (α) for the interaction were obtained by Schild analysis using the relationship EC50ratio – 1 = ([SB269,652] × (1 − α))/(α[SB269,652] + K), where α is the cooperative factor between dopamine and SB269,652 and K is the apparent affinity of SB269,652 for the dopamine receptors (Ehlert, 1988). All values presented in the text are expressed as mean ± S.E.M.
Results
Binding Profile of SB269,652 at D3 and D2 Receptors Expressed in CHO Cells.
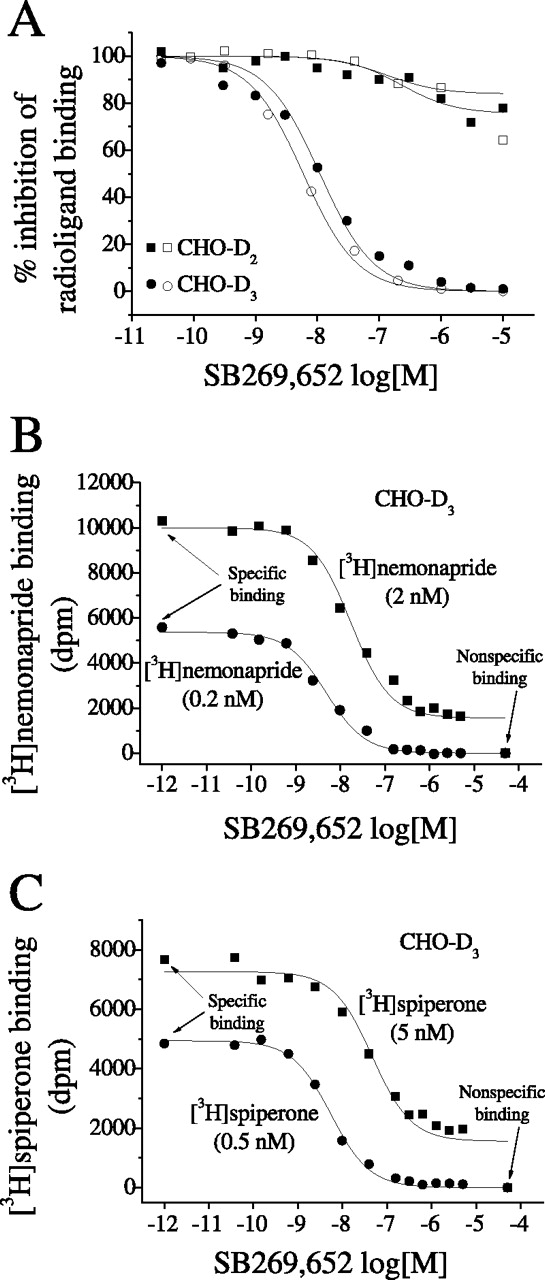
In competition binding studies, SB269,652 concentration-dependently and potently inhibited the binding of [3H]nemonapride and [3H]spiperone to D3 receptors expressed in CHO cells (Fig. 2A). IC50corr values of 1.86 ± 0.10 and 2.51 ± 0.18 nM were found with [3H]nemonapride and [3H]spiperone, respectively.

Inhibition isotherms by SB269,652 of radioligand binding to D3 and D2 receptors. Influence of SB269,652 upon the binding of 0.2 nM [3H]nemonapride (filled symbols) and 0.5 nM [3H]spiperone (open symbols) to D2 and D3 receptors stably transfected in CHO cells (A). Influence of SB269,652 upon the binding of 0.2 and 2 nM concentrations of [3H]nemonapride (B) and 0.5 and 5 nM concentrations of [3H]spiperone (C) to D3 receptors. In the last two experiments, the total amount of protein in each sample was ∼10 μg. Isotherms are representative of single experiments, each of which was undertaken four times and performed in triplicate.
At D2 receptors expressed in CHO cells, SB269,652 inhibited [3H]nemonapride and [3H]spiperone binding by approximately 20 to 30%, over a concentration range of 0.01 to 10 μM (Fig. 2A). IC50corr values for the partial inhibition of [3H]nemonapride and [3H]spiperone binding at D2 receptors were 33.9 ± 17.4 and 43.5 ± 24.2 nM, respectively. Inhibition binding experiments with CHO-D3 cells using a 10-fold higher concentration of [3H]nemonapride (2 nM) and [3H]spiperone (5 nM) showed that SB269,652 could inhibit binding by only ∼80% (Fig. 2, B and C). IC50corr values were similar to values calculated with the lower concentrations of the radioligands: 1.79 ± 0.41 and 3.08 ± 0.95 nM for [3H]nemonapride and [3H]spiperone, respectively.
In another set of experiments, we measured Kd and Bmax values of [3H]nemonapride in the presence or absence of fixed concentrations of SB269,652. As shown in Table 1 and Supplemental Fig. S1, A and B, SB269,652 reduced the Bmax values at both D3 and D2 receptors without altering Kd values. Collectively, these equilibrium binding experiments suggest that SB269,652 behaves as an allosteric compound at dopamine D3 and D2 receptors.
Equilibrium and kinetic binding experiments of [3H]nemonapride in the presence and absence of a fixed concentration of SB269,652 in stable transfected CHO-D2 and CHO-D3 cells
All values are the mean ± S.E.M. of three experiments, each of which was performed in triplicate. The concentration of [3H]nemonapride in association binding experiments was 0.65 nM. Data are the mean ± S.E.M. of two experiments each performed in triplicate. The Koff values reported in the control columns for D2 and D3 receptors are the averages of values specified in the text for dissociations performed in the presence of haloperidol or sulpiride.
Effect of SB269,652 on the Rate of Radioligand Dissociation from Dopamine D3 and D2 Receptors.
To acquire further insight into the allosteric nature of SB269,652, we performed binding kinetics. [3H]Nemonapride dissociation from D3 receptors in the presence of haloperidol and sulpiride was best fit by a one-phase exponential decay and off rates were similar: Koff = 0.059 ± 0.006 min−1 and 0.064 ± 0.004 min−1, respectively (Fig. 3A). Dissociation kinetics were clearly decreased by SB269,652 at D3 receptors compared with haloperidol and sulpiride; [3H]nemonapride dissociation in the presence of SB269,652 was best fit by a one-phase exponential decay with Koff = 0.007 ± 0.0003 min−1 (Fig. 3A). At D2 receptors, off rates of [3H]nemonapride (Fig. 3B) in the presence of haloperidol and sulpiride were similar, Koff = 0.023 ± 0.002 min−1 and 0.022 ± 0.001 min−1, respectively. In accordance with D3 receptors, dissociation kinetics of [3H]nemonapride were clearly decreased by SB269,652, with Koff = 0.007 ± 0.0003 min−1. Similar results were obtained with [3H]spiperone (Fig. 3C): Koff = 0.033 ± 0.003 min−1 and 0.036 ± 0.003 min−1, respectively, for haloperidol and supiride, and Koff = 0.011 ± 0.0002 min−1 with SB269,652.

Effect of haloperidol, sulpiride, and SB269,652 on the rate of radioligand dissociation from dopamine D3 and D2 receptors expressed in CHO cells. Dissociation rate kinetics were measured for [3H]nemonapride (A and B) binding to membranes containing D3 and D2 receptors, respectively, and for [3H]spiperone (C) binding to membranes containing the D2 receptors. The dissociation rates were measured by first equilibrating dopamine receptors with radioligands at 30°C followed by 100-fold dilution and addition of excess (10 μM) nonisotopic antagonist (haloperidol, sulpiride, or SB269,652). Kinetics are from representative experiments, each of which was undertaken five (A), four (B), or two (C) times and performed in triplicate.
Such a marked increase in the off rate would imply a major increase in affinity of the radioligand in the presence of SB269,652 unless there were a corresponding decrease in the on rate. To test this, we performed association kinetics with and without a fixed concentration of SB269,652. As shown in Table 1 and Supplemental Fig. S2, A and B, SB269,652 greatly reduced the Kon at D3 receptors and, albeit less so, D2 receptors. The Kd values measured by the ratio Koff/Kon were similar in the presence and absence of SB269,652 (10 μM) at both D3 and D2 receptors.
Structural Determinants of SB269,652 Binding Determined in Chimeric D2/D3 Receptors Expressed in COS-7 Cells.
Chimeric D2/D3 receptors (Table 2) were used to delineate receptor domains engaged in SB269,652 binding. As illustrated in Table 2, [3H]nemonapride showed no differences in affinity for any of the tested chimeras. It is worth noting that in COS-7 cells, the Kd of [3H]nemonapride at wild-type dopamine D3 and D2 receptors was considerably lower than in stably transfected CHO-D3 and CHO-D2 cells.
Affinity constants (IC50corr) of SB269,652 for wild-type D3 and D2 and chimeric dopamine D3/D2 receptors in transiently transfected COS-7 cells
The amount of cDNA transiently transfected into COS-7 cells was 4 μg in each case. The concentration of [3H]nemonapride in displacement experiment was 2 nM. IC50corr = IC50 corrected for [3H]nemonapride occupancy. Data are the mean ± S.E.M. of at least three experiments, each performed in triplicate.
At chimera A, which contains the first transmembrane segment (TMS) of D3 and the last six TMSs of D2, only high concentrations of SB269,652 modestly inhibited the binding of [3H]nemonapride (Table 2 and Supplemental Fig. S3A). Introduction of the second TMS together with extracellular loop (EL)-I and the proximal part of the third TMS of D3 in the D2 receptor results in chimera B. SB269,652 displaced by approximately 44% the binding of [3H]nemonapride to this chimera, and its affinity was 5.92 ± 0.63 nM (Table 2 and Supplemental Fig. S3A). Further addition of the distal part of TMS-III, as in chimera C, and of TMS-VI, as in chimera D, did not result in any significant increase in binding affinity or percentage of inhibition (Table 2 and Supplemental Fig. S3A). In sharp contrast, introduction of EL-II together with TD-V of D3 in D2 receptors, as in chimera E, substantially increased the potency of SB269,652 in displacing [3H]nemonapride binding (∼20-fold), and the percentage of inhibition reached 91.6% (Table 2 and Supplemental Fig. S3A). The addition of the third cytoplasmic loop together with the proximal part of TD-VI, as in chimera F, failed to produce any further enhancement of SB269,652 binding (Table 2 and Supplemental Fig. S3A). Chimera G was constructed by inserting EL-II of D3 in the D2 receptor. SB269,652 bound to this chimera with an affinity similar to that when it bound to D2 but attained a significantly higher (31.3%) inhibition of [3H]nemonapride binding (Table 2 and Supplemental Fig. S3B). In contrast, at chimera H, in which EL-II of D2 was inserted in the D3 receptor, SB269,652 inhibited [3H]nemonapride binding to an extent similar to its inhibition at D3 receptors, but with ∼10-fold lower potency (Table 2 and Supplemental Fig. S3B). Finally, the insertion of TD-I of D2 in D3 receptor as in chimera I did not modify the profile of SB269,652 compared with the wild-type D3 (Table 2 and Supplemental Fig. S3B).
Influence of SB269,652 upon Dopamine-Stimulated Coupling to Gαi3 at D3 and D2 Receptors.
In line with previous work (Millan et al., 2004b), dopamine potently enhanced [35S]GTPγS binding to Gαi3 at CHO-expressed D3 receptors. SB269,652 given alone had no effect (not shown), but it potently and fully suppressed stimulation of Gαi3 by dopamine 1 μM, with a pKB of 9.44 ± 0.20 (Fig. 4A). Consistent with a noncompetitive mode of inhibition by SB269,652, when the concentration of dopamine was raised to 10 μM, concentrations of SB269,652 up to 10 μM maximally reduced [35S]GTPγS binding by only approximately 60%. The preferential D2 antagonist 3-[4-(4-chlorophenyl)-4-hydroxypiperidin-l-yl]methyl-1H-indole (L741,626), ineffective alone (not shown), was much less potent than SB269,652, displaying a pKB of 7.06 ± 0.05.

Antagonism by SB269,652 and L741,626 of D3 and D2 receptor-coupled Gαi3 activation as determined by antibody-capture/scintillation proximity assays. Studies were performed at D3 and D2 receptors stably expressed in CHO cells. A, concentration-dependent antagonism by SB269,652 of dopamine (1 μM; DA) induced Gαi3 activation at CHO cell-expressed D3 receptors. Partial inhibition was attained when dopamine concentration was raised to 10 μM. B, concentration-dependent, but partial, attenuation by SB269,652 of dopamine (1 and 10 μM) induced Gαi3 activation at CHO cell-expressed D2 receptors. Data are mean ± S.E.M. of three independent experiments, each performed in triplicate.
In CHO-expressed D2 receptors, dopamine enhanced [35S]GTPγS binding to Gαi3 with a pEC50 of 6.17 ± 0.04. At D2 sites, SB269,652 (inactive alone) reduced the enhancement of [35S]GTPγS binding induced by 10 μM dopamine by approximately 30%, with a pKB of 7.67 ± 0.14 (Fig. 4B). It is noteworthy that when the concentration of dopamine was lowered to 1 μM, SB269,652 reduced [35S]GTPγS binding by approximately 50%. In distinction, the competitive antagonist L741,626 completely prevented the enhancement of [35S]GTPγS binding at D2 receptors induced by 10 μM dopamine with a pKB of 8.74 ± 0.14 (Fig. 4B). These results demonstrate that functional inhibition of the SB269,652 plateau above basal level is strictly dependent on the agonist concentration, supporting the allosteric nature of this ligand in functional assays in addition to binding procedures.
Influence of SB269,652 on Quinpirole Induced D2 Activation of Gαqi5 Detected with the Aequorin Assay.
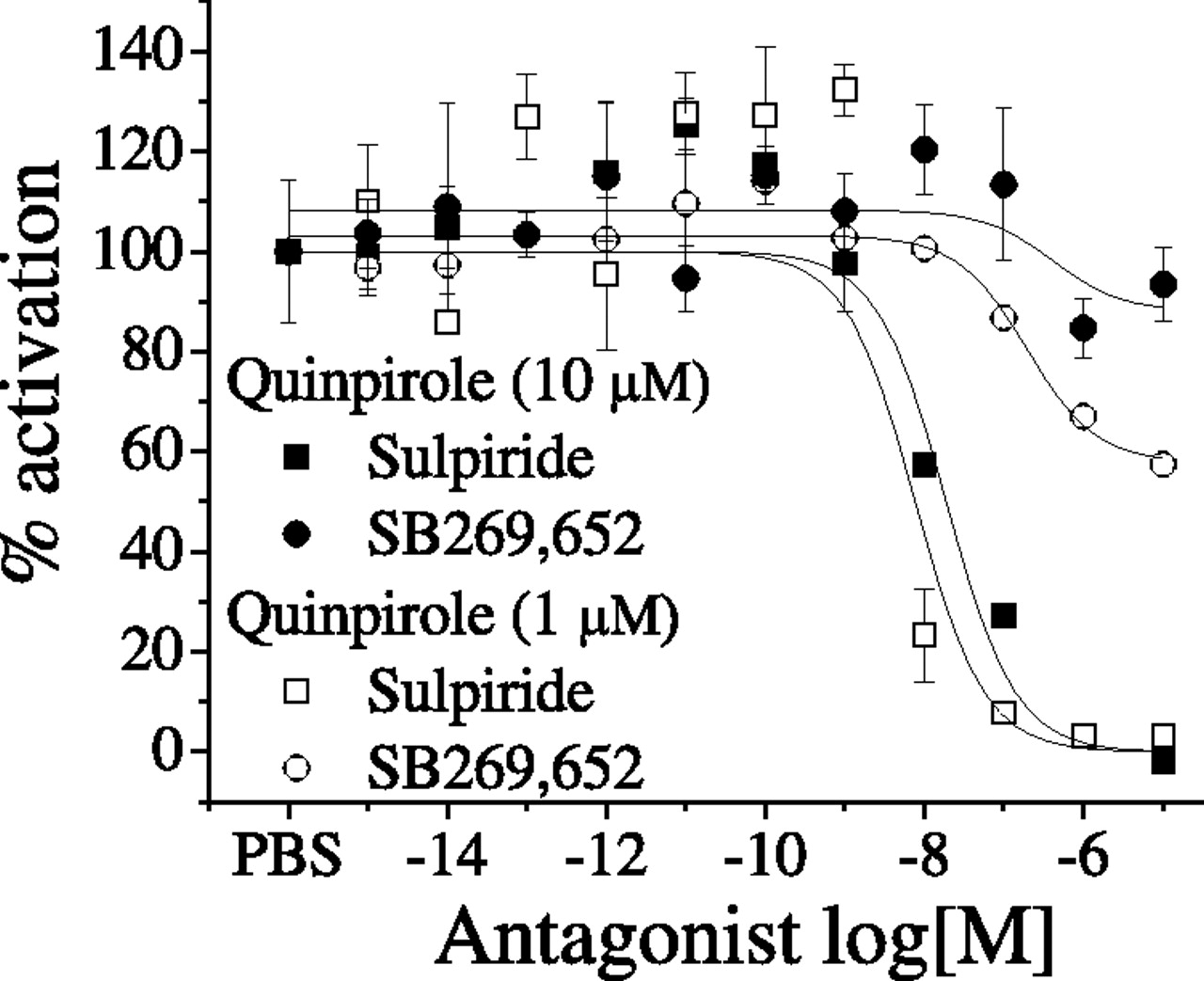
In Flp-In T-REx 293 cells, activation of D2, transfected alone, does not lead to luminescence, but cotransfection with a pertussis toxin-resistant Gαqi5 that can signal from Gαi-coupled receptors leads to activation-induced luminescence. In cells transfected with D2 and Gαqi5, quinpirole strongly stimulated the luminescence signal (data not shown). The competitive antagonist sulpiride completely inhibited the effect of 1 and 10 μM quinpirole with IC50 values of 11.5 ± 1.4 and 5.9 ± 1.6 nM, respectively (Fig. 5). In contrast, SB269,652 only submaximally suppressed D2 receptor-mediated stimulation of Gαqi5, and the extent of inhibition was dependent on the concentration of the agonist, with approximately 20 and 45% of inhibition at 10 and 1 μM quinpirole, respectively. The IC50 value of SB269,652 calculated in the presence of the two concentrations of quinpirole were very similar, 421 ± 16 and 212 ± 1.67 nM at 10 and 1 μM quinpirole, respectively. This novel approach to studying G protein activation strengthens the findings of [35S]GTPγS binding experiments in confirming the partial inhibitory effect of SB269,652 on D2 receptors.

Influence of SB269,652 and sulpiride on quinpirole-induced D2 activation of Gαqi5 detected with an aequorin assay. Antagonist effect of sulpiride and SB269,652 in presence of two different concentrations of quinpirole in Flp-In T-REx 293 cells cotransfected with D2L and Gαqi5 protein. Data are mean ± S.E.M. of three independent experiments, each performed in triplicate. PBS, phosphate-buffered saline.
Influence of SB269,652 upon Dopamine-Induced Phosphorylation of ERK1/2 at D3 and D2 Receptors.
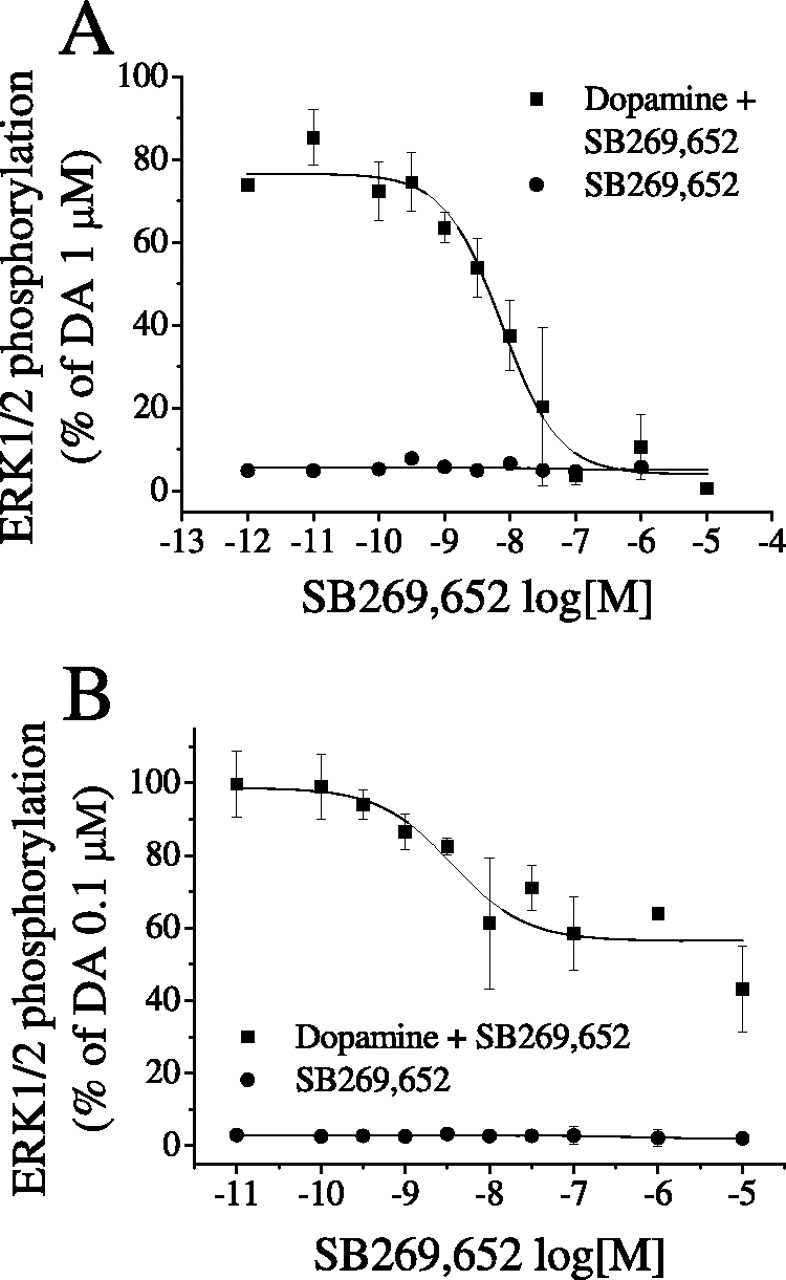
In another set of experiments, we tested how SB269,652 affects downstream G protein-dependent signaling. As described in previous studies (Cussac et al., 1999; Millan et al., 2004a), in CHO cells transfected with D3 receptors, dopamine triggered the transient phosphorylation of ERK1/2. In contrast to dopamine, SB269,652 failed to increase levels of the phosphorylated forms of ERK1/2 (Fig. 6A). Furthermore, SB269,652 concentration-dependently abolished activation by dopamine of ERK1/2 at D3 receptors with a pKB of 9.26 ± 0.19 (Fig. 6A). Dopamine also elicited a transient phosphorylation of ERK1/2 via D2 receptors. SB269,652 alone exerted no effect, but at concentrations up to 10 μM, it reduced the dopamine-induced phosphorylation of ERK1/2 by only 50% with a pKB value of 8.57 ± 0.21 (Fig. 6B). These results clearly demonstrate that SB269,652 inhibits ERK1/2 phosphorylation in the same way that it inhibits G protein activation.

Influence of SB269,652 upon D3 and D2 receptor-coupled ERK1/2 phosphorylation, as determined by immunoblot assays. Studies were performed at D3 and D2 receptors stably expressed in CHO cells. A, concentration-dependent antagonism by SB269,652 of dopamine (DA) induced ERK1/2 phosphorylation at CHO cell-expressed D3 receptors. B, concentration-dependent but partial attenuation by SB269,652 of dopamine-induced ERK1/2 phosphorylation at CHO cell-expressed D2 receptors. The concentration of dopamine was 1 μM and 0.1 μM for D3 and D2 receptors, respectively. Data are mean ± S.E.M. of three independent experiments, each performed in triplicate.
Influence of SB269,652 upon Dopamine-Induced Recruitment of β-Arrestin2 to the D2 Receptor.
These experiments were undertaken to test whether recruitment of β-arrestin2 was affected by SB269,652 in the same manner as G protein coupling. Agonist-induced recruitment of β-arrestin2 to D2 receptor was determined by a BRET assay. The addition of dopamine to cells coexpressing β-arrestin2-mVenus and D2L-Rluc8 induced a concentration-dependent increase in the BRET signal (data not shown). Sulpiride or SB269,652 given alone did not alter the BRET signal (Fig. 6). The effect of dopamine was completely prevented by preincubation with the competitive antagonist sulpiride with a pIC50 of 7.68 ± 0.03 nM, whereas it was reduced approximately 36% by SB269,652 with a pIC50 of 5.28 ± 0.03 μM (Fig. 7). These results show that, at least at D2 receptors, SB269,652 affects recruitment of β-arrestin2 in the same way that it affects G protein coupling.

Recruitment of β-arrestin2 to the dopamine D2 receptor detected by BRET assay. Flp-In T-REx 293 cells were stably transfected with human β arrestin2-mVenus as acceptor and Flag-D2L-Rluc8 as donor (see Materials and Methods). Cells were treated with sulpiride or SB269,652 at room temperature for 30 min, incubated with 5 μM coelenterazine h for 5 min, and then treated with 10 μM dopamine (DA). The BRET signal was determined at 5 min by calculating the ratio of the light emitted by mVenus (510–540 nm) over the light emitted by Rluc8 (485 nm). Data are mean ± S.E.M. of three independent experiments, each performed in triplicate.
Schild Analysis of the Actions of SB269,652 at D3 and D2 Receptors in [35S]GTPγS Binding Assays.
This last set of experiments was performed to further underpin the allosteric nature of SB269,652 and to estimate its affinity and cooperativity at dopamine D3 and D2 receptors. In CHO-D3 cells, increasing concentrations of SB269,652 induced a nonproportional rightward shift of the dose-response curve of dopamine with a significant decrease of agonist efficacy (Fig. 8A). Plotting the data by the Schild method of linear regression gave a slope significantly less than 1 (0.27). The predicted affinity of SB269,652 for the putative allosteric site at D3 receptors, calculated by the method of Ehlert (1988) for curvilinear Schild plots, was 0.17 ± 0.05 nM, and the cooperative factor α was 0.002 ± 0.0002 (Fig. 8B).
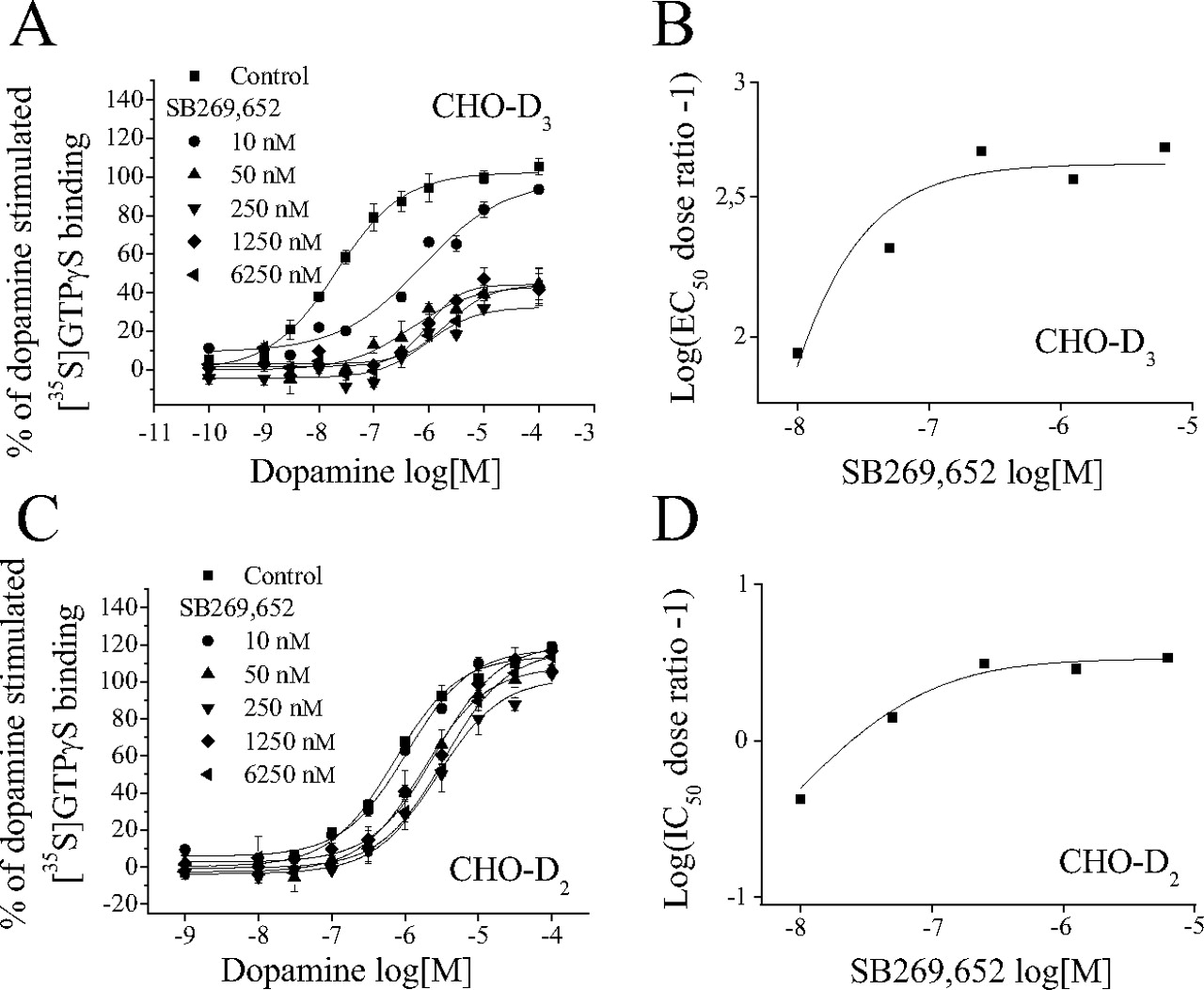
Schild analysis of the influence of SB269,652 upon dopamine-induced [35S]GTPγS binding to Gαi3 at D3 and D2 receptors expressed in CHO cells. A, concentration-response curves of dopamine for activation of Gαi3 at D3 receptors evaluated in the absence (■) or presence of incremental concentrations of SB269,652. B, Schild plot transformation of data in A. C, concentration-response curves of dopamine for activation of Gαi3 at D2 receptors evaluated in the absence (■) or presence of incremental concentrations of SB269,652. D, Schild plot transformation of data in C. In B and D, the points are fitted according to the allosteric model of Ehlert (1988) and correspond to the behavior expected for a negative allosteric modulator. Graphs are from representative experiments, each of which was undertaken three times and performed in triplicate.
As for D3 receptors, the decrease in potency of dopamine-mediated [35S]GTPγS binding to Gαi3 was not proportional to the increase in SB269,652 concentrations at D2 receptors (Fig. 8C). Schild analysis yielded a slope significantly less than 1 (0.3). The predicted affinity of SB269,652 for the putative allosteric site on D2 receptors was 13.1 ± 4.3 nM, and the cooperative factor α was 0.23 ± 0.01 (Fig. 8D).
Discussion
Binding Profiles of SB269,652 at Dopamine D3 and D2 Receptors.
At cloned D3 receptors, SB269,652 potently and completely inhibited the binding of two chemically distinct radiolabeled antagonists. However, increasing the concentration of the radioligands to 10 times their Kd unmasked the limited ability of maximal concentrations of SB269,652 to inhibit specific binding. In contrast to D3 receptors, SB269,652 less potently suppressed the binding of [3H]nemonapride and [3H]spiperone to D2 receptors, and inhibition of radioligand binding by SB269,652 reached a plateau at submicromolar concentrations. Furthermore, saturation binding experiments with and without a fixed concentration of SB269,652 showed a decrease in Bmax with no change in Kd, a typical feature of noncompetitive antagonism. This distinctive binding profile of SB269,652 at D3 and D2 receptors differentiates SB269,652 both from the preferential D2 antagonist L741,626 and from other ligands, including antipsychotics (e.g., sulpiride and haloperidol) and highly “selective” D3 receptor antagonists [e.g., (+)-trans-3,4,4a,5,6,10b-hexahydro-9-carbamoyl-4-propyl-2H-naphth [1,2-b]-1,4-oxazine (S33084) and N-{trans-4-[2-(6-cyano-3,4-dihydroisoquinolin-2(1H)-yl)ethyl]cyclohexyl}quinoline-4-carboxamide (SB277,011)] (Millan et al., 2000a,b; Reavill et al., 2000).
These results raise the possibility that SB269,652 is not competing for radioligands at orthosteric D3 and D2 binding sites but instead affecting their binding through an allosteric binding site. Our studies with chimeric D3/D2 receptors indicated an essential role for both the first and second extracellular loops in the higher potency of SB269,652 at D3 receptors, consistent with an allosteric site more extracellular than the orthosteric site located in a hydrophobic core created by the TMSs.
The muscarinic acetylcholine receptors, and in particular the M2 subtype, are among the best-characterized family A G protein-coupled receptors with respect to allosteric modulation by small molecules. The location of allosteric sites on these receptors have been studied in detail, and EL-II and -III and the proximal part of the adjacent TMSs have been implicated in binding allosteric compounds (Matsui et al., 1995; Prilla et al., 2006). Single residues, such as Tyr177 in EL-II and Trp422 in TM-VII of M2 can account for the selectivity of structurally different allosteric agents (Prilla et al., 2006). Further experiments with 1) chimeric D3/D2 receptors in which domain exchanges are less extensive (Alberts et al., 1998) and 2) receptors sustaining point mutations (Ballesteros et al., 2001; Shi and Javitch, 2004) would be instructive for more detailed characterization of the proposed binding sites of SB269,652 on D3 and D2 receptors. Strong negative cooperativity for [3H]nemonapride and [3H]spiperone would explain why SB269,652 abolishes the binding of lower concentrations of these radioligands at D3 sites, whereas weak negative cooperativity for these radioligands would explain why “saturating concentrations” of SB269,652 only partially affect binding at D2 sites. As discussed below, the hypothesis of an allosteric effect of SB269,652 at D3 and D2 site is corroborated by a series of functional experiments.
Antagonism of G-Protein Signaling, ERK1/2, and β-Arrestin Recruitment.
SB269,652 abolished dopamine-induced activation of Gαi3 in CHO-D3 cells, an inhibitory effect that became partial when the dopamine concentration was raised to 10 μM. As described previously in CHO cells, Gαi3 is a major link between D3 receptors and downstream cellular cascades such as ERK1/2 phosphorylation (Ahlgren-Beckendorf and Levant, 2004; Neve et al., 2004). The recruitment of ERK1/2 by D3 receptors is a pertussis toxin-sensitive phenomenon involving Gβγ subunits, an atypical protein kinase C, and the Phosphoinositide 3-kinase (Cussac et al., 1999; Newman-Tancredi et al., 1999; Beom et al., 2004; Neve et al., 2004). In the present study, as expected from experiments exploring the effects of SB269,652 upon G-protein coupling to D3 receptors, phosphorylation of ERK1/2 and protein kinase B (Akt) (Supplemental data) (Beaulieu et al., 2009) stimulated by dopamine was potently abolished by SB269,652 in CHO-D3 cells underpinning its antagonist properties at D3 receptors.
Contrasting with D3 receptors, SB269,652 only partially antagonized the recruitment of Gαi3 and Gαqi5 by dopamine in CHO-D2 and Flp-In T-REx 293 cells, respectively. In addition, the dopamine-mediated ERK1/2 phosphorylation in CHO-D2 cells was also partially inhibited by SB269,652. As shown in Supplemental Data, a series of functional assays (phosphorylation of Akt and measurement of cAMP accumulation) confirmed the partial effect of SB269,652 at D2 receptors. All together, these results suggest that pertussis toxin-sensitive G-protein-mediated signaling is only weakly and submaximally affected by SB269,652 at D2 transfected cells.
Initially, the recruitment of β-arrestins was thought to be involved mainly in receptor desensitization (DeWire et al., 2007; Kendall and Luttrell, 2009). Recent data indicate that β-arrestins can also recruit signaling cascades independently of G protein activation by forming multiprotein scaffolds that bring components of specific signaling pathways into close proximity (DeWire et al., 2007; Kendall and Luttrell, 2009). The BRET assay showed that SB269,652 only partially antagonized the recruitment of β-arrestin2 at D2 receptor expressing cells. These results indicate that both G proteins and β-arrestin-mediated signaling are similarly modulated by SB269,652, reinforcing the hypothesis of the allosteric nature of this compound.
Allosteric Interaction of SB269,652 with D3 and D2 Receptors: Schild and Kinetic Analysis.
The above-discussed binding and functional data collectively suggest that SB269,652 does not compete with radiolabeled ligands at the orthosteric site of D3 and D2 receptors but instead acts via an allosteric effect. Indeed, negative allosterism would explain why “saturating concentrations” of SB269,652 only partially compete with binding of radiolabeled ligands. Whereas a competitive ligand will decrease the bound radioligand down to nonspecific levels, the maximal inhibition produced by an allosteric antagonist will depend upon the magnitude of the cooperativity factor, α, and the concentration of the radioligand. The lower the degree of negative cooperativity and the higher the concentration of the radioligand, the less the allosteric antagonist will inhibit. For example, the prototypical allosteric modulator of muscarinic M2 receptors gallamine, which is characterized by negative cooperativity, completely inhibits specific binding of radioligands when evaluated at their Kd values, but increasing the concentration of the radioligand to 10 times the Kd unmasks the limited ability of the negative allosteric modulator to inhibit specific binding (Lazareno and Birdsall, 1995; Christopoulos and Kenakin, 2002; May et al., 2007). Herein, this occurred with D3 receptors in both binding and functional assays where increasing the concentration of the radioligand and the agonist, respectively, unmasked the limited ability of SB269,652 to inhibit binding and function.
Two further arguments favor an allosteric interaction of SB269,652 with D3 and D2 receptors. First, in CHO cells expressing D2 receptors, increasing concentrations of SB269,652 elicited a nonproportional rightward shift of the dopamine concentration-response curve in [35S]GTPγS binding assay without a significant change of agonist efficacy. This observation is compatible with the influence of an allosteric modulator upon orthosteric ligand affinity. In addition, a notable deviation from linearity was observed in the Schild analysis, a phenomenon occurring with allosteric antagonism and reflecting the saturable nature of the antagonism (Christopoulos and Kenakin, 2002).
Concerning D3 receptors, a nonproportional rightward shift of the dopamine concentration-response curve was also observed in [35S]GTPγS binding assay. In this functional assay, high concentrations of SB269,652 elicited a strong decrease of agonist efficacy at D3 sites compatible with an allosteric compound. In addition, the displacement of the dopamine concentration-response curves by SB269,652 also reached a plateau, as reported for D2 receptors; suggesting a negative allosteric effect (Christopoulos and Kenakin, 2002). In line with this allosteric profile of SB269,652, the Schild regression deviated from a straight line with a slope less than 1.
Second, at D2 and D3 receptors, binding kinetics of [3H]nemonapride and [3H]spiperone were clearly modified in the presence of SB269,652 compared with the two orthosteric antagonists haloperidol and sulpiride. It is noteworthy that at both D3 and D2 receptors, dissociation kinetics was decreased by SB269,652. This finding could be interpreted as SB269,652 physically occluding the orthosteric site so that competitive antagonists are unable to readily leave (or enter) the site if the allosteric modulator is present (Christopoulos and Kenakin, 2002; May et al., 2007). To compensate for this reduction in off rate, a proportional on-rate decrease left the ratio between the dissociation and association rate constants unchanged.
Collectively, then, four complementary lines of evidence strongly indicate that SB269,652 behaves as a negative allosteric modulator at both D2 and D3 receptors. Two recently developed compounds, (−)-OSU6162 and ACR16, were also suggested to act allosterically at D2 receptors, although data were complex and supporting evidence limited (Tamminga and Carlsson, 2002; Rung et al., 2008). The stimulation of dopamine-induced [35S]GTPγS incorporation by low (−)-OSU6162 concentrations was attributed to an allosteric site, whereas higher concentrations of (−)-OSU6162 antagonized dopamine by an action at the orthosteric site. A mixed competitive/allosteric interaction was also proposed for amiloride and its nitrogen-substituted derivatives on the basis of kinetic and equilibrium binding data (Hoare and Strange, 1996). In contrast to (−)-OSU6162, amiloride, and its nitrogen-substituted derivatives, SB269,652 seems to be a “pure” allosteric compound at D2 and D3 receptors.
Concluding Remarks.
To summarize, the chemically distinct tetrahydroisoquinoline derivative, SB269,652, behaves as a highly potent allosteric antagonist at recombinant D3 receptors, whereas it less potently and submaximally interferes with D2 receptors. As such, SB269,652 should prove of unique utility for the definition of actions mediated via D3 compared with D2 receptors, in both cellular and in vivo procedures. Furthermore, SB269,652 could prove useful as a lead for the design of novel allosteric ligands at D2 and or D3 receptors with potential advantages relative to conventional orthosteric agents in the treatment of psychiatric and neurological disorders.
Footnotes
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.This work was supported in part by Fondo per gli Investimenti della Ricerca di Base (FIRB) [Grant RBIN04CKYN]; Programmi di Ricerca Scientifica di Rilevante Interesse Nazionale (PRIN) [Grant 20085PPEK7]; the National Institutes of Health National Institute on Drug Abuse [Grant DA022413]; the National Institutes of Health National Institute of Mental Health [Grant MH54137]; the Lieber Center for Schizophrenia Research and Treatment; and IDR Servier (to E.S.).
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.110.065755.
-
ABBREVIATIONS:
- OSU6162
- (3S)-3-[3-(methylsulfonyl)phenyl]-1-propylpiperidine hydrochloride
- ACR16
- pridopidine
- SB269,652
- 1H-indole-2-carboxylic acid {4-[2-(cyano-3,4-dihydro-1H-isoquinolin-2-yl)-ethyl]-cyclohexyl}-amide
- CHO
- Chinese hamster ovary
- BRET
- bioluminescence resonance energy transfer
- ERK
- extracellular signal-regulated kinase
- TMS
- transmembrane segment
- EL
- extracellular loop
- L741,626
- 3-[4-(4-chlorophenyl)-4-hydroxypiperidin-l-yl]methyl-1H-indole
- GTPγS
- guanosine 5′-O-(3-thio)triphosphate
- S33084
- (+)-trans-3,4,4a,5,6,10b-hexahydro-9-carbamoyl-4-propyl-2H-naphth[1,2-b]-1,4-oxazine]
- SB277
- 011N-{trans-4-[2-(6-cyano-3,4-dihydroisoquinolin-2(1H)-yl)ethyl]cyclohexyl}quinoline-4-carboxamide.

- Received April 21, 2010.
- Accepted July 27, 2010.
- Copyright © 2010 The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}