


Abstract
The compound 4-(5-(4-bromophenyl)-3-(6-methyl-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid (DQP-1105) is a representative member of a new class of N-methyl-d-aspartate (NMDA) receptor antagonists. DQP-1105 inhibited GluN2C- and GluN2D-containing receptors with IC50 values that were at least 50-fold lower than those for recombinant GluN2A-, GluN2B-, GluA1-, or GluK2-containing receptors. Inhibition was voltage-independent and could not be surmounted by increasing concentrations of either coagonist, glutamate or glycine, consistent with a noncompetitive mechanism of action. DQP-1105 inhibited single-channel currents in excised outside-out patches without significantly changing mean open time or single-channel conductance, suggesting that DQP inhibits a pregating step without changing the stability of the open pore conformation and thus channel closing rate. Evaluation of DQP-1105 inhibition of chimeric NMDA receptors identified two key residues in the lower lobe of the GluN2 agonist binding domain that control the selectivity of DQP-1105. These data suggest a mechanism for this new class of inhibitors and demonstrate that ligands can access, in a subunit-selective manner, a new site located in the lower, membrane-proximal portion of the agonist-binding domain.
Introduction
The N-methyl-d-aspartate (NMDA) receptors belong to the family of ionotropic glutamate receptors that mediate the majority of excitatory neuronal transmission within the central nervous system (Traynelis et al., 2010). NMDA receptors comprise four subunits that combine to form functional ion channels. Each subunit folds into four semiautonomous domains, including the amino-terminal domain, the ligand-binding domain, the transmembrane domain, and the carboxyl-terminal domain. Functional NMDA receptors are formed by the assembly of two GluN1 subunits that bind the coagonist glycine together with two GluN2 subunits that bind the coagonist glutamate. Four independent genes encoding GluN2 subunits have been identified (GluN2A–D) and account for differences in functional properties of the NMDA receptors, including deactivation time course, agonist potency, mean open time, and open probability (Dingledine et al., 1999; Chen and Wyllie, 2006; Paoletti and Neyton, 2007; Traynelis et al., 2010). The GluN2 subunit also controls the sensitivity of the receptor to Mg2+ block, proton inhibition, modulation by Zn2+, as well as potency and efficacy of the endogenous agonists glutamate and glycine (Traynelis et al., 2010).
The spatial and temporal expression of the various GluN2-containing NMDA receptors has led to hypotheses concerning specific roles for the various NMDA receptor subunits in certain pathophysiological conditions, including Parkinson's disease and schizophrenia (Hallett and Standaert, 2004; Chen and Lipton, 2006). Despite considerable progress in our understanding of ionotropic glutamate receptor structure and function, the lack of subunit-selective pharmacological tools targeting NMDA receptors has impeded advances in understanding the roles of different GluN2-containing NMDA receptor subtypes both in normal brain function and in various disease states. Here we report a new structural class of antagonists, typified by 4-(5-(4-bromophenyl)-3-(6-methyl-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid (DQP-1105), which is approximately 50-fold selective for GluN2C or GluN2D subunit-containing NMDA receptors over GluN2A- or GluN2B-containing receptors. We determined the mechanism of action and identified the structural determinants of subunit-selectivity for DQP-1105. Identification of the structure-activity relationship for DQP-1105, the structural determinants of subunit selectivity, and the mechanism of action of this new class of NMDA receptor antagonist will facilitate the development of pharmacological probes with increased selectivity among GluN2 subunits.
Materials and Methods
Molecular Biology.
cDNAs for rat wild-type NMDA receptor subunits GluN1-1a (GenBank accession number U08261; hereafter GluN1), GluN2A (GenBank accession number D13211), GluN2B (GenBank accession number U11419), GluN2C (GenBank accession number M91563), GluN2D (GenBank accession number L31611; see Monyer et al., 1994), GluA1 (GenBank accession number X17184), and GluK2 (GenBank accession number Z11548) were provided by Drs. S. Heinemann (Salk Institute for Biological Sciences, La Jolla, CA), S. Nakanishi (Kyoto University, Kyoto, Japan), and P. Seeburg (University of Heidelberg, Heidelberg, Germany). cDNAs encoding the full open reading frames of human NMDA receptor subunits were assembled from pieces of cDNA obtained from the I.M.A.G.E. Consortium (http://image.hudsonalpha.org/) and a commercial source (Origene, Rockville, MD). High GC-content in the open reading frames of human GluN2C and GluN2D subunits was reduced to increase expression in Xenopus laevis oocytes. The final cDNAs encode protein sequences corresponding to wild-type human NMDA receptor subunits GluN1-1a (GenBank accession number NP_015566), GluN2A (GenBank accession number NP_000824), GluN2B (GenBank accession number NP_000825), and GluN2D (GenBank accession number NP_000827.1). The cDNA encoding human GluN2C contained a Glu-Pro-Pro insertion at amino acid position 1048 (as in GenBank accession number NP_000826.1) and an arginine residue at amino acid position 1212 (as in GenBank accession number NP_000826.2) and is therefore an intermediate between GenBank accession numbers NP_000826.1 and NP_000826.2. Chimeric receptor subunits were constructed as described previously (Table 5) (Mullasseril et al., 2010; Hansen and Traynelis, 2011). Site-directed mutagenesis was accomplished using the QuikChange approach (Agilent Technologies, Santa Clara, CA). All DNA constructs were verified by sequencing (SeqWright, Houston, TX).
Cell Culture.
HEK 293 cells (American Type Culture Collection, Rockville, MD; hereafter HEK cells) were plated on glass coverslips (5 mm diameter; Warner Instruments, Hamden, CT) coated with 0.1 mg/ml poly-d-lysine. HEK cells were maintained in 5% humidified CO2 at 37°C in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum, 10 units/ml penicillin, and 10 μg/ml streptomycin. HEK cells were transiently transfected using the FuGENE 6 transfection reagent (Roche Diagnostics, Indianapolis, IN) with cDNAs encoding green fluorescent protein, GluN1, and GluN2A or GluN2D at a ratio of 1:1:1 and 0.5 μg/well total cDNA for 16 to 24 h before whole-cell voltage-clamp recordings were conducted, as described previously (Yuan et al., 2009). After transfection, cells were incubated in media supplemented with NMDA receptor antagonists dl-2-amino-5-phosphonovalerate (200 μM) and 7-chlorokynurenic acid (200 μM). BHK-21 cells (American Type Culture Collection; hereafter BHK cells) were maintained in Dulbecco's modified Eagle's medium (Invitrogen), 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 1 mg/ml G418, 10 μg/ml blasticidin, 200 μM 7-chlorokynurenic acid, and 200 μM d-(−)-2-amino-5-phosphonopentanoic acid.
Ca2+ Imaging from BHK Cells.
Experiments were performed as described previously (Hansen et al., 2008) with the following modifications. One day before the experiment, the cells were seeded in 20 μl of media at 6 × 105 cells/ml in black, clear-bottomed 384-well plates (CellBind; Corning Life Sciences, Lowell, MA) using a Multidrop 384 Reagent Dispenser (Thermo Fisher Scientific, Waltham, MA). On the day of the experiment, the media was gently aspirated by a 384-format Biomek liquid handler (Beckman Coulter, Fullerton, CA). The cells were then loaded with Fluo-4 “no wash” (Invitrogen) dissolved in HEPES-buffered saline (Invitrogen) with 2.5 mM (1%) probenecid and 30 μM 7-chlorokynurenic acid for 60 min at 37°C in the dark. Cells were gently washed again with 30 μl/well using the same buffer without Fluo-4 dye, and placed in 20 μl/well buffer. The cell plates were then loaded into a functional drug-screening system (FDSS7000; Hamamatsu Corporation, Bridgewater, NJ), and real-time recordings of changes in Fluo-4 emission were performed (excitation at 480 nm and emission at 540 nm) at room temperature (20–22°C). After 10 s of baseline recordings, 10 μl/well of 3× concentrated test compound, controls, or assay buffer in HEPES-buffered saline, pH 7.4, and 1 mM glycine (final concentration) were added. The high glycine concentration will displace the 7-chlorokynurenic acid (Hansen et al., 2010b). After 2 min, an additional 10 μl/well of a 4× concentrated EC80 solutions of NMDA (110 μM for GluN2A, 100 μM for GluN2C, or 30 μM for GluN2D) were added; an EC100 concentration of NMDA (1 mM) was used for GluN2B. Changes in fluorescence were subsequently recorded for 2 min. For determination of the concentration-response relationships, test compounds were 3-fold serially diluted over 10 concentration steps. Responses [fluorescence units (FU)] were normalized to the first recording and expressed as percentage of NMDA activation (no inhibition) and NMDA plus 100 μM dizocilpine maleate (MK801) as 100% inhibition according to

The IC50 value was determined by nonlinear least-squares fitting as described below.
Whole-Cell and Single-Channel Patch-Clamp Recordings.
Whole-cell voltage-clamp recordings were conducted on transiently transfected HEK cells using an Axopatch 200B amplifier (Molecular Devices, Union City, CA). Current responses were digitized at 40 kHz by pClamp10 software (Molecular Devices). Recordings were filtered at 8 kHz using an eight-pole Bessel filter (-3 dB; Frequency Devices, Haverhill, MA). Thin-walled borosilicate glass capillary tubes (World Precision Instruments, Sarasota, FL) were used to form recording micropipettes for whole-cell currents; thick-walled borosilicate capillary tubes (Warner Instruments) were used to form micropipettes for single-channel recording. All recording micropipettes were filled with an internal solution containing 110 mM d-gluconic acid, 110 mM CsOH, 30 mM CsCl, 5 mM HEPES, 5 mM 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA), 4 mM NaCl, 2 mM MgCl2, 2 mM NaATP, 0.5 mM CaCl2, and 0.3 mM NaGTP, pH 7.35. Cells were bathed at 23°C in external solution that contained 150 mM NaCl, 10 mM HEPES, 3 mM KCl, 0.5 mM CaCl2, 0.01 mM EDTA, and 30 mM d-mannitol at pH 7.4. All recording solutions were made from external solution, and recordings were performed at holding potentials of −60 mV unless otherwise stated. Rapid solution exchange for macroscopic recordings was accomplished with a two-barrel θ-shaped glass pipette controlled by a piezoelectric translator (Burleigh Instruments, Fishers, NY); 10-to-90% open tip solution exchange times were under 1 ms, and solution exchange around the whole cell was under 5 ms (Vance et al., 2011).
Outside-out patches containing multiple GluN1/GluN2D channels were excised from transfected HEK cells and recorded at a holding potential of −80 mV at pH 8.0. Recombinant NMDA receptors were activated by a maximally effective concentration of glutamate (1 mM) and glycine (50 μM) for 2 min, after which glutamate and glycine were coapplied with increasing concentrations (3–30 μM) of DQP-1105 for 2 min. Response was also measured after removal of DQP-1105.
Perforated Patch-Clamp Recordings.
Perforated whole-cell patch-clamp recording from transfected HEK cells under voltage clamp (holding potential, −60 mV) was performed with an Axopatch 200B amplifier. The recording chamber was continually perfused with the recording solution described above. Recording electrodes (3–4 MΩ) were filled with 150 mM CsMeSO4, 10 mM NaCl, 0.5 mM CaCl2, 10 mM HEPES, and 25 to 50 μg/ml gramicidin D (pH adjusted to 7.3 with CsOH and osmolality adjusted to 310 mOsmol/kg with sucrose). Once a gigaohm seal was achieved, the cell was lifted and held in the bath until electrical access to the intracellular compartment was achieved by gramicidin (Ebihara et al., 1995). It took 10 to 30 min to achieve perforation with series resistance ranging from 30 to 60 MΩ. Rapid solution exchange was achieved with a two-barreled θ-shaped glass pipette controlled by a piezoelectric translator as described above. The cell was first exposed to 100 μM glutamate and 100 μM glycine, followed by a jump to 100 μM glutamate and 100 μM glycine with various concentrations of DQP-1105 (0.3–30 μM) for 3 s, and a subsequent wash-out of antagonist with 100 μM glutamate and 100 μM glycine. All electrophysiological data from HEK cells in this study were collected at room temperature (23°C).
Two-Electrode Voltage-Clamp Recording from X. laevis Oocytes.
Two-electrode voltage-clamp recordings were performed in X. laevis oocytes expressing recombinant rat GluN1/GluN2A, GluN1/GluN2B, GluN1/GluN2C, GluN1/GluN2D, GluA1, or GluK2. In some experiments, recordings were performed in oocytes expressing recombinant human GluN1/GluN2A, GluN1/GluN2B, GluN1/GluN2C, or GluN1/GluN2D. Oocytes were isolated from X. laevis as approved by the Emory University Institutional Animal Care and Use Committee and treated according to methods described previously (Dravid et al., 2007). The pipettes for cRNA injection were filled with mineral oil and attached to an automatic injector (Nanoject II; Drummond Scientific, Broomall, PA). The cRNA was transcribed in vitro using the mMessage Machine kit (Ambion, Austin, TX), diluted with nuclease-free water, and injected at a ratio of 1:2 GluN1/GluN2 (5–10 ng of total cRNA). The oocytes were kept in Barth's solution composed of 88 mM NaCl, 5 mM Tris-HCl, 2.4 mM NaHCO3, 1 mM KCl, 0.84 MgSO4, 0.41 mM CaCl2, 0.33 mM Ca(NO3)2, 0.1 mg/ml gentamicin sulfate, 1 U/ml penicillin, and 1 μg/ml /streptomycin at pH 7.4 at 15–17°C for 2 to 5 days before experiments.
Oocytes were placed into a perfusion chamber and continually washed with recording solution containing 90 mM NaCl, 1.0 mM KCl, 0.5 mM BaCl2, 0.005 mM EDTA, and 10 mM HEPES at pH 7.4 (23°C). Glass electrodes with a tip resistance of 0.5 to 2.5 MΩ were pulled from thin-walled glass capillary tubes (World Precision Instruments). Voltage electrodes were filled with 0.3 M KCl, and current electrodes were filled with 3.0 M KCl. Voltage clamp recordings were conducted at a holding potential of −40 mV using an amplifier (OC-725; Warner Instrument Co). For some experiments, the current-voltage relationship was evaluated by stepping the membrane potential in 10-mV increments from −60 to +30 mV. All compounds were made as 20 mM stock solutions in DMSO and dissolved to reach the final concentration in recording solution. Final DMSO content was 0.05 to 0.5% (v/v). Oocytes expressing GluK2 were pretreated with 10 μM concanavalin-A for 10 min before recording. Some oocytes expressing GluN1/GluN2A were pretreated with 50 μM BAPTA-acetoxymethyl ester for 10 min or injected with 50 nl of 2 mM K-BAPTA to prevent gradual run-up of the current response.
Data Analysis.
Antagonist IC50 values were determined by nonlinear least-squares fitting of the equation
 to concentration-response data from individual experiments normalized to the response in the absence of inhibitor (100%), where IC50 is the concentration that inhibits the response half-maximally and nH is the Hill slope; inhibition was assumed to be complete at saturation. Some IC50 values were determined by fitting eq. 2 to mean composite data (HEK patch experiments, data describing the structure-activity relationship for multiple ligands). Agonist concentration-response data were fitted by the equation
to concentration-response data from individual experiments normalized to the response in the absence of inhibitor (100%), where IC50 is the concentration that inhibits the response half-maximally and nH is the Hill slope; inhibition was assumed to be complete at saturation. Some IC50 values were determined by fitting eq. 2 to mean composite data (HEK patch experiments, data describing the structure-activity relationship for multiple ligands). Agonist concentration-response data were fitted by the equation
 where EC50 is the concentration of agonist that produces a half-maximally effective response and nH is the Hill slope. In patch recordings from transfected HEK cells, the GluN1/GluN2D current responses to coapplication of agonist and DQP-1105 relaxed with a single exponential time course that we interpret to reflect the time course of association of drug with receptor. We fitted this response time course to an exponential function
where EC50 is the concentration of agonist that produces a half-maximally effective response and nH is the Hill slope. In patch recordings from transfected HEK cells, the GluN1/GluN2D current responses to coapplication of agonist and DQP-1105 relaxed with a single exponential time course that we interpret to reflect the time course of association of drug with receptor. We fitted this response time course to an exponential function
 where τONSET describes the onset of DQP-1105 inhibition. The association rate (kON) of DQP-1105 was calculated by plotting 1/τONSET versus DQP-1105 concentration. Linear regression analysis was calculated for
where τONSET describes the onset of DQP-1105 inhibition. The association rate (kON) of DQP-1105 was calculated by plotting 1/τONSET versus DQP-1105 concentration. Linear regression analysis was calculated for
 where kON is the slope and describes the association rate of the compound, kOFF is the y-intercept and describes the dissociation rate, and [inhibitor concentration] is the concentration of the DQP-1105 compound. The dissociation constant KD was determined from kOFF/kON.
where kON is the slope and describes the association rate of the compound, kOFF is the y-intercept and describes the dissociation rate, and [inhibitor concentration] is the concentration of the DQP-1105 compound. The dissociation constant KD was determined from kOFF/kON.
Channel recordings were filtered after acquisition using a digital 4-kHz Bessel (eight-pole) filter and idealized using time course fitting of openings [SCAN, provided by Dr. David Colquhoun, University College London, London, UK (http://www.ucl.ac.uk/Pharmacology/dcpr95.html#scan)]. Open and closed duration histograms were constructed using EKDIST (http://www.ucl.ac.uk/Pharmacology/dcpr95.html#ekdist) with an open-time resolution of 53 μs and a shut-time resolution of 31 μs (Colquhoun and Sigworth, 1995). Open periods were defined as the duration of all contiguous openings, regardless of conductance level. That is, openings that showed direct transition between two conductance levels were treated as having a single duration. Although all patches contained several active channels, only a minority of openings (∼1%) were due to simultaneous opening of two or more channels. Thus, multiple simultaneous channel openings were easily identified and excluded from open- and closed-duration histograms and for calculation of mean open time, mean shut time, and POPEN. POPEN was calculated according to the equation
 where MOT is the mean open time, MST is the mean shut time, NumberOPEN is the number of openings, and NumberCLOSE is the number of closed durations.
where MOT is the mean open time, MST is the mean shut time, NumberOPEN is the number of openings, and NumberCLOSE is the number of closed durations.
Similar results for POPEN were found by integrating the idealized trace to obtain the average current level of the recording and dividing by the average single channel current. Open-period distributions and shut-duration distributions were fitted with mixtures of exponential components using maximum likelihood estimation with EKDIST. Open point amplitude distributions were fitted by the sum of two Gaussian components (EKDIST).
Computational Modeling.
The comparative homology modeling program MODELLER 9v1 (Sali and Blundell, 1993) was used to generate homology models for the GluN1/GluN2D and GluN1/GluN2A containing NMDA receptors. A protein family alignment was generated using the rat sequences for the NMDA receptors (GluN1, GluN2A-D), AMPA receptors (GRIA1–4), and kainate receptors (GRIK1–5). The MUSCLE alignment program was used to align the sequences and was subsequently used in model construction (Edgar, 2004). The crystal structure of the AMPA receptor GluA2 (PDB entry 3KG2; 3.60-Å resolution) was used as template, which served as a scaffold to build an NMDA receptor that includes the amino-terminal domain, the ligand binding domain, and the transmembrane domain. The symmetry used in the construction of the models is the same as that suggested by Sobolevsky et al. (2009): Chain A,C = GluN1; Chain B,D = GluN2. Because of a low overall homology between GluA2 and the NMDA receptors, multiple templates were used (19% sequence identity between GluA2 and GluN2D and 23% sequence identity between GluA2 and GluN1). The resolved structure for the monomeric ATD of GluN2B (Karakas et al., 2009; PDB entry 3JPY; 3.21-Å resolution) was used as a template to model the amino-terminal domain of both GluN1 and GluN2 (sequence identities of 14 and 23%, respectively). The structure of the ligand binding domain of the GluN1 and GluN2A complex (PDB entry 2A5T; 2.00-Å resolution; GT-linker removed) was used as a template to model the respective domain in the GluN1/GluN2D models (Furukawa et al., 2005). The transmembrane region was modeled based on the AMPA coordinates, the unresolved regions being omitted. The model was subjected to quality analysis using PDB sum generator (http://www.ebi.ac.uk/pdbsum; Laskowski, 2009). The analysis revealed some discrepancy in the overall G factors between the crystal structure (0.32) and the GluN1/GluN2D (−0.29) model, which is expected for models built on low-resolution structures with low homology; scores above −0.5 are considered acceptable.
Reagents.
The dihydroquinolone-pyrazoline analog DQP-997 (3; 4-(3-(6-bromo-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4-bromophenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid) was obtained from Asinex (Moscow, Russia). DQP-1105, DQP-1209 (9; 4-(3-(6-bromo-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(2-chlorophenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid), DQP-1210 (22; 4-(3-(6-bromo-4-phenylquinolin-3-yl)-5-(4-bromophenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid), DQP-1308 (18; 4-(3-(6-methyl-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-phenyl-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid), and DQP-1309 (21; 6-bromo-3-(5-(4-bromophenyl)-1-propionyl-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one) were obtained from Pharmeks (Moscow, Russia); DQP-1177 (25; 4-(3-(4-bromophenyl)-5-(6-methyl-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid), DQP-1178 (8; 4-(3-(6-bromo-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(2-fluorophenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid), DQP-1183 (1; 4-(3-(6-bromo-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-phenyl-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid), DQP-1184 (2; 4-(3-(6-bromo-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4-fluorophenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid), DQP-1185 (5; 4-(3-(6-bromo-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(p-tolyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid), and DQP-2003 (7; 4-(3-(6-bromo-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(2,5-dimethoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid) were obtained from Life Chemicals Inc. (Kiev, Ukraine). DQP-1250 (24; 3-(1-acetyl-5-(4-bromophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-6-methyl-4-phenylquinolin-2(1H)-one) and DQP-2005 (20; 4-(5-(2-methoxyphenyl)-3-(6-methyl-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid) were obtained from Enamine (Kiev, Ukraine). DQP-2033 (4; 4-(3-(6-bromo-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4-nitrophenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid) was obtained from Chembridge (Moscow, Russia). DQP-1128 (6; 4-(3-(6-bromo-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid) was obtained from SPECS (Wakefield, RI). All compounds were more than 90% pure as determined by the chemical suppliers. Glutamate, glycine, and all other chemicals were from Sigma-Aldrich (St. Louis, MO). DQP-1105 had a molecular weight of 558.42, a maximum solubility at 1 h in oocyte recording buffer plus 0.1% DMSO of 27 μM, and maximum solubility at 24 h in phosphate-buffered saline (no DMSO) of 6.5 μM (Supplemental Information). DQP-1105 is also known as DC060015 and was identified in a virtual screen as a low-potency (KD, 18.6 μM) inhibitor of SARS-Coronavirus 3CL, where it was reported to have a CLogP value of 6.55 (Chen et al., 2006).
Results
Structure Activity Relationship for Dihydroquinolone-Pyrazolines.
The dihydroquinilone-pyrazoline scaffold was identified as an inhibitor of NMDA receptors from a multiwell Ca2+-based screen of approximately 105 compounds from ChemDiv and Asinex diversity libraries against GluN1/GluN2C- or GluN1/GluN2D-expressing BHK cell lines (Hansen et al., 2010b). Evaluation of a subset of analogs sharing this scaffold led to the identification of DQP-1105 (Fig. 1A), which is the primary focus of this study. Various substitution patterns of the dihydroquinolone-pyrazoline-containing scaffold (Table 1) affect potency and selectivity and reveal several key features of the ligand-receptor interactions. Beginning from the initial screening hit (compound 3), we evaluated the IC50 value at four different recombinant NMDA receptors for analogs that altered substituents at five positions as a starting point for assessing the structure-activity relationship. The IC50 values of compound 3 ranged from 2.7 μM at GluN2D-containing receptors to >75 μM at GluN2A when evaluated at recombinant receptors expressed in X. laevis oocytes. Evaluation of various substitutions on the C-ring while the A-ring R1 bromine was held constant showed that the R4-bromo (compound 3) is the most favorable configuration. Substituting R1 of the A-ring with a methyl group led to compound 19, referred hereafter to as DQP-1105, which shows the most favorable combination of potency and selectivity for GluN2D over GluN2A and GluN2B of all of the analogs tested. We also evaluated core structural perturbations to better understand the requirements of activity (Table 2). Beginning with analogs of the initial screening hit (compound 3), truncation of the acyl-chain off the pyrazoline nitrogen from an acid to an ethyl group (compound 21) eliminated antagonist activity. Likewise, removal of the succinate group and substituting with R1-methyl analog (compound 24) abolishes activity. Removing the oxo-functionality from the quinoline (compound 22), which removes a resonance form, also eliminates activity (Table 2). Thus, the position of the imine within the pyrazoline compound as well as the connectivity, length, and charge of the acyl chain are important features for potency and selectivity. We selected DQP-1105 (compound 19) as the compound with the best combination of potency and selectivity to study in terms of mechanism of action.

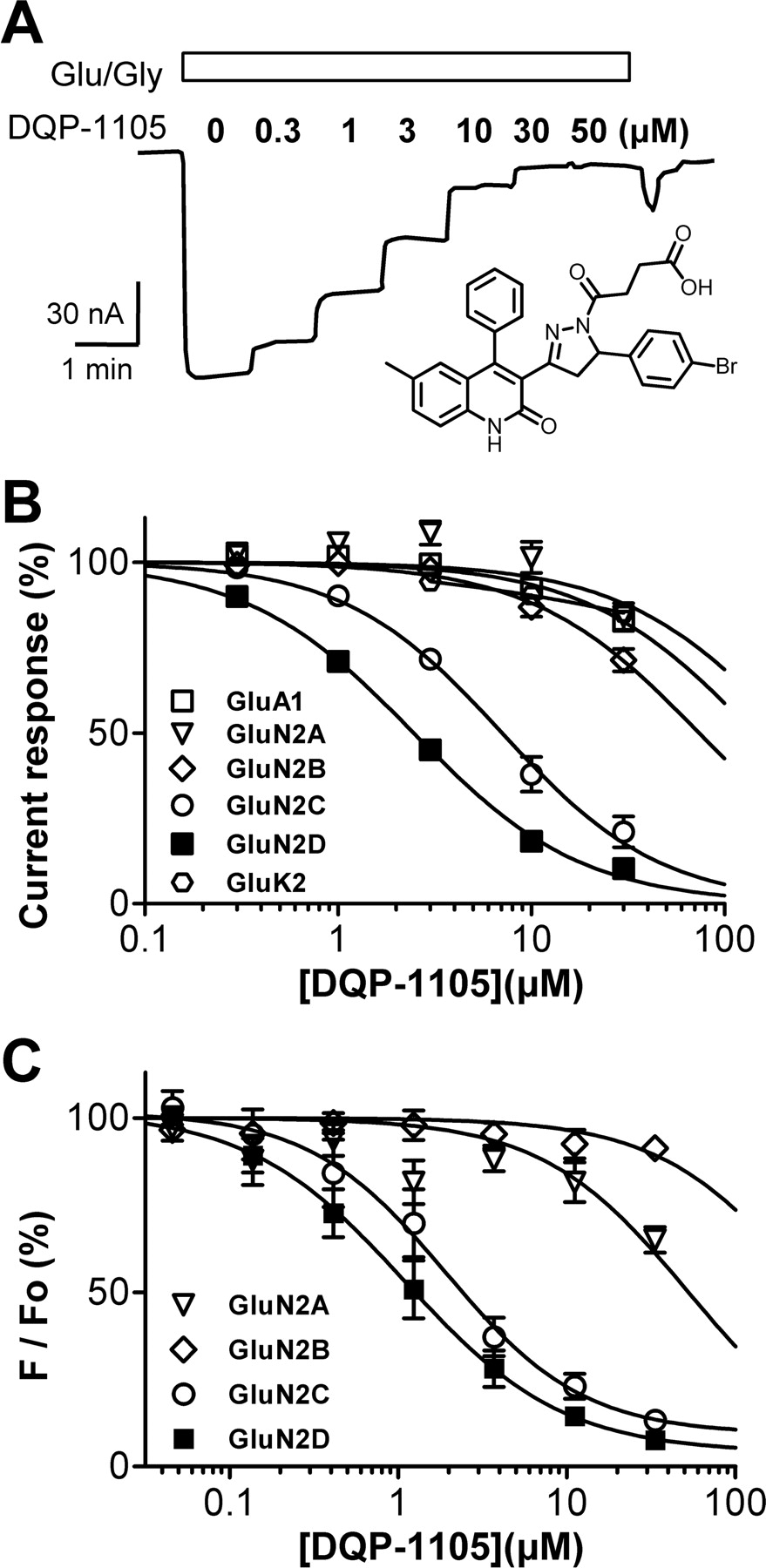
Subunit selectivity of DQP-1105. A, representative current response of GluN1/GluN2D receptors expressed in X. laevis oocyte recorded during coapplication of 100 μM glutamate, 30 μM glycine (Glu/Gly), and the designated concentration of DQP-1105 (structure shown in inset). B, composite concentration-effect curves determined using two-electrode voltage-clamp electrophysiology for DQP-1105 against recombinant AMPA, kainate, and NMDA receptors expressed in X. laevis oocytes. C, concentration-effect curves determined using fluorescence-based measurements of intracellular calcium expressed as a percent of baseline fluorescence (F/Fo) for DQP-1105 inhibition of recombinant GluN1/GluN2A, GluN1/GluN2B, GluN1/GluN2C, or GluN1/GluN2D stably expressed in BHK cells (see Materials and Methods). IC50 values are listed in Table 3.
Structure-activity relationship for DQP analogs
Fitted IC50 values were determined for the indicated combinations of recombinant GluN1/GluN2 receptors expressed in X. laevis oocytes and activated by 100 μM glutamate plus 30 μM glycine. Compounds that produced less than 25% inhibition at 30 μM were considered to have no effect. IC50 values were determined from composite concentration-effect curves as described under Materials and Methods. Hill slopes were in the range 0.6 to 1.2. Experiments were performed on 6 to 22 oocytes from two to five frogs for all receptors. The asterisk in the molecule indicates the chiral center.
Backbone modification DQP analogs
The fitted IC50 values were determined for the indicated combinations of recombinant GluN1/GluN2 receptors expressed in X. laevis oocytes and activated by 100 μM glutamate plus 30 μM glycine. Compounds that produced less than 25% inhibition at 30 μM were considered to have no effect. IC50 values were determined from composite concentration-effect curves as described under Materials and Methods. Experiments were performed on 6 to 22 oocytes from two to five frogs for all receptors. Compounds 3 and DQP-1105 in Table 2 are shown here to facilitate comparison.
Subunit Selectivity of DQP-1105 Inhibition.
We have evaluated the actions of DQP-1105 against current responses from recombinant NMDA, AMPA, and kainate receptors expressed in X. laevis oocytes (Fig. 1B; Table 3) to determine its potency and selectivity across the glutamate receptor ion channel family. When coapplied in solutions containing maximally effective concentrations of glutamate and glycine, DQP-1105 inhibited recombinant NMDA receptors containing the GluN1/GluN2C and GluN1/GluN2D subunits with IC50 values that were lower than that for inhibition of GluN1/GluN2A (Table 3). For example, in X. laevis oocytes, DQP-1105 inhibited GluN1/GluN2D receptors with an IC50 value of 2.7 ± 0.2 μM (mean ± S.E.M.; n = 44), whereas it had minimal effects on GluN1/GluN2A. The IC50 value at GluN1/GluN2A determined by fitting each concentration-effect curve was at a minimum 206 ± 36 μM, and might be higher, because responses from some oocytes showed no inhibition and thus could not be fitted and included in this analysis (n = 13; Fig. 1B; Table 3). IC50 values determined at recombinant human and rat NMDA receptors were similar (Table 3). DQP-1105 had minimal effects on current responses of homomeric GluA1 AMPA receptors or homomeric GluK2 kainate receptors (Table 1), and no effect on the leak currents in uninjected X. laevis oocytes (n = 4; data not shown), or on the leak currents from oocytes expressing NMDA receptors (n = 4; data not shown).
Concentration-response data for DQP-1105 at ionotropic glutamate receptors
Mean IC50 values (± S.E.M.) for inhibition of responses in oocytes to 100 μM glutamate and 30 μM glycine were determined as described under Materials and Methods. Oocyte experiments were performed in 3 to 44 oocytes from one to four frogs. Oocytes assayed using NMDA as agonist were activated using 50 to 1000 μM NMDA and 50 μM glycine. BHK cell imaging results are from three or more independent experiments (Materials and Methods). For HEK perforated-patch experiments, responses were evoked by 100 μM glutamate and 30 μM glycine in six to eight cells. From HEK whole-cell experiments using conventional dialyzed patches, data are from six to eight cells activated by 100 μM glutamate or 1000 μM NMDA plus 30 μM glycine. Mean fitted Hill slopes for GluN1/GluN2C and GluN1/GluN2D were between 0.87 and 1.14; Hill slope was fixed to 1.0 for GluN1/GluN2A, GluN1/Glun2B, GluA1, and GluK2.
To determine whether DQP-1105 is selective for GluN2C/D- over GluN2A/B-containing NMDA receptors expressed in mammalian cells, we evaluated the actions of this compound on recombinant NMDA receptor responses in BHK cells. IC50 values for DQP-1105 inhibition of the fluorescent signal in BHK cell lines stably expressing GluN1/GluN2A, GluN1/GluN2B, GluN1/GluN2C, or GluN1/GluN2D loaded with the Ca2+-sensitive dye Fluo4 were determined (Hansen et al., 2008, 2010b). DQP-1105 inhibited responses of GluN1/GluN2C and GluN1/GluN2D receptors activated by an EC80 concentration of NMDA plus glycine with IC50 values that were slightly more potent than those obtained from oocyte recordings (Table 3; Fig. 1C). GluN1/GluN2A-containing NMDA receptor responses were inhibited to only 65 ± 3% of control by 33 μM DQP-1105, which allowed us to estimate a mean IC50, whereas GluN1/GluN2B responses were negligibly inhibited (Table 3; Fig. 1C). Thus, DQP-1105 exhibited a similar level of selectivity for GluN2C/D in intact mammalian cells as was observed in X. laevis oocytes.
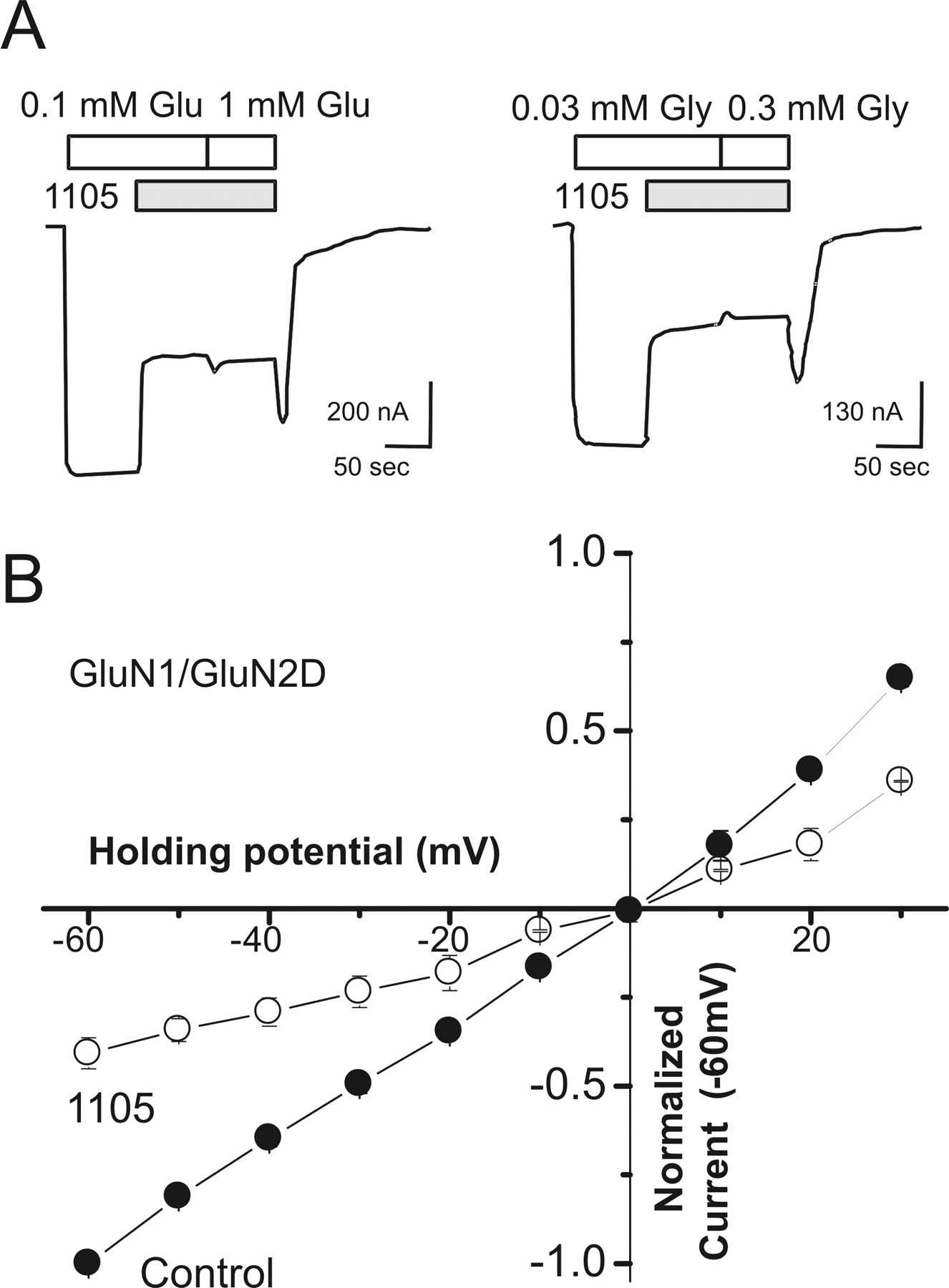
We also recorded recombinant NMDA receptor responses from transiently transfected HEK cells using the whole-cell patch-clamp recording configuration. These experiments confirmed that DQP-1105 inhibited the current response at GluN1/GluN2D receptors with potency similar to those of the other expression systems (Fig. 2; Table 3). We were surprised to find that DQP-1105 showed little selectivity for GluN2D-containing receptors over GluN2A-containing receptors in HEK cells recorded in the whole-cell configuration (Fig. 2D; Table 3). Whereas DQP-1105 inhibited GluN2D with similar potency in all three preparations, it was more potent at inhibiting GluN2A in whole-cell patch recordings than in oocytes or BHK cells studied using Ca2+-sensitive fluorescent dyes (Table 3). Because we had observed selectivity in both oocytes and BHK cells in which the intracellular milieu had not been disrupted by dialysis, we reasoned that the whole-cell patch recording configuration, which is known to enhance NMDA receptor desensitization (Sather et al., 1990) (Fig. 2, compare A and B), may have artificially increased the potency of DQP-1105 on GluN2A receptors. To test this idea, we used perforated patch recordings in which electrical access to the cell was obtained through gramicidin pores (Ebihara et al., 1995). This configuration allows measurements of whole-cell currents without changing the intracellular composition. As predicted, we observed considerable selectivity between glutamate-activated GluN2D-containing (IC50, 2.1 μM) over GluN2A-containing (IC50 54 μM) NMDA receptors when we recorded using perforated patches (Table 3). This observation suggests that dialysis of the cytoplasm and perhaps the associated enhancement of desensitization can increase the affinity of DQP-1105 for its binding site. We interpret the differences between the observed IC50 values for GluN2A with the different patch configurations to be due to a diffusible factor that is lost during dialysis of the intracellular compartment. We also interpret the lack of effect of dialysis on the DQP-1105 IC50 for GluN1/GluN2D to suggest that this receptor is not sensitive to loss of the same diffusible factor. Table 3 summarizes the potency and selectivity of DQP-1105 for GluN2C/D-containing NMDA receptors under the various conditions used in this study.

DQP-1105 inhibits recombinant NMDA receptors expressed in HEK cells. A, representative traces of conventional whole cell voltage-clamp recordings of currents evoked from recombinant GluN1/GluN2A receptors by 100 μM glutamate and 100 μM glycine. Coapplication of 30 μM DQP-1105 with glutamate and glycine attenuates the steady-state current response to a new steady-state level that is 24 ± 6% (n = 5) of control. B, representative gramicidin perforated-patch, whole-cell voltage-clamp recordings from recombinant GluN1/GluN2A receptors of currents evoked by 100 μM glutamate and 100 μM glycine. DQP-1105 (30 μM) attenuated the steady-state current response to 65 ± 3.1% (n = 6) of control. C, representative perforated whole-cell voltage-clamp recordings of current responses from GluN1/GluN2D evoked by 100 μM glutamate and 100 μM glycine. DQP-1105 (30 μM) inhibited the steady-state current response upon coapplication with glutamate and glycine, with a final steady-state response of 0.2 ± 0.1% (n = 3) compared with the glutamate and glycine control. Data are from six to eight cells. D, DQP-1105 composite concentration-response curves of the steady-state current responses activated by glutamate and glycine from dialyzed whole-cell patch recordings give a fitted IC50 value of 3.2 μM GluN1/GluN2D receptors and 12 μM for GluN1/GluN2A receptors (data are from six cells). E, composite concentration-response curves of the steady-state current responses to glutamate and glycine from perforated whole-cell patch recordings give an IC50 of 2.1 μM GluN1/GluN2D receptors and 54 μM for GluN1/GluN2A receptors, suggesting that dialysis alters IC50 of DQP-1105 at GluN1/GluN2A (data are from six to eight cells).
Mechanism of Action of DQP-1105.
To begin to determine the mechanism of DQP-1105 inhibition, we evaluated whether the actions of DQP-1105 were dependent on the agonist concentration for GluN1/GluN2D receptor responses recorded in X. laevis oocytes. Submaximal inhibition of GluN1/GluN2D receptor responses by 5 μM DQP-1105 could not be surmounted by 10-fold increases in either glutamate or glycine concentration, suggesting that DQP-1105 acts by a noncompetitive mechanism (Fig. 3A). Furthermore, the EC50 values for glutamate and glycine at GluN1/GluN2D receptors expressed in X. laevis oocytes were not significantly altered in the presence of an IC75 concentration of DQP-1105 (5 μM). Glutamate EC50 values in 30 μM glycine were 0.30 ± 0.02 and 0.27 ± 0.03 μM in the absence and presence of DQP-1105, respectively (p > 0.05, unpaired t test, n = 10–11). Glycine EC50 values in 100 μM glutamate were 0.11 ± 0.01 and 0.11 ± 0.01 μM in the absence and presence of DQP-1105, respectively (p > 0.05, unpaired t test, n = 4). To determine whether DQP-1105 inhibition involved the permeation pathway, we evaluated the inhibition at various membrane potentials. The degree of inhibition produced by DQP-1105 was the same at all voltages tested (p = 0.86, one-way ANOVA with Tukey's test; n = 5), suggesting that DQP-1105 inhibition is voltage-independent. In addition, the mean reversal potential (VREV) was not significantly different between control response to glutamate plus glycine (VREV, −0.31 ± 1.4 mV; n = 5) and the response to glutamate and glycine plus 3 to 5 μM DQP-1105 (VREV, −0.81 ± 3.3 mV; n = 5; p = 0.76; t test; Fig. 3B). We interpret these data to suggest that DQP-1105 does not inhibit receptor function by competing with glutamate or glycine at the agonist binding domain or by interacting with the channel pore in a manner that is influenced by the transmembrane electric field.

DQP-1105 inhibits recombinant GluN1/GluN2D receptors through a noncompetitive and voltage-independent mechanism. A, GluN1/GluN2D responses to 100 μM glutamate and 30 μM glycine were inhibited by coapplication of glutamate, glycine, and 5 μM DQP-1105, and the glutamate concentration was subsequently increased 10-fold to 1000 μM (left panel). GluN1/GluN2D responses elicited by 100 μM glutamate and 30 μM glycine were inhibited by coapplication of 5 μM DQP-1105, and the glycine concentration was subsequently increased 10-fold to 300 μM glycine (right). The increase of neither glutamate nor glycine altered the level of inhibition, suggesting a noncompetitive mechanism. B, the mean current-voltage relationship of recombinant GluN1/GluN2D receptors was determined in the absence and presence of 3–5 μM DQP-1105 (10-mV steps from −60 to +30 mV; n = 5). Error bars are S.E.M. and shown when larger than symbol size.
We subsequently examined the effects of DQP-1105 on the NMDA receptor response time course in whole-cell voltage-clamp recordings of GluN1/GluN2D receptors expressed in HEK cells. Current responses were evoked by rapid solution exchange from glycine alone to glycine plus glutamate or NMDA (Fig. 4A). The current response of GluN1/GluN2D undergoes virtually no desensitization during agonist application, as described previously (Vicini et al., 1998; Wyllie et al., 2008). Coapplication of agonist plus 0.3 to 30 μM DQP-1105 caused concentration-dependent relaxation of the current response (Fig. 4), with virtually complete inhibition of GluN1/GluN2D responses at 30 μM. Concentration-effect curves for the steady-state current responses to the agonist NMDA show that the IC50 for DQP-1105 inhibition on GluN1/GluN2D receptors was in excellent agreement with that obtained in the other expression systems (Fig. 4B; Table 3). Coapplication of DQP-1105 with NMDA and glycine induced a relaxation of the current response that could be fitted by a single exponential function. Coapplication of different concentrations of DQP-1105 with NMDA resulted in different time constants (τONSET), which we interpret to reflect the time course of association of DQP-1105 with the receptor. In this case, the reciprocal of τONSET should be linearly related to DQP-1105 concentration with a slope equal to kON and an intercept equal to kOFF. Consistent with this idea, values of 1/τONSET were significantly correlated with DQP-1105 concentration (r = 0.996; Pearson's test, p < 0.005) for GluN1/GluN2D (Fig. 4C). Regression analysis of this relationship for NMDA-activated GluN1/GluN2D gives values of 4.5 × 105 M−1 · s−1 for kON and 0.65 s−1 for kOFF, from which KD can be calculated as 1.4 μM, in agreement with the fitted IC50 value (Fig. 4C; Table 3). Inhibition by DQP-1105 also was evaluated when GluN1/GluN2D receptors were activated by 100 μM glutamate and 30 μM glycine. Analysis of the glutamate-activated GluN1/GluN2D response relaxation time course as a function of DQP-1105 concentration yielded similar values for kON (4.4 × 105 M−1 · s−1) and kOFF (1.44 s−1; n = 6 cells). The resulting KD for DQP-1105 inhibition of glutamate-evoked currents (3.3 μM) was similar to the independently determined IC50 for glutamate-evoked responses (Table 3). In addition, the KD and IC50 values for DQP-1105 inhibition were typically less than 2-fold lower for the partial agonist NMDA-evoked than for glutamate-evoked currents, suggesting the affinity of DQP-1105 is only modestly influenced by agonist efficacy.

Determination of DQP-1105 KD for inhibition of recombinant GluN1/GluN2D NMDA receptors expressed in HEK cells. A, conventional whole cell voltage-clamp recordings of currents evoked from recombinant GluN1/GluN2D receptors by 2-s applications of 1000 μM NMDA and 50 μM glycine. Coapplication of DQP-1105 with NMDA and glycine caused a concentration-dependent attenuation of the steady-state current response, with a final steady-state response of 3.4 ± 1.1% (n = 8) at 30 μM DQP-1105 compared with control. B, the composite concentration-response relationship of the steady-state current responses to 1000 μM NMDA plus 30 μM glycine shows that DQP-1105 inhibits GluN1/GluN2D receptors with an IC50 value of 1.9 μM. C, there is a linear relationship between 1/τONSET and DQP-1105 concentration, suggesting that the time-dependent inhibition of NMDA-activated GluN1/GluN2D reflects the association of DQP-1105 with its binding site. From this linear relationship, KD can be calculated from kON and kOFF, and was 1.4 μM for GluN1/GluN2D receptors, similar to the measured IC50 value.
We next evaluated whether the relaxation of the current response during DQP-1105 application was dependent upon the binding of NMDA or glycine. GluN1/GluN2D receptors expressed in HEK cells were recorded under voltage-clamp and activated by rapid 5-s application of 200 μM NMDA in the continuous presence of 100 μM glycine. The peak and steady-state current responses then were compared with the currents evoked by application of 200 μM NMDA after preincubation with 100 μM glycine and 3 μM DQP-1105 (Fig. 5A). Even when DQP-1105 was preapplied, the NMDA-evoked currents exhibited a pronounced relaxation of the current response, with a peak response of 84 ± 1.9% compared with control and a steady-state response of 21 ± 1.6% compared with control (Fig. 5, A and C; n = 7), suggesting that DQP-1105 did not bind with the same affinity in the absence of NMDA. The time course could be described by a single exponential function with a time constant (580 ± 33 ms) that was not significantly different from that obtained with no preapplication of DQP-1105 (530 ± 27 ms; p = 0.72, unpaired t test; compare Figs. 4A and 5A). By contrast, activation of GluN1/GluN2D by a rapid application of 300 μM glycine after preincubation with 20 μM NMDA and 3 μM DQP-1105 (Fig. 5B) did not show a pronounced relaxation in current response time course. Rather, the peak response was inhibited to 29 ± 0.9% of control, with a steady-state response of 24 ± 1.6% of control (Fig. 5, B and C; n = 5), suggesting that DQP-1105 can access its binding site with a similar affinity when NMDA is present, as observed during steady-state responses to NMDA plus glycine. We interpret this result to suggest that the binding of DQP-1105 is dependent upon binding of NMDA but not glycine. This use dependence upon activation of the GluN2D subunit is reflected in the significant difference in the peak responses of GluN1/GluN2D receptors activated by 5-s application of NMDA or glycine in the presence of DQP-1105 (p < 0.05; paired t test). The maximal, steady-state inhibition by DQP-1105 is not significantly influenced by preincubation with either NMDA or glycine (Fig. 5, C and D). These results are similar to that for quinazolin-4-one GluN2C/D modulators (Hansen and Traynelis, 2011).

Inhibition by DQP-1105 is dependent upon binding of NMDA to the GluN2D subunit. Current responses were evoked by agonist application to HEK cells transiently expressing GluN1/GluN2D receptors that were recorded under voltage clamp at −60 mV. A, GluN1/GluN2D receptors were activated by a 5-s application of 200 μM NMDA in the continuous presence of 100 μM glycine and subsequently were activated by 5-s applications of 200 μM NMDA in the continuous presence of both 100 μM glycine and 3 μM DQP-1105. These recordings displayed a pronounced relaxation of the current response after NMDA binding, with an 84 ± 1.9% peak response compared with control and a steady-state response of 21 ± 1.6% compared with control (n = 7). B, GluN1/GluN2D receptors were activated by a 5-s application of 300 μM glycine in the continuous presence of 20 μM NMDA, and subsequently activated by a 5-s application of 300 μM glycine in the continuous presence of both 20 μM NMDA and 3 μM DQP-1105. These recordings did not show a prominent relaxation of response, with a peak response of 29 ± 0.9% compared with control and a steady-state response of 24 ± 1.6% of the control response amplitude (n = 5). C, although the peak responses of GluN1/GluN2D receptors activated by NMDA or glycine after pretreatment with 3 μM DQP-1105 were significantly different (p < 0.05; paired t test), the steady-state responses (D) were not significantly different (paired t test). These data suggest that DQP-1105 shows higher potency after NMDA (but not glycine) binding to the receptor.
We subsequently recorded the single channel activity of GluN1/GluN2D in excised outside-out patches in response to a maximally effective concentration of glutamate (1 mM) and glycine (50 μM) plus DQP-1105 (3 and 30 μM). Figure 6 shows typical channel activity from patches containing several channels in the absence and presence of DQP-1105. Time course fitting of the open and closed durations in four patches revealed that DQP-1105 increased the mean shut time in a concentration-dependent manner but did not significantly affect the mean open time or either of the two time constants describing the open duration histogram (Table 4). The open probability was decreased to 43 ± 12% (Table 4) by 3 μM DQP-1105, a value that agrees well with the extent of inhibition of macroscopic currents in X. laevis oocytes by 3 μM DQP-1105 (35 ± 3.6% of control, n = 11), suggesting inhibition of channels in excised outside-out patches quantitatively matches inhibition observed in intact cells. In addition, 30 μM DQP-1105 decreased the open probability of GluN1/GluN2D receptors to 2.0 ± 0.6% of control (Fig. 6). DQP-1105 had minimal effects on chord conductance values (Table 4). These data suggest DQP-1105 does not affect the pore conductance or the stability of the open state of the receptor but rather acts to prevent the channel from opening by raising the activation energy for a critical conformational change.

DQP-1105 increases mean shut time and decreases open probability of GluN1/GluN2D receptors. A, unitary currents recorded from outside-out patches from HEK cells expressing GluN1/GluN2D were activated by glutamate (1 mM) and glycine (50 μM) at −80 mV B, after a 2-min recording in control conditions, 3 μM DQP-1105 was coapplied with agonists to the patch and currents were recorded for 2 min. C, 30 μM DQP-1105 was then coapplied to the same patch and currents recorded for 2 min. For A to C, the boxed region in the top trace is shown on an expanded time scale in the three lower traces. Individual openings and closings were idealized by time course fitting (see Materials and Methods) and composite open duration (D) and shut duration (E) histograms were constructed from idealized data from four patches. The open-period distribution was fitted with the sum of two exponential components (Table 4) and the shut time distribution was fitted with the sum of five exponential components with τ (percentage area) of 0.026 (27%), 0.16 (11%), 1.4 (9%), 7.2 (43%), and 22 ms (10%) for control (7547 intervals), 0.016 (36%), 0.13 (12%), 3.3 (9%), 14 (34%), and 55 ms (9%) for 3 μM DQP-1105 (5617 intervals), and 0.026 (27%), 0.23 (13%), 11 (22%), 85 ms (18%), and 1500 (20%) for 30 μM DQP-1105 (952 intervals).
DQP-1105 decreases open probability of GluN1/GluN2D receptor channels
GluN1/GluN2D receptors were expressed in HEK cells and activated by glutamate (1 mM) and glycine (50 μM) in outside-out patches held under voltage clamp at −80 mV. Responses were recorded first to glutamate plus glycine (7862 open periods), and subsequently to glutamate plus glycine coapplied with 3 μM (5724 open periods) or 30 μM DQP-1105 (954 open periods) to the same patch. All values are mean ± S.E.M. (n = 4 patches). Because all patches contained multiple channels, POPEN was normalized to the value for the patch in the absence of DQP-1105 for statistical testing.
Structural Determinants of DQP-1105 Activity.
DQP-1105 shows sufficient selectivity for GluN2D over GluN2A to allow use of a chimeric strategy in which domains of subunits are swapped to identify the divergent structural elements of the GluN2D subunit that might account for the observed selectivity. We previously constructed a series of GluN2A and GluN2D chimeric receptors that transferred different portions of GluN2D into GluN2A to test for gain of function for novel GluN2C/D preferring modulators (Mullasseril et al., 2010; Hansen and Traynelis, 2011) (Table 5). We used this set of chimeric receptors to evaluate the structural determinants of DQP-1105 action and found that transferring the ATD, S1 region of the ligand-binding domain, or the transmembrane elements by themselves (including two transmembrane helices and a reentrant loop) did not render GluN2A containing receptors sensitive to DQP-1105 (Fig. 7, A and B). By contrast, transferring either the S1-S2 regions or the S2 domain alone from GluN2D to GluN2A completely transferred the DQP-1105 sensitivity to GluN2A (Fig. 7, A and B), suggesting structural determinants that define subunit selectivity may be contained within the S2 region of GluN2D. This result is surprisingly similar to the structural determinants for neurosteroids (Jang et al., 2004; Horak et al., 2006) and the structurally unrelated quinazolin-4-ones (Hansen and Traynelis, 2011). Consistent with this result, experiments in which the GluN2D subunit ATD has been removed show that the ATD is not involved in the selectivity of DQP-1105 for GluN2D over GluN2A (Fig. 7, A and B).
Amino acid numbering and composition for chimeric receptors
Amino acid numbering and composition for chimeric GluN2A-GluN2D subunits. Amino acid numbering is according to the full-length GluN2 subunits, including the signal peptide (initiating methionine is 1).

Identification of structural determinants of GluN2D-selective inhibition using chimeric GluN2A-GluN2D receptors. A, indicated regions of the polypeptide were exchanged between wild-type GluN2A and GluN2D. Concentration-effect curves at GluN1/GluN2D, GluN1/GluN2A, GluN1/GluN2A(2D-S1S2), GluN1/GluN2A(2D-S1), and GluN1/GluN2A(2D-S2), B, GluN1/GluN2A(2D-M1M2M3), GluN1/GluN2A(2D-ATD), and GluN1/GluN2A(ΔATD) receptors were analyzed, and the mean IC50 values (± S.E.M.) are shown. These data suggest that the S2 region transfers DQP-1105 sensitivity to GluN2A. C, linear representation of the S2 regions of GluN2A(2D-S2) in which regions of GluN2D-S2 have been reverted to that of GluN2A to probe for loss of inhibition of DQP-1105. D, the inhibition produced by 5 μM DQP-1105 in the presence of 1000 μM glutamate and 300 μM glycine is shown as a percent of control. Three chimeric receptors show significantly reduced inhibition compared with GuN2A(S2) chimera (p < 0.05, one-way ANOVA with Tukey's test, n = 5–13 oocytes per chimeric receptor). E, site-directed mutagenesis of residues within GluN2D S2 chimeric regions identified Gln701 and Leu705 (asterisks in B) as key structural determinants for antagonist activity of DQP-1105 at GluN2D receptors (p < 0.05, one-way ANOVA with Tukey's test; n = 4–11 oocytes per mutant receptor).
To identify specific divergent structural elements within the S2 region that might define the subunit selectivity, we used 12 additional chimeric receptors (Hansen and Traynelis, 2011) that revert nonconserved S2 residues within the GluN2A(2D-S2) chimera back to wild-type GluN2A residues (see Fig. 7, C and D). This strategy allowed us to examine whether clusters of residues that are divergent between GluN2D and GluN2A affect DQP-1105 selectivity. We found that the S2a, S2b, and S2c chimeric receptors exhibited reduced sensitivity to DQP-1105 when tested at a single concentration, suggesting that some of the nine divergent residues between GluN2A and GluN2D within these three regions control DQP-1105 potency at the receptor. Subsequent mutation of each of these divergent residue in GluN2D to that observed in Glun2A identified two amino acids (GluN2D Gln701 and Leu705) that significantly reduced inhibition (Fig. 7E). Concentration-effect curves showed that GluN2D(Q701Y) and GluN2D(L705F) decreased the potency of DQP-1105 more than 6-fold to 19 ± 2 μM (n = 8) and 17 ± 2 μM (n = 8), respectively. These data suggest that DQP-1105 shares structural determinants with quinazolin-4-ones (Hansen and Traynelis, 2011) and perhaps neurosteroids (Horak et al., 2006), identifying this region as a site at which structurally diverse endogenous and exogenous ligands can interact.
Discussion
We have identified a novel class of GluN2C/D-preferring noncompetitive antagonists that inhibit NMDA receptors by blocking a conformational change necessary for channel opening. Inhibition occurs without actions on the stability of the open state or the channel closing rate. Rather, binding of DQP-1105, presumably to the GluN2 subunit, reduces the frequency of channel openings without other actions. We also show, unexpectedly, that the structural determinants of selectivity for DQP-1105 reside within a portion of the S2 region of GluN2D that overlaps closely with previously identified structural determinants important in the selectivity of quinazolin-4-ones, another class of GluN2C/D-preferring antagonists (Hansen and Traynelis, 2011). In addition, the shared reliance of quinazolin-4-ones, dihydroquinolone-pyrazolines, and neurosteroids on the lower lobe of the ligand-binding domain clamshell indicate that this region is an important site at which channel gating can be controlled (Jang et al., 2004). These data also show that the allosteric binding site(s) in this region can accommodate a wide range of structurally diverse, negative modulators, which increases the likelihood that well tolerated and potentially therapeutically useful subunit-selective NMDA receptor antagonists can be developed. Furthermore, the findings presented here provide support for a previously unknown antagonist binding site that does not involve the amino-terminal domain, the well conserved agonist recognition sites, or residues within the channel pore. That is, this new site is distinct from GluN2B-selective negative allosteric modulators (Hansen et al., 2010a), competitive antagonists, and channel blockers.
Several studies have recently identified positive and negative allosteric modulators that act with enhanced potency at the GluN2C/D subunits (Costa et al., 2010; Mosley et al., 2010; Mullasseril et al., 2010). Of these, only one other class of noncompetitive GluN2C/D antagonist has been reported with potency in the low micromolar range and greater than 20-fold selectivity over NMDA receptors composed of other GluN2 subunits, AMPA receptors, and kainate receptors. That class, which contains the previously identified AMPA antagonist quinazolin-4-one backbone (Mosley et al., 2010), is chemically distinct from DQP-1105 yet shares similar structural determinants of action and a similar mechanism, in that glutamate/NMDA binding enhances antagonist potency (Hansen and Traynelis, 2011). Structural determinants within NMDA receptor subunits have not been defined for other GluN2C/D-preferring antagonists, apart from the suggestion that the S2 region of the ligand binding domain is important in their actions (Horak et al., 2006; Costa et al., 2010). Thus, it is presently unclear whether the neurosteroids and phenanthrene derivatives share structural determinants with DQP-1105 and the quinazolin-4-ones.
It is known that the ligand binding domain undergoes a significant conformational change upon binding of agonist (Furukawa and Gouaux, 2003). The data from our single channel analysis, as well as the structural determinants that were identified, support the idea that DQP-1105 could interfere with a crucial gating step that leads to an increase in the energy barrier for the transition from the closed to the open state for DQP-1105-bound receptors. Moreover, we have previously proposed that the GluN2C/D ligand binding domain closure around glutamate can increase the potency for quinazolin-4-one antagonists (Hansen and Traynelis, 2011), which share structural determinants of selectivity with DQP-1105. In this model, residues implicated by site-directed mutagenesis as being important for the actions of inhibitors, including the residues identified in this study, are close enough to the linkers (S1-M1 and M3-S2) connecting the transmembrane helices to the proximal region of the ligand binding domain to influence their movement (Fig. 8). Numerous studies have shown that structural determinants near or within the linker regions modulate receptor gating (Krupp et al., 1998; Yuan et al., 2005; Talukder et al., 2010). Most recently, specific residues within the S1-M1, M3-S2, and S2-M4 linker regions were suggested to serve as potential targets for allosteric modulators (Talukder et al., 2010). Our identification of residues in the vicinity of the extracellular end of the transmembrane linkers raises the possibility that DQP-1105 and related analogs could interact with both the ligand binding domain and portions of the linker regions to negatively modulate the receptors.
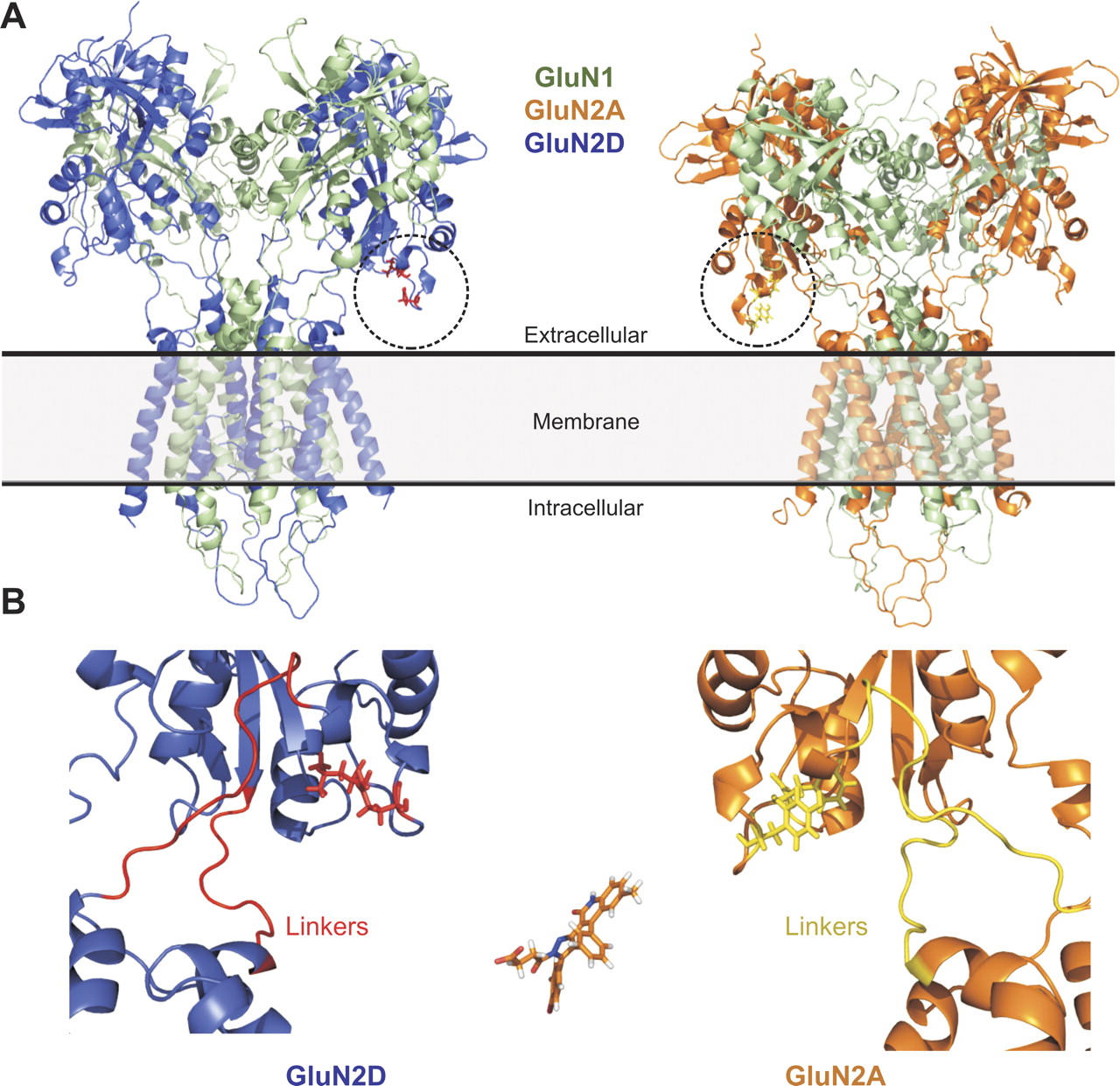
Homology models of GluN1-GluN2 receptors shows that residues identified in lower lobe of the clamshell shaped ligand binding domain are localized near each other in three-dimensional space and in close proximity to the linkers that connect the ligand binding domain to the transmembrane helices. A, left, a GluN1-GluN2D receptor homology model (ATD omitted); right, shows a GluN1-GluN2A homology model (ATD omitted). B, the GluN2D-subunit is expanded (left) to show the lower portion of the ligand-binding domain containing residues Gln701 and Leu705 (red), which are critical divergent structural determinants for the antagonist activity of DQP-1105. The S1-M1 and M3-S2 linker regions are also shown in red. A GluN2A-subunit is expanded (right) to show the same region with the residues corresponding to GluN2D-Gln701(Tyr in GluN2A, yellow) and GluN2D-Leu705 (Phe in GluN2A yellow); linkers are also shown in yellow. Compound DQP-1105 is shown in the lower panel on the same scale.
Although the residues implicated by mutagenesis data in DQP-1105 selectivity are not immediately adjacent on the polypeptide chain, homology models of GluN1/GluN2 place these residues close in three dimensional space (Fig. 8). However, no crystallographic data exist for GluN2C, and the region identified in chimeric receptors shows a high degree of chain flexibility in the crystallographic structure of the isolated GluN2D ligand binding domain (Vance et al., 2011). Moreover, whereas we conclude that GluN2D residues Gln701 and Leu705 control the selectivity between GluN2A and GluN2D, the determinants of GluN2C selectivity have yet to be explored in depth. One of the residues important for GluN2D selectivity (Leu705) is a conserved Phe in GluN2A/B/C, suggesting this residue does not contribute to selectivity for GluN2C over GluN2A. Nevertheless, it is tempting to speculate that these residues may form or influence part of the binding pocket for DQP-1105 and related analogs. The finding that the DQP class of molecules shares structural determinants with the previously identified quinazolin-4-ones suggests that potent and selective molecules incorporating functionality from both groups might be developed through medicinal chemistry efforts.
Authorship Contributions
Participated in research design: Acker, Yuan, Hansen, Vance, Ogden, Jensen, Burger, Mullasseril Snyder, Liotta, and Traynelis.
Conducted experiments: Acker, Yuan, Hansen, Vance, Ogden, and Mullasseril.
Performed data analysis: Acker, Yuan, Hansen, Vance, Ogden, Jensen, Burger, Mullasseril and Traynelis.
Wrote or contributed to the writing of the manuscript: Acker, Yuan, Hansen, Vance, Ogden, Jensen, Burger, Mullasseril Snyder, Liotta, and Traynelis.
Acknowledgments
We thank Kimberly Vellano, Phuong Le, Charlotte Fredsøe Hundahl, Deborah Culver, and Sara Dawit for excellent technical assistance.
Footnotes
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.This work was supported by the Michael J. Fox Foundation; the National Institutes of Health National Institutes of Neurological Disorders and Stroke [Grants R01-NS036654, R01-NS065371, F31-NS071802]; the National Institutes of Health National Institute of General Medical Sciences [Grant T32-GM008602]; the National Institutes of Health National Institute on Drug Abuse [Grant T32-DA01504006]; the National Institutes of Health National Institute of Environmental Health Sciences [Grant T32-ES012870]; the Villum Kann Rasmussen Foundation; the Lundbeck Foundation; Research Grants to Emory University from Pfizer Inc. and Lundbeck AS; and the Emory Chemistry Biology Discovery Center.
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.111.073239.
-
ABBREVIATIONS:
- NMDA
- N-methyl-d-aspartate
- HEK
- human embryonic kidney
- MK801
- dizocilpine maleate
- BAPTA
- 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- PDB
- Protein Data Bank
- AMPA
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- DMSO
- dimethyl sulfoxide
- DQP
- dihydroquinolone-pyrazoline-containing
- DQP-997
- 3; 4-(3-(6-bromo-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4-bromophenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid
- DQP-1105
- 19; 4-(5-(4-bromophenyl)-3-(6-methyl-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid
- DQP-1209
- 9; 4-(3-(6-bromo-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(2-chlorophenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid
- DQP-1210
- 22; 4-(3-(6-bromo-4-phenylquinolin-3-yl)-5-(4-bromophenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid
- DQP-1308
- 18; 4-(3-(6-methyl-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-phenyl-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid
- DQP-1309
- 21; 6-bromo-3-(5-(4-bromophenyl)-1-propionyl-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one
- DQP-1177
- 25; 4-(3-(4-bromophenyl)-5-(6-methyl-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid
- DQP-1178
- 8; 4-(3-(6-bromo-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(2-fluorophenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid
- DQP-1183
- 1; 4-(3-(6-bromo-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-phenyl-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid
- DQP-1184
- 2; 4-(3-(6-bromo-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4-fluorophenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid
- DQP-1185
- 5; 4-(3-(6-bromo-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(p-tolyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid
- DQP-2003
- 7; 4-(3-(6-bromo-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(2,5-dimethoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid
- DQP-1250
- 24; 3-(1-acetyl-5-(4-bromophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-6-methyl-4-phenylquinolin-2(1H)-one) and
- DQP-2005
- 20; 4-(5-(2-methoxyphenyl)-3-(6-methyl-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid
- DQP-2033
- 4; 4-(3-(6-bromo-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4-nitrophenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid
- DQP-1128
- 6; 4-(3-(6-bromo-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid.

- Received April 25, 2011.
- Accepted August 1, 2011.
- Copyright © 2011 The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}