


Abstract
Multiple sclerosis (MS) therapies modulate T-cell autoimmunity in the central nervous system (CNS) but may exacerbate latent infections. Fingolimod, a nonselective sphingosine-1-phosphate (S1P) receptor agonist that induces sustained lymphopenia and accumulates in the CNS, represents a new treatment modality for MS. We hypothesized that sustained lymphopenia would not be required for efficacy and that a selective, CNS-penetrant, peripherally short-acting, S1P1 agonist would show full efficacy in a mouse MS model. Using daily treatment with 10 mg/kg 2-(4-(5-(3,4-diethoxyphenyl)-1,2,4-oxadiazol-3-yl)-2,3-dihydro-1H-inden-1-yl amino)ethanol (CYM-5442) at the onset of clinical signs in myelin oligodendrocyte glycoprotein MOG35–55- induced experimental allergic encephalomyelitis (EAE), we assessed clinical scores, CNS cellular infiltration, demyelination, and gliosis for 12 days with CYM-5442, vehicle, or fingolimod. CYM-5442 levels in CNS and plasma were determined at experiment termination, and blood lymphopenia was measured 3 and 24 h after the last injection. Plasma levels of cytokines were assayed at the end of the protocol. Changes in S1P1-enhanced green fluorescent protein expression on neurons and astrocytes during active EAE and upon CYM-5442 treatment were quantified with flow cytometry and Western blotting by using native-locus enhanced green fluorescent protein-tagged S1P1 mice. S1P1 agonism alone reduced pathological features as did fingolimod (maximally lymphopenic throughout), despite full reversal of lymphopenia within each dosing interval. CYM-5442 levels in CNS but not in plasma were sustained. Neuronal and astrocytic S1P1 expression in EAE was suppressed by CYM-5442 treatment, relative to vehicle, and levels of key cytokines, such as interleukin 17A, were also significantly reduced in drug-treated mice. S1P1-selective agonists that induce reversible lymphopenia while persisting in the CNS may be effective MS treatments.
Introduction
MS is a chronic inflammatory disease of the CNS that is characterized by demyelination, axonal severing, axonal loss, and astrogliosis and causes severe neurological dysfunction. Although the direct pathogenesis is unknown, establishment of a chronic autoimmune response in the CNS requires that T cells that are autoreactive to myelin components escape negative selection and peripheral suppression to enter the CNS, initiating a cascade of inflammation, demyelination, phagocytosis of myelin debris, and astrogliosis. These processes culminate in the sclerotic plaques that are pathognomonic for MS (Compston and Coles, 2008).
Several treatments for MS inhibit lymphocyte entry into the CNS. Tysabri, a monoclonal antibody against very late antigen 4, prevents integrin-mediated invasion of T cells into the CNS (Yednock et al., 1992) but is associated with the potentially fatal recrudescence of JC virus infection known as progressive multifocal leukoencephalopathy (Kleinschmidt-DeMasters and Tyler, 2005). Fingolimod (FTY720, Gilenya; Novartis, Basel, Switzerland), whose active phosphate acts on the S1P receptors S1P1, S1P3, S1P4, and S1P5 (Mandala et al., 2002) and prevents the egress of lymphocytes from secondary lymphoid organs, was approved as an oral treatment (Brinkmann et al., 2010). Fingolimod induction of lymphocyte sequestration is entirely dependent on its actions on S1P1 (Mehling et al., 2008; Gräler, 2010). However, S1P receptors are expressed on a wide variety of cells other than lymphocytes, including neurons (Kimura et al., 2007), oligodendrocytes (Miron et al., 2008), macrophages (Singer et al., 2005), microglia (Durafourt et al., 2011), and astrocytes (Malchinkhuu et al., 2003). S1P receptors in primary CNS cells directly affect morphological features, survival, proliferation, and differentiation (Brinkmann et al., 2010). Astrocytes, which migrate to S1P through S1P receptors (Mullershausen et al., 2007), may play a critical role in the pathogenesis of MS, inasmuch as mice lacking S1P1 on astrocytes do not develop symptoms as severe in MOG35–55-induced EAE (Choi et al., 2011). Because of the lack of selectivity of fingolimod phosphate, insights into the specific receptors involved in its efficacy in treating MS have been limited.
Here we explore the role of S1P1 in the amelioration of EAE symptoms by using the selective S1P1 agonist 2-(4-(5-(3,4-diethoxyphenyl)-1,2,4-oxadiazol-3-yl)-2,3-dihydro-1H-inden-1-yl amino)ethanol (CYM-5442) (Gonzalez-Cabrera et al., 2008) in combination with S1P1-eGFP knock-in mice (Cahalan et al., 2011), which allow direct analysis of receptor expression, signaling, and subcellular localization. We demonstrate that daily CYM-5442 administration significantly reduces MOG35–55-induced EAE in mice despite the cyclical return of blood lymphocyte levels to normal within each dosing interval. In addition, we show that S1P1-eGFP expression during daily CYM-5442 treatment leads to degradation of S1P1-eGFP on neurons and astrocytes but not peripheral lymphocytes. Therefore, cyclical restoration of normal lymphocyte recirculation during each dosing interval, with a selective S1P1 agonist that persists within the CNS, provides a foundation of adequate efficacy that may preserve the ability of the host to maintain sufficient lymphocyte surveillance.
Materials and Methods
Mice.
Six- to 8-week-old female C57BL/6J mice and congenic Edg1eGFP/eGFP (S1P1-eGFP) mice were used (Cahalan et al., 2011). All procedures were approved by the animal care and use committee at The Scripps Research Institute (La Jolla, CA).
EAE Induction and Clinical Scoring.
Mice, lightly anesthetized with isofluorane (Isothesia, Butler, IL), received 200-μl intradermal lumbar inoculations of immunogen (2 mg/ml MOG35–55 peptide; Cedarlane, Burlington, NC) dissolved in water and emulsified 1:1 in incomplete Freund's adjuvant (Difco, Detroit, MI) supplemented with 4 mg/ml heat-inactivated Mycobacterium tuberculosis (strain H37 RA; Difco). Immediately after immunization and on day 2 after immunization, mice received 0.1-ml i.p. injections of PTX (2 mg/ml) in PBS. Animals were weighed daily, and neurological signs were scored as follows (Miller et al., 2010): 0, no symptoms; 1, complete loss of tail tone or hind limb weakness; 2, loss of tail tone plus hind limb weakness; 3, partial hind limb paralysis; 4, full hind limb paralysis; 5, moribund. Animals with scores of 5 were euthanized and were included in the clinical scoring.
S1P1 Agonist Administration.
CYM-5442 was synthesized as described previously (Gonzalez-Cabrera et al., 2008) and was dissolved in sterile water. Mice were divided randomly into 2 groups at the onset of clinical symptoms (10–13 days after immunization), received daily injections of S1P1 agonists, dosed at 10 mg/kg i.p. (CYM-5442) and 3 mg/kg i.p. (fingolimod; Cayman Chemical, Ann Arbor, MI), or equal volumes of vehicle for an additional 8 to 12 days, and then were euthanized for additional studies.
Histological Assessment and Immunofluorescence.
Histological examinations of brain and spinal cord specimens from vehicle- and drug-treated groups were performed at the end of the study and on days 18 to 19 after EAE induction, when significant therapeutic differences in clinical scoring results between drug-treated and vehicle-treated mice typically were observed. After euthanasia, animals were perfused with PBS and ice-cold 4% paraformaldehyde, and spinal cords and brains were carefully removed and incubated for 1 h on ice in 4% paraformaldehyde, followed by 72 h at 4°C in 30% sucrose. Cellular infiltration and anatomic features were assessed in paraffin-embedded CNS tissue sections that had been cut at 10 μm and stained with hematoxylin and eosin. Luxol fast blue (LFB) and FluoroMyelin Red (Invitrogen, Carlsbad, CA) staining was performed with spinal cords, for assessment of demyelination. Phase-contrast images of hematoxylin and eosin- and LFB-stained sections were acquired by using a microscope (BX51; Olympus (Tokyo, Japan).
Colocalization studies in spinal cords of S1P1-eGFP mice were performed with frozen sections from tissues processed as described in the previous paragraph. Tissues were embedded in Tissue-Tek compound (Sakura Finetek USA, Inc., Torrance, CA), frozen in a dry ice/2-methylbutane bath, and sectioned at 10 μm by using a cryostat. Slides underwent blocking at room temperature for 1 h in PBS containing 1% fetal bovine serum, 1% normal goat serum, 0.01% fish gelatin, and 0.1% Tween 20. Tissues were then incubated overnight at 4°C with antibodies against green fluorescent protein (1:10,000; Thermo Fisher Scientific, Waltham, MA), the vascular endothelial marker CD31/platelet endothelial cell adhesion molecule (1:50; BD Pharmingen, San Diego, CA), the neuronal marker microtubule-associated protein 2 (1:10,000; Abcam, Cambridge, MA), the astrocyte marker glial fibrillary acidic protein (1:1000; Abcam), or the oligodendrocyte marker myelin basic protein (1:100; Millipore, Billerica, MA). Slides were washed three times with PBS containing 0.1% Tween 20 and then were incubated for 1 h at room temperature with secondary antibodies conjugated to 546-nm or 633-nm Alexa fluor dyes (Invitrogen). Slides were washed with PBS containing 0.1% Tween 20, incubated with 0.5 μg/ml 4′,6-diamidino-2-phenylindole in PBS containing 0.1% Tween 20, rinsed with PBS, and mounted with Vectashield mounting medium (Vector Labs, Burlingame, CA). Staining of spinal cord sections with the S1P1 carboxyl terminus-recognizing antibody (clone H-60, 1:50; Santa Cruz Biotechnology, Santa Cruz, CA) or an isotype control was performed in paraffin-embedded tissue.
Measurement of Blood Lymphocyte Counts.
Cardiac blood was obtained from mice in each treatment group and was left to rotate for 2 h in EDTA-containing tubes on a Clay Adams Nutator (BD Biosciences, San Jose, CA). Red blood cell lysis was performed with two washes with 1 M Tris/azide/calcium chloride buffer for 15 min at 37°C. Samples were resuspended in 900 μl of fluorescence-activated cell-sorting buffer, counted with a Coulter counter (Beckman Coulter, Fullerton, CA), and stained with APC-CD4, peridinin-chlorophyll protein-Cy5.5-CD8, and PE-CD19 antibodies (BioLegend, San Diego, CA), for determination of T cell and B cell numbers. Analysis was performed with FlowJo software (Treestar, Ashland, OR).
Cellular Isolation and Flow Cytometry.
Brains from wild-type or S1P1-eGFP mice were manually dissociated in Hanks' buffered salt solution containing 1% fetal bovine serum, 500 μM EDTA, and 25 mM HEPES, and myelin was removed from the samples by using a myelin removal kit (Miltenyi Biotec, Auburn, CA), according to the manufacturer's instructions. Lymph nodes were manually dissociated in the same buffer. Samples were then stained with one or more of the following antibodies: PE-F4/80 (BioLegend), APC-GLAST-1 (Miltenyi), APC-Cy7-CD11b (BD Biosciences), PE-Cy7-CD4 (eBioscience, San Diego, CA), peridinin chlorophyll protein complex-Cy5.5-CD8 (BD Biosciences), and/or Pacific Blue-B220 (BD Biosciences), and data were collected by using an LSR II flow cytometer (BD Biosciences). Calculation of mean (neurons and astrocytes) or modal (B220+ and CD4+ cells) fluorescence intensity was performed with the method and for the statistical reasons described in detail in the supplemental materials of the original description of S1P1-eGFP mice (Cahalan et al., 2011).
Protein Electrophoresis and Western Blotting.
Processing of whole-brain and spinal cord specimens from mice with EAE for electrophoresis was performed as described previously (Cahalan et al., 2011). After electrophoresis, gels were scanned by using a Typhoon in-gel scanner (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK) with a fluorescein isothiocyanate filter for identification of S1P1-eGFP. The H-60 anti-S1P1 antibody (Santa Cruz Biotechnology) was used to confirm S1P1 expression in the brains of S1P1-eGFP mice. Detection of S1P1-eGFP ubiquitinylation in CNS samples was performed as described previously (Gonzalez-Cabrera et al., 2007).
Statistical Analysis.
All analyses were performed by using either unpaired, two-tailed, Student's t tests or analyses of variance. The data bars and error bars indicate mean ± S.E.M.
Results
Modulation of EAE by CYM-5442.
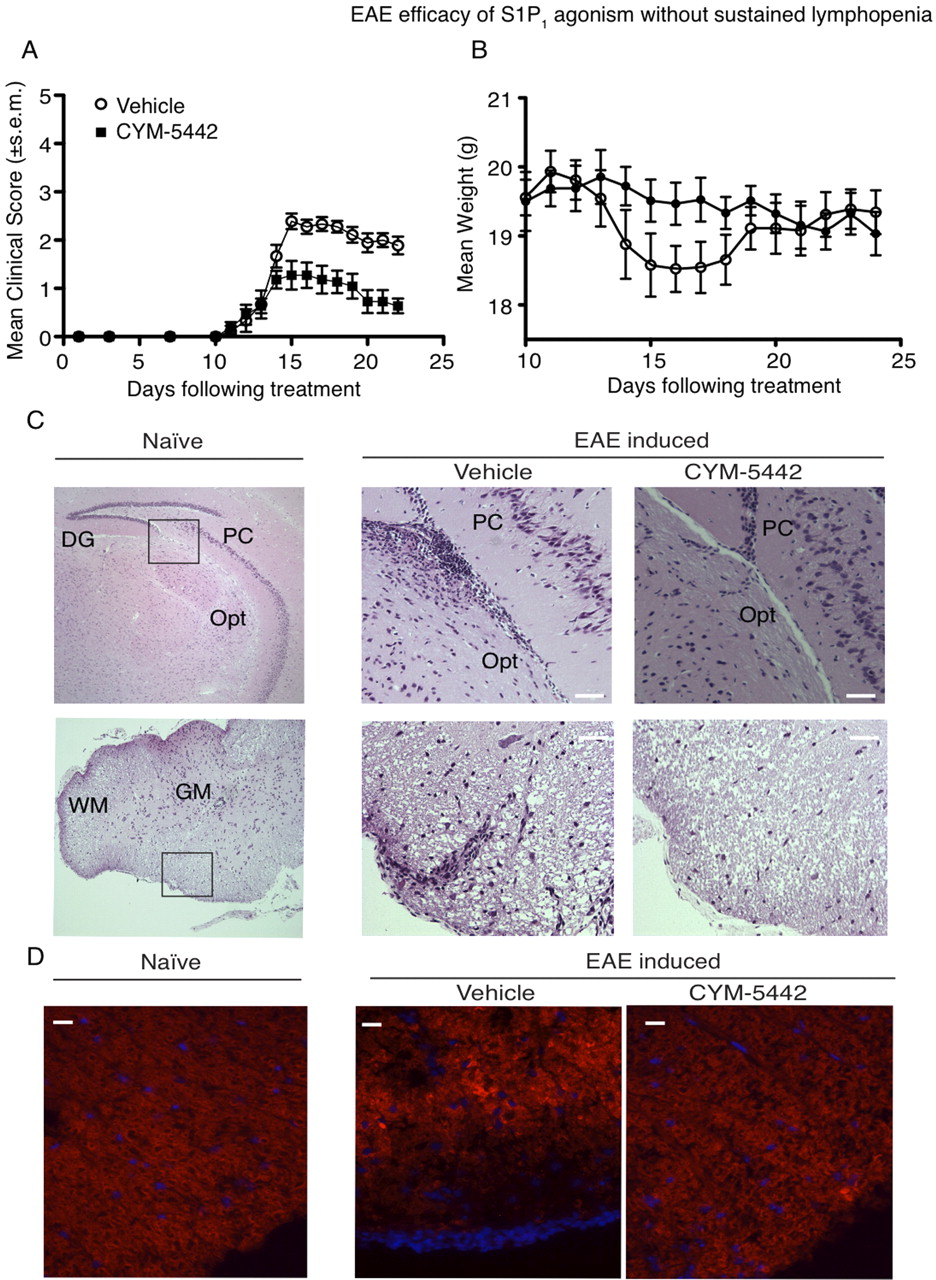
Because long-lived nonselective S1P receptor agonist prodrugs such as fingolimod ameliorate EAE and selective S1P1 agonists inhibit the development of experimental autoimmune neuritis (Zhang et al., 2009), a similar demyelinating disease, we sought to determine whether the short-acting S1P1 agonist CYM-5442 could modulate EAE severity in mice with clinical signs. Daily treatment of mice with 10 mg/kg CYM-5442 from the onset of clinical signs after EAE induction with MOG35–55 peptide significantly attenuated both clinical signs and weight loss (Fig. 1, A and B). Maximal clinical score differences between vehicle- and CYM-5442-treated mice were observed at day 16 after immunization (vehicle, 2.38 ± 0.16, n = 9; CYM-5442, 1.11 ± 0.29, n = 9; p < 0.0001). A significant reduction in infiltrating lymphocytes in the brain parenchyma of CYM-5442-treated mice but not vehicle-treated mice tracked with improved clinical scores. This was particularly evident in the optic tract and within the perivasculature feeding the white matter of the lumbar and thoracic spinal cord sections (Fig. 1C). Spinal cords of mice treated with CYM-5442 showed preservation of myelination within the outmost white matter, as demonstrated with FluoroMyelin red staining (Fig. 1D) and LFB staining (Supplemental Fig. 1A). LFB staining in brains of mice with EAE showed that CYM-5442 treatment also protected the myelinated tracts lining the optic tract (Supplemental Fig. 1B) and inhibited astrogliosis (Supplemental Fig. 1C). Efficacy was also seen in severe EAE with high mortality rates (Supplemental Fig. 2), as measured with clinical scores (vehicle, 4.25 ± 0.25, n = 4; CYM-5442, 2.28 ± 0.36, n = 7; p = 0.0043). Therefore, CYM-5442 administration after disease onset inhibited cellular infiltration, neuronal damage, and astrogliosis during MOG35–55-induced EAE, which was reflected in improved clinical scores, decreased CNS parenchymal cell infiltration, reduced demyelination, and reduced mortality rates.

Daily CYM-5442 treatment alleviates EAE. A, mean clinical scores for the indicated treatment in EAE are plotted. Mice underwent EAE induction with a MOG35–55/incomplete Freund's adjuvant/M. tuberculosis emulsification. Mice received daily injections of 10 mg/kg i.p. CYM-5442 in water or an equal volume of water (vehicle). B, CYM-5442 prevents weight loss in mice with induced EAE. The graph indicates the mean weights of both animal groups from the onset of treatment. C, CYM-5442 treatment inhibits inflammatory cell infiltration into the optic tract (Opt) of the brain and into perivascular cuffs of the spinal cords of mice with EAE. DG, dentate gyrus; PC, pyramidal cell; GM, gray matter; WM, white matter. Scale bar, 40 μm. Images are of hematoxylin and eosin-stained sagittal sections of brains and spinal cords. The left images are from a naive mouse. Scale bar, 200 μm. D, CYM-5442 ameliorates demyelination in the spinal cord of mice with EAE. FluoroMyelin red staining of spinal cord sections obtained at the end of the protocol shows that outermost demyelination is reduced in CYM-5442-treated mice. Blue staining, 4′,6-diamidino-2-phenylindole staining. Scale bar, 20 μm. All curves and images are representative of three experiments with at least six mice per group per experiment.
Cyclical Lymphopenia with CYM-5442 Sufficient for Efficacy in EAE.
CYM-5442 has a high CNS-to-plasma ratio and a short half-life in circulation (Gonzalez-Cabrera et al., 2008). Similar to CYM-5442, fingolimod displays a high CNS to plasma ratio (Foster et al., 2007); however, fingolimod has been shown to induce sustained lymphopenia with a single dose (Luo et al., 1999). Rapid clearance of CYM-5442 from the circulation led to short-duration lymphopenia, blood lymphocyte counts reaching a nadir 4 h after treatment and returning to pretreatment levels 16 to 24 h after treatment (Fig. 2A). A single low dose of fingolimod (0.2 mg/kg i.p.) induced maximal lymphopenia for at least 48 h (Fig. 2B). Therefore, we compared CYM-5442 and fingolimod to assess whether efficacy driven by transient lymphopenia would be similar to that achieved with prolonged lymphopenia.

CYM-5442 induces short-term lymphocyte sequestration. A, dosing of naive mice once with 10 mg/kg CYM-5442 induces both T and B lymphocyte sequestration from the blood, which recovers by 24 h. Graphs are representative of two experiments, with four mice per group per experiment. B, fingolimod induces sustained maximal lymphopenia for 48 h in naive mice treated with a single 0.2 mg/kg i.p. dose. The graph represents one experiment with at least six animals per group. C, mean clinical scores for the indicated treatment in EAE are plotted. Mice underwent EAE induction with a MOG35–55/incomplete Freund's adjuvant/M. tuberculosis emulsification. Mice received daily intraperitoneal injections of 10 mg/kg CYM-5442 in water, 3 mg/kg fingolimod in water, or an equal volume of water. The graph is representative of two experiments with at least three mice per group per experiment. D, blood from mice with induced EAE was removed at day 23, either 3 h or 24 h after the last fingolimod or CYM-5442 dose, and demonstrated that, in mice with EAE, blood lymphocyte counts recover only in the CYM-5442 group, even after many daily doses of CYM-5442. The graphs represent one experiment with at least three animals per group. ns, not significant. **, p < 0.01; ***, p < 0.001. E, cytokine levels are reduced in plasma of CYM-5442- and fingolimod-treated mice on day 23 of EAE. *, p < 0.05; **, p < 0.01. The graph represents one experiment with at least three animals per group. TNF, tumor necrosis factor; RANTES, regulated on activation, normal T expressed and secreted; MCP, monocyte chemotactic protein; IL, interleukin.
Mice with EAE that were treated daily with either 10 mg/kg CYM-5442 or 3 mg/kg fingolimod from the onset of symptoms showed equivalent attenuation in mean clinical scores relative to vehicle treatment (Fig. 2C). Maximal clinical score differences between vehicle and CYM-5442 treatment and between vehicle and fingolimod treatment were observed at day 21 after immunization (vehicle, 2.33 ± 0.10, n = 6; CYM-5442, 0.86 ± 0.14, n = 7; fingolimod, 0.83 ± 0.42, n = 6; p < 0.001).
Lymphocyte sequestration kinetics for CYM-5442 and fingolimod on the last day of EAE treatment are shown in Fig. 2D. The drugs showed similar B- and T-lymphocyte sequestration at 3 h after the last injection, compared with vehicle, but only CYM-5442 treatment showed significant recovery of lymphocyte counts to near untreated levels by 24 h. In contrast, fingolimod maintained nadir lymphopenia between 3 h and 24 h. These data suggest that sustained lymphocyte sequestration through S1P1 agonism is not required for efficacy in MOG35–55-induced EAE in mice. Additional analyses of the plasma of animals with EAE, at 24 h after the last injection, indicated that both CYM-5442 and fingolimod led to significant reductions in the concentrations of interleukin 17 and interleukin 1β, two proinflammatory cytokines involved in the progression of EAE (Fig. 2E). Because fingolimod phosphate uptake into CNS has been associated with cellular targeting and amelioration of EAE (Foster et al., 2007), we determined the kinetics of CYM-5442 during EAE amelioration. CYM-5442 accumulated in the CNS, relative to plasma (Table 1), high concentrations being found 3 h after the last CYM-5442 treatment (brain, 5.91 ± 0.17 μmol/mg of tissue; spinal cord, 4.32 ± 0.16 μmol/mg of tissue). CYM-5442 persisted in the CNS in significant concentrations 24 h after the last CYM-5442 treatment (brain, 352 ± 32 nmol/mg of tissue; spinal cord, 360 ± 72 nmol/mg of tissue). In contrast, CYM-5442 was cleared from plasma below the concentration required to maintain lymphopenia by 24 h (3 h, 320 ± 30 nM; 24 h, 10 ± 1 nM). Therefore, the persistent presence of CYM-5442 in the CNS may play an important role in the amelioration of EAE.
CYM-5442 retention in brain and spinal cord
Values are representative of two experiments, with three mice per group per experiment.
S1P1-eGFP Expression in CNS.
S1P1 is thought to be expressed on several cell types involved in the pathogenesis of MS, including lymphocytes that invade and attack the CNS, endothelial cells that normally provide a barrier to entry of these lymphocytes into the CNS parenchyma (Cahalan et al., 2011), neurons that are targeted for destruction by autoreactive lymphocytes (Nishimura et al., 2010), and astrocytes, which play a major role in inflammation and CNS scarring associated with MS (Sorensen et al., 2003). We analyzed S1P1-eGFP protein expression on these different cell types through immunofluorescence assays using S1P1-eGFP mice, which retain full physiological and pharmacological functions from the native S1P1 locus (Cahalan et al., 2011). S1P1-eGFP expression within the spinal cord was localized primarily in gray matter (Fig. 3A). Within the spinal cord white matter, S1P1-eGFP was expressed on CD31-expressing endothelial cells and microtubule-associated protein 2-expressing neurons, whereas expression on myelin basic protein-expressing oligodendrocytes within the white matter was below the limits of detection (Fig. 3B). S1P1-eGFP expression in the brain was widespread over cell bodies and neurites, with the highest levels in the cerebellum as well as the cortex, hippocampus, and caudate putamen (Supplemental Fig. 3).

S1P1-eGFP is highly expressed within the spinal cord. A, fluorescence microscopic images of spinal cord specimens from wild-type and Edg1eGFP/eGFP mice. S1P1-eGFP is preferentially expressed within the gray matter (GM) in the spinal cord. Scale bar, 100 μm. B, S1P1-eGFP is expressed on endothelial cells and neurons but not oligodendrocytes within the spinal cord. Spinal cord sections from Edg1eGFP/eGFP mice were stained with antibodies against CD31 (endothelial cells; scale bar, 20 μm), myelin basic protein (MBP) (oligodendrocytes; scale bar, 20 μm), or microtubule-associated protein 2 (MAP2) (neurons; scale bar, 100 μm). WM, white matter.
CYM-5442-Induced S1P1-eGFP Degradation During EAE Treatment.
We induced EAE in S1P1-eGFP mice to examine the effects of EAE induction, and subsequent daily treatment with CYM-5442, on the expression of S1P1-eGFP. CYM-5442 administration for 8 days reduced EAE clinical scores for S1P1-eGFP mice relative to vehicle treatment (vehicle, 3.75 ± 0.25, n = 4; CYM-5442, 2.20 ± 0.2, n = 5; p = 0.0017) (Supplemental Fig. 4A). A single 10 mg/kg CYM-5442 dose induced similar maximal lymphocyte sequestration in both C57BL/6J and S1P1-eGFP mice under naive conditions (no EAE) (Supplemental Fig. 4B), whereas lymphocyte subsets were significantly reduced in S1P1-eGFP mice 3 h after the last CYM-5442 injection in active EAE (Supplemental Fig. 4C). Because CYM-5442 accumulates in the brain and numerous S1P1 agonists cause cellular S1P1 internalization and subsequent ubiquitin-dependent S1P1 down-regulation (Gräler and Goetzl, 2004; Gonzalez-Cabrera et al., 2007), we examined the ability of CYM-5442 to modulate S1P1-eGFP expression within the brain during active EAE, at 3 h after the last CYM-5442 treatment. Treatment with CYM-5442 caused significant loss of S1P1-eGFP in the spinal cords and brains of mice with EAE (Fig. 4). This loss was clearly evident in immunofluorescence examinations at 488 nm of frozen sections of spinal cords from agonist-treated S1P1-eGFP mice and in assays using an antibody against the C terminus of S1P1 in paraffin sections of spinal cords (Fig. 4, B and C). The concomitant increase in polyubiquitinylated receptors in brain from S1P1-eGFP mice with EAE (Fig. 4C) indicated sustained agonist-induced S1P1 internalization and subsequent S1P1 down-regulation in the CNS. S1P1-eGFP expression also seemed to increase in the gray matter after induction of EAE (Fig. 4, A and B); however, we found that injecting PTX alone over the span of 2 days, to resemble the EAE protocol, was able to up-regulate S1P1-eGFP expression in the brains and spinal cords of naive mice (Supplemental Fig. 5).

Daily CYM-5442 treatment leads to degradation of S1P1-eGFP specifically in the CNS. A, expression of S1P1-eGFP in the spinal cord is significantly reduced after daily CYM-5442 treatment. Expression of S1P1 within the spinal cord was assessed with immunofluorescence assays (488 nm) using S1P1-eGFP mice (scale bar, 20 μm) or immunohistochemical staining of wild-type tissue sections with an antibody against the carboxyl terminus of S1P1 (C-t-S1P1) (scale bar, 100 μm). The left images are from a naive mouse. GM, gray matter; WM, white matter. B, S1P1-eGFP is degraded in the CNS of mice with induced EAE that were treated daily with 10 mg/kg CYM-5442. Both astrocytes and neurons up-regulate S1P1-eGFP after EAE induction and degrade S1P1-eGFP after daily CYM-5442 treatment. Gray, wild-type mouse; green, naive S1P1-eGFP mouse; red, vehicle-treated S1P1-eGFP mouse with EAE; blue, CYM-5442-treated S1P1-eGFP mouse with EAE. Astrocytes were identified as FSC-AloSSC-AhiCD11b−GLAST-1+ cells, whereas neurons were identified as FSC-AloSSC-AhiCD11b−GLAST-1− cells. Lymphocytes in the lymph node demonstrate negligible S1P1-eGFP degradation after daily CYM-5442 treatment. Mean (neurons and astrocytes) or modal (B cells and CD4+ T cells) fluorescence intensities are indicated for each group. C, brain S1P1-eGFP degradation with CYM-5442 is associated with increased S1P1-eGFP polyubiquitinylation. Receptor expression was measured through in-gel fluorescence imaging of brain lysates and densitometry or Western blotting using the antibody against the carboxyl terminus of S1P1 (C-t-S1P1). Polyubiquitinylation was measured through immunoprecipitation (IP) of brain lysates with green fluorescent protein, followed by immunoblotting (IB) for ubiquitin (P4D1). GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
We used flow cytometry for single-cell analysis of S1P1-eGFP expression on specific cell types within the brain and lymph nodes 2 weeks after EAE induction, with or without daily CYM-5442 dosing. After dissociation of myelin, astrocytes (identified as FSC-AloSSC-AhiCD11b−GLAST-1+ cells) and neurons (identified as FSC-AloSSC-AhiCD11b−GLAST-1− cells) up-regulated S1P1-eGFP after induction of EAE, probably because of the direct PTX effect. In addition, both of these cell types showed S1P1-eGFP down-modulation with daily CYM-5442 treatment. In contrast, CYM-5442 treatment did not decrease S1P1-eGFP expression in lymphocytes isolated from lymph nodes 3 h after the last injection of CYM-5442 and EAE induction did not induce changes in S1P1-eGFP expression in lymphocytes (Fig. 4B), despite the significant lymphocyte sequestration seen with CYM-5442 at this time point. Supplemental Fig. 6 shows internalization of membrane-associated S1P1-eGFP into cytoplasmic vesicles with an acute 30-min CYM-5442 treatment in neuronal progenitor cells isolated from day 13 S1P1-eGFP embryos.
Discussion
Fingolimod has ushered in a new era of orally administered disease-modifying MS treatments based on endpoints of clinical score progression, annualized relapse rates, and the accrual of gadolinium-enhancing lesions in magnetic resonance imaging. All current MS treatments have significant adverse effects, some of which may be life-threatening. Natalizumab (Tysabri; Elan Pharmaceuticals, South San Francisco, CA), for example, yields a significantly increased risk of progressive multifocal leukoencephalopathy, which is the result of inadequate control of preexisting JC virus infection stemming from deficits in immunosurveillance caused by natalizumab inhibition of memory T cells' entry into tissues through postcapillary venules (Kleinschmidt-DeMasters and Tyler, 2005). The long-term safety of the nonselective S1P receptor prodrug fingolimod, which suppresses lymphocyte recirculation for 4 to 6 weeks after withdrawal (Johnson et al., 2010) as a result of its 1-week half-life, has yet to be determined. However, it does not sequester circulating effector T cells efficiently from the circulation (Xie et al., 2003) and thus may allow for effective immunosurveillance.
Understanding the molecular basis of S1P receptor agonist efficacy in CNS inflammatory disease provides important insights into approaches that might improve the safety/efficacy window for patients. Administration of shorter-acting agents, with which lymphopenia can return toward normal levels during the dosing interval (Gonzalez-Cabrera et al., 2008; Cahalan et al., 2011), may offer some advantages over intermittent administration of long-acting agents, which cause sustained lymphocyte sequestration. Here we demonstrate this by using a short-acting selective CNS-penetrant S1P1 receptor agonist and S1P1-eGFP knock-in mice in a murine EAE model, which permits tracking of ligand and physiologically and pharmacologically active receptors in both peripheral and CNS tissues. This has allowed an accounting of cell- and tissue-specific receptor distributions under physiological, disease, and treatment conditions.
The detailed quantitative distribution of S1P1 in the CNS of normal, inflamed, and CYM-5442-treated mice is important, because agonism of S1P1 alone shows significant efficacy across all measures of EAE. Although expression of S1P receptors in the CNS has been explored by using mRNA profiling (Chae et al., 2004) and sometimes by using antibody detection of S1P1 protein (Akiyama et al., 2008; Sinha et al., 2009), S1P1-eGFP mice allow a high-resolution view of S1P1 expression, at the protein level, within the CNS. This enables the documentation of significant up-regulation of S1P1-eGFP on neurons and astrocytes in mice with EAE. Because in vitro S1P enhances neurite extension (Toman et al., 2004) and can dampen neuronal excitability through Gi-coupled suppression of intracellular cAMP (Zhang et al., 2006), S1P1 up-regulation may be a compensatory mechanism to limit excitatory neuronal damage in the inflammatory processes of EAE.
We showed here that S1P1 agonism alone is sufficient to ameliorate EAE. CYM-5442 is an important pharmacological tool for understanding EAE because its pharmacokinetic features produce strong lymphopenia that persists for 6 to 8 h and returns peripheral blood lymphocyte numbers to basal levels within 24 h after treatment. Given the association of progressive multifocal leukoencephalopathy with the use of natalizumab and the reports of some CNS herpetic infections with the use of fingolimod, it is helpful to understand the boundary conditions for the degree of lymphopenia that is essential for efficacy. The full recovery of lymphocyte recirculation within a dosing interval, as observed for CYM-5442, might result in lower efficacy compared with longer-duration agonists that induce full sequestration for days. This was not the case for CYM-5442 treatment, for which the treatment outcome was indistinguishable from that of fingolimod in both mild and severe models of EAE. Peripheral lymphopenia, although a convenient surrogate marker for efficacy, is clearly not the sole contributor to the effective amelioration of both demyelination and leukocyte infiltration in the brain and the spinal cord parenchyma during EAE.
Although lymphocyte recirculation was restored during each dosing interval of CYM-5442 treatment, the expression of S1P1-eGFP in the CNS after daily treatment was down-regulated, consistent with the higher maintained levels of CYM-5442 within the CNS. Although we observed decreased but not absent expression of S1P1-eGFP on neurons and astrocytes, evidence for receptor down-regulation and degradation demonstrates long-term alterations in S1P1 signaling tone in the CNS and does not definitively demonstrate functional antagonism by CYM-5442 in these cells in vivo. Despite lower total S1P1 expression, the agonist present may lead to continual S1P1 signaling within the CNS, where any receptor that reaches the cell surface is rapidly activated and internalized, consistent with the polyubiquitinylation of S1P1-eGFP detected in mice treated with CYM-5442. Degradation of S1P1-eGFP in the CNS was in contrast to findings observed with peripheral lymphocytes, which reflects the preferential distribution of CYM-5442 to CNS tissue compared with the relatively rapid clearance from plasma and secondary lymphoid organs. The lack of S1P1-eGFP degradation on peripheral lymphocytes observed with CYM-5442 is also in contrast to results found with either fingolimod phosphate (Oo et al.2011) or the recently described short-duration agonist N-[4-[5-[3-cyano-4-(1-methylethoxy)phenyl]-1,2,4-oxadiazol-3-yl]-2,3-dihydro-1H-iden-1-yl]-β-alanine (RP-001) (Cahalan et al., 2011), a compound closely related to CYM-5442. This disparity demonstrates that agonist-induced changes in S1P1 expression are dependent on both the cell type on which S1P1 is expressed and the specific agonist used; therefore, the properties of pharmacological probes must be examined in detail.
Although we demonstrated that S1P1 agonism is sufficient to alleviate EAE, the relative quantitative contributions of lymphocyte sequestration, sustained S1P1 signaling, and S1P1 down-regulation require additional study. Cell-specific deletion of S1P1 on astrocytes suggested that astrocytic S1P1 contributes to fingolimod efficacy in models of EAE, at the peak of disease severity (Choi et al., 2011). Continued development of additional selective S1P1 agonists with differences in tissue distribution, potency, and capacity to down-regulate receptors would allow for separation of these components. Of particular interest would be identification of agonists that do not cross the blood-brain barrier and evaluation of the ability of such agonists to reverse EAE when administered either systemically, which could exclude the role of CNS-expressed S1P1, or intracranially, which could exclude the role of peripherally expressed S1P1. The combination of chemical and genetic approaches to systematic mapping of S1P receptor signaling under physiological, pathological, and therapeutic conditions probably would facilitate the development of better therapeutic agents for the treatment of MS, which could achieve clinical efficacy while best preserving host defenses against latent viruses within the CNS.
Authorship Contributions
Participated in research design: Gonzalez-Cabrera, Cahalan, Kago, and Rosen.
Conducted experiments: Gonzalez-Cabrera, Cahalan, Nguyen, Sarkisyan, Leaf, Cameron, and Kago.
Performed data analysis: Gonzalez-Cabrera, Cahalan, Nguyen, Leaf, and Cameron.
Wrote or contributed to the writing of the manuscript: Gonzalez-Cabrera, Cahalan, and Rosen.
Acknowledgments
We thank Margaret Chadwell for histological expertise.
Footnotes
H.R. is a cofounder of Receptos, Inc.
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.This work was supported by the National Institutes of Health National Institute of Allergy and Infectious Diseases [Grants U01-AI074564, R01-AI055509] and the National Institutes of Health National Institute of Mental Health [Grant U54-MH084512].
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
ABBREVIATIONS:
- MS
- multiple sclerosis
- CNS
- central nervous system
- PBS
- phosphate-buffered saline
- PE
- phycoerythrin
- APC
- antigen-presenting cell
- S1P
- sphingosine-1-phosphate
- MOG
- myelin oligodendrocyte glycoprotein
- EAE
- experimental allergic encephalomyelitis
- eGFP
- enhanced green fluorescent protein
- PTX
- pertussis toxin
- LFB
- Luxol fast blue
- CYM-5442
- 2-(4-(5-(3,4-diethoxyphenyl)-1,2,4-oxadiazol-3-yl)-2,3-dihydro-1H-inden-1-yl amino)ethanol
- RP-001
- N-[4-[5-[3-cyano-4-(1-methylethoxy)phenyl]-1,2,4-oxadiazol-3-yl]-2,3-dihydro-1H-iden-1-yl]-β-alanine.

- Received September 27, 2011.
- Accepted October 26, 2011.
- Copyright © 2012 The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}