


Abstract
The nuclear receptor peroxisome proliferator–activated receptor (PPAR)α is known primarily as a regulator of fatty acid metabolism, energy balance, and inflammation, but evidence suggests a wider role in regulating the biotransformation of drugs and other lipophilic chemicals. We investigated whether PPARα directly regulates the transcription of cytochrome P450 3A4, the major human drug-metabolizing enzyme. Using chromatin immunoprecipitation in human primary hepatocytes as well as electrophoretic mobility shift and luciferase reporter-gene assays, we identified three functional PPARα-binding regions (PBR-I, -II, and -III) within ∼12 kb of the CYP3A4 upstream sequence. Furthermore, a humanized CYP3A4/3A7 mouse model showed in vivo induction of CYP3A4 mRNA and protein by [4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio]acetic acid (WY14,643) in liver but not in intestine, whereas hepatic occupancy of PBRs by PPARα was ligand independent. Using lentiviral gene knock-down and treatment with WY14,643 in primary human hepatocytes, PPARα was further shown to affect the expression of a distinct set of CYPs, including 1A1, 1A2, 2B6, 2C8, 3A4, and 7A1, but not 2C9, 2C19, 2D6, or 2E1. Interestingly, the common phospholipid 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphocholine (16:0/18:1-PC), previously proposed to reflect nutritional status and shown to be a specific endogenous ligand of PPARα, induced CYP3A4 (up to 4-fold) and other biotransformation genes in hepatocytes with similar selectivity and potency as WY14,643. These data establish PPARα as a direct transcriptional regulator of hepatic CYP3A4. This finding warrants investigation of both known and newly developed PPARα-targeted drugs for their drug-drug interaction potential. Furthermore, our data suggest that nutritional status can influence drug biotransformation capacity via endogenous phospholipid signaling.
Introduction
Genes encoding enzymes and transporters that protect higher animals from toxic effects of lipophilic xenobiotics have evolved not only to optimize their substrate selectivity and catalytic properties but also their regulation to allow flexible responses to environmental changes. In particular, the lipid- and xenobiotics-sensing nuclear receptors pregnane X-receptor (PXR; NR1I2), the constitutive androstane receptor (CAR; NR1I3), and the aryl hydrocarbon receptor, together with liver-enriched transcription factors hepatocyte nuclear factor (HNF)4α (NR2A1), CAATT/enhancer binding protein (C/EBP), and others form networks of transcription factors that coordinately regulate hepatic expression of most of the drug-metabolizing cytochromes P450 (P450), phase II enzymes, and transporters in response to xenobiotic exposure (Handschin and Meyer, 2005; Pascussi et al., 2008; Pelkonen et al., 2008). Recent research elucidated extensive crosstalk between these receptors and other nuclear receptors and transcription factors, linking xenobiotic metabolism to the homeostasis of lipids, bile acids, glucose, and other endogenous processes (Moreau et al., 2008; Gao and Xie, 2012). Such insight into the connections between xenobiotic metabolism and endogenous homeostasis can therefore enhance our mechanistic understanding of drug toxicity as well as that of pathophysiological conditions of the liver, such as dyslipidemia, nonalcoholic fatty liver disease, and others (Anderson and Borlak, 2008; Buechler and Weiss, 2011).
The most important class of nuclear receptors for lipid homeostasis are the peroxisome proliferator–activated receptor (PPAR)α (NR1C1), β/δ (NR1C2), and γ (NR1C3), which act as lipid sensors to control the expression of gene networks involved in lipid and energy homeostasis, adipocyte differentiation, and inflammatory responses (Lalloyer and Staels, 2010; Wahli and Michalik, 2012). PPARα expression is high in tissues with active fatty acid catabolism, including liver, intestine, heart, muscle, and others, where it primarily regulates intracellular trafficking and metabolism of lipids, including mitochondrial and peroxisomal β-oxidation, microsomal ω-hydroxylation, and also cholesterol and bile acid biosynthesis (Pyper et al., 2010). Activating ligands include synthetic fibrates used to treat dyslipidemia and the investigative specific agonist WY14,643 [4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio acetic acid], as well as various endogenous lipids (Forman et al., 1997; Kliewer et al., 1997; Chakravarthy et al., 2009). Elucidation of PPARα target genes by treatment of human and mouse hepatocytes with WY14,643 revealed that regulation of hepatic lipid metabolism is generally well conserved, whereas other regulated genes were largely divergent (Rakhshandehroo et al., 2009, 2010). For example, CYP4 family members, which function as microsomal fatty acid ω-hydroxylases, are more inducible in rodents compared with humans (Waxman 1999; Richert et al., 2003; Hsu et al., 2007), whereas drug-metabolizing CYPs of families 1–3, including CYP3A4, CYP2B6, CYP2C8, and CYP1A2, appeared to be inducible by fibrates in humans only (Richert et al., 2003; Prueksaritanont et al., 2005; Rakhshandehroo et al., 2009). Although functional PPREs (peroxisome proliferator response elements) were identified in CYP4 genes (Johnson et al., 1996), as well as in several phase II enzymes, including members of the UDP-glucuronosyltransferases (Barbier et al., 2003; Senekeo-Effenberger et al., 2007), the basis for PPARα-dependent hepatic expression of human CYPs of families 1–3 remained unclear, in particular whether regulation occurs directly or indirectly, for example, by involvement of other nuclear receptors such as PXR (Aouabdi et al., 2006). Further evidence suggesting an involvement of PPARα as a regulator of CYP3A4, the most important drug-metabolizing P450 of human liver and intestine (Zanger and Schwab, 2013), includes a systems biology approach that analyzed time-dependent expression profiles of primary human hepatocytes (PHH) treated with statins (Schröder et al., 2011) and a pharmacogenomics study, which identified PPARα polymorphisms as a predictive marker of hepatic CYP3A4 expression and function (Klein et al., 2012).
Here we used PHH and a CYP3A4/3A7-humanized mouse model to investigate whether PPARα directly regulates transcription of CYP3A4. Our data suggest a previously unrecognized role of PPARα as a direct transcriptional regulator of drug-metabolizing CYPs of families CYP1–3. We also tested whether a specific phospholipid that was recently shown to act as endogenous ligand of PPARα and that is believed to serve as an indicator of nutritional status (Chakravarthy et al., 2009) is able to induce CYP3A4 and other ADME (absorption, distribution, metabolism, and excretion) genes. Our data suggest intricate interactions between lipid homeostasis, nutritional status, and drug biotransformation capacity that may be of relevance under diseased conditions such as obesity and hepatic steatosis. Furthermore, our findings suggest a largely uninvestigated potential for PPARα-dependent drug-drug interactions.
Materials and Methods
Primary Human Hepatocytes and Treatments
The use of human hepatocytes for research was approved by the local ethics committees of Berlin and Regensburg, and written informed consent was obtained from all patients. Hepatocytes were isolated and cultured essentially as described (Klein et al., 2012) except that 12-well plastic dishes precoated with collagen were used (OMNILAB, Bremen, Germany). Cells were treated for 24 hours with the indicated substances in 0.1% dimethylsulfoxide, rifampicin (5,6,9,17,19,21-hexahydroxy-23-methoxy- 2,4,12,16,18,20,22-heptamethyl-8-[N-(4-methyl- 1-piperazinyl)formimidoyl]-2,7-(epoxypentadeca- [1,11,13]trienimino)-naphtho[2,1-b]furan- 1,11(2H)-dion-21-acetat) (R3501), CITCO (6-(4-Chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime) (C6240), or WY14,643 (C7081) from Sigma-Aldrich (Steinheim, Germany) or in 4% bovine serum albumin in phosphate-buffered saline (for phospholipids) purchased from Avanti Polar Lipids (Hamburg, Germany).
Chromatin Immunoprecipitation (ChIP) Assay
ChIP was performed using MAGnify Chromatin Immunoprecipitation Kit (Invitrogen, Darmstadt, Germany) according to the manufacturer’s description. DNA was sheared by sonication to an average length of ∼300 bp using Bioruptor (diagenode, Liege, Belgium) and incubated with 10μg of anti-PPARα antibody (PP-H0723-00; R&D Systems, Wiesbaden, Germany), previously bound to 10 µl of magnetic beads at 4°C for 2 hours, and DNA was purified using DNA purification beads and eluted in 150 µl of elution buffer. Promoter occupation was analyzed with 10 μl of immunoprecipitated DNA by Sybr-Green polymerase chain reaction (PCR). Primers are listed in Supplemental Table 1. The results were normalized to HMGCR (3-hydroxy-3-methyl-glutaryl-CoA reductase) promoter region (−1970/−2200 bp) used as positive control (van der Meer et al., 2010) and the untranscribed region Untr-5 as negative control (Hariparsad et al., 2009). For ChIP analysis of fresh-frozen mouse livers, we applied the same protocol using specific manufacturer recommendations for tissue handling. Binding to Gys2 (glycogen synthase 2) promoter was assessed in parallel as a positive control for binding of mouse Pparα (Mandard et al., 2007).
Plasmids
Previously Described Plasmids.
Human PPARα expression plasmid pcDNA 3-h PPARα was kindly provided by T. Tanaka, University of Tokyo, Japan (Tanaka et al., 2002); pcDhsRXRA, encoding human retinoid X receptor (RXR)α, was described elsewhere (Mathäs et al., 2012); CYP3A4 luciferase reporter gene constructs were described previously (Tegude et al., 2007). PPREX3-TK-luc construct, containing three PPREs upstream of the firefly luciferase gene under control of a minimal thymidine kinase promoter, was purchased from Addgene (Cambridge, MA) (#1015) (Kim et al., 1998). Mutations in the identified PPREs were generated using QuickChange II Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA). The wild-type and contextual mutation constructs (12–17) were generated from self-designed gene fragments that were synthesized by GeneArt (Life Technologies, Darmstadt, Germany) and cloned into construct 1.
Electrophoretic Mobility Shift Assays (EMSAs)
Human PPARα and RXRα proteins were synthesized using expression plasmids and TNT T7 Quick Coupled Transcription/Translation System (Promega, Madison, WI). Complementary single-stranded oligonucleotides (Supplemental Table 2) were annealed and radiolabeled as described (Tegude et al., 2007). Binding reactions and gel electrophoresis were performed as described (Geick et al., 2001). Retarded complexes were quantified using BAS1800 II phosphor-storage scanner (Fuji, Kanagawa, Japan) and AIDA software, version 4.19 (Raytest, Straubenhardt, Germany).
In Silico Analysis
For predictions of potential PPARα binding sites, NUBIScan version 2.0 (cutoff Z-score 7; Podvinec et al., 2002) at www.nubiscan.unibas.ch and PPRESearch (defaults settings; Venkatachalam et al., 2009) at www.classicrus.com/PPRE were used.
Luciferase Reporter Gene Assays
Human hepatocarcinoma HepG2 cells were transfected with 250 ng of reporter plasmid and 50 ng of pcDNA 3-h PPARα expression vector using Turbofect (Fermentas, St. Leon-Rot, Germany). pcDNA3 [plasmid (p) with cytomegalovirus (c) promoter] plasmid DNA was added to adjust the total amount of transfected DNA to 300 ng. To control for transfection efficiency, 25 ng of pGL3-TK-Renilla (Promega) was used. Cells were harvested in reporter “passive” lysis buffer (Promega) after 24 hours of incubation, and cell extracts were analyzed for firefly and renilla luciferase activities using Enspire Multimode Plate-Reader (PerkinElmer, Rodgau, Germany) and Luciferase Assay System from P.J.K. GmbH (Kleinblittersdorf, Germany).
Animal Experiments
Experiments with humanized huCYP3A4/3A7 mice (Hasegawa et al., 2011) were performed at TaconicArtemis GmbH (Cologne, Germany). Animals were maintained in a temperature-controlled room (23 ± 1°C) with a 12-hour light/dark cycle. All experiments were approved by the Local Ethical Review Committee and conducted in compliance with the United Kingdom “Animals (Scientific Procedures) Act” (1986). Fifty milligrams of WY14,643 in corn oil per kilogram of body weight was orally administered (with syringe) to five male huCYP3A4/3A7 mice at approximately 3 months of age daily for 4 days. Control animals received corn oil vehicle only. Mice were then anesthetized and exsanguinated, and tissues were removed and flash-frozen in liquid nitrogen.
Quantitative Reverse Transcriptase-PCR Analysis
Isolation of total RNA, quantification, and complementary DNA synthesis were performed as described (Klein et al., 2012). The assays for the detection of PPARα mRNA (Klein et al., 2012), CYP1A1 (400 nM of fw:5′-TGG TCA AGG AGC ACT ACA AAA CC-3′, and rev:5′-TCA TCT GAC AGC TGG ACA TTG G-3′ primers, and 200nM of probe MGB: 6-FAM-GAT GTG GCC CTT CTC-MGB), and CYP3A4 (Wolbold et al., 2003) were self-designed and oligonucleotides were from Applied Biosystems (Darmstadt, Germany). For all other genes, predesigned Taqman assays were purchased from Applied Biosystems. For comprehensive quantitative PCR profiling, we used Fluidigm’s Biomark high-throughput quantitative PCR chip platform (Fluidigm Corporation, San Francisco, CA) with 96 gene expression assays (Applied Biosystems; list available on request) following manufacturer’s instructions (Spurgeon et al., 2008). For final evaluation of fold changes, ribosomal gene RPLP0 (human) and beta-actin (mouse) were used for normalization.
Western Blot Analysis
CYP3A4 protein was detected in mouse liver homogenates using 50 μg of protein per lane, as described earlier (Wolbold et al., 2003). β-actin was detected using mouse monoclonal antibody from Sigma-Aldrich A5441 in 1:5000 dilutions.
Lentivirus-Mediated RNAi in Human Primary Hepatocytes
Treatment of PHH with lentivirus encoding PPARα-specific small hairpin RNAs (shRNA) was performed as described (Klein et al., 2012). Further details can be found at http://seek.virtuelle-leber.de/sops/13.
Measurement of P450 Enzyme Activities
P450 enzyme activities were determined in culture supernatants using a liquid chromatography with tandem mass spectrometry–based substrate cocktail assay, as reported previously (Feidt et al., 2010).
Statistical Methods
For pairwise analysis of statistical significance, the Student’s paired t test (two-tailed) was applied and statistical significance was defined as P < 0.05. To identify statistically significant differences, one-way analysis of variance with Student-Newman-Keuls posttest was performed with the mean values of at least three independent experiments done in triplicate. For correlations, the Spearman rank correlation coefficient (rs) was calculated. All calculations were performed using GraphPad Prism (GraphPad Software Inc., San Diego, CA).
Results
ChIP Reveals Occupation of CYP3A4 Promoter by PPARα in PHH.
In silico analysis of the CYP3A4 promoter and upstream region (20 kb) identified a vast number of putative direct repeat (DR)1 and DR2 motifs with different degrees of homology to the consensus AGGTCA nuclear receptor half site; however, it did not identify any 100% consensus motif. Thus, we systematically screened ∼12 kb of upstream region by ChIP of isolated chromatin from PHH using a total of 40 primer pairs. As evident from Fig. 1, three regions, designated as PPARα-binding regions (PBR-I, −2600/−3100 bp; PBR-II, −7200/−7800 bp; PBR-III, −8500/−8900 bp) showed significant enrichment of promoter binding by PPARα compared with HMGCR and unoccupied intermediary gene regions. Pretreatment of hepatocytes with WY14,643 did not change enrichment profiles (unpublished data). Region PBR-II overlapped partially with the well described xenobiotic-responsive enhancer module (XREM), which includes binding sites for several transcriptional regulators of CYP3A4, including PXR, HNF4α, and farnesoid X receptor.
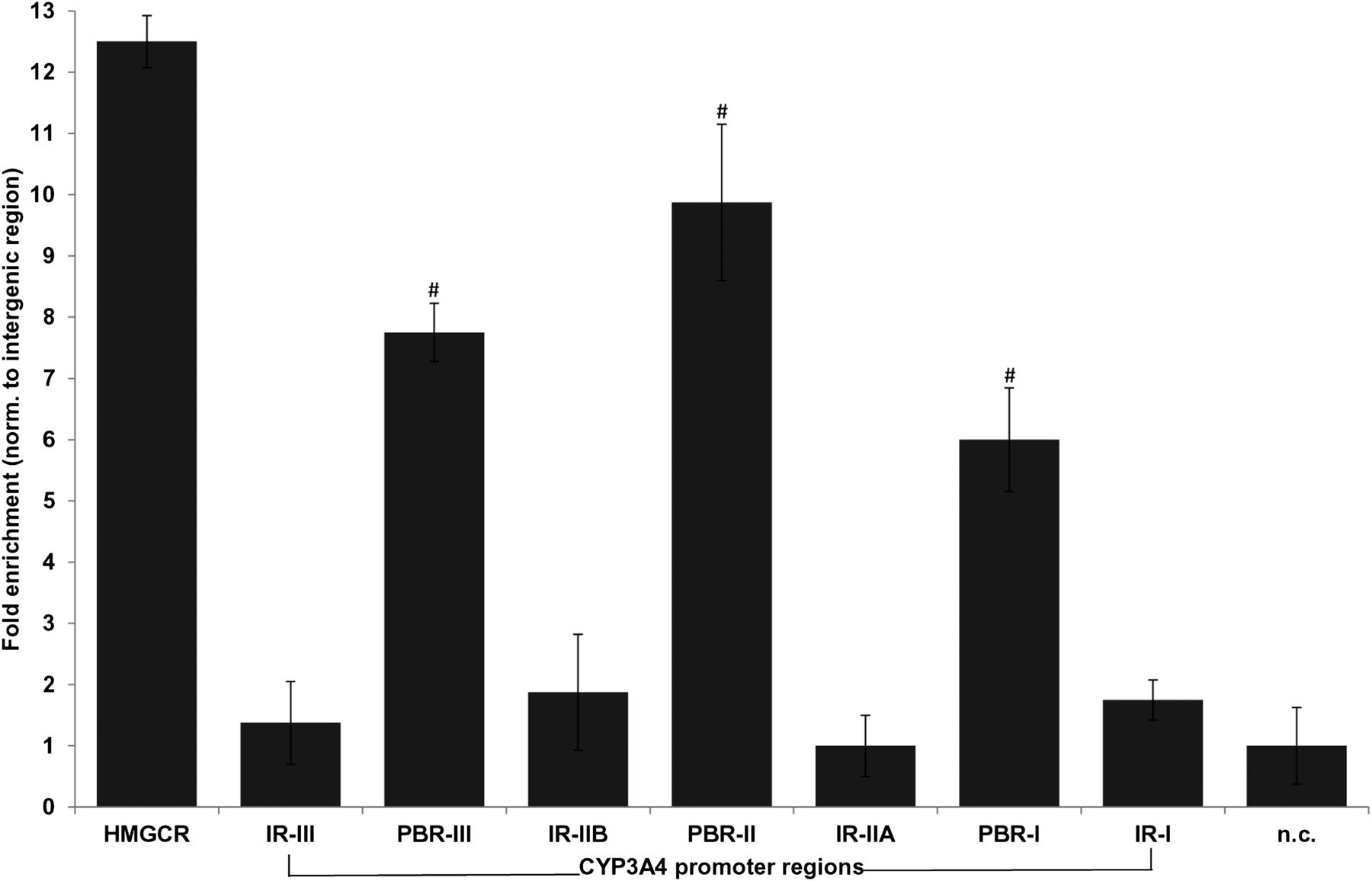
Binding of PPARα to the CYP3A4 promoter in vivo. Human primary hepatocytes were fixed with formaldehyde in suspension, and DNA was sheared by sonication. Sheared chromatin was incubated with PPARα antibody coupled to magnetic beads. Protein-DNA complexes were precipitated, and bound protein was digested by protein kinase K. Precipitated DNA was purified and was used, together with input DNA, as template for Sybr-Green PCR using a total of 40 primer pairs spanning approximately 10 kb of the CYP3A4 promoter region. Raw Ct (cycle threshold) values were normalized to input DNA to calculate the percentage of DNA immunoprecipitated. Primers encompassing the PPRE of the human HMGCR gene were used as positive control. Means relative to intergenic binding (n.c.) ± S.D. of three independent donors (two male, one female) are shown. Significant enrichment was assessed using ANOVA (analysis of variance) analysis and is indicated by hashes. IR, intermediary region; n.c., negative control.
PPARα Directly Binds to Distinct Motifs within the Identified PPARα Binding Regions.
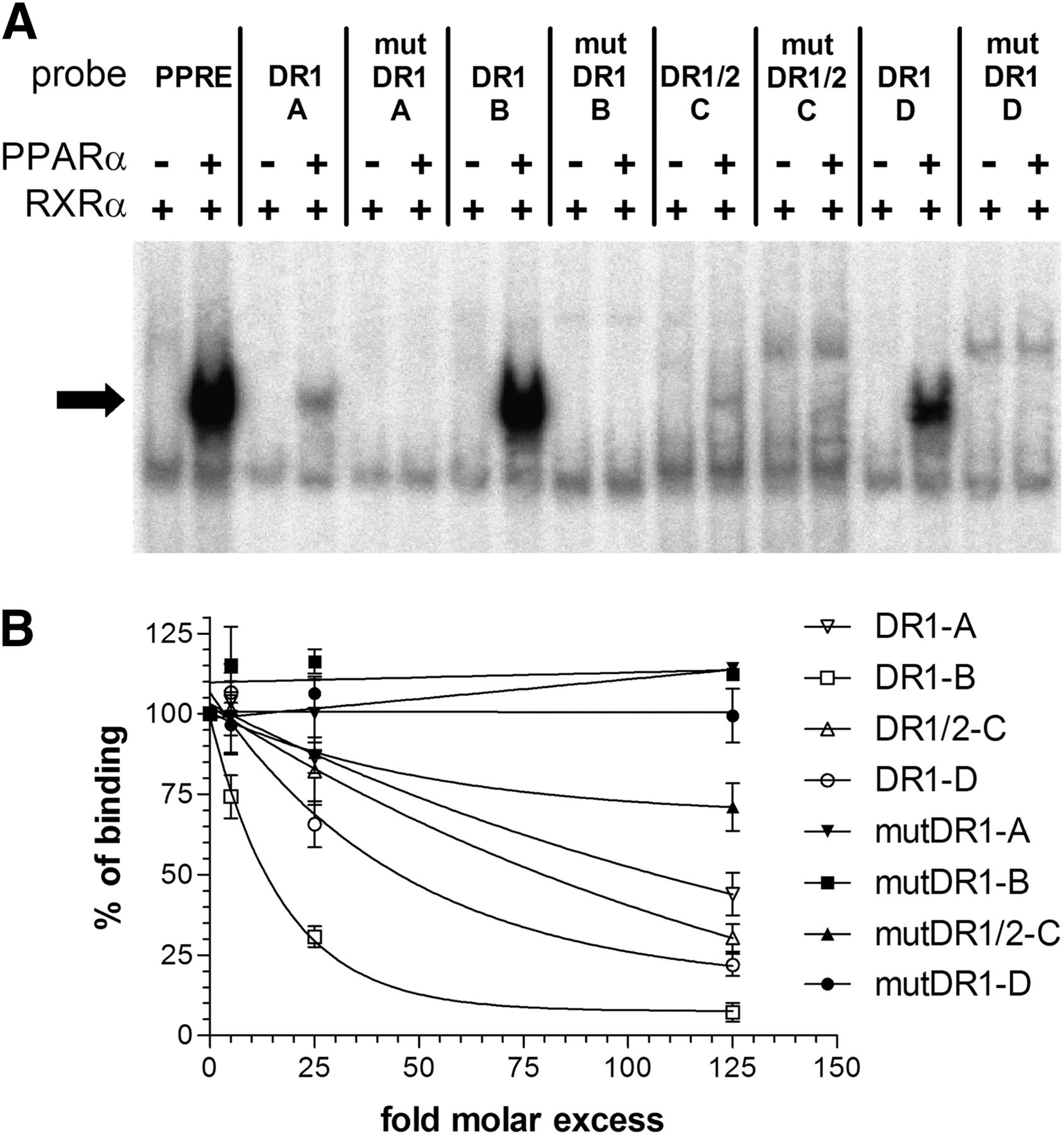
We next investigated whether PPARα can directly bind to PPREs in the identified PBRs. PPARα is known to bind as PPARα/RXRα heterodimer to DR1 and DR2 motifs and in silico analysis of PBRs I to III identified several high-score putative binding motifs (Table 1). PPARα/RXRα specifically bound to three DR1-type motifs and to one DR1/DR2 motif within the identified PBRs (Fig. 2A). Competition EMSA experiments determined relative in vitro binding affinities of PPARα/RXRα in the order DR1-B > DR1-D ≥ DR1/DR2-C > DR1-A (Fig. 2B).
PPREs identified in the CYP3A4 upstream region

PPARα specifically binds to distinct DR1/DR2 motifs with differential affinities. (A) Electrophoretic mobility shift assays using in vitro translated proteins bound to radiolabeled double-stranded oligonucleotide probes corresponding to the indicated wild-type or mutated (mut) motifs of CYP3A4. As a positive control for binding, the known PPARΑ binding site of the rat ACOX1 gene (36), here indicated as PPRE, was used. Binding reactions contained (+) or lacked (−) the indicated proteins. Complexes of PPARα /RXRα heterodimers with the oligonucleotides are marked by an arrow. (B) Competition electrophoretic mobility shift assays with radiolabeled wild-type motif DR1-B as probe and n-fold molar excess of indicated unlabeled motifs as competitors were performed. Retarded complexes were quantified and expressed as the percentage of the complex obtained in the absence of competitors. Data represent means of two independent experiments for each competitor ± S.D.
Functional Redundancy and Cooperativity of CYP3A4 PPREs in Transcriptional Regulation by PPARα.
Results of HepG2 cell cotransfection experiments performed in the absence and presence of WY14,643 are shown in Fig. 3. Deletion analysis of the CYP3A4 5′ upstream promoter region showed that only constructs comprising the region between −2.0 kb and −8.8 kb were transcriptionally activated by PPARα (Fig. 3A). The −8.8kb construct showed 1.9-fold induction of PPARα activation by WY14,643. The presence of the PBR-I region comprising motifs DR1-A and DR1-B was sufficient for PPARα-dependent transactivation (construct 4), which was further enhanced if regions PBR-II and -III with motifs DR1/DR2-C and DR1-D were also present (construct 5). All three distal upstream PBRs acted as PPARα-dependent enhancers as they distance-independently conferred activation by the receptor to the proximal CYP3A4 promoter (Fig. 3A, constructs 6–11). To dissect the contribution of individual PPREs, contextual mutational analysis was performed. Because construct 5 was too large for manipulation by mutagenesis, an artificial construct 12 was created, which comprises the three PBRs and 360 bp of proximal promoter sequence (Fig. 3B). Although mutation of DR1-A and DR1-D did not show any significant effect, mutation of motifs DR1-B and DR1/DR2-C completely abolished constitutive and inducible PPARα-dependent transactivation.

The PPARα binding regions in the CYP3A4 promoter show differential and redundant functions. Luciferase reporter gene constructs [A (A, 1–11); (B, 12–17)] containing the indicated sequences of the CYP3A4 promoter (in kilobase pairs, kb) were cotransfected with a PPARα expression plasmid and renilla-luciferase expression vector into HepG2 cells. Cells were treated with either vehicle [DMSO (dimethylsulfoxide), gray bars] or 50µM WY14,643 (black bars) for 24 hours before measurement of firefly/renilla luciferase activities. Firefly luciferase activities were normalized to renilla luciferase activities. Data are means ± S.D. of three independent experiments, each performed in triplicates; •, mutated sites; #, statistically significant (P < 0.05) compared with construct 1; *, statistically significant (P < 0.05) compared with untreated. The TK gene promoter fused to three copies of the ACOX1-PPRE was used as positive control.
PPARα Regulates CYP3A4 In Vivo in a Humanized CYP3A4/CYP3A7 Mouse Model.
Since it is was shown that mouse cyp3a genes were not responsive to the WY14,643 treatment (Rakhshandehroo et al., 2009), we used a CYP3A4-humanized mouse model to investigate whether CYP3A4 is regulated by mouse Pparα in vivo. Indeed, as shown in Fig. 4, oral administration of WY14,643 for 4 days resulted in the significant induction of the mouse Acox1 (4-fold) and Cyp4a10 (27-fold) genes, as well as approximately 5-fold induction of the human CYP3A4 in the liver (Fig. 4A). Western blot analysis confirmed about 2-fold induction of CYP3A4 protein (Fig. 4C). However, no induction was seen in the intestine, although the mouse Pparα target genes were still induced, although at lower fold changes (Fig. 4B).

Induction of CYP3A4 by WY 14,643 in humanized huCYP3A4/3A7 mice. Humanized CYP3A4/3A7 mice were fed with WY14,643 or vehicle (corn oil) for 4 days, and mRNA expression of target genes was analyzed by real-time reverse transcriptase-PCR in liver (A) and intestine (B) tissue lysates normalized to β-actin. The bars indicate means ± S.D. relative to vehicle control set at 1, with five animals in each group. # indicates significant changes in the expression of target genes. (C) Western blot analysis of liver lysates of the huCYP3A4/3A7 mice treated with vehicle (oil, n = 5) or WY14,643 (WY, n = 5). An exemplary Western blot is shown. CYP3A4 protein was quantitated and normalized against β-actin. (D) ChIP analysis of Pparα binding to the CYP3A4 promoter in huCYP3A4/3A7 mice. ChIP assay was performed with liver tissues (n = 3 in each group) as described in Fig. 1. The promoter of glycogen synthase 2 (Gys2) was analyzed as a positive control for binding of mouse Pparα. Statistical significance (P < 0.05, ANOVA) was assessed for expression changes of WY-treated group versus oil (A-C) or for Pparα binding enrichment of PBRs versus negative control (n.c.). IR, intermediary region.
Because induction of CYP3A4 by WY14,643 treatment of the humanized mice could also be due to indirect mechanisms or to mouse regulatory sequences farther upstream of the human CYP3A4 promoter, we analyzed promoter occupancy by ChIP assay in the resected liver tissues of the humanized mice. As shown in Fig. 4D, screening of the 5′-flanking sequence confirmed human CYP3A4 promoter occupancy by mouse Pparα at binding regions PBR-III and PBR-II, whereas PBR-I did not demonstrate binding. As already observed with human hepatocytes, the enrichment profile was essentially identical between untreated and WY14,643-treated animals.
PPARα Regulates a Selective Set of CYP Genes in Human Primary Hepatocytes.
To investigate whether regulation of CYP3A4 by PPARα was characteristic of a broader scheme, we applied lentiviral vectors for shRNA-mediated downregulation of PPARα to PHH. As shown in Fig. 5A, knock-down of PPARα with two different PPARα-directed shRNA vectors strongly decreased mRNA expression not only of ACOX and CYP3A4 but also of CYPs 1A1, 1A2, 2B6, 2C8, and 7A1, whereas CYPs 2A6, 2C9, 2C19, 2E1, and 3A5 were only marginally influenced or remained unchanged. Measurement of corresponding enzyme activities after PPARα gene silencing or treatment with WY14,643 resulted in consistent changes of enzyme activities (Fig. 5B).

PPARα gene silencing and activation regulates the expression and activity of a selective set of P450s. (A) Two shRNA-encoding lentiviral vectors targeted to PPARα were used to silence PPARα gene expression in primary human hepatocytes obtained from three donors (light bars, shRNA1; dark bars, shRNA2). mRNA levels were measured by quantitative PCR 5 days after infection and compared with mRNA levels measured in cells treated with nontargeting shRNA control vector set at 1.0. Data of three independent donors (all male) are shown. (B) Enzyme activities measured by liquid chromatography-tandem mass specotrometry cocktail assay in the culture supernatants of human hepatocytes following PPARα gene silencing (means of both shRNAs) (dark bars) or treatment with WY14,643 (light bars). CYP activities measured: CYP1A2, phenacetin O-deethylation; CYP2B6, bupropion 4-hydroxylation; CYP2C8, amodiaquine N-desethylation; CYP2C9, tolbutamide 4’-hydroxylation; CYP2C19, S-mephenytoin 4’-hydroxylation; CYP2D6, propafenone 5-hydroxylation; CYP3A4, atorvastatin o-hydroxylation. Data are means of three independent donors (A) and of two donors (B). Statistical significance for RNAi experiments in comparison with control shRNA is indicated by # (P < 0.05, paired t test). Two independent experiments were made for CYP activity measurements; error bars indicate the two measurements.
Inducibility of ADME Genes by Endogenous PPARα Phospholipid-Ligands.
To investigate whether the lipid sensor function of PPARα is of relevance for the regulation of ADME genes, we compared several phospholipids with WY14,643 for inducing potency (Fig. 6A). Indeed, treatment of PHH from three donors with 16:0/18:1PC, but not several other phospholipids tested, resulted in significant induction of ACOX1 (1.2- to 1.7-fold), CYP3A4 (1.8- to 3.1-fold), CYP2C8 (3.1- to 4.2-fold), CYP1A2 (0.8- to 2.15-fold), and CYP2B6 (0.95- to 2.2-fold) genes, whereas the PPARα target genes IL1-β and CYP7A1 were strongly downregulated by both synthetic and natural ligands. The PPARα-activating properties of 16:0/18:1PC thus closely resembled those of WY14,643 in terms of target gene selectivity and potency of induction. This was also shown with a broader selection of target genes (Fig. 6B; Supplemental Table 2). The most highly induced genes were, as expected, metabolic genes (CPT1A, HMGCS2, PDK4, up to 5-fold), whereas several CYPs and other ADME genes (SULT, PXR) were induced by 86% (CYP3A5) up to 280% (CYP2C8). The high similarity in WY14,643 or 16:0/18:1PC-induced gene expression changes was reflected in a strong correlation (rs = 0.83, P < 0.0001, n = 91). WY14,643 treatment of PHH further closely mirrored the gene silencing effects as reflected in a negative correlation (rs = −0.56, P < 0.0001, n = 91).

Endogenous phospholipid ligands of PPARα induce ADME gene expression. Primary human hepatocytes of two independent donors (male) were treated for 24 hours with WY14,643 (50 µM) or with different phospholipids (50 µM): 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (16:0/18:1 GPC); 1-hexadecanoyl-sn-glycero-3-phosphocholine (16:0 GPC); 1-(9Z-octadecenoyl)-sn-glycero-3-phosphocholine (18:1 GPC); 1-stearoyl-2-oleoyl-sn-glycero-3-phosphocholine (18:0/18:1 GPC). (A) Expression of selected genes determined by quantitative reverse transcriptase-PCR and expressed relative to vehicle (DMSO; dimethylsulfoxide) control, which was set as 1. Data represent means ± S.D. of two independent experiments measured in triplicate. Statistical significance (P < 0.05, paired t test) is indicated by #. (B) Heat map showing mRNA expression changes (fold) in human hepatocytes from three independent donors after PPARα gene silencing or after treatment with WY14,643 (50 µM) or 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (16:0/18:1 GPC; 50 µM). Fold changes were calculated in relation to control shRNA (RNAi, means of both shRNAs shown) or vehicle treatment (WY14,643, 16:0/18:1PC). The color scale from blue (downregulation) to red (upregulation) spans from −2.5 to 2.5 (log2-scale). Right panel, corresponding Spearman rank correlation analysis of expression profiles of 91 genes analyzed (see Supplemental Table 3).
Comparative Transcriptional Profiling of PPARα and the Xenosensors PXR and CAR.
The broad effects of PPARα activation on ADME gene expression, which resembled those of the classic xenosensors pregnane X receptor (PXR) and CAR, prompted us to compare these three nuclear receptors more completely. We treated PHH from three different donors with the most specific available agonists (CITCO for CAR, rifampicin for PXR, and WY14,643 for PPARα) to analyze their gene regulation patterns in a wider selection of target genes. Figure 7 presents a gene expression heat map of the three donors. Induction of PXR and CAR resulted in the expected distinct upregulation of ADME genes and only slight changes in genes of energy metabolism. Activation of PPARα revealed a similar pattern of ADME gene upregulation and a stronger influence on metabolic-relevant gene expression.

Comparison of CAR, PXR, and PPARα transcriptional regulation. Heat map showing color-coded mRNA expression differences in human hepatocytes from three donors (two male, one female) after induction of CAR (CITCO, 1 µM), PXR (rifampicin, 10 µM) and PPARα (WY14,643, 50 µM). The individual donor gene expression patterns are shown on the left (1–3), and the mean values are represented on the right (1–3). The upper panel presents selected ADME genes, the lower panel selected genes of energy metabolism. The color scale from blue (downregulation) to red (upregulation) spans from −2.5 to 2.5 (log2-scale).
Discussion
Direct regulation of CYP3A4 expression by PPARα is supported by three lines of evidence: first, by ChIP of PHH, we elucidated three PBRs of 400–∼600 bp length; second, four PPREs were identified by EMSA; and third, reporter gene analysis demonstrated that two of the PPREs (DR1-B and DR1/DR2-C) are essential for transcriptional activation by PPARα. The DR1-A and DR1-B sites are located closely together in a previously unremarkable region at approximately −3kb. The DR1-B site displayed the strongest binding affinity of all four sites, in agreement with its strong functional role, as demonstrated by mutational analysis. It is of interest that the PBR-I region was less occupied in huCYP3A4/3A7 mouse liver as well as in PHHs from two male donors diagnosed with adiposity and diabetes in comparison with one nondiabetic female donor (unpublished data). As we can exclude DNA sequence variants at the identified PPARα binding sites (unpublished data), the basis for this difference remains unclear. The DR1/DR2-C site located within the well documented XREM partially overlaps with an HNF4α-binding site, which cooperatively interacts with adjacent PXR sites to promote inducibility by PXR ligands (Tirona et al., 2003). Although the DR1/DR2-C site appeared to be transcriptionally highly active and inducible, its affinity by EMSA was lower compared with the DR1-B site. The presence of several nearby and overlapping transcription factor binding sites may lead to protein-protein interactions that could explain the apparent discrepancy. The DR1-D site is located in a previously undescribed region of the CYP3A4 5′ upstream region and apparently functions independently, although in the context of all PPREs it was not essential. Taken together, our experiments suggest redundancy as well as cooperativity of at least three functional PPARα-dependent enhancers in CYP3A4, which mediate both constitutive and inducible transactivation. Interestingly, pretreatment of hepatocytes with WY14,643 did not change the enrichment profiles of PPARα binding within the upstream regions of CYP3A4. Based on our data, we hypothesize that upstream binding regions of CYP3A4 are continuously occupied by PPARα and the cofactor environment determines repression or activation of CYP3A4 transcription following binding of a ligand. Therefore, the addition of a chemical ligand does not lead to the increased binding but rather to the reinforced release of corepressors and thereby increase in transcription.
We investigated a possible involvement of PPARα in the regulation of other drug biotransformation genes by gene silencing experiments. Our data show a pattern of selectively regulated CYPs, including 1A1, 1A2, 2B6, 2C8, 3A4, and 7A1 but not CYPs 2C9, 2C19, 2D6 or 2E1, that is well in agreement with previous studies using PPARα agonists such as WY14,643 (Rakhshandehroo et al., 2009) or fibrates (Prueksaritanont et al., 2005). Beyond that, our data imply an involvement of PPARα in the expression of these genes not only by induction but also at the constitutive level, although we have not formally addressed direct regulation of these genes. Of note, two functional PPREs were found to be responsible for the regulation of CYP1A1 in intestinal CaCo2 cells (Villard et al., 2011). It is possible that these PPREs are involved in the regulation of both CYP1A1 and CYP1A2 because they are located within the bidirectional promoter between the two CYPs. Preliminary bioinformatic analyses suggest the presence of potential high-score DR1/DR2-type binding sites in the genes regulated by PPARα compared with unregulated genes.
Direct transcriptional activation by PPARα of other drug biotransformation genes has already been reported for several UDP-glucuronosyltransferases (Barbier et al., 2003; Senekeo-Effenberger et al., 2007), some drug transporters (Cheng et al., 2005; Moffit et al., 2006), as well as the rate-limiting enzyme of heme biosynthesis, aminolevulinic acid synthase (34). The regulation of hepatic P450s by PPARα can thus be regarded as a “missing link” within the drug biotransformation network. Our direct comparison of ligand-induced gene expression changes mediated by PPARα and the two prototypic xenosensors PXR and CAR further emphasize the similar target profiles of these three nuclear receptors with respect to the regulation of drug biotransformation genes in the liver.
With respect to the pronounced species difference concerning the regulation of human CYP1-3 and orthologous mouse genes by PPARα (Rakhshandehroo et al., 2009), our experiment with huCYP3A4/3A7 mice suggests that this difference must be due to the lack of functional PPRE sites within the mouse Cyp genes because mouse Pparα a was able to bind to the CYP3A4 PBRs II and III and activate hepatic transcription. Therefore, the huCYP3A4/3A7 mouse appears to be a useful model to study CYP3A4 regulation by PPARα and other factors (Hasegawa et al., 2011). The lack of Pparα-dependent CYP3A4 induction in mouse intestine is probably related to the similar observation in regard to the XREM HNF4α-binding site that regulates CYP3A4 in human liver (Tirona et al., 2003) but not in intestinal cells, where a specific additional factor was proposed to be missing (Tegude et al., 2007). Lower inducibility of CYP3A4 in the duodenum compared with liver has been described in this and another humanized mouse model before (Hasegawa et al., 2011). Interestingly, rat CYP3A and 2C genes were found to be inducible by the fibrate gemfibrozil (Liu et al., 2011).
The current data on direct P450 induction through PPARα suggest a link between endogenous lipids and the regulation of drug biotransformation in human liver. We tested this hypothesis by treating PHH with phospholipids because a recent study identified POPC (1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphocholine choline) as an authentic endogenous ligand bound to PPARα (Chakravarthy et al., 2009). Indeed, we could show induction of CYP3A4 and numerous other CYPs and drug biotransformation enzymes specifically by POPC but not by related phospholipid species. To our knowledge, this is the first observation that a phospholipid acts as a selective inducer of drug biotransformation genes in human hepatocytes. As described by Chakravarthy and colleagues (2009), intracellular availability of POPC as a PPARα ligand is limited by fatty acid synthase and choline-ethanolamine phosphotransferase-1, suggesting that metabolic perturbations in this system (e.g., in obesity and hepatic steatosis) may not affect only hepatic lipid metabolism but also drug biotransformation. The fact that externally administered POPC leads to enzyme induction in PHH suggests that dietary phospholipid components may also influence drug biotransformation gene expression. As a variety of dietary and endogenous lipids, including saturated and unsaturated fatty acids, phospholipids, eicosanoids, and many derivatives and metabolites, have been implicated in PPARα activation, our findings suggest an intricate interplay between intermediary metabolism, nutritional status, and biotransformation.
In conclusion, we have elucidated the mechanistic basis for constitutive and inducible transcriptional regulation of CYP3A4 by PPARα, and we provided evidence for a broader range of similarly regulated drug-metabolizing P450s. Since fibrate hypolipidemic drugs are used worldwide in the treatment of dyslipidemia, there is clearly a need to readdress the potential for drug-drug interactions, which may depend on nutritional status. This is of particular importance for newly developed PPARα ligands to target obesity, insulin resistance, and diabetes (Lalloyer and Staels, 2010). Our results furthermore demonstrate the direct influence of phospholipids on CYP regulation, indicating a physiologic link between lipid homeostasis and drug biotransformation in the liver, which is likely of importance in diseased states such as obesity and hepatic steatosis.
Acknowledgments
The authors gratefully acknowledge Sonja Seefried and Ute Hofmann for P450 activity measurements, and Igor Liebermann and Karina Abuazi Rincones for expert technical assistance. The authors also thank Nico Scheer (TaconicArtemis) for help with designing the mouse experiments, and Bruce Spiegelman (Dana-Farber Cancer Institute) for providing the PPREX3-TK-luc construct. Furthermore, the authors are indebted to the Charitable Foundation Human Tissue and Cell Research, Regensburg, for making human tissue available for research.
Authorship Contributions
Participated in research design: Thomas, Burk, Kandel, Damm, Weiss, Klein, Zanger.
Conducted experiments: Thomas, Burk, Kandel, Klumpp.
Performed data analysis: Thomas, Burk, Kandel, Klumpp.
Wrote or contributed to the writing of the manuscript: Thomas, Burk, Schwab, Zanger.
Footnotes
This study was supported by the German Federal Ministry of Education and Research [Virtual Liver Network Grants 0315755, 0315753, and 0315741]; and by the Robert Bosch Foundation, Stuttgart, Germany.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- ACOX1
- acyl-CoA oxidase 1
- ADME
- absorption, distribution, metabolism and excretion
- CAR
- constitutive androstane receptor
- ChIP
- chromatin immunoprecipitation
- DR
- direct repeat
- EMSA
- electrophoretic mobility shift assays
- HMGCR
- 3-hydroxy-3-methyl-glutaryl-CoA reductase
- HNF
- hepatocyte nuclear factor
- PBR
- PPARα-binding region
- PCR
- polymerase chain reaction
- P450
- cytochrome P450
- PHH
- primary human hepatocytes
- PPARα
- peroxisome proliferator–activated receptor alpha
- POPC
- 16:0/18:1-PC, 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphocholine choline
- PPRE
- peroxisome proliferator response element
- PXR
- pregnane X receptor
- RXR
- retinoid X receptor
- shRNA
- small hairpin RNA
- WY14,643
- 4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio acetic acid
- XREM
- xenobiotic-responsive enhancer module
- Received September 19, 2012.
- Accepted January 7, 2013.
- Copyright © 2013 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}