


Abstract
The glucagon-like peptide-1 receptor (GLP-1R) is a major therapeutic target for the treatment of type 2 diabetes due to its role in glucose homeostasis. Despite the availability of peptide-based GLP-1R drugs for treatment of this disease, there is great interest in developing small molecules that can be administered orally. The GLP-1R system is complex, with multiple endogenous and clinically used peptide ligands that exhibit different signaling biases at this receptor. This study revealed that small molecule ligands acting at this receptor are differentially biased to peptide ligands and also from each other with respect to the signaling pathways that they activate. Furthermore, allosteric small molecule ligands were also able to induce bias in signaling mediated by orthosteric ligands. This was dependent on both the orthosteric and allosteric ligand as no two allosteric-orthosteric ligand pairs could induce the same signaling profile. We highlight the need to profile compounds across multiple signaling pathways and in combination with multiple orthosteric ligands in systems such as the GLP-1R where more than one endogenous ligand exists. In the context of pleiotropical coupling of receptors and the interplay of multiple pathways leading to physiologic responses, profiling of small molecules in this manner may lead to a better understanding of the physiologic consequences of biased signaling at this receptor. This could enable the design and development of improved therapeutics that have the ability to fine-tune receptor signaling, leading to beneficial therapeutic outcomes while reducing side effect profiles.
Introduction
Type 2 diabetes mellitus and its associated obesity are predominantly characterized by a decrease in peripheral tissue response to insulin in association with impaired pancreatic β-cell function that results in an increase in fasting glycemia (DeFronzo, 1992). The incretin hormone, glucagon-like peptide-1 (GLP-1) has well established effects on pancreatic β-cell insulin secretion and, despite a reduction in secreted levels of this hormone in diabetic patients, it retains its potent insulinotropic activity. This action combined with a number of other important effects, including reduction in glucagon secretion, delayed gastric emptying, induction of satiety, and increasing pancreatic β-cell mass, have attracted significant interest in GLP-1 and related analogs for the treatment of type 2 diabetes mellitus (Drucker and Nauck, 2006).
GLP-1 exerts its effects by binding to the GLP-1 receptor (GLP-1R), which belongs to the family B subclass of the G protein-coupled receptor (GPCR) superfamily. In recent years, it has become clear that individual GPCRs can exist in multiple receptor conformations and can elicit numerous functional responses, both G protein- and non-G protein-mediated. This has led to the discovery that different ligands can stabilize distinct subsets of receptor conformations that can “traffic” stimulus to diverse functional outputs with varying prominence, a concept referred to as biased agonism (also known as functional selectivity, stimulus bias or ligand-directed signaling) (Kenakin, 2011). The GLP-1R is predominantly expressed in pancreatic β-cells and mediates its effects through coupling primarily to Gαs, resulting in an increase in cAMP, cell depolarization and an increase in cytosolic calcium that ultimately promotes insulin secretion (Drucker et al., 1987; Holz et al., 1993). Although cAMP formation is a critical component of GLP-1R-mediated signaling required for insulin secretion, there are also roles of other signaling pathways in augmentation of insulin responses. In addition to cAMP formation, activated GLP-1Rs can promote epidermal growth factor receptor transactivation (Buteau et al., 2003), phosphatidylinositol 3 kinase activity, insulin receptor substrate-2 signaling (Park et al., 2006), extracellular signal-regulated kinase 1 and 2 (ERK1/2) activity (Montrose-Rafizadeh et al., 1999), mobilization of intracellular calcium (iCa2+) (Baggio and Drucker, 2007), as well as nuclear translocation of protein kinase C to mediate β-cell proliferation and differentiation and promote insulin gene transcription (Buteau et al., 2003). Recent studies also support an essential role of β-arrestins in downstream GLP-1R-mediated insulin secretion (Sonoda et al., 2008; Quoyer et al., 2010). Although some of these pathways have been linked to therapeutically relevant outputs, such as insulin secretion and β-cell survival, the underlying GLP-1R-mediated signaling required for therapeutically beneficial effects, such as delaying gastric emptying and inducing satiety, are not fully understood.
Currently, approved therapeutics acting at the GLP-1R are peptide-based; however, there is substantial interest in development of small molecule drugs. In recent years, an increasing number of reports have shown discovery of structurally diverse small molecule agonists of the GLP-1R (Willard et al., 2012a). These include (but are not limited to) a series of quinoxalines, the best characterized being Compound 2 (6.7-dichloro-2-methylsulfonyl-3-tert-butylaminoquinoxaline), a series of pyrimidines, the best characterized being BETP (4-(3-benzyloxyphenyl)-2-ethylsulfinyl-6-(trifluoromethyl)pyrimidine), substituted cyclobutanes such as Boc5 (1,3-bis [[4-(tert-butoxy-carbonylamino)benzoyl]amino]-2,4-bis[3-methoxy-4-(thiophene-2-carbonyloxy)-phenyl]cyclobutane-1,3-dicarboxylic acid), and a series of compounds reported in patents by Transtech Pharma. In addition to displaying agonism in their own right, small molecule compounds that bind allosterically to the GLP-1R have the potential to modulate the function of endogenous hormones, allowing fine control of receptor function and/or spatial and temporal elements of endogenous orthosteric peptide signaling. There are many orthosteric peptide agonists of the GLP-1R, including multiple endogenous ligands, as well as several peptides that are used therapeutically or are in clinical trials (Baggio and Drucker, 2007). All peptide agonists studied to date preferentially activate cAMP over ERK1/2 and iCa2+ mobilization in vitro (Koole et al., 2010). However, the relative degree of bias is variable between ligands, with truncated GLP-1 peptides and exendin-4 having greater bias toward cAMP than full-length GLP-1 peptides and oxyntomodulin (Koole et al., 2010). In addition, allosteric ligands can differentially alter the signaling profile mediated by these endogenous peptides and can therefore induce biased signaling in a peptide-specific manner.
While most of the small molecules developed to date are not drug-like compounds, they may represent pharmacophores that can be further optimized for clinical evaluation. They also provide us with a range of useful research tools that can be used to help understand the mechanism by which these small molecules bind and exert their physiologic effects. In this study, we used an analytical approach, investigating the signaling of the GLP-1R across multiple signaling pathways to assess and quantify stimulus bias for a range of low molecular weight ligands (both peptide and nonpeptide). The ability of these small ligands to act allosterically to modulate the responses and bias of distinct orthosteric peptide ligands was also assessed.
Materials and Methods
Materials.
Small molecule GLP-1 ligands BETP (Sloop et al., 2010), Compound 2 (Knudsen et al., 2007), Boc5 (Chen et al., 2007), [(2S)-2-[[(8S)-7-benzoyl-3-[4-[(3,4-dichlorophenyl)methoxy]phenyl]-2-oxo-1,6,8,9-tetrahydropyrido[4,3-g][1,4]benzoxazine-8-carbonyl]amino]-3-[4-(4-cyanophenyl)phenyl]propanoic acid] (TT15) (Rao, 2009), and BMS21 (Mapelli et al., 2009) were synthesized according to literature and standard methods (see Supplemental Data, experimental procedure for more details). GLP-1(7–36)NH2, GLP-1(1–36)NH2, exendin-4, and oxyntomodulin were purchased from American Peptide Company (Sunnyvale, CA). Dulbecco’s modified Eagle’s medium and Fluo-4 AM were purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum (FBS) was purchased from Thermo Electron Corporation (Melbourne, VIC, Australia). AlphaScreen reagents, 96-well UniFilter GF/C filter plates, 384-well Proxiplates, and Microscint 40 scintillant were purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA). SureFire ERK1/2 reagents were obtained from TGR Biosciences (Adelaide, SA, Australia). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO) or BDH Merck (Melbourne, VIC, Australia) and were of an analytical grade.
Transfections and Cell Culture.
Human GLP-1Rs were isogenically integrated into FlpIn-Chinese hamster ovary (Flp-In-CHO) cells (Invitrogen) and selection of receptor-expressing cells accomplished by treatment with 600 μg/ml hygromycin-B as previously described (May et al., 2007). Transfected and parental Flp-In-CHO cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% heat-inactivated FBS and incubated in a humidified environment at 37°C in 5% CO2.
Radioligand Binding Assay.
Flp-In-CHO GLP-1R cells were seeded at a density of 3 × 104 cells/well into 96-well culture plates and incubated overnight at 37°C in 5% CO2, and radioligand binding carried out as previously described (Koole et al., 2011). For each cell line in all experiments, total binding was defined by 0.5 nM 125I-exendin(9–39) alone, and nonspecific binding was defined by 1 μM exendin(9–39). For analysis, data are normalized to the specific binding for each individual experiment.
cAMP Accumulation Assay.
Flp-In-CHO wild-type and mutant human GLP-1R cells were seeded at a density of 3 × 104 cells/well into 96-well culture plates and incubated overnight at 37°C in 5% CO2, and cAMP detection carried out using the PerkinElmer AlphaScreen kit, as previously described (Koole et al., 2010). All values were converted to concentration of cAMP using a cAMP standard curve performed in parallel, and data were subsequently normalized to the response of 100 μM forskolin.
pERK1/2 Assay.
Flp-In-CHO GLP-1R cells were seeded at a density of 3 × 104 cells/well into 96-well culture plates and incubated overnight at 37°C in 5% CO2. Receptor-mediated pERK1/2 was determined using the AlphaScreen ERK1/2 SureFire protocol as previously described (May et al., 2007). Initial pERK1/2 time course experiments were performed over 1 hour to determine the time at which agonist-mediated pERK1/2 was maximal. Subsequent experiments were then performed at the time required to generate a maximal pERK1/2 response (7 minutes). Data were normalized to the maximal response elicited by 10% FBS determined at 6 minutes (peak FBS response).
Intracellular Ca2+ Mobilization Assay.
Flp-In-CHO GLP-1R cells were seeded at a density of 3 × 104 cells/well into 96-well culture plates and incubated overnight at 37°C in 5% CO2, and receptor-mediated iCa2+ mobilization determined as previously described (Werry et al., 2005). Fluorescence was determined immediately after ligand addition, with an excitation wavelength set to 485 nm and an emission wavelength set to 520 nm, and readings taken every 1.36 seconds for 120 seconds. Peak magnitude was calculated using five-point smoothing, followed by correction against basal fluorescence. The peak value was used to create concentration-response curves. Data were normalized to the maximal response elicited by 100 μM ATP.
β-Arrestin Recruitment Assays.
Flp-In-CHO cell lines stably expressing GLP-1 receptor-Rluc8 and either β-arrestin (β-Arr)1- or β-Arr2-Venus were generated using gateway technology as previously described (Willard et al., 2012b). Cells were seeded in 96-well white culture plates at a density of 40,000 cells/well and cultured for 24 hours. Cells were rinsed once with Hanks’ balanced salt solution to remove traces of phenol red and incubated in fresh Hanks’ balanced salt solution for a further 15 minutes. The Rluc substrate coelenterazine-h was added to reach a final concentration of 5 μM. After a 5-minute incubation, the corresponding agonist was added and bioluminescence resonance energy transfer (BRET) readings were collected using a LumiSTAR Omega instrument (BMG Labtech, Ortenberg, Germany) that allows sequential integration of signals detected in the 465–505- and 515–555-nm windows using filters with the appropriate band pass. The BRET signal was calculated by subtracting the ratio of 515–555-nm emission over 465–505-nm emission for a vehicle-treated cell sample from the same ratio for the ligand-treated cell sample. In this calculation, the vehicle-treated cell sample represents background, and results are expressed as ligand-induced BRET. This eliminates the requirement for measuring a donor-only control sample. Initial time course experiments were performed over 20 minutes to determine the time at which β-Arr1 and β-Arr2 recruitment was maximal for each ligand in the absence and presence of BETP. Coaddition of ligands was performed for interaction assays and BRET signals were collected at this peak time point.
Data Analysis.
Data were analyzed using Prism 5.03 (GraphPad, La Jolla, CA) using the three-parameter logistic equation or an operation model of allosteric agonism.
Allosteric modulator-inhibition binding data were fitted to the following allosteric-ternary complex model. In this case, nondepletion of ligands was assumed (Avlani et al., 2008): (1)where
(1)where (2)where Y represents radioligand binding, Bmax denotes maximal binding site density, and NS denotes the fraction of nonspecific binding. [A] and KAPP denote the concentration of radioligand and equilibrium dissociation constant for the radioligand, respectively. [B] and KB denote the concentration of allosteric ligand and equilibrium dissociation constant for the allosteric ligand, respectively. [I] and KI denote the concentration of peptide agonist used in competition with the radioligand and the equilibrium dissociation constant for the peptide agonist, respectively. α and α′ represent cooperativity factors, which are defined as the allosteric interaction of the modulator with the radioligand, and modulator with the peptide agonist, respectively. Values of α or α′ > 1 are indicative of an allosteric-mediated increase in binding activity, while values of 0 < α or α′ < 1 are indicative of an allosteric-mediated decrease in binding affinity.
(2)where Y represents radioligand binding, Bmax denotes maximal binding site density, and NS denotes the fraction of nonspecific binding. [A] and KAPP denote the concentration of radioligand and equilibrium dissociation constant for the radioligand, respectively. [B] and KB denote the concentration of allosteric ligand and equilibrium dissociation constant for the allosteric ligand, respectively. [I] and KI denote the concentration of peptide agonist used in competition with the radioligand and the equilibrium dissociation constant for the peptide agonist, respectively. α and α′ represent cooperativity factors, which are defined as the allosteric interaction of the modulator with the radioligand, and modulator with the peptide agonist, respectively. Values of α or α′ > 1 are indicative of an allosteric-mediated increase in binding activity, while values of 0 < α or α′ < 1 are indicative of an allosteric-mediated decrease in binding affinity.
To compare agonist profiles and quantify stimulus bias (functional selectivity) between the different ligands, agonist concentration-response curves were fitted to the following form of the operational model of agonism ((Black and Leff, 1983; Koole et al., 2010; Evans et al., 2011), (3)where Em is the maximal possible response of the system; basal is the basal level of response; KA denotes the equilibrium dissociation constant of the agonist (A); τ is an index of the signaling efficacy of the agonist and is defined as RT/KE, where RT is the total number of receptors and KE is the coupling efficiency of each agonist-occupied receptor; and n is the slope of the transducer function that links occupancy to response. The analysis assumes that the maximal system responsiveness (Em) and the transduction machinery used for a given cellular pathway are the same for all agonists, such that the Em and transducer slope (n) are shared between agonists. The ratio, τ/KA (determined as a logarithm, i.e., log (τ/KA)) is referred to herein as the “transduction coefficient” (Kenakin et al., 2012), as this composite parameter is sufficient to describe agonism and bias for a given pathway, i.e., stimulus-biased agonism can result from either a selective affinity (KA−1) of an agonist for a given receptor state(s) and/or a differential coupling efficacy (τ) toward certain pathways. To cancel the impact of cell-dependent effects on the observed agonism at each pathway, the log (τ/KA) values were then normalized to that determined for the endogenous agonist, GLP-1(7–36)NH2, at each pathway to yield a “normalized transduction coefficient,” Δlog (τ/KA), i.e., Δlog (τ/KA) = log (τ/KA)test − log (τ/KA)GLP-1(7–36)NH2. Finally, to determine the actual bias of each agonist for different signaling pathways, the Δlog (τ/KA) values were evaluated statistically between the pathways. The ligand bias of an agonist for one pathway, j1, over another, j2, is given as
(3)where Em is the maximal possible response of the system; basal is the basal level of response; KA denotes the equilibrium dissociation constant of the agonist (A); τ is an index of the signaling efficacy of the agonist and is defined as RT/KE, where RT is the total number of receptors and KE is the coupling efficiency of each agonist-occupied receptor; and n is the slope of the transducer function that links occupancy to response. The analysis assumes that the maximal system responsiveness (Em) and the transduction machinery used for a given cellular pathway are the same for all agonists, such that the Em and transducer slope (n) are shared between agonists. The ratio, τ/KA (determined as a logarithm, i.e., log (τ/KA)) is referred to herein as the “transduction coefficient” (Kenakin et al., 2012), as this composite parameter is sufficient to describe agonism and bias for a given pathway, i.e., stimulus-biased agonism can result from either a selective affinity (KA−1) of an agonist for a given receptor state(s) and/or a differential coupling efficacy (τ) toward certain pathways. To cancel the impact of cell-dependent effects on the observed agonism at each pathway, the log (τ/KA) values were then normalized to that determined for the endogenous agonist, GLP-1(7–36)NH2, at each pathway to yield a “normalized transduction coefficient,” Δlog (τ/KA), i.e., Δlog (τ/KA) = log (τ/KA)test − log (τ/KA)GLP-1(7–36)NH2. Finally, to determine the actual bias of each agonist for different signaling pathways, the Δlog (τ/KA) values were evaluated statistically between the pathways. The ligand bias of an agonist for one pathway, j1, over another, j2, is given as (4)where
(4)where
 (5)
(5)A lack of functional selectivity will thus result in bias values not substantially different from the value of 1 between pathways and, hence, log (bias) values not significantly different from zero. To account for the propagation of error associated with the determination of composite parameters, the following equation was used.
 (6)
(6)In cell-signaling ligand interaction studies, data were fitted to the following two forms of an operational model of allosterism and agonism to derive functional estimates of modulator affinity and cooperativity (Leach et al., 2007; Aurelio et al., 2009) (7)
(7) (8)where Em is the maximum attainable system response for the pathway under investigation, [A] and [B] are the concentrations of orthosteric agonist and allosteric modulator/agonist, respectively, KB is the dissociation constant of the allosteric modulator, EC50 is the concentration of orthosteric (full) agonist yielding 50% of the response between minimal and maximal receptor activation in the absence of allosteric ligand, n is a transducer slope factor linking occupancy to response, α is the binding cooperativity factor, β is an empirical scaling factor describing the allosteric effect of the modulator on orthosteric agonist signaling efficacy, respectively, and τA and τB are operational measures of the ligands’ respective signaling efficacies that incorporate receptor expression levels and efficiency of stimulus-response coupling. Equation 4 was used in interaction studies performed between allosteric ligand (BEPT) and a full agonist (in cAMP and pERK1/2 assays), while eq. 5 was used when the BEPT was interacted with a partial agonist (in iCa2+, β-Arr1, and β-Arr2 assays). This is so because eq. 4 is only valid in cases where the orthosteric agonist has high efficacy (τ >> 1) such that KA is >> [A].
(8)where Em is the maximum attainable system response for the pathway under investigation, [A] and [B] are the concentrations of orthosteric agonist and allosteric modulator/agonist, respectively, KB is the dissociation constant of the allosteric modulator, EC50 is the concentration of orthosteric (full) agonist yielding 50% of the response between minimal and maximal receptor activation in the absence of allosteric ligand, n is a transducer slope factor linking occupancy to response, α is the binding cooperativity factor, β is an empirical scaling factor describing the allosteric effect of the modulator on orthosteric agonist signaling efficacy, respectively, and τA and τB are operational measures of the ligands’ respective signaling efficacies that incorporate receptor expression levels and efficiency of stimulus-response coupling. Equation 4 was used in interaction studies performed between allosteric ligand (BEPT) and a full agonist (in cAMP and pERK1/2 assays), while eq. 5 was used when the BEPT was interacted with a partial agonist (in iCa2+, β-Arr1, and β-Arr2 assays). This is so because eq. 4 is only valid in cases where the orthosteric agonist has high efficacy (τ >> 1) such that KA is >> [A].
Statistics.
All data are represented as mean ± S.E.M. and were compared using analysis of variance followed by Dunnett’s test. Repeated measures analysis of variance was used to assess the statistical significance between time courses. The null hypothesis was rejected at P < 0.05.
Results
Small Molecules Ligands and Peptides Differentially Couple the GLP-1R to Cellular Effectors.
The ability of a GPCR to couple to multiple intracellular signaling components is a requirement for stimulus bias. Like most GPCRs, the GLP-1R couples to different classes of heterotrimeric G proteins, including Gαs, Gαq, and Gαi, as well as various other signaling and regulatory proteins such as the β-Arrs. In this study, the selective GLP-1R small molecules, BETP (Sloop et al., 2010), Compound 2 (Knudsen et al., 2007), TT15 (Rao, 2009), Boc5 (Chen et al., 2007), and a modified GLP-1 analog (BMS21) (Mapelli et al., 2009) (Fig. 1) were assessed for their ability to activate various intracellular signaling pathways. These included cAMP (as a surrogate of canonical Gαs coupling), iCa2+ mobilization (as a measure of Gαq, and to some extent Gαi coupling), pERK1/2 [as a downstream measure of various convergent pathways (G protein and non-G protein-mediated)], and recruitment of the regulatory proteins β-Arr1 and β-Arr2.

Small molecule ligand structures. Structures of small molecule ligands used in this study.
GLP-1(7–36)NH2 can activate all five of these signaling/regulatory pathways in the Flp-In-CHO GLP-1R cell line selected for this study; however, none of the small molecules or the 11-mer peptide (BMS21) tested were able to fully mimic the actions of the native peptide ligand (Fig. 2; Table 1). BMS21 had a much lower potency than GLP-1(7–36)NH2; however, this ligand displayed higher efficacy for cAMP signaling with an increased Emax (Fig. 2; Table 1). Interestingly, this small peptide displayed a similar potency in pERK1/2 and iCa2+-mobilization assays as in the cAMP assay; however, in these instances the observed Emax was dramatically lower than that of GLP-1(7–36)NH2. In addition, BMS21 was unable to recruit β-Arrs within the tested concentration range (Fig. 2; Table 1) suggesting that although this peptide is similar to the N-terminal portion of the native ligand, this in itself is insufficient to mimic the functions of full length GLP-1(7–36)NH2.

Signaling profiles of GLP-1R ligands. Dose response curves for cAMP accumulation (A), pERK1/2 (B), iCa2+ mobilization (C), β-Arr1 recruitment (D), and β-Arr2 (E) recruitment for GLP-1(7–36)NH2, BMS21, Boc5, TT15, BETP, and Compound 2. Data are normalized to the response elicited by GLP-1(7–36)NH2 and analyzed using a three-parameter logistic equation. All values are means ± S.E.M. of three to four independent experiments conducted in duplicate.
Differential effects of peptide/small molecule agonists of the human GLP-1R in cAMP accumulation, iCa2+ mobilization, pERK1/2, and β-arrestin1 and β-arrestin2 recruitment in Flp-In-CHO cells stably expressing the human GLP-1R
pEC50 values are the negative logarithm of the concentration of agonist that produces half the maximal response. Emax represents the maximal response normalized to that of GLP-1(7–36)NH2. All values are mean ± S.E.M. of three to five independent experiments conducted in duplicate.
In agreement with previous studies, the nonpeptidic compound Boc5 was able to increase cAMP with a lower potency and efficacy than GLP-1(7–36)NH2 and BMS21 (Fig. 2A; Table 1). Boc5 also had similar efficacy in pERK1/2 and iCa2+-mobilization assays. No β-Arr recruitment could be detected for this ligand. TT15 displayed a similar potency but a marginally higher Emax for cAMP signaling compared with Boc5; however, it displayed a weaker pERK1/2 response and no iCa2+ mobilization was detectable (Fig. 2; Table 1). Unfortunately, this ligand nonspecifically interfered with BRET assay for β-Arr recruitment and therefore characterization of TT15 for β-Arr recruitment could not be performed. Compound 2 and BETP are low potency agonists for cAMP accumulation with BETP displaying weak partial agonism and Compound 2 strong partial agonism. Both compounds also displayed weak partial agonism in pERK1/2; however, in the case of BETP this was barely detectable within the concentration range assessed. Compound 2 displayed no detectable iCa2+ response; however, BETP was an agonist for this pathway with an EC50 similar to that observed for its cAMP response, and with an Emax of 42 ± 10% of that of GLP-1(7–36)NH2. However, both ligands were weak agonists for β-Arr1 and β-Arr2 recruitment with Emax estimates of 30–40% of the response of GLP-1(7–36)NH2 (Fig. 2; Table 1).
These effects on ligand bias can be readily observed in bias plots, which display the response observed to equimolar conentrations of ligand for one pathway relative to another (Fig. 3). More importantly, this relative bias can be quantified by calculation of bias factors to compare relative bias to the reference ligand, in this case the primary endogenous ligand GLP-1(7–36)NH2 (Table 2). It is apparent for all of the small molecule ligands that the GLP-1R shows less preference for coupling to cAMP over other pathways in comparison with activation by GLP-1(7–36)NH2. However some ligands heavily change the relative bias. The most dramatic changes in bias are observed with activation by BETP, whereby signaling is biased toward iCa2+ mobilization and β-Arr1 and β-Arr2 recruitment over cAMP and pERK1/2 compared with the reference agonist (Fig. 3, B–E, H, and J; Table 2). However, little change in the relative bias between iCa2+ and arrestin recruitment was observed (Fig. 3F; Table 2). In contrast BMS21 biases the receptor toward pERK1/2 and cAMP over arrestin recruitment and iCa2+ mobilization relative to GLP-1(7–36)NH2 (Fig. 3, C–E; Table 2). In addition, compared with GLP-1(7–36)NH2, compound 2 biases the receptor conformations toward β-Arr1 and β-Arr2 recruitment relative to iCa2+ (where no response was observed) and cAMP (Fig. 3, F and G; Table 2).

Synthetic ligands display stimulus bias relative to the endogenous ligand GLP-1(7–36)NH2. Bias plots of cAMP versus pERK1/2 (A), cAMP versus iCa2+ mobilization (B), cAMP versus β-Arr1 (C), cAMP versus β-Arr2 (D), iCa2+ versus pERK1/2 (E), iCa2+ versus β-Arr1 (F), iCa2+ versus β-Arr2 (G), β-Arr1 versus pERK1/2 (H), β-Arr1 versus β-Arr2 (I), and β-Arr2 versus pERK1/2 (J). Data for each ligand in each pathway are normalized to the maximal response elicited by GLP-1(7–36)NH2, and analyzed with a three-parameter logistic equation with 150 points defining the curve.
Stimulus bias exhibited by ligands relative to the reference agonist GLP-1(7–36)NH2
Data were analyzed using an operational model of agonism as defined in eq. 4 to estimate log τc/KA ratios. Changes in log τc/KA ratios were calculated to provide a measure of the degree of stimulus bias exhibited between different signaling pathways relative to that of the reference agonist (GLP-1(7–36)NH2). Values are expressed as means ± S.E.M. of three to five independent experiments conducted in duplicate. Data were analyzed with one-way analysis of variance and Dunnett’s post test.
BETP and Compound 2 Selectively Modulate the Affinity of Agonists at the GLP-1R.
In agreement with our previous study, Compound 2 displayed probe dependence in that it caused a concentration-dependent increase in affinity of oxyntomodulin, but not of GLP-1(7–36)NH2, exendin-4, or GLP-1(1–36)NH2. BETP also displayed the same probe dependence with potentiation of oxyntomodulin affinity and no effect on the other three peptides (Supplemental Fig. 2). The other small molecules did not alter the competition binding profile of 125I-exendin(9–39) in the presence of any peptide ligand tested (Supplemental Fig. 1).
BETP and Compound 2 Differentially Alter Peptide-Mediated GLP-1R Signaling Bias.
Analysis of the interaction between BETP and orthosteric peptide ligands with the allosteric operational model revealed BETP differentially modulated GLP-1R agonist intrinsic efficacy in a ligand and pathway-dependent manner. (Figs. 4–7; Table 3). Combined affinity-efficacy (αβ) estimates for cAMP were consistent with affinity cooperativity estimates from the binding studies (Fig. 4; Supplemental Fig. 2; Table 3). Thus, exendin-4, GLP-1(7–36)NH2, and GLP-1(1–36)NH2 displayed neutral cooperativity for both binding and cAMP accumulation, whereas BETP potentiated oxyntomodulin affinity and cAMP responses (Fig. 4; Supplemental Fig. 2). In contrast, BETP showed significant negative cooperativity with exendin-4, GLP-1(7–36)NH2, and GLP-1(1–36)NH2 for coupling to pERK1/2 and neutral/weak negative cooperativity with oxyntomodulin for this pathway. In iCa2+-mobilization assays, BETP displayed positive cooperativity with exendin-4 and to a lesser extent GLP-1(7–36)NH2; however, neutral cooperativity with oxyntomodulin was observed (Fig. 6). Assessment of β-Arr recruitment revealed neutral cooperativity between BETP and exendin-4 for both β-Arr1 and β-Arr2 and neutral cooperativity for GLP-1(7–36)NH2 in recruiting β-Arr1 (Fig. 7; Table 3). However, weak potentiation of β-Arr2 and of both β-Arr1 and β-Arr2 recruitment was observed for GLP-1(7–36)NH2 and oxyntomodulin, respectively, in the presence of BETP (Fig. 7; Table 3). These data indicate that BETP can engender stimulus bias at the level of the signaling pathway in a ligand-dependent manner.

BETP displays positive allosteric effects on GLP-1R-mediated cAMP accumulation in an agonist-dependent manner. Concentration response curves were generated for exendin-4 (A), GLP-1(7–36)NH2 (B), oxyntomodulin (C), or GLP-1(1-36)NH2 (D) in the absence and presence of increasing concentrations of BETP in Flp-In-CHO cells stably expressing the human GLP-1R. The curves represent the best global fit of an operational model of allosterism (eq. 4). All values are mean ± S.E.M. of three to four independent experiments performed in duplicate. Panel C reproduced from Willard et al. (2012b).

BETP displays negative allosteric effects on GLP-1R-mediated pERK1/2 by peptide ligands. Concentration response curves were generated for exendin-4 (A), GLP-1(7–36)NH2 (B), oxyntomodulin (C) or GLP-1(1–36)NH2 (D) in the absence and presence of increasing concentrations of BETP in Flp-In-CHO cells stably expressing the human GLP-1R. The curves represent the best global fit of an operational model of allosterism (eq. 5). All values are mean ± S.E.M. of three to four independent experiments performed in duplicate. Panel C reproduced from Willard et al. (2012b).

BETP positively modulates GLP-1R-mediated iCa2+ mobilization by peptide ligands. Concentration response curves were generated for exendin-4 (A), GLP-1(7–36)NH2 (B) or oxyntomodulin (C) in the absence and presence of increasing concentrations of BETP in Flp-In-CHO cells stably expressing the human GLP-1R. The curves represent the best global fit of an operational model of allosterism (eq. 5). All values are mean ± S.E.M. of three to four independent experiments performed in duplicate. Panel C reproduced from Willard et al. (2012b).

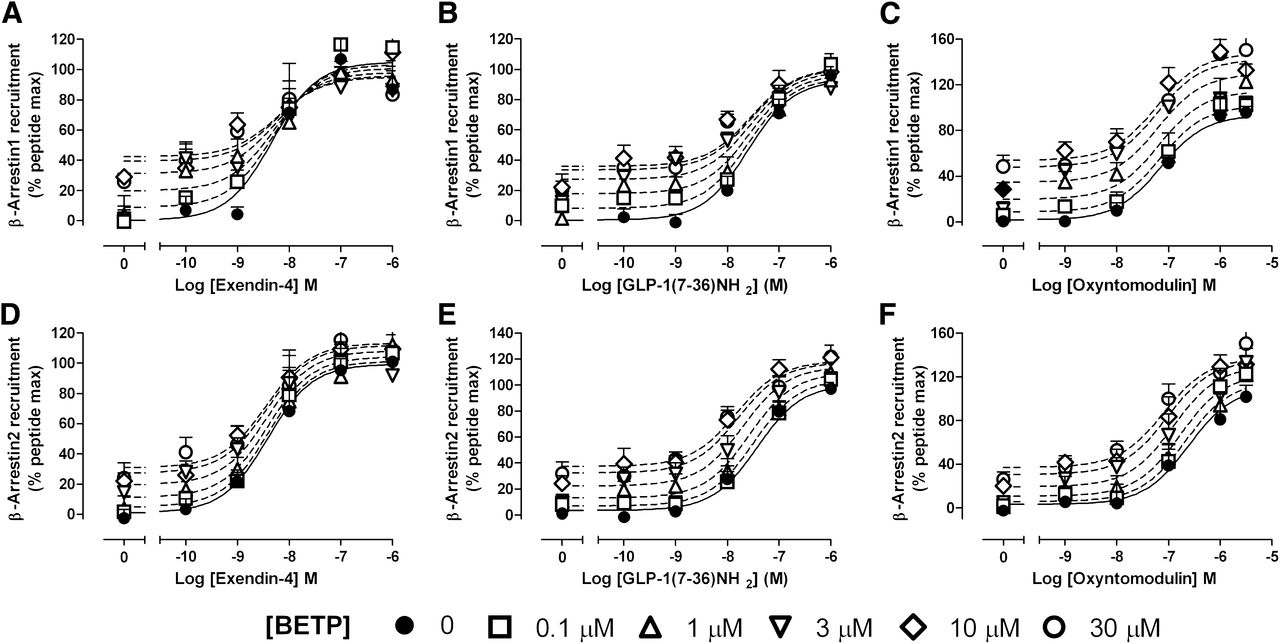
BETP does not significantly alter GLP-1R-mediated recruitment of β-arrestins by peptide ligands. Concentration response curves were generated for exendin-4 (A and D), GLP-1(7–36)NH2 (B and E), or oxyntomodulin (C and F) in the absence and presence of increasing concentrations of BETP for β-Arr1 (A–C) and β-Arr2 (D–F) recruitment. The curves represent the best global fit of an operational model of allosterism (eq. 5). All values are mean ± S.E.M. of four to five independent experiments performed in duplicate. Panel C reproduced from Willard et al. (2012b).
Functional cooperativity estimates for the interaction between BETP or Compound 2 and GLP-1R peptide ligands
Data derived from analysis of interaction concentration-response curves with an operational model of allosterism as defined in eqs. 4 and 5. pKb values are the negative logarithms for the functional affinity of the allosteric ligands; log αβ represents the composite cooperativity between the allosteric ligand and the orthosteric peptide ligand. Antilogarithms are shown in parentheses. Values represent the mean ± S.E.M. of four to six independent experiments performed in duplicate. Data were analyzed with one-way analysis of variance and Dunnett’s post test.
Functional interaction assays for cAMP accumulation and iCa2+ mobilization between each peptide ligand and Compound 2 confirmed previous findings (Koole et al., 2010); Compound 2 potentiated oxyntomodulin-induced cAMP responses but not intracellular calcium mobilization (Supplemental Figs. 3 and 4). In contrast, neutral cooperativity was observed between Compound 2 and the other three peptides in both pathways. Interaction assays for the pERK1/2 experiments included higher concentrations of Compound 2 than previously published, which revealed significant negative cooperativity of Compound 2 on exendin-4-mediated pERK1/2 responses (Supplemental Fig. 5). A similar trend was observed for both the full-length and truncated GLP-1 peptides (and to a lesser extent oxyntomodulin), although this negative cooperativity did not reach statistical significance. In contrast, Compound 2 displayed positive cooperativity with exendin-4, GLP-1(7–36)NH2, and oxyntomodulin for recruitment of both β-Arr1 and β-Arr2. The estimated cooperativity factors (αβ) revealed that this potentiation was greater for GLP-1(7–36)NH2 and oxyntomodulin than that of exendin-4 (Fig. 8). Like BETP, Compound 2 can also generate stimulus bias in a probe-dependent manner; however, it is important to note that these two allosteric ligands engender significantly different signaling profiles that only manifest when multiple signaling pathways are explored.

Compound 2 potentiates GLP-1R-mediated recruitment of β-arrestins by peptide ligands. Concentration response curves were generated for exendin-4 (A and D), GLP-1(7–36)NH2 (B and E) or oxyntomodulin (C and F) in the absence and presence of increasing concentrations of Compound 2 for β-Arr1 (A–C) and β-Arr2 (D–F) recruitment. The curves represent the best global fit of an operational model of allosterism (eq. 5). All values are mean ± S.E.M. of four to five independent experiments performed in duplicate.
GLP-1(1–36)NH2 did not display agonism in either iCa2+-mobilization assays or in recruitment of β-Arrs either in the presence or absence of either BETP or Compound 2.
In contrast to BETP and Compound 2, the small molecules TT15, Boc5, and the BMS21 peptide did not modulate any signaling pathway mediated by any of the GLP-1 peptide agonists (Supplemental Figs. 6–8). These compounds at high concentrations (particularly evident with BMS21) have characteristics consistent with a competitive mode of action with GLP-1- and GLP-1-related peptide agonists, which suggests these small ligands may share at least a partially overlapping binding site with the orthosteric pocket.
BETP and Compound 2 Can Potentiate Responses to BMS21, TT15, and Boc5.
Consistent with the evidence above indicating at least a partial overlap in binding interactions formed by TT15 and BMS21 with orthosteric ligands, these two ligands when tested for interaction with each other in a cAMP assay displayed behavior consistent with a competitive interaction (Supplemental Fig. 9). In addition, BETP and Compound 2 strongly potentiated cAMP responses mediated by both TT15 and the small peptide, BMS21 (Fig. 9; Table 4). Interestingly, BETP also potentiated Boc5-mediated cAMP responses (Fig. 9; Table 4); however, only weak modulation was observed using Compound 2 (Fig. 9; Table 4). This is particularly interesting as Boc5, when interacted in a cAMP assay with either TT15 or BMS, had a profile consistent with competitive behavior between the two ligands (Supplemental Fig. 9). This indicates that although both ligands may bind in a site partially overlapping the orthosteric site, the cooperativity between the site of Compound 2 binding and Boc5 is different from that of TT15 and BMS21. In addition, the differential degrees of cooperativity induced by the two structurally distinct modulators, BETP and Compound 2, indicate that these two compounds interact differentially with the GLP-1R.

Compound 2 and BETP potentiate GLP-1R-mediated cAMP accumulation by BMS21, Boc5 and TT15. Concentration response curves were generated for BMS21 (A and B), Boc5 (C and D), or TT15 (E and F) in the absence and presence of increasing concentrations of either Compound 2 (A, C, and E) or BETP (B, D, and F) in Flp-In-CHO cells stably expressing the human GLP-1R. The curves represent the best global fit of an operational model of allosterism (eq. 5). All values are mean ± S.E.M. of three independent experiments performed in duplicate.
Functional cooperativity estimates for the interaction between BETP or Compound 2 and Boc5, TT15, or BMS21
Data derived from analysis of interaction dose-response curves with an operational model of allosterism as defined in eq. 4. Log αβ represents the composite cooperativity between the allosteric ligand and the orthosteric peptide ligand. Antilogarithms are shown in parentheses. Values represent the mean ± S.E.M. of four to six independent experiments performed in duplicate. Data were analyzed with one-way analysis of variance and Dunnett’s post test.
Discussion
The GLP-1R is a major therapeutic target for the treatment of type 2 diabetes, however, despite the success of natural or modified GLP-1R-binding peptides for clinical treatment, low molecular weight, orally active compounds are still pursued as the preferred therapeutic approach. Traditionally, these types of molecules were designed to mimic the properties of the natural ligand by targeting the orthosteric site and this approach has been successful for many GPCR targets (Black, 1989). However, there are many cases where this has been unsuccessful, in particular for non-family A GPCRs.
Orthosteric peptide ligands for family B GPCRs bind predominantly to the large N-terminal domain prior to initiating receptor activation (Hoare, 2005). This is mechanistically different from many family A GPCRs, whose ligands primarily make contact within the transmembrane domain. Due to the size of peptide ligands and their mechanism of receptor activation, the discovery of surrogate small molecule agonists that mimic these actions has been difficult. However, several groups have recently reported small molecule nonpeptide and smaller peptide fragments that act as GLP-1R agonists or positive allosteric modulators. In this study we have revealed significant signaling bias induced by these compounds when compared with the predominant endogenous peptide, indicating that small ligands may not be able to fully mimic the actions of larger peptide hormones. In addition, we show that allosteric modulation is complex, with pathway-dependent modulation of receptor response that is determined by the combination of orthosteric ligand and allosteric ligand used. This emphasizes the need for broad elucidation of mechanism of action when developing allosteric compounds.
Activation by peptide ligands predominantly couples the GLP-1R to GαS-proteins, leading to an increase in cAMP. This is the best studied pathway of the GLP-1R and is crucial for enhancing glucose-dependent insulin secretion (Baggio and Drucker, 2007). However, like many GPCRs, the GLP-1R elicits signals via diverse pathways, including iCa2+ mobilization and pERK1/2, in addition to coupling to regulatory proteins such as β-Arrs that can activate other effectors (Montrose-Rafizadeh et al., 1999; Sonoda et al., 2008). Each of these pathways has been linked to physiologic effects of GLP-1. iCa2+ mobilization can significantly modulate the magnitude of insulin secretion, and β-Arr1 also has a role in insulin secretion, although the molecular mechanism of this regulation is poorly understood. Sustained effects on gene transcription and the preservation of β-cell mass involve multiple signaling pathways; both cAMP-dependent and -independent; the latter include activation of mitogen-activated kinases, such as ERK1/2. It is clear that the physiological response downstream of GLP-1R activation is a composite of the interplay of various signaling pathways, but even for those that have been identified, the extent and magnitude to which these effectors contribute to the physiological signaling profile and the ideal combination of these that lead to a therapeutically beneficial output has yet to be established.
Evaluation of signaling across five pathways (cAMP, pERK1/2, iCa2+ mobilization, β-Arr1, and β-Arr2 recruitment) demonstrated that, in comparison with the reference ligand [GLP-1(7–36)NH2], all of the small ligands, with the exception of BETP, coupled most strongly to cAMP production. In addition, for BMS21, TT15, and Boc5, the relative order of efficacy for the five pathways was similar to GLP-1(7–36)NH2 (Fig. 1; Table 1). Despite this, each of the ligands showed elements of signal bias, with all three having less preference for cAMP relative to pERK1/2, but no significant change when comparing the preference between all other pathways (Table 2). However, Compound 2 displayed significant signal bias with less preference for cAMP signaling relative to iCa2+ mobilization, β-Arr1, or β-Arr2. Interestingly BETP displayed a very different profile to GLP-1(7–36)NH2, as this compound heavily biased GLP-1R signaling to β-Arr1, β-Arr2, and iCa2+ mobilization relative to cAMP and pERK1/2. The response was also biased toward β-Arr1 recruitment and iCa2+ mobilization over β-Arr2 (Table 2). The ability of individual ligands to differentially activate the GLP-1R to produce distinct functional profiles may provide a unique opportunity in drug development, with the potential to sculpt receptor signaling to target physiologically important responses and exclude those that do not provide beneficial outputs.
This concept also extends to allosteric modulation of orthosteric ligand responses. In addition to small molecules displaying differential intrinsic efficacy profiles, if they bind allosterically, they can also differentially modulate peptide (both endogenous and exogenous) responses in a pathway-specific manner. Therefore, determining the modulatory profile of small molecule ligands in numerous functional outputs and using multiple orthosteric ligands is important, especially when the endogenous systems involve the interplay of many natural ligands and several signaling pathways to elicit physiological consequences. Compound 2 engendered significant bias in the response mediated by oxyntomodulin with selective enhancement of cAMP, β-Arr1, and βArr2; however, for GLP-1(7–36)NH2, only β-Arr responses were enhanced. BETP also engendered significant stimulus bias in a probe-dependent manner, with selective enhancement of oxyntomodulin-mediated cAMP responses and to a smaller extent β-Arr-1 and-2, but only iCa2+ mobilization and β-Arr2 responses were weakly enhanced when GLP-1(7–36)NH2 was cobound, while a strong negative effect on pERK1/2 was observed. When considering the clinically used exendin-4, the bias was again different; in this case only iCa2+ mobilization was significantly enhanced, with negative cooperativity seen for pERK1/2. This revealed that GLP-1R conformations induced by the cobinding of an allosteric modulator and orthosteric ligand can vastly alter the combined signaling profile of the receptor such that no two combinations of allosteric-orthosteric ligand pair were able to produce the same profile of behavior. From these studies, it is unclear whether Compound 2 and BETP share a common binding pocket, and further elucidation to identify their binding site(s) will be required. However, even if they do occupy the same pocket, the specific interactions formed between these compounds and the receptor is clearly different as they induce very distinct bias in their efficacy and modulatory properties.
This type of behavior, where ligands can alter one pathway while having very different effect on another pathway and differential probe-dependent effects at both acute and regulatory signaling pathways, may provide a therapeutic advantage by allowing fine-tuning of receptor response. However, this also presents a significant challenge, as currently it is not clear what will be the key pathway/combination of pathways that need to be manipulated to provide an ideal therapeutic response. Understanding the activity profiles of small ligands may be key for drug discovery programs. These types of compounds, which display differential efficacy and modulatory profiles, provide us with tools that could potentially be used in an in vivo/ex vivo setting to explore the physiological consequences of biased signaling. Further research is required to fully understand these concepts and ascertain the preferred signaling profile for new and better therapeutics.
The final part of this study identified that Compound 2 and BETP were able to strongly modulate cAMP responses of BMS21 and TT15 at the GLP-1R. Boc5 could also be potentiated but to a lesser extent. Data from our interaction assays also suggest that these compounds behave in a competitive manner with peptide ligands and each other. BMS21 was designed to mimic the N-terminal region of GLP-1, which is proposed to bind to the top of the transmembrane bundle and extracellular loop regions of the receptor. It is also possible that TT15 may bind in a similar region. Boc5 has also been proposed to bind in the extracellular regions of the receptor; however, its binding site may be distinct from that of BMS and TT15 as weaker cooperativity was observed with BETP and Compound 2. These observations could also represent an opportunity to aid in drug optimization. For example, ligands like BMS21, TT15, and Boc5 are less biased agonists than Compound 2 and BETP, and if mimicking the actions of GLP-1(7–36)NH2 rather than altering the bias of the natural hormone was identified as the best therapeutic approach, then elucidation of the binding sites for these ligands could aid in development of higher affinity drug-like molecules that bind to the same binding pocket. Alternatively, all small ligands identified to date display weak affinity for the GLP-1R that could arguably be due to the limited number of contacts they can form with the receptor (compared with peptide ligands). The ability of one small molecule to enhance the signaling induced by another (and vice versa) may indicate some therapeutic potential for small molecule therapies to be used in combination.
In conclusion, we have demonstrated that small molecule ligands induce biased signaling at the GLP-1R and also bias in the signaling profile of orthosteric ligands. Further work is required to delineate the extent to which such bias exists in a native cellular environment and the in vivo consequences. In recent years, the pace of identifying small molecule GLP-1R ligands has increased and this should aid in the types of studies that may lead to the discovery and development of compounds with the potential to sculpt therapeutics that show greater selectivity and improved therapeutic outcomes.
Acknowledgments
The authors thank Dan Kohlman, Mark Tebbe, Mike Coghlan, and Jorge Alsina-Fernandez for technical assistance and support.
Authorship Contributions
Participated in research design: Wootten, Willard, Sloop, Christopoulos, Sexton.
Conducted experiments: Wootten, Savage.
Contributed new reagents or analytic tools: Willard, Bueno, Sloop.
Performed data analysis: Wootten, Savage.
Wrote or contributed to the writing of the manuscript: Wootten, Willard, Bueno, Sloop, Christopoulos, Sexton.
Footnotes
- Received December 17, 2012.
- Accepted January 24, 2013.
This work was funded by National Health and Medical Research Council project [Grants 1002180 and 519461].
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- β-Arr
- beta arrestin
- BETP
- 4-(3-benzyloxyphenyl)-2-ethylsulfinyl-6-(trifluoromethyl)pyrimidine
- Boc5
- 1,3-bis [[4-(tert-butoxy-carbonylamino)benzoyl]amino]-2,4-bis[3-methoxy-4-(thiophene-2-carbonyloxy)-phenyl]cyclobutane-1,3-dicarboxylic acid
- BRET
- bioluminescence resonance energy transfer
- BSA
- bovine serum albumin
- CHO
- Chinese hamster ovary
- Compound 2
- 6.7-dichloro-2-methylsulfonyl-3-tert-butylaminoquinoxaline
- ERK1/2
- extracellular signal-related kinases 1 and 2
- FBS
- fetal bovine serum
- Forskolin
- (3R,4aR,5S,6S,6aS,10S,10aR,10bS)-6,10,10b-trihydroxy-3,4a,7,7,10a-pentamethyl-1-oxo-3-vinyldodecahydro-1H-benzo[f]chromen-5-yl acetate
- GLP-1
- glucagon-like peptide
- GLP-1R
- glucagon-like peptide-1 receptor
- GPCR
- G protein-coupled receptor
- iCa2+
- intracellular calcium
- TT15
- (2S)-2-[[(8S)-7-benzoyl-3-[4-[(3,4-dichlorophenyl)methoxy]phenyl]-2-oxo-1,6,8,9-tetrahydropyrido[4,3-g][1,4]benzoxazine-8-carbonyl]amino]-3-[4-(4-cyanophenyl)phenyl]propanoic acid
- Copyright © 2013 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}