


Abstract
Development of acquired antihormone resistance exposes a vulnerability in breast cancer: estrogen-induced apoptosis. Triphenylethylenes (TPEs), which are structurally similar to 4-hydroxytamoxifen (4OHT), were used for mechanistic studies of estrogen-induced apoptosis. These TPEs all stimulate growth in MCF-7 cells, but unlike the planar estrogens they block estrogen-induced apoptosis in the long-term estrogen-deprived MCF7:5C cells. To define the conformation of the TPE:estrogen receptor (ER) complex, we employed a previously validated assay using the induction of transforming growth factor α (TGFα) mRNA in situ in MDA-MB 231 cells stably transfected with wild-type ER (MC2) or D351G ER mutant (JM6). The assays discriminate ligand fit in the ER based on the extremes of published crystallography of planar estrogens or TPE antiestrogens. We classified the conformation of planar estrogens or angular TPE complexes as “estrogen-like” or “antiestrogen-like” complexes, respectively. The TPE:ER complexes did not readily recruit the coactivator steroid receptor coactivator-3 (SRC3) or ER to the PS2 promoter in MCF-7 and MCF7:5C cells, and molecular modeling showed that they prefer to bind to the ER in an antagonistic fashion, i.e., helix 12 not sealing the ligand binding domain (LBD) effectively, and therefore reduce critical SRC3 binding. The fully activated ER complex with helix 12 sealing the LBD is suggested to be the appropriate trigger to initiate rapid estrogen-induced apoptosis.
Introduction
17β-Estradiol (E2) is a key stimulus of growth for estrogen receptor (ER)–positive breast cancer. Endocrine therapy has been the gold standard of treatment in ER-positive breast cancer (Jordan, 2009), but acquired antihormone resistance to long-term antihormone therapy is a continuing clinical dilemma. Discovery of the evolution of acquired resistance exposed a vulnerability of cells by paradoxically triggering apoptosis with physiologic E2 treatment (Jordan, 2004). Laboratory evidence demonstrates that E2 is capable of inducing apoptosis in long-term estrogen-deprived MCF-7 cells (Lewis et al., 2005a,b). Similarly, tamoxifen-stimulated tumors (Osborne et al., 1987; Gottardis and Jordan, 1988) that develop in athymic nude mice in about 1 year will undergo regression after 5 years of tamoxifen if exposed to physiologic E2 (Yao et al., 2000).
Clinical data support the use of estrogens in the treatment of ER-positive postmenopausal breast cancer. Synthetic high-dose estrogens induced regression of tumors in postmenopausal women with advanced breast cancer in the first ever reported cancer chemical therapy (chemotherapy)–mediated clinical study (Haddow et al., 1944). Clinical trials now exploit the concept for patients with metastatic breast cancer who develop resistance to endocrine therapy, which shows that estrogens induce a partial to complete response in about 30% of postmenopausal breast cancer patients who had previous exhaustive antihormone therapy (Lønning et al., 2001; Ellis et al., 2009). A reanalysis (Anderson et al., 2012) of the Women Health Initiative estrogen alone trial (Anderson et al., 2004), which compared conjugated equine estrogen therapy with placebo in hysterectomized postmenopausal women, showed a significant decrease in the incidence and mortality from breast cancer in these patients. The success of estrogen therapy in postmenopausal women depends on the menopausal status of the patients. Women who are more than 5 years postmenopausal, i.e., long-term estrogen deprived, have better tumor remission rate as well as prevention from breast cancer (Obiorah and Jordan, 2013).
The ER is the key signal transduction pathway for breast cancer growth and apoptosis based on studies with competitive inhibition of E2 action with antiestrogen (Lewis et al., 2005a; Maximov et al., 2011). The question to be addressed is how a series of estrogens with planar or angular structures can reprogram the estrogen–ER complex to be either a survival signal in breast cancer or to trigger apoptosis. We previously classified estrogens (Jordan et al., 2001) based on reported data on the crystallization of the ligand binding domain (LBD) of the ER with estrogens (E2, diethylstilbestrol) and antiestrogens (4-hydroxytamoxifen [4OHT] and raloxifene) (Brzozowski et al., 1997; Shiau et al., 1998) (see Fig. 1). The planar estrogens are sealed within the LBD by helix 12, thus activating the AF2 domain, which leads to coactivator binding and subsequent interaction of AF1 and AF2 (Tzukerman et al., 1994) to initiate growth and protein synthesis. In contrast, the bulky side chain of nonsteroidal antiestrogen causes displacement of helix 12 and prevents coactivator binding to the AF2 resulting in antiestrogenic action. Tamoxifen, a substituted triphenylethylene (TPE) derivative possesses estrogen-like activity (Harper and Walpole, 1966; Levenson et al., 1998; MacGregor and Jordan, 1998). We previously discovered that the surface amino acid D351 within the LBD is critical for the estrogenic actions of 4OHT (MacGregor Schafer et al., 2000; Jordan et al., 2001). Unlike raloxifene, which is less estrogenic and possesses an antiestrogen side chain that shields and neutralizes D351, the side chain in 4OHT is too short (Liu et al., 2002).

Functional test: Putative conformations of the complex with ligand in LBD for Type II estrogen to be “antiestrogenic” with regard to helix 12 positioning. The assay discriminates between ligands (A), which allow helix 12 to seal the LBD or not (B and C). Sealing of helix 12 over the LBD is important for the ability of the ligands to trigger apoptosis.
To interrogate the relationship of structure of an estrogenic ligand to program the conformation of the ER complex, we synthesized a range of estrogenic TPEs (Maximov et al., 2010), which are structurally similar to 4OHT. We and others hypothesize that the structure of the ligand governs the external surface of the ER complex with either planar estrogens or the TPEs (McDonnell et al., 1995; Jordan et al., 2001). As a result of the ligand shape, the estrogens can program the conformation of the estrogen–ER complex to modulate rapid or delayed apoptosis. The growth response of the ER-positive breast cancer cells is very sensitive to a wide range of estrogenic ligands. This is to ensure cancer cell survival in austere estrogen environments. This may not be true for estrogen-induced apoptosis, and the ligand shape may be required to be more specific to trigger cell death. The estrogen-deprived cancer cell is protected.
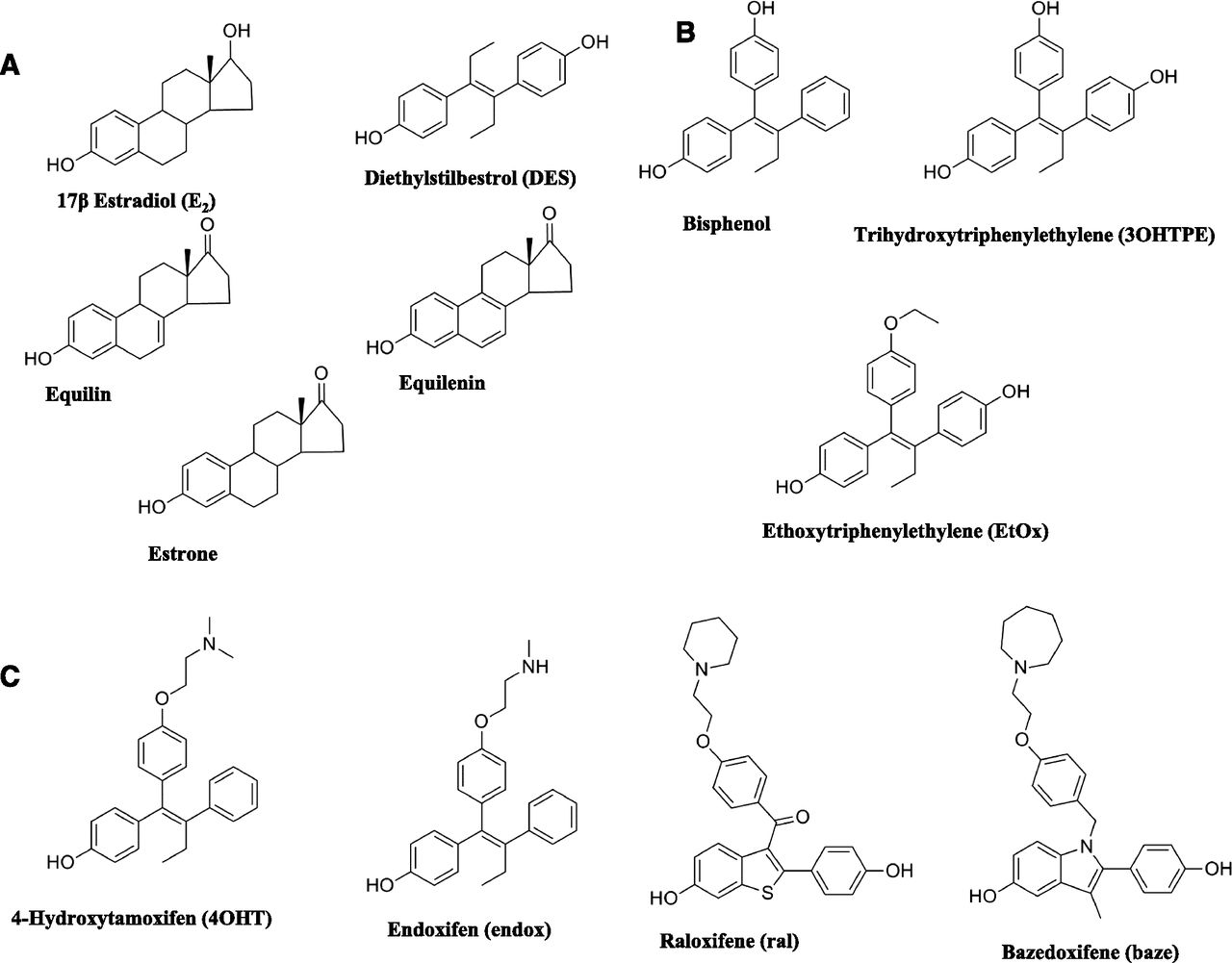
We investigated the actions of clinically relevant planar estrogens (E2, diethylstilbestrol, equilin, estrone, and equilenin), antiestrogens (4OHT, endoxifen, raloxifene, and bazedoxifene), and model TPEs (bisphenol, trihydroxytriphenylethylene, and ethoxytriphenylethylene) on growth in MCF-7 cells and apoptosis in MCF7:5C cells. To understand the biologic activity of the TPE:ER, we employed a validated ER engineered assay using induction of the mRNA for the transforming growth factor α (TGFα) gene in situ in MDA-MB231 cells stably transfected with wild-type ER or mutant D351G:ER (Jordan et al., 2001) (Fig. 1). We classified the structure of the ligands based on their ability to initiate TGFα mRNA synthesis through the ER complex. The biologic assay predicts two extremes of the ligand ER complex based on known X-ray crystallography (Brzozowski et al., 1997; Shiau et al., 1998): an “estrogen-like” shape and an “antiestrogen-like” shape. We find that the TPE:ER complex is antiestrogen-like, which explains the delayed apoptosis in MCF7:5C cells compared with the estrogenic complex formed by the planar estrogens.
Materials and Methods
Cell Culture and Reagents.
Cell culture media were purchased from Invitrogen (Grand Island, NY) and fetal calf serum (FCS) was obtained from HyClone Laboratories (Logan, UT). Compounds E2, diethylstilbesterol (DES), equilin, estrone, equilenin, ICI 182,780 (7α,17β-[9-[(4,4,5,5,5-pentafluoropentyl)sulfinyl]nonyl]estra-1,3,5(10)-triene-3,17-diol), and 4OHT were obtained from Sigma-Aldrich (St. Louis, MO), and chemical structures are as previously described (Brzozowski et al., 1997; Shiau et al., 1998; Sawicki et al., 1999; Howell et al., 2000; Wang et al., 2009). Raloxifene (ral) was a gift from Eli Lilly (Indianapolis, IN), and bazedoxifene (baze) was synthesized as previously described (Lewis-Wambi et al., 2011). The TPEs were synthesized as previously described (Maximov et al., 2010). MCF7:5C were derived from MCF-7 cells obtained from the Drs. Bill McGuire and Dean Edwards (San Antonio, TX), as reported previously (Jiang et al., 1992; Lewis et al., 2005b). MC2 and JM6 were obtained as previously described (MacGregor Schafer et al., 2000). MCF7:WS8 cells were derived from the original MCF-7 wild-type and were maintained in RPMI media supplemented with 10% FCS, 6 ng/ml bovine insulin, and penicillin and streptomycin. These have been maintained for >20 years. The MCF-7 cells were cultivated in phenol red-free media containing 10% charcoal dextran-treated FCS for 3 days prior to the start of the experiment. MCF7:5C cells were maintained in phenol-red free RPMI media containing 10% charcoal dextran treated FCS, 6 ng/ml bovine insulin, and penicillin and streptomycin. MC2 and JM6 cells were maintained in phenol red-free minimal essential medium supplemented with 5% 33 dextran-coated, charcoal-treated calf serum, 2 mM glutamine, 6 ng/ml bovine insulin, 100 units/ml penicillin/100 mg/ml streptomycin, nonessential amino acids, and 500 mg/ml G418. The cells were treated with indicated compounds for the specified time and were subsequently harvested for tissue culture experiments.
Cell Growth Assay.
The cell growth was monitored by measuring the total DNA content per well in 24-well plates. Fifteen thousand cells were plated per well, and treatment with indicated concentrations of compounds was started after 24 hours, in triplicates. Media containing the specific treatments were changed every 48 hours. On day 7, the cells were harvested using hypotonic buffer solution and were subsequently sonicated. The DNA content was assessed using a fluorescent DNA quantitation kit (Cat # 170-2480; Bio-Rad, Hercules, CA) and was performed as previously described (Lewis et al., 2005a).
Annexin V Analysis of Apoptosis.
The annexin V–fluorescein isothiocyanate (FITC)–labeled Apoptosis Detection Kit I (Pharmingen, San Diego, CA) was used to detect and quantify apoptosis by flow cytometry according to the manufacturer’s instructions. In brief, MCF7:5C cells (1 × 106 cells/ml) were seeded in 100-mm dishes and cultured overnight in estrogen-free RPMI 1640 medium containing 10% stripped fetal serum. The next day, cells were treated with <0.1% ethanol vehicle (control), estradiol (1 nM), TPEs (1 μM), or selective estrogen receptor modulators (SERMs) (1 μM) for 72 hours and then harvested in cold phosphate-buffered saline (Invitrogen) and collected by centrifugation for 10 minutes at 500g. Cells were then resuspended and stained simultaneously with FITC-labeled annexin V and propidium iodide. Cells were analyzed using a fluorescence-activated cell sorter flow cytometer (Becton Dickinson, San Jose, CA).
RNA Isolation and Real-Time Polymerase Chain Reaction.
Total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA) and RNAeasy kit according to the manufacturer’s instructions. Real-time polymerase chain reaction (RT-PCR) was performed as previously described (Sengupta et al., 2010). Briefly, high-capacity cDNA reverse transcription kits (Applied Biosystems, Foster City, CA) was used to generate cDNA using 1 μg of total RNA in a total volume of 20 μl. The cDNA was subsequently diluted to 500 μl, and RT-PCR was performed using ABI Prism 7900 HT Sequence Detection System (Applied Biosystems). In each well, a 20-μl reaction volume included 10 μl SYBR green PCR master mix (Applied Biosystems), 125 nM each of forward and reverse primers, and 5 μl of diluted cDNA. RT-PCR was performed using specific primers as previously described (Sengupta et al., 2013), and the change in expression of transcripts was determined and the ribosomal protein 36B4 mRNA was used as the internal control.
Transforming Growth Factor Assay.
Three hundred thousand MDA-MB231 cells stably transfected with wild-type ER (MC2) or mutant D351ER were seeded in six-well plates and treated with either vehicle control or various concentrations of planar estrogens, triphenylethylenes, or antiestrogens after 24 hours. After 24 hours, the cells were harvested for mRNA, and RT-PCR was performed to quantify TGFα mRNA levels as mentioned previously. The assay elucidates the putative conformation of ligand–receptor complex in relation to apoptotic-inducing action of the ligands (Fig. 1).
Immunoblotting.
Proteins were extracted in cell lysis buffer (Cell Signaling Technology, Beverly, MA) supplemented with Protease Inhibitor Cocktail (Roche, Indianapolis, IN) and Phosphatase Inhibitor Cocktail Set I and Set II (Calbiochem, San Diego, CA). Total protein content of the lysate was determined by a standard bicinchoninic acid assay using the reagent from Bio-Rad. Twenty-five micrograms of total protein was separated on 10% SDS polyacrylamide gel and transferred to a nitrocellulose membrane. The membrane was probed with primary antibodies followed by incubation with secondary antibody conjugated with horseradish peroxidase and reaction with Western Lighting plus-ECL enhanced chemiluminescent substrate (PerkinElmer, Waltham, MA). Protein bands were visualized by exposing the membrane to X-ray film.
Chromatin Immunoprecipitation Assay.
Chromatin immunoprecipitation (ChIP) assay was performed as described previously (Maximov et al., 2011). Briefly, cells were treated with indicated compounds for 45 minutes and cross-linked using 1.25% paraformaldehyde for 15 minutes; cross-linking was subsequently stopped with 2 M glycine. Cells were collected, followed by nuclei isolation by centrifugation. Isolated nuclei were resuspended in SDS-lysis buffer followed by sonication and centrifugation at 14,000g for 20 minutes at 4°C. The supernatants were diluted 1:10 with ChIP dilution buffer. Normal rabbit IgG and Magna ChIP protein A magnetic bead (Upstate Cell Signaling Solutions, Temecula, CA) were used to immunoclear the supernatant followed by immunoprecipitation with antibodies against ERα (1:1 mixture of cat# sc-543 and sc-7207; Santa Cruz Biotechnology Inc., Dallas, TX) and steroid receptor coactivator-3 (SRC3) (cat# 13066; Santa Cruz Biotechnology, Inc.). Immunocomplexes were pulled down using protein A magnetic beads and a magnet. The beads bound to immunocomplexes were washed using different buffers as described (Maximov et al., 2011). Precipitates were finally extracted twice using freshly made 1% SDS and 0.1 M NaHCO3 followed by decross-linking. The DNA fragments were purified using QiaQuick PCR purification kit (Qiagen, Valencia, CA). RT-PCR was performed using 2 μl isolated DNA, using primers specific for PS2 promoter (Maximov et al., 2011). The data are presented as percent input of starting chromatin input after subtracting the percent input pull down of the negative control (normal rabbit IgG).
Molecular Modeling.
The molecular modeling study was performed using the available X-ray crystallographic structures of ERα in the agonist and antagonist conformations. The three-dimensional coordinates of ERα cocrystallized with E2 (1GWR) and 4OHT (3ERT) were extracted from RCSB Protein Data Bank (Data Supplements) (Berman et al., 2000). The ligand was prepared for docking using the LigPrep utility (LigPrep 2.5; Schrodinger, LLC, Portland, OR). Protein preparation workflow (Schrodinger, LLC) was employed to prepare the proteins for molecular docking. The residues well known to be important for biologic activity, D351 and E353, were kept charged in both receptors, the free rotation of hydroxyl group for T347 was allowed, and H524 residue was protonated at the epsilon nitrogen atom based on the available literature data. Glide software (Glide 5.7; Schrodinger, LLC) was used for molecular docking, and the best docking poses were selected based on the composite score, Emodel, which accounts not only for the binding affinity but also for the energetic terms, such as ligand strain energy and interaction energy
Results
Growth Effects of Estrogens and Antiestrogens in MCF-7 Cells.
To study the biologic activity of the planar estrogens (Fig. 2A), which include E2, DES, equilin, estrone, and equilenin), and triphenylethylenes (Fig. 2B), namely, ethoxytriphenylethylene (EtOX), trihydroxytriphenylethylene (3OHTPE), and bisphenol, we tested their ability to induce cell proliferation in wild-type MCF-7cells. As controls we used SERMs: 4OHT, endoxifen (endox), ral, and baze (Fig. 2C), which are known antiestrogens. MCF-7 cells were grown in estrogen-free media for 3 days and treated with various concentrations of the indicated compounds, and their effects were compared with E2. All planar estrogens (Fig. 3A) were able to induce cell proliferation in a concentration-dependent manner to the same maximum level as E2. DES, equilin, and estrone induced cell proliferation with maximum stimulation occurring at 0.1 nM, whereas equilenin reached maximal stimulation at 1 nM compared with 0.01 nM for E2. Similarly, the triphenylethylenes tested were able to induce cell growth to the same maximum level as E2, although their agonistic potency was less than E2 (Fig. 3B). Bisphenol, EtOX, and 3OHTPE all induced cell proliferation in a concentration-dependent manner with maximum stimulation at 1–10 nM compared with 0.01 nM for E2. Nonetheless, the TPEs all were potent estrogen agonists in this assay. On the other hand, as expected, the SERMs, 4OHT, endox, ral, and baze (Fig. 3C), which are antiestrogens, did not induce cell growth.

Chemical structures of the compounds used in the experiments: planar estrogens (A), triphenylethylenes (B), and selective estrogen receptor modulators (C).
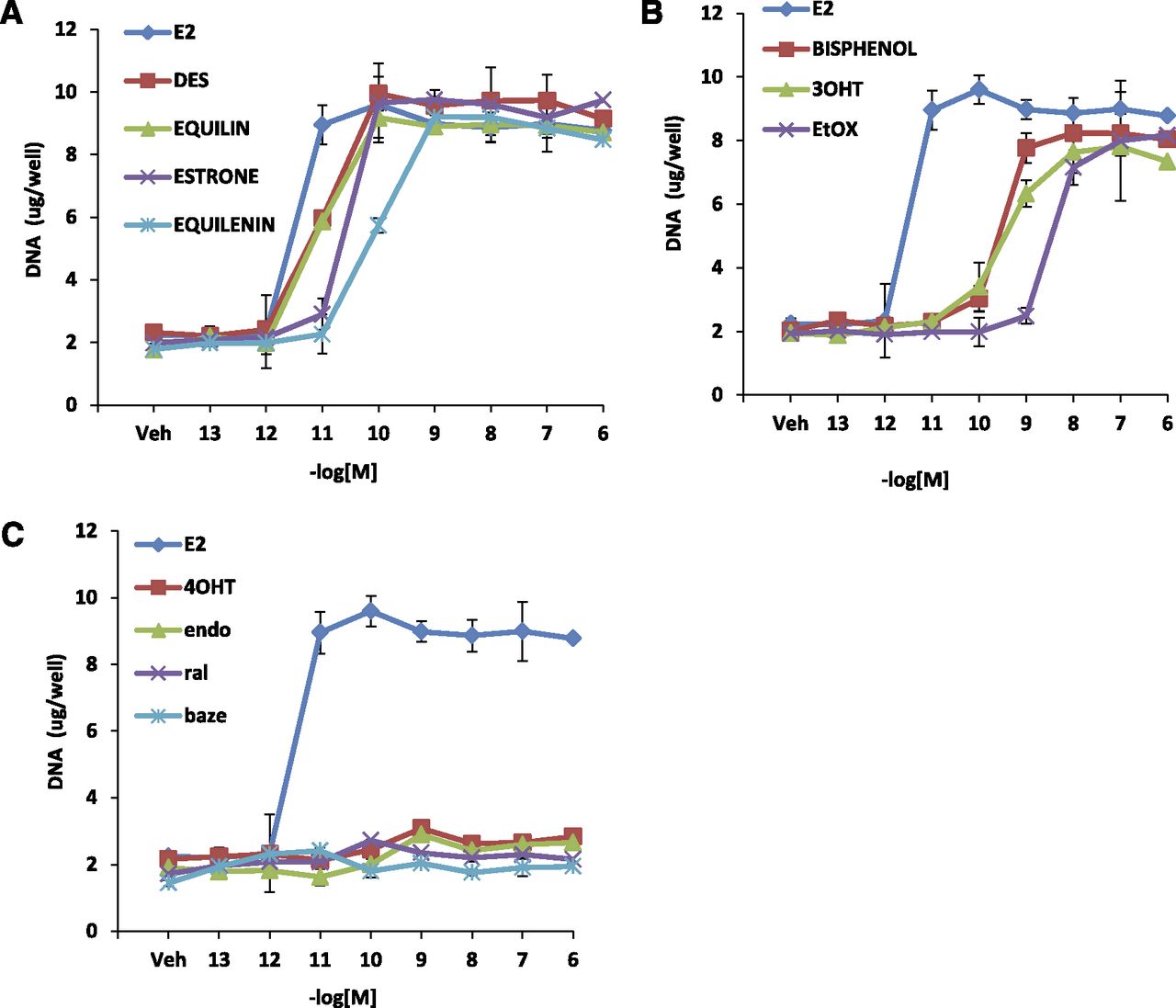
Growth characteristics of planar estrogens and triphenylethylenes in MCF7:WS8 cells. MCF7:WS8 cells were seeded in 24-well plate and treated with planar estrogens (A) over a range of doses for 7 days. Cell growth was assessed as DNA content in each well. Induction of cell growth by triphenylethylenes (B) and SERMs (C) was assessed in comparison with E2. Each data point is average ± S.D. of three replicates.
Effects of Planar Estrogens, TPEs, and SERMS on Apoptosis in MCF7:5C Cells.
We tested whether TPEs and SERMS were able to induce apoptosis in long-term estrogen-deprived MCF7:5C breast cancer cells as effectively as E2. All planar estrogens were able to cause growth inhibition as effectively as E2 (Fig. 4A). All the planar estrogens achieved maximal growth inhibition in the range of 1 nM compared with E2, which achieved maximal growth at 0.1 nM. To confirm that the decrease in cell proliferation was due to apoptosis, MCF7:5C cells were treated with ethanol vehicle (control), E2 (1 nM) or DES (1 nM), equilin (1 nM), estrone (1 nM), and equilenin (1 nM) for 72 hours, and annexin V–FITC and propidium iodide fluorescence was determined by flow cytometry. In the control-treated group, only 5.9% stained positive for apoptosis, whereas, in the E2-treated group (Supplemental Fig. 1A), cells that stained positive for apoptosis increased by 3-fold. Interestingly, the estrogenic triphenylethylenes did not inhibit the growth of MCF7:5C cells even at higher concentrations (Fig. 4B) at the end of a 7-day assay. Compared with E2, bisphenol, 3OHTPE, and EtOX did not show any effective apoptosis even at micromolar concentration (Supplemental Fig. 1B) and were comparable to that of the SERMs (Fig. 4C). Furthermore, the TPEs were able to block E2-mediated apoptosis in a similar manner to the SERMs (Fig. 4, D–E). However, the TPEs were able to induce apoptosis after 14 days of treatment (Fig. 4F), whereas the SERMs still did not induce apoptosis in the MCF7:5C cells (Supplemental Fig. 1C).

Differential effect of planar estrogens and triphenylethylenes in MCF7:5C cells. Dose dependent effect of planar estrogens (A), triphenylethylenes (B), and SERMs (C) on apoptosis of MCF7:5C cells treated for 7 days as indicated. Cells were treated with 1 nM E2 in presence of increasing concentration of indicated TPEs (D) and SERMs (E). (F) Effect of TPE in MCF7:5C cells after 14 days of treatment. Each data point is average ± S.D. of three replicates.
Regulation of TGFα Gene by Planar and Nonplanar Estrogens in MDA:MB-231 Cells Stably Transfected with Wild-Type ERα or D351G Mutant ERα.
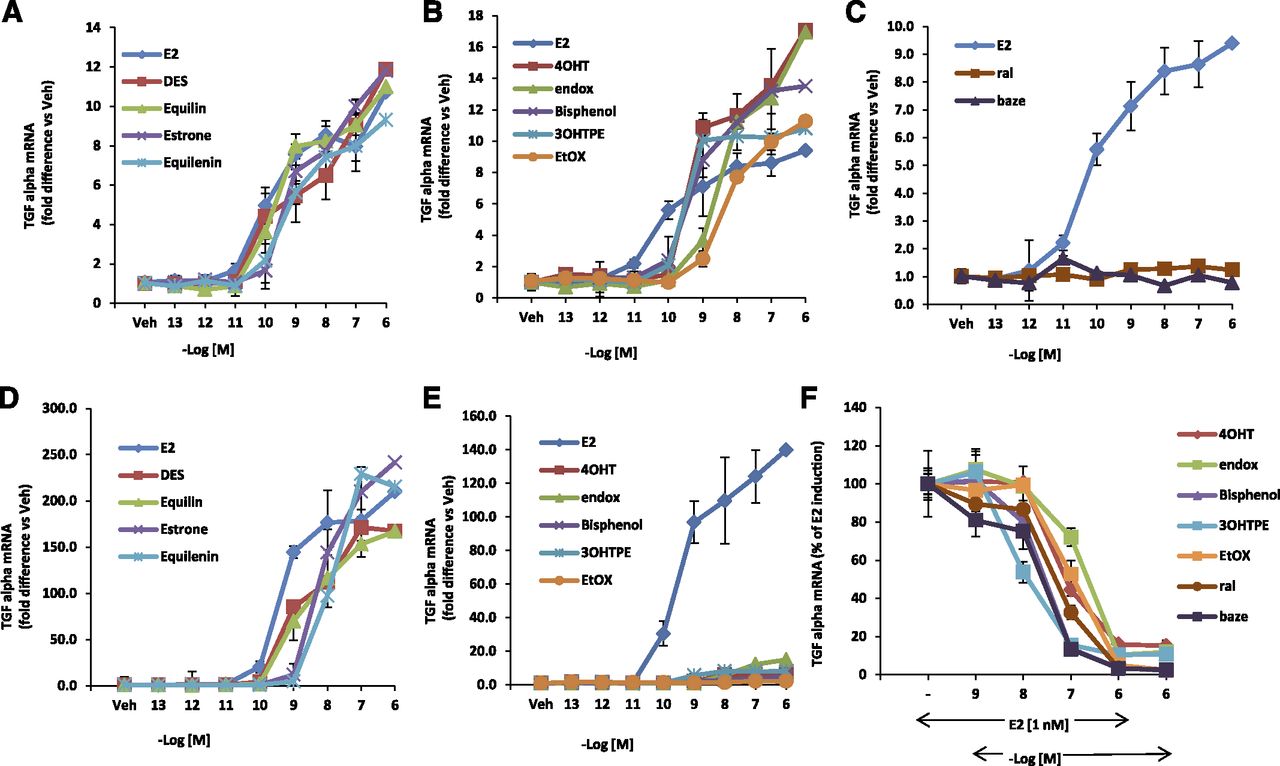
The TGFα gene is induced by 4OHT as effectively as E2 in MDA:MB-231 cells stably transfected with wild-type ERα (MC2 cells). In contrast, in MDA:MB-231 cells stably transfected with a mutant D351ER (JM6 cells), 4OHT fails to induce expression of the TGFα gene, but E2 retains its ability to induce the TGFα gene. We determined if the TPEs (3OHTPE, EtOX, and bisphenol) and the planar estrogens (DES, equilin, estrone, and equilenin) resembled E2 or 4OHT in inducing the TGFα gene expression by using the assay system summarized in Fig. 1. As expected, all the planar estrogens were able to induce TGFα gene expression in a concentration-dependent manner in both wild-type ERα (MC2) (Fig. 5A) and D351G mutant ERα (JM6) cells (Fig. 5D). On the other hand, the TPEs and tamoxifen metabolites 4OHT and endox were able to induce TGFα gene expression in MC2 cells (Fig. 5B) in a concentration-dependent manner, whereas ral and baze do not activate the TGFα gene in this cell line (Fig. 5C). By contrast, the TPEs 4OHT and endox distinctly failed to induce TGFα gene expression (Fig. 5E) in JM6 cells, which express the D351G mutant form of the ERα, rather they block E2-mediated TGFα induction (Fig. 5F). Similarly, ral and baze are antiestrogenic in the mutant stable transfectant. These findings indicate that the TPEs possess antiestrogenic properties and bind with ERα in a manner that is distinctly different from the planar estrogens but strikingly resembles 4OHT and endox.
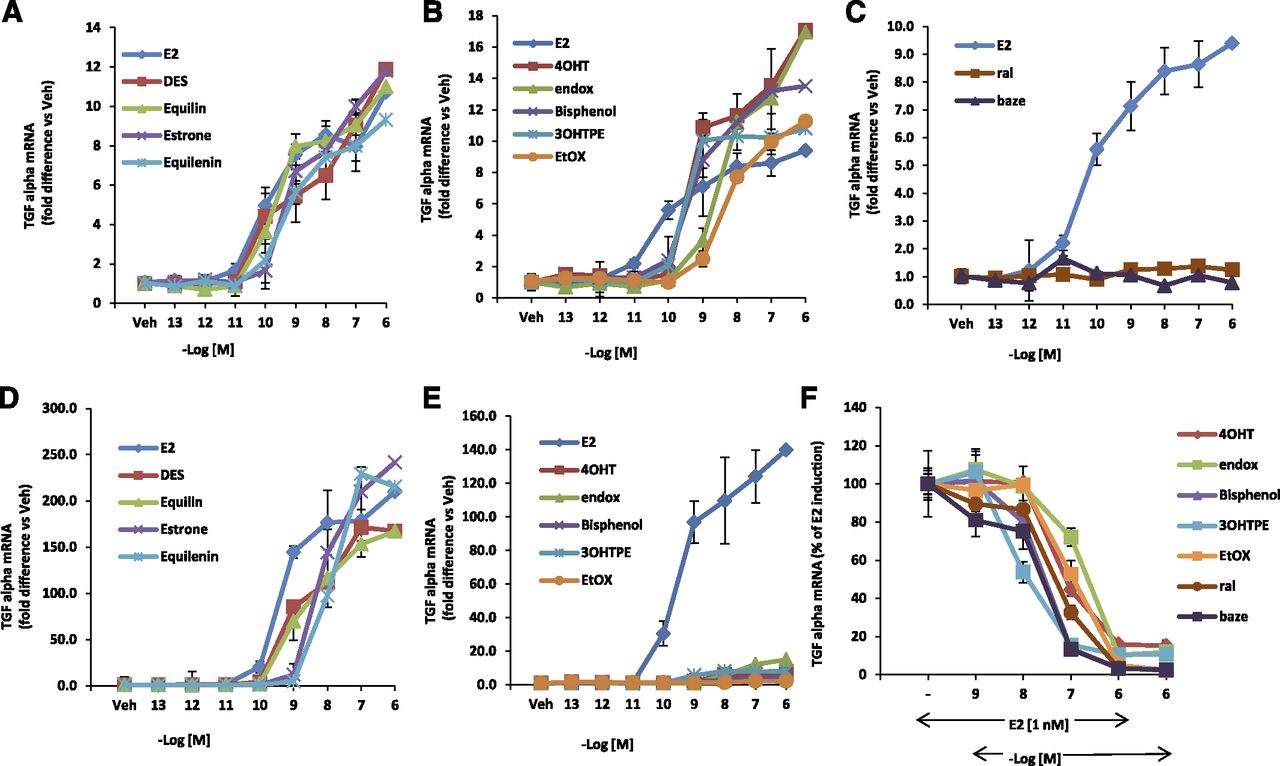
The concentration-dependent action of test compounds using wild-type (MC2) and mutant D351G ER (JM6) stable transfectants. MC2 cells were treated with planar estrogens (A) and the TPEs 4OHT and endox (B) for 24 hours at indicated concentrations, and expression of TGFα RNA was measured using quantitative real-time PCR. (C) MC2 cells treated with ral and baze in a dose-responsive manner. JM6 cells were treated with planar estrogens (D) and E2 (E), TPEs, active metabolites of tamoxifen (4OHT and endox) for 24 hours with various concentrations and expression of TGFα RNA was measured using quantitative real-time PCR. (F) JM6 cells were treated with 1 nM E2 alone or in presence of increasing concentration of indicated TPEs and SERMs. A–E are represented as fold difference versus vehicle-treated cells. Each data point is average ± S.D. of three replicates.
Recruitment of ERα and SRC3 at the Proximal Promoter of PS2 Gene after Treatment with Triphenylethylenes.
To further understand the ER-mediated mechanism involved in the regulation of the model estrogen responsive gene PS2 by the TPEs in MCF7:WS8 and MCF7:5C cells, we determined the recruitment of the ERα and SRC-3 protein at the proximal promoter of PS2 gene, which has a classic estrogen responsive element (Fig. 6A), using ChIP assay after 45 minutes of treatment with TPEs (1 µM) and compared it with E2 (1 nM) and 4OHT (1 µM). The whole assay was repeated two more times with similar results occurring in each cell line (Supplemental Figs. 4 and 5). In MCF7:WS8 cells, E2 was able to recruit very high level of ERα at the PS2 promoter (Fig. 6B), where more than 8% of input PS2 promoter region was occupied by ERα. On the other hand TPEs were ∼50% as efficient as E2 treatment in terms of recruiting ERα, whereas a very low level (∼20% of E2) of ERα recruitment was observed after 4OHT treatment. Recruitment of the coactivator SRC3, which is critical in inducing the estrogen responsive gene, was not observed at all after 4OHT treatment at the PS2 promoter. All the TPEs tested recruited only about 15–20% of SRC3 compared with E2 treatment, which showed 0.9% of input PS2 promoter region was occupied by SRC3 protein. Interestingly, in MCF7:5C cells treated with E2, around 5% of input PS2 promoter region was occupied by ERα (Fig. 6C). In MCF7:5C, cells treated with TPEs had 50% less ERα occupancy, and ∼80% less SRC3 occupancy was observed compared with E2 treatment in MCF7:5C cells, whereas no SRC3 recruitment was observed after 4OHT treatment. These ChIP data concur with the PS2 mRNA induction level in MCF7:WS8 and MCF7:5C cells with their respective treatments (Supplemental Fig. 2, A and B).

Recruitment of ERα and SRC3 (AIB1) at PS2 proximal promoter region containing estrogen responsive element (ERE) in MCF7:WS8 and MCF7:5C cells. (A) Depiction of PS2 proximal promoter region and the ERE region relative to transcription start site (TSS). (B) MCF7:WS8 cells treated for 45 minutes with E2 (1 nM), 3OHTPE (1 µM), EtOX (1 µM), bisphenol (1 µM), and 4OHT (1 µM), and ChIP assay was performed as described in Materials and Methods. (C) MCF7:5C cells were treated as mentioned above, and ChIP assay was performed under identical conditions. Data are represented as percent input of the starting chromatin used for the ChIP. Veh, vehicle.
Induction of ERα Expression by Planar and Nonplanar Estrogens.
To test whether the structure the compounds create with the ER affects the ERα expression levels, 4 breast cancer cell lines, which include MCF7:WS8, MCF7:5C, MC2, and JM6 cells, were treated with planar estrogens (1 nM), TPEs (1 µM), and SERMs (1 µM) for 24 hours, and ERα levels were determined by Western blotting. ICI was included as a positive control. All planar estrogens and ICI caused decrease in the ERα protein levels in MDA-MB231 cells stably transfected with either wild-type ER (MC2) (Fig. 7A) or with the mutant receptor (JM6) (Fig. 7B). On the other hand, the TPEs do not decrease the ERα protein levels in the MC2 cells, whereas 4OHT and endox cause accumulation of the receptor, whereas ral and baze cause moderate downregulation of the ER. In the JM6 cells, all TPEs and SERMs did not dramatically affect the ERα protein expression. As expected, all planar estrogens and ICI cause a decrease of ERα protein levels in MCF7:WS8 (Fig. 7C) and MCF7:5C (Fig. 7D) cells, whereas the tamoxifen metabolites caused increase in ERα protein expression. Interestingly the TPEs cause moderate decrease of ERα in MCF7:WS8 and MF7:5C cells compared with E2, and the reduction is more dramatic in the MCF7:5C cells. In contrast to the tamoxifen metabolites, ral and baze also cause a reduction in the protein levels of ERα in both MCF-7–derived breast cancer cell lines. ERα protein levels of all breast cancer cell lines used in the study are compared in Supplemental Fig. 3.
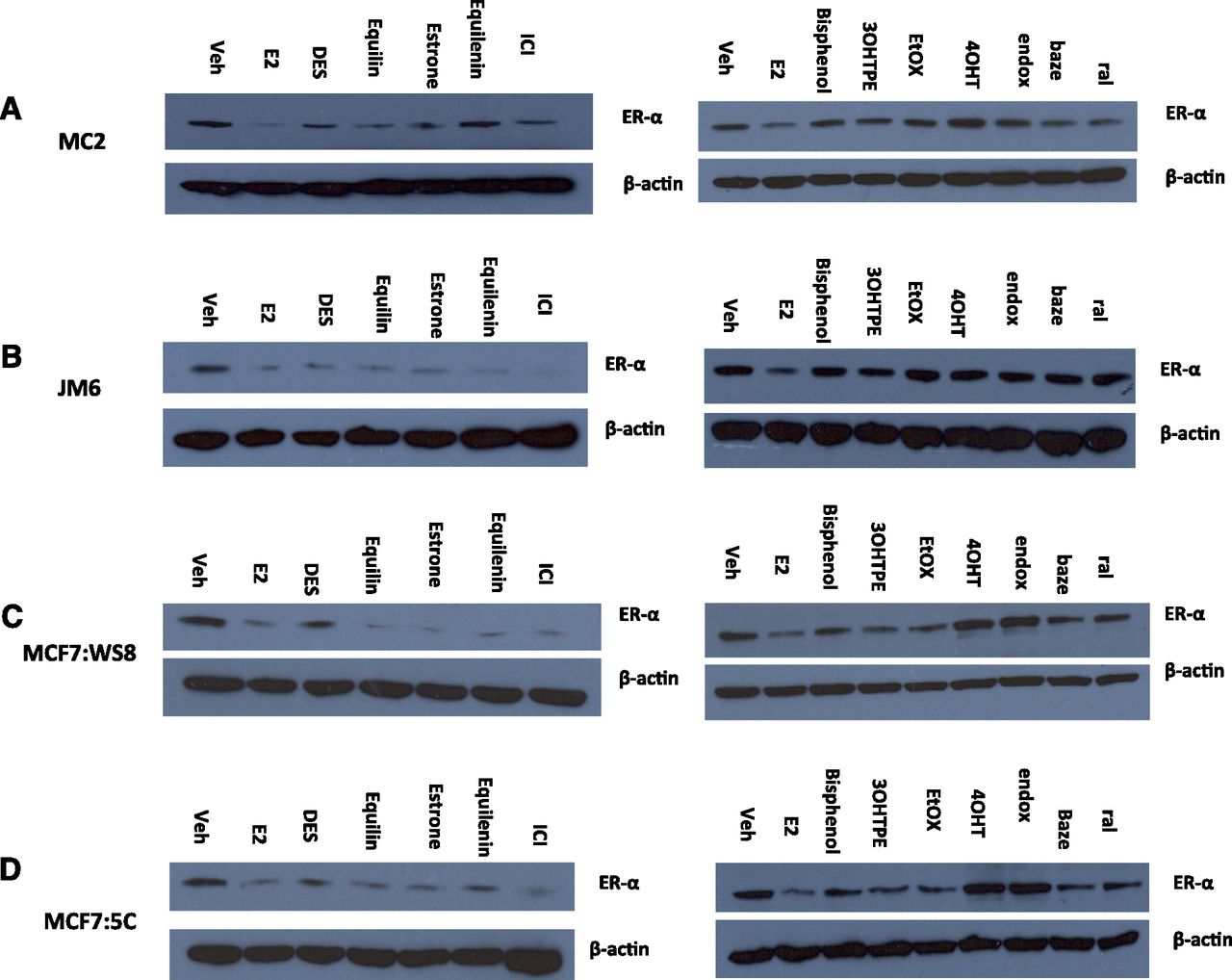
Differential regulation of the ERα protein by planar and nonplanar estrogens. MC2 (A), JM6 (B), MCF7:WS8 (C), MCF7:5C (D) cells were treated with E2 (1 nM), DES (1 nM), equilin (1 nM), estrone (1 nM), equilenin (1 nM), 3OHTPE (1 µM), EtOX (1 µM), bisphenol (1 µM), 4OHT (1 µM), endox (1 µM), baze (1 µM), ral (1 µM), and cell lysates were analyzed by Western blotting by anti-ERα antibody. Blot was reprobed by anti-actin antibody.
Binding of Bisphenol to the LBD of ERα.
Next, the binding mode of the TPEs was investigated by the molecular docking of bisphenol to the LBD of ERα. Thus, the flexible docking of bisphenol into the LBD of the receptor cocrystallized with E2 and 4OHT (Fig. 8, A and B) was performed. The superimposition of the top-ranked docking pose of the ligand onto the E2 cocrystallized with ERα, the agonist conformation of the receptor, shows some incompatibility (Fig. 8C). Hence the resulting model revealed sterical clashes between bisphenol and “Leu crown,” mostly with the side chains of Leu525 and Leu540. Because of this steric hindrance, it is most unlikely for bisphenol to bind in a conformation of ERα that is similar to that of E2. On the other hand, when bisphenol is docked into the binding site of 4OHT cocrystallized with ERα (Fig. 8D), the binding mode is similar to that of 4OHT. Namely, the same alignment of the ligand in the binding pocket is noticed, having the propensity to form the same hydrophobic contacts with the amino acids lining the binding cavity and to recapitulate the complex H-bond network involving E353, R394, and a highly ordered water molecule. Taken together, these data show that bisphenol and extrapolating TPEs would most likely bind to the ERα in the antagonist conformation of the receptor.

The binding site of ERα with different ligands. The ligands are depicted with their corresponding grid molecular surfaces colored in gray. Also, Leu525 and Leu540 are depicted as grid molecular surfaces colored in blue. (A) Agonist conformation of ERα with E2 (magenta; PDB ID 1GWR). (B) Antagonist conformation of ERα with 4OHT (green; PDB ID 3ERT). (C) Docking of bisphenol in agonist conformation (cyan; PDB ID 1GWR). (D) Docking of bisphenol in antagonist conformation (cyan: PDB ID 3ERT).
Discussion
Estrogens are potent mitogens for the proliferation of breast cancer cells. In contrast, planar estrogens (class 1) can induce apoptosis of long-term estrogen-deprived MCF-7 cells (MCF7:5C) in a paradoxical manner. 4OHT has no effect in the MCF:5C cells but rather blocks E2-mediated apoptosis (Maximov et al., 2011). TPEs, which are structurally similar to 4OHT, possess estrogenic properties in the MCF-7 cells at comparable concentrations to the planar estrogens. The TPEs (class II angular estrogens) do not rapidly trigger estrogen-induced apoptosis in MCF7:5C cells, but block class 1 planar estrogen-induced apoptosis. However, prolonged treatment with the TPEs leads to an eventual induction of apoptosis in the MCF7:5C cells, whereas the cells continue to be resistant to the actions of the SERMs, which are known antiestrogens (Supplemental Fig. 1C). As a result of these aforementioned findings, we initially proposed a hypothesis (Maximov et al., 2011) that the TPE–ER complex mimics an antiestrogen-ER complex and this may be responsible for the delay of apoptosis by the TPEs. We addressed the hypothesis in four ways: utilizing our validated functional assay to classify estrogens using the induction of the TGFα gene (Jordan et al., 2001) (Fig. 1), binding of ER and recruitment of SRC3 to the promoter region of a model estrogen response gene (PS2) (Fig. 6), ligand-bound ER accumulation, or reduction and putative ER docking experiments (Fig. 8).
We previously demonstrated the critical importance of D351 in modulating the SERM:ER complex (Levenson et al., 2001) for the estrogen-like actions of the 4OHT by removing the exposed surface charge by engineering a mutant ER D351G, which causes a conversion of the 4OHT:ER from being estrogenic to completely antiestrogenic at the TGFα gene (Levenson et al., 1998; MacGregor Schafer et al., 2000). The anchoring role of D351 in the activation of the helix 12–mutated ER has recently (Merenbakh-Lamin et al., 2013; Toy et al., 2013) been illustrated in tissue from metastatic breast cancer resistant to antihormones. Mutations of Y537 in helix 12 are shown to anchor to D351 to accomplish sealing of the unoccupied LBD by helix 12. This provides evidence of the clinical relevance of our assay system.
To determine whether the conformation of the ER complex determines the triggering of apoptosis in long-term estrogen-deprived ER-positive breast cancer cells, MCF7:5C, we employed (Jordan et al., 2001) an assay using induction of the mRNA for the TGFα gene in situ in MDA-MB-231 cells stably transfected with cDNA wild-type (MC2) or D351G ER (JM6). As expected, all planar estrogens cause activation of the TGFα gene in the MC2 and JM6 cells. The planar estrogens are not affected by the mutation on D351, because upon binding to the ER, they are sealed within the LBD by helix 12, allowing for coactivator binding on the surface of helix 12 (AF-1) and gene activation. The TPEs induce TGFα gene at comparable concentrations to the tamoxifen metabolites, 4OHT and endox in the MC2 cells (Fig. 5B), but lose this estrogen-like action in the JM6 cells (Fig. 5E) and block E2 induction of TGFα (Fig. 5F). The results of the TGF assay imply that TPEs adopt a 4OHT-like conformation with the ER with helix 12 pushed back and D351 exposed. By inference, the “antiestrogenic conformation” of the TPE:ER complex is responsible for the initial inhibition of E2-induced apoptosis. The short aminoethoxy side chain of the tamoxifen metabolites (MacGregor Schafer et al., 2000) and the absence of this side chain in the TPEs prevent adequate shielding of the charged D351, whereas the antiestrogenic side chain of ral and baze provides effective interaction and neutralization of this charge (Liu et al., 2002) (Fig. 5C). Thus, this prevents the induction of the TGFα gene by ral and baze.
SRC3 has been shown to be extremely important in estradiol-induced growth in breast cancer cells (Font de Mora and Brown, 2000; List et al., 2001; Lahusen et al., 2009). Additionally, SRC3 knockdown was found to reduce apoptosis induced by E2 in MCF7:5C cells (Hu et al., 2011). Using ChIP assays we show that TPEs are able to recruit ERα but less efficiently compared with E2, and this was further observed with SRC3 (Fig. 6). The ER:TPE complex binds to the promoter with about 50% of E2, but SRC3 binding is <25% of E2. This suggests that treatment with TPEs influences the conformation of the liganded–ERα complex such that efficiency of ERα binding to estrogen responsive element region is moderately inhibited, whereas binding of SRC3 is severely inhibited compared with E2, which is a planar estrogen. This may also explain why bisphenol is a partial agonist at the prolactin gene and exhibits antiestrogen properties (Jordan and Lieberman, 1984; Jordan et al., 1984). Of notable importance, the magnitude of SRC3 recruitment by the TPEs is far less in MCF7:5C cells (Fig. 6C) compared with MCF7:WS8 cells (Fig. 6B) and may play a crucial role in manifesting the functional role of the TPEs in these cells. This observation may contribute to the robust cell replication in MCF-7 with TPEs but delayed apoptosis in MCF7:5C.
Estradiol induces downregulation of the ER in breast cancer cells (Borras et al., 1994, 1996; Reid et al., 2003), and this process is inhibited by 4OHT, thereby causing accumulation of ERα (Wijayaratne et al., 1999). Similarly in all our cell lines, the planar estrogens all downregulate the ER, whereas tamoxifen metabolites 4OHT and endox do not (Fig. 7). The Western blot analysis shows that the TPEs do not readily decrease ERα protein levels when compared with the planar estrogens. This illustrates the fact that the TPE:ER complex appears to be “antiestrogen-like” compared with 4OHT and endox (Fig. 7). However, in the MCF7:5C cells, their ability to downregulate ERα protein levels is more apparent. The ER complex resembles the vehicle (control) rather than the extremes of E2 or 4OHT. Ral and baze also cause moderate decrease in ERα levels, which concurs with previous studies done on these compounds (Lewis-Wambi et al., 2011). Bourgoin-Voillard et al. (2010) determined that class II ligands such as bisphenol had less tendency to promote recruitment of coactivators containing LxxLL motif, and this appeared to be a requirement for the downregulation of the ER in MCF-7 cells. Bourgoin-Voillard et al. (2010) also illustrated the accumulation of the bisphenol:ER complex in MCF-7 cells using immunocytochemistry.
The molecular modeling data (Fig. 8) provide evidence that the TPEs bind to the ERα in a manner similar to that observed with 4OHT using X-ray crystallography. The bulky phenyl ring of the TPEs prevent helix 12 from sealing the LBD and will result in an initial steric hindrance when attempting to bind in the E2–ERα conformation, resulting in their blockade of E2-induced apoptosis. However, continuous treatment of the MCF7:5C with the TPEs for 14 days results in induction of apoptosis similar to the planar estrogens. This suggests that the antiestrogenic conformation the TPEs create with the ER prevents immediate coactivator binding, causing a delay in the trigger for apoptosis, but this delay disappears with prolonged treatment. This conclusion correlates with the Haddow clinical study (Haddow et al., 1944), where postmenopausal women with advanced breast cancer were treated with TPE-like estrogens, leading to an about 30% response rate during breast cancer therapy. The planar estrogens form a compact estrogen–ER complex with excellent SRC3 binding and recruitment, and it appears that this event is necessary to induce apoptosis in the MCF7:5C cells. On the other hand, angular TPEs form an antiestrogen-like ER complex with less SRC3 binding and recruitment, thereby leading to delayed apoptosis, whereas the SERMS do not recruit SRC3 so this results in no apoptosis.
In conclusion, we have advanced the hypothesis that TPE–ER conformation is initially similar to that of tamoxifen metabolites 4OHT and endox, and our molecular classification assay indicates that helix 12 is pushed back in the TPE–ER complex. The antiestrogenic conformation of the TPE–ER complex appears to be responsible for the initial blocking of apoptosis and reduction in coactivator recruitment observed with the TPEs in the MCF7:5C cells. It is important to stress that the evidence we present suggests that the TPE:ER complex conformation may in fact be in between the extreme structures of E2:ER and 4OHT:ER ligand binding domain (Brzozowski et al., 1997; Shiau et al., 1998). Because prolonged treatment with TPEs causes triggering of ER-mediated apoptosis similar to that of the planar estrogens but 4OHT does not, an intermediate conformation of the TPE:ER complex may be responsible for these observations.
Acknowledgments
The authors thank Russell McDaniel and Phillip Maximov for their input in this project.
Authorship Contributions
Participated in research design: Obiorah, Sengupta, Jordan.
Conducted experiments: Obiorah, Sengupta, Curpan.
Performed data analysis: Obiorah, Sengupta, Curpan, Jordan.
Wrote or contributed to the writing of the manuscript: Obiorah, Curpan, Jordan.
Footnotes
- Received August 30, 2013.
- Accepted March 7, 2014.
This work was supported by the Department of 1090 Defense Breast Program [Grant W81XWH-06-1-0590] Center of Excellence; the Susan G. Komen for the Cure Foundation [Grant SAC100009]; the Lombardi Comprehensive Cancer 1095 Center Support Grant (CCSG) Core Grant; the National Institutes of Health National Cancer Institute [Grant P30-CA051008]; and National Research Council (CNCS)-Executive Agency for Higher Education, Research, Development and Innovation (UEFISCDI) (Romania) [Grant PN II-RU PD_502/2010].
The views and opinions of the authors do not reflect those of the US Army or the Department of Defense.
This work was presented as a poster at the following conferences: Obiorah I, Sengupta S, Curpan R, Jordan VC (2012) Defining the conformation of the estrogen receptor complex that controls estrogen-induced apoptosis in breast cancer. Lombardi Research Days at Georgetown University Medical Center; 2012 March 7; Washington, DC; and Obiorah I, Sengupta S, Curpan R, Jordan VC (2012) Defining the conformation of the estrogen receptor complex that controls estrogen-induced apoptosis in breast cancer. AACR Annual Meeting; 2012 April 6–13; Chicago, IL.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- baze
- bazedoxifene
- ChIP
- chromatin immunoprecipitation
- DES
- diethylstilbestrol
- E2
- 17β-estradiol
- endox
- endoxifen
- ER
- estrogen receptor
- EtOX
- ethoxytriphenylethylene
- FCS
- fetal calf serum
- FITC
- fluorescein isothiocyanate
- ICI 182,780
- 7α,17β-[9-[(4,4,5,5,5-pentafluoropentyl)sulfinyl]nonyl]estra-1,3,5(10)-triene-3,17-diol
- LBD
- ligand binding domain
- 4OHT
- 4-hydroxytamoxifen
- 3OHTPE
- trihydroxytriphenylethylene
- ral
- raloxifene
- RT-PCR
- real-time polymerase chain reaction
- SERM
- selective estrogen receptor modulator
- SRC3
- steroid receptor coactivator-3
- TPE
- triphenylethylene
- TGFα
- transforming growth factor α
- Copyright © 2014 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}