


Abstract
Chemokines and chemokine receptors are key modulators in inflammatory diseases and malignancies. Here, we describe the identification and pharmacologic characterization of nanobodies selectively blocking CXCR2, the most promiscuous of all chemokine receptors. Two classes of selective monovalent nanobodies were identified, and detailed epitope mapping showed that these bind to distinct, nonoverlapping epitopes on the CXCR2 receptor. The N-terminal–binding or class 1 monovalent nanobodies possessed potencies in the single-digit nanomolar range but lacked complete efficacy at high agonist concentrations. In contrast, the extracellular loop-binding or class 2 monovalent nanobodies were of lower potency but were more efficacious and competitively inhibited the CXCR2-mediated functional response in both recombinant and neutrophil in vitro assays. In addition to blocking CXCR2 signaling mediated by CXCL1 (growth-related oncogene α) and CXCL8 (interleukin-8), both classes of nanobodies displayed inverse agonist behavior. Bivalent and biparatopic nanobodies were generated, respectively combining nanobodies from the same or different classes via glycine/serine linkers. Interestingly, receptor mutation and competition studies demonstrated that the biparatopic nanobodies were able to avidly bind epitopes within one or across two CXCR2 receptor molecules. Most importantly, the biparatopic nanobodies were superior over their monovalent and bivalent counterparts in terms of potency and efficacy.
Introduction
Chemokines provide directional cues for leukocyte migration and tissue colonization. The chemokine receptor CXCR2 is considered a key molecular target for the diagnosis and treatment of a variety of acute and chronic inflammatory diseases such as chronic obstructive pulmonary disease, asthma, fibrosis, psoriasis, multiple sclerosis, cystic fibrosis, rheumatoid arthritis, inflammatory bowel disease, allograft rejection, and angiogenesis and also in cancer metastasis (Stadtmann and Zarbock, 2012; Hertzer et al., 2013; Sharma et al., 2013). CXCR2 is a G protein–coupled receptor (GPCR) that is expressed on many different cells and tissues, including neutrophils, mast cells, CD8+ T cells, epithelial, endothelial, smooth muscle, and a variety of cell types in the central nervous system (Chapman et al., 2009). Several high-affinity ligands have been identified, CXCL1 (growth-related oncogene α [GRO-α]), CXCL8 (interleukin-8), and CXCL5 (ENA-78) as well as lower affinity ligands CXCL2 (GRO-β), CXCL3 (GRO-γ), CXCL6 (GCP-2), and CXCL7 (NAP-2), making it the most promiscuous of all CXC chemokine receptors. Targeting of the CXCR2 receptor with small-molecule antagonists has been pursued for pharmacologic treatment of various disease states in which cells of myeloid lineage are thought to contribute to disease pathophysiology (Chapman et al., 2009).
GPCRs are one of the most important classes of targets for small-molecule drug discovery, but many GPCRs of current interest are proving intractable to small-molecule discovery and may be better approached with biotherapeutics. Inhibition of CXCR2 with small molecules has shown promising therapeutic benefits in a variety of acute and chronic inflammatory diseases (Chapman et al., 2009), but many have failed during clinical trials (Schall and Proudfoot, 2011). Although some of these are likely to be due to the target biology, others have failed due to unacceptable pharmacologic properties, insufficient selectivity, poor pharmacokinetics, or off-target toxicity, and an antibody approach may prove to be better treatment.
Nanobodies are a novel class of antibody-derived therapeutic proteins based on immunoglobulin single-variable domains (Van Bockstaele et al., 2009; Kolkman and Law, 2010). Nanobody is a trademark of Ablynx N.V. (Ghent, Belgium). These domains are derived from the variable domains of heavy chain-only antibodies (HCAb), which are naturally occurring in camelids (Hamers-Casterman et al., 1993). In contrast to the heterotetrameric conventional antibodies, HCAbs are homodimers consisting of two heavy chains that lack the CH1 domain. To compensate for the reduced antigen-binding interface (formed by both the heavy and light chain variable domains in conventional Abs), HCAbs and nanobodies derived from these have evolved toward longer complementary determining regions in their variable domains. This, combined with their small size (12–15 kDa), enables nanobodies to recognize unique antigenic sites with high affinity and specificity that are normally not recognized by conventional antibodies and engineered Fab and sc-Fv fragments (Unciti-Broceta et al., 2013). Additional characteristics such as high solubility, low immunogenicity, and flexible formatting make them excellent candidates as standalone biotherapeutics in addition to use for targeted delivery of biologically active components (Muyldermans, 2001; Siontorou, 2013).
Therapeutic nanobodies have been generated against cancer-specific drug targets such as the receptor tyrosine kinases epidermal growth factor receptor (Omidfar et al., 2004; Roovers et al., 2007), human epidermal growth factor receptor 2 (Vaneycken et al., 2011), c-Met (Slørdahl et al., 2013), and vascular endothelial growth factor receptor 2 (Behdani et al., 2012). In addition, there have been studies demonstrating the successful generation of inhibitory nanobodies directed against GPCRs and specifically the chemokine receptors CXCR4 (Jähnichen et al., 2010) and CXCR7 (Maussang et al., 2013). Here, we describe the identification and pharmacologic characterization of inhibitory nanobodies directed against the CXCR2 receptor. Two different classes of monovalent nanobodies were identified, based on binding to two distinct nonoverlapping epitopes within the CXCR2 receptor. Bivalent and biparatopic nanobodies were generated, respectively combining monovalent nanobodies from the same or different classes via glycine/serine linkers. Interestingly, receptor mutation and competition studies demonstrated that the biparatopic nanobodies were able to avidly bind epitopes within one or across two CXCR2 receptor molecules. Most importantly, the biparatopic nanobodies were superior over their monovalent and bivalent counterparts in terms of potency and efficacy.
Materials and Methods
Cell Culture and Membrane Preparation
All cell culture reagents purchased from Life Technologies (Carlsbad, CA). All cells were grown adherently at 37°C, 5% CO2, and 100% humidity and subcultured when 90% confluent using trypsin/EDTA solution. All Chinese hamster ovary (CHO) cells were grown in Ham’s F-12 medium with Glutamax, supplemented with 10% (v/v) fetal bovine serum (FBS), although the selection agent differed; CHO-CXCR2 (Euroscreen, Gosselies, Belgium) included 400 µg/ml geneticin; CHO-CXCR2-W15A, W112A, and D274A included 500 µg/ml hygromycin; CHO-CXCR2-CCR9-ECL2 (extracellular loop 2) included 300 µg/ml Zeocin and 10 µg/ml blasticidin; and CHO-W15A-CXCR2:CXCR2-CCR9-ECL2 cells included 500 µg/ml hygromycin, 300 µg/ml Zeocin, and 10 µg/ml blasticidin. L2071-CXCR1 cells (Richard Ye, University of Illinois) were grown in Dulbecco’s modified Eagle medium without sodium pyruvate, with glucose and pyridoxine HCL, supplemented with 10% (v/v) FBS and 0.3 mg/ml geneticin. Membranes were prepared from CHO-CXCR2 cells as previously described elsewhere (Salchow et al., 2010), and the protein concentration was determined using the method described by Bradford (1976).
Immunizations, Phage Display Selection, and Production of Nanobodies
Llamas were immunized essentially as described by Maussang et al. (2013) with CXCR2-expressing cells (four animals) and/or pVAX1-human CXCR2 DNA (four animals) or pVAX1-human CXCR2 Δ1–17 DNA (four animals), in which the sequence encoding the first 17 N-terminal amino acids was deleted. After immunization, immune blood and lymph node samples were taken, and total RNA was extracted. Nanobody-encoding cDNA was amplified by reverse-transcription polymerase chain reaction and ligated into phagemid vector pAX50 to generate nanobody phage libraries (size ∼107) where the phage particles express individual nanobodies with C-terminal c-Myc and His6 tags in fusion with the Gene-III protein, as previously described elsewhere (Roovers et al., 2007).
Phage display selections were performed using either a biotinylated peptide consisting of the first 1 to 19 amino acids of the CXCR2 receptor (Bachem, Bubendorf, Switzerland), cells expressing CXCR2 receptor, or cell membranes prepared from CHO-CXCR2 cells. After two to three rounds of selection on various combinations of these target formats, the resulting phage outputs were transfected into Escherichia coli strain TG1, and individual colonies were grown in 96-deep-well plates. The expression of monoclonal nanobodies was induced by the addition of isopropyl β-d-1-thiogalactopyranoside, and the periplasmic extract containing the nanobodies was prepared by freeze-thawing the bacterial pellets in phosphate-buffered saline (PBS) and subsequent centrifugation to remove cell debris.
Small to medium scale productions and purifications of monovalent, bivalent, and biparatopic c-Myc and His6-tagged nanobodies were performed essentially as described elsewhere (Maussang et al., 2013). The flexible Gly-Ser linkers used to combine the monovalent building blocks in the bivalent and biparatopic constructs are referred to as 9GS (GGGGSGGGS) or 35GS ([GGGGS]7).
Off-Rate Determinations for Nanobodies Binding to Biotinylated CXCR2 1–19 Peptide
Off-rate analysis of the nanobodies was performed using surface plasmon resonance technology, using the ProteOn XPR36 instrument (Bio-Rad Laboratories, Hercules, CA). ProteOn PBS/Tween (phosphate buffered saline, pH 7.4, 0.005% Tween20) was used as running buffer, and the experiments were performed at 25°C. Biotinylated peptide representing the N-terminus (CXCR2 1-19) of human CXCR2 was captured on a ProteOn NLC Sensor Chip, injected at 50 mM in ProteOn PBS/Tween. Periplasmic extracts of the nanobodies were diluted 10 times in ProteOn PBS/Tween and injected for 2 minutes at 45 µl/min and allowed to dissociate for 600 seconds. Between different samples, the surfaces were regenerated with a 50-second injection of sodium hydroxide (50 mM) at 30 µl/min. Analysis was performed with ProteOn Manager 2.1.0.38, version 2.0.1 (Bio-Rad Laboratories). Data were double referenced by subtraction of reference ligand lane and a blank buffer injection. Processed curves were evaluated via fitting a standard 1:1 ligand-binding model (Langmuir model provided by ProteOn software).
CXCL1 Binding Assay
CHO-CXCR2 cells (2 × 105) were incubated with purified anti-CXCR2 nanobodies in fluorescence-activated cell sorter (FACS) buffer [PBS, 10% (w/v) FBS] for 30 minutes at 4°C. Fluorescence microvolume assay technology Blue-labeled CXCL1 (labeled CXCL1) (Applied Biosystems, Foster City, CA) (3 nM) diluted in FACS buffer was added to the cell mix and incubated for a further 30 minutes at 4°C in the dark. The cells were then washed 3 times in FACS buffer. Dead cells were stained with propidium iodide before the samples were analyzed on the FACSarray (BD Biosciences, San Jose, CA) to detect the amount of labeled CXCL1 bound to the cells.
[35S]GTPγS Binding Assay
[35S]GTPγS binding to CHO-CXCR2 membranes was determined using the method described previously elsewhere (Bradley et al., 2009). However, human serum albumin replaced bovine serum albumin in the assay buffer. To determine IC50 values for anti-CXCR2 nanobodies, the activity of either recombinant human CXCL1 or CXCL8 (Life Technologies) (EC50 concentration) was measured in the presence or absence of a range of concentrations of purified anti-CXCR2 nanobodies. For Schild analysis experiments, dilution series were prepared for CXCL1 and incubated with a range of concentrations of anti-CXCR2 nanobody; the rest of the protocol was as previously described. Finally, to investigate the effect of nanobodies on basal levels of [35S]GTPγS binding, the assay was performed as detailed for IC50 determinations with the exception that assay buffer was added in the place of agonist.
Nanobody Binding Competition FACS Assay
CHO-CXCR2 (105/well) cells were incubated for 1.5 hours at 4°C with a mixture of serially diluted c-Myc–tagged nanobodies and a fixed concentration (EC30) of 3x-Flag–tagged nanobody. Detection was performed using anti-Flag M2 antibody (Sigma-Aldrich, St. Louis, MO) for 30 minutes at 4°C followed by the addition of phycoerythrin-labeled goat anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA) for a further 30 minutes at 4°C. Cells were washed 3 times with FACS buffer between each incubation, and dead cells were stained with TO-PRO-3 (Molecular Probes/Life Technologies). The amount of anti-FLAG antibody bound to the cells was detected on a FACSArray machine (BD Biosciences).
CXCR1 Receptor Selectivity Assay
L2071 cells expressing human CXCR1 receptor were seeded in 96-well plates and incubated overnight at 37°C. On the day of the experiment, the cells were loaded with Fluo-4 dye for 30 minutes at 37°C followed by 30 minutes of incubation with purified monovalent and biparatopic nanobodies. Finally, the addition of CXCL8 (EC80–100 concentration) was performed using a Fluorometric Imaging Plate Reader (Molecular Devices, Sunnyvale, CA) followed by the detection of a fluorescent signal, corresponding to the release of intracellular calcium. The Schering Plough low-molecular-weight antagonist Sch527123 (2-hydroxy-N,N-dimethyl-3-{2-[[(R)-1-(5-methyl-furan-2-yl)-propyl]amino]-3,4-dioxo-cyclobut-1-enylamino}-benzamide) was used as a control (Gonsiorek et al., 2007).
Human Neutrophil Whole Blood Shape Change Assay
Human whole blood was collected from healthy volunteers and anticoagulated with 52 mM EDTA. A range of concentrations of purified nanobodies were incubated with whole blood for 10 minutes at room temperature. CXCL1 (EC70 concentration) was then added to the blood and incubated at 37°C for a further 5 minutes with gentle shaking. The tubes were then placed on ice while ice-cold optimized CellFix solution (BD Biosciences) was added to each tube. The tubes were incubated for a further 5 minutes with gentle shaking, after which time the ammonium chloride solution was added to each of the tubes to lyse the red blood cells. This was incubated for 20 minutes before the samples were analyzed using flow cytometry (FACSCalibur; Becton Dickinson, Franklin Lakes, NJ). Specifically, cells were gated on forward scatter/side scatter (FSC/SSC) parameters, followed by forward scatter/fluorescence-channel 2 (FSC/FL-2) gating using the granulocyte gates from the first analysis; 5000 events were counted per sample.
Human Neutrophil Chemotaxis Assay
The chemotaxis of human neutrophils to CXCL1 (EC80 concentration) after preincubation with either assay buffer or a range of concentrations of purified anti-CXCR2 nanobody was determined using the method previously described elsewhere (Bradley et al., 2009).
Epitope Mapping of CXCR2 Nanobody Binding Sites
A CXCR2 receptor mutation library was created by random mutagenesis using the shotgun mutagenesis platform from Integral Molecular (Paes et al., 2009). A total of 714 clones were generated with all residues mutated twice, with one conservative and one nonconservative mutation including one alanine substitution. All clones were expressed in mammalian cells, and cell surface expression was measured using a commercially available anti-CXCR2 polyclonal antibody. Nanobodies bound to CXCR2 mutants were detected using an anti–c-Myc antibody (Integral Molecular, Philadelphia, PA) followed by a goat anti-mouse–horseradish peroxidase conjugate (Jackson ImmunoResearch). Residues involved in the nanobody epitope were identified as those that fell within the binding thresholds (<30% nanobody reactivity and >60% polyclonal anti-CXCR2 antibody), and that included an alanine residue substitution and were located in the extracellular loops.
Generation of Mutated CXCR2 Receptor Cell Lines
Single-Point Mutations.
CXCR2-W15A mutant with an N-terminal FLAG-tag and the CXCR2-W112A mutant with an N-terminal 3xHA-tag were produced by gene synthesis and inserted into a pMA vector. The sequences were subcloned into the pcDNA5/FRT vector using restriction digestion. The D274A mutant was produced by site-directed mutagenesis of the HA-tagged wild-type CXCR2 using a Quickchange II kit (Agilent, Santa Clara, CA) according to the manufacturer’s protocol. CHO Flp-In cells (Life Technologies) were grown overnight without antibiotics and the next day were transfected with using Lipofectamine 2000. CXCR2 constructs were mixed with pOGG44 vector in a ratio of 1:9 in Opti-Mem. Lipofectamine 2000 was diluted in Opti-Mem (1 in 25 dilution) and then added to the DNA/Opti-Mem mix in a 1:1 ratio and incubated at room temperature for 20 minutes before being added to the cells. The cells were then incubated overnight at 37°C, and transfected cells were selected using 500 µg/ml of hygromycin.
Chimeric CXCR2-CCR9-ECL2.
Human CXCR2 cDNA incorporated in a pcDNA3.1(+) vector was mutated whereby the extracellular loop 2 (ECL2) of CXCR2 was replaced with the ECL2 from the CCR9 receptor creating a CXCR2-CCR9 chimeric cDNA sequence with a triple HA tag engineered at the N-terminus (Missouri S&T cDNA Resource Center, Rolla, MO). This chimeric HA-tagged cDNA was subsequently subcloned into pcDNA4/TO at HindIII(5′) and XhoI(3′) sites, and the correct recombinant plasmids were identified by restriction enzyme analysis. The HA-CXCR2-CCR9-ECL2 cell line was produced using Nucleofector Kit T (Amaxa Biosystems, Cologne, Germany). We transfected 1 × 106 CHO T-Rex cells [this is a tetracycline-regulated expression (T-Rex) cell line] with 5 µg of plasmid DNA according to the kit instructions. Transfected cells were treated with 300 µg/ml of Zeocin 48 hours after transfection. Positive clones were selected after 24 hours of incubation with 2 µg/ml of doxycycline to induce receptor expression and 60 minutes of incubation with 5 µg/ml mouse anti-HA fluorescein isothiocyanate–conjugated antibody (Sigma-Aldrich) before measurement of fluorescence using FACs.
Double Mutant Receptor Cell Line.
CHO-CXCR2-W15A cells were grown overnight in the absence of antibiotics before the addition of CXCR2-CCR9-ECL2 plasmid (16 µg) using an Opti-MEM/Fugene (Promega, Madison, WI) mixture (ratio 33:1), which had been incubated at room temperature for 20 minutes before being added to the cells. Selection of positive transformants was done with medium containing both 500 µg/ml of hygromycin and 300 µg/ml of Zeocin. Single-cell sorting was performed using the FACS Aria (Becton Dickinson) made more efficient with the use of the fluorescein isothiocyanate–conjugated anti-HA tag antibody, which enabled only those cells expressing the HA-tagged receptor to be selected.
Alexa647–Anti-his Tag Nanobody Binding Assays Using the IN Cell Analyzer
All cells were plated at 3000 cells/well in media in 96-well black clear-bottom plates (Corning Life Sciences, Corning, NY) and incubated overnight at 37°C. Dilution series were prepared for 127D1, 163E3, and 127D1-163E3 in blocking buffer [PBS with 20% (v/v) FBS] and added to the cell plate. This plate was further incubated for 60 minutes at 37°C before the plate was washed 2 times with wash buffer [PBS, 0.1% (w/v) human serum albumin]. All nanobodies used in this assay have a His6-tag, so anti-His Alexa647-conjugated antibody (ABD Serotec, Oxford, UK) diluted (1 in 20) in blocking buffer containing 2 µM Hoechst (Molecular Probes/Life Technologies, Carlsbad, CA) was added and incubated for 15 minutes at room temperature before the plate was washed again before the measurement of fluorescence on the IN Cell Analyzer 2000 (GE Healthcare, Fairfield, CT).
Data Analysis and Statistical Procedures
All values reported in the text and tables are mean ± S.E.M. for three separate experiments, unless indicated otherwise. All experiments were analyzed by nonlinear regression using Prism 6.0 (GraphPad Software, San Diego, CA). For statistical procedures, one-way or two-way analysis of variance (stated in table legends) was performed, followed by Tukey’s honest significant difference post hoc test.
Results
Identification of Monovalent CXCR2 Binding Nanobodies.
Phage display selections identified 24 distinct nanobody families (derived from different B-cell lineages) binding specifically to CXCR2, as determined by inhibition of labeled CXCL1 binding to CXCR2 expressing cells. Eight of these families were found to bind the N-terminal of CXCR2 by ELISA (not shown) or off-rate analysis against a peptide constituting residues 1–19. The libraries derived from the animals immunized with CXCR2-expressing cells exclusively delivered nanobodies showing binding to the N-terminal peptide, whereas libraries derived from the llamas immunized with DNA also delivered nanobodies that inhibited CXCL1 binding but did not bind to the N-terminal peptide.
Six nanobodies, representing different families, 2B2, 54B12, 127D1, 97A9, 163E3, and 163D2 were analyzed in more detail. All of these nanobodies showed concentration-dependent inhibition of labeled CXCL1 binding to CHO-CXCR2 cells (Table 1). Periplasmic extracts of nanobodies 2B2, 54B12, and 127D1 showed binding to the biotinylated CXCR2 1–19 peptide with off-rates shown in Table 1. Interestingly, none of these three peptide-binding nanobodies were able to fully inhibit the binding of CXCL1 to CXCR2. In contrast, nanobodies 97A9, 163E3, and 163D2, which did not show binding to the CXCR2 1–19 peptide, were able to fully inhibit the binding of labeled CXCL1 to CXCR2 but were significantly less potent than nanobodies 2B2, 54B12, and 127D1 (Table 1).
Activity of representative nanobodies: off-rate determinations on biotinylated CXCR2 1–19 peptide (single experiment using periplasmic extracts) and inhibition of labeled CXCL1 binding to CHO-CXCR2 cells (purified nanobodies)
Number is given in parentheses.
All 24 nanobody families that were identified fell into one of two distinct classes. Class 1 nanobodies were those that were able to bind the CXCR2 1–19 peptide but were unable to fully inhibit the labeled CXCL1 binding to CHO-CXCR2 cells; in contrast, class 2 nanobodies were unable to bind to CXCR2 1–19 peptide but were able to fully inhibit labeled CXCL1 binding to CHO-CXCR2 cells. In addition, class 1 nanobodies were more potent than the class 2 nanobodies at inhibiting labeled CXCL1 binding to CHO-CXCR2 cells, despite lack of full efficacy.
Pharmacologic Characterization of Monovalent Nanobodies.
Further characterization of these nanobodies showed inhibition of CXCR2 agonist-mediated signaling in CHO-CXCR2 membranes and neutrophils (two examples from class 1: 2B2 and 127D1; and three examples from class 2: 97A9, 163E3, and 163D2). Table 2 shows the pIC50 values and the percentage of inhibition of the maximum agonist response for monovalent CXCR2 nanobodies determined with CXCL1- and CXCL8-mediated [35S]GTPγS binding to CHO-CXCR2 membranes. Per class, the nanobodies were able inhibit both CXCL1 and CXCL8 with similar pIC50 values and the percentage of inhibition of maximum response. In addition, all monovalent nanobodies showed inhibition of whole blood neutrophil shape change and chemotaxis with a range of potencies, as shown in Table 2. It is of note that the class 1 nanobodies (2B2 and 127D1) that were unable to fully inhibit the binding of labeled CXCL1 were also unable to fully inhibit CXCL1 mediated [35S]GTPγS binding. In whole blood neutrophil shape change, 2B2 was also unable to fully inhibit the CXCL1 response.
Potency and efficacy values of monovalent, bivalent, and biparatopic nanobodies in a range of functional assays
As a result of these observations, further studies were performed, using representatives from each class, to assess the mechanism of action of these nanobodies. Concentration effect curves were generated for each nanobody in the presence of increasing concentrations of CXCL1. The influence of agonist concentration on nanobody function was investigated in two different assays. As shown in Fig. 1A, the ability of 127D1 to fully inhibit [35S]GTPγS binding (to CHO-CXCR2 membranes) is totally dependent on the concentration of CXCL1 used and at 150 nM CXCL1 (10 × EC50) 127D1 is almost inactive. This observation was not restricted to experiments performed on recombinant cells: Fig. 1B shows that in the presence of 20 nM CXCL1 (approximately 20 × EC50) 2B2 is completely inactive in a neutrophil shape change assay. In contrast, as shown in Fig. 1C, 163E3 is able to fully inhibit [35S]GTPγS binding at all of the CXCL1 concentrations tested.

Effect of increasing CXCL1 concentration on anti-CXCR2 nanobody inhibition of CXCL1-stimulated [35S]GTPγS binding assay using CHO-CXCR2 cell membranes (A and C) and of human whole blood neutrophil shape change assay (B). Data presented for (A) and (C) are mean ± S.E.M. from three separate experiments. Data presented for (B) are representative of three experiments performed once.
To further understand the antagonist mechanism of action of these two classes of CXCR2 nanobodies, we performed a Schild analysis. Figure 2A shows that 30 nM 127D1 can produce a rightward shift in the CXCL1 concentration effect curve, but further increasing concentrations of 127D1 have no additional effect. This pattern of behavior is not consistent with simple competitive behavior. In contrast, Fig. 2B shows that 163E3 produces concentration-dependent rightward shifts in the CXCL1 concentration effect curve indicative of a competitive mechanism of action, and the Schild plot (Fig. 2C) yielded a straight line with a mean slope of 0.97 ± 0.04, in line with a competitive mode of action and a mean pKB of −7.78 ± 0.11. As the data for 127D1 did not indicate a competitive mode of action, a Schild plot to determine a pKB was not generated.

Schild experiments performed using CXCL1-stimulated [35S]GTPγS binding assay, with CHO-CXCR2 cell membranes, in the presence of a range of concentrations of (A) 127D1 and (B) 163E3 anti-CXCR2 nanobodies. (C) Schild plot for 163E3 derived from data shown in (B). Data are presented as the mean ± S.E.M. of three separate experiments.
Competition experiments using a representative FLAG-tagged monovalent nanobody from each class (127D1 for class 1 and 163E3 for class 2) showed that nanobodies within the same class compete with each other, whereas nanobodies from different classes do not compete with each other, as shown in Fig. 3. Mean pIC50 values were determined to be 7.33 ± 0.04 and 7.59 ± 0.18, for 163E3 and 163D2 (in competition with 163E3) and 9.13 ± 0.03 and 8.08 ± 0.02 for 127D1 and 2B2 (in competition with 127D1), respectively. It is therefore possible for two monovalent nanobodies from different classes to simultaneously bind epitopes within one receptor subunit.

Inhibition of FLAG-tagged nanobody binding to CHO-CXCR2 cells by monovalent nanobodies (A) using FLAG-tagged 163E3 and (B) using FLAG-tagged 127D1. Data presented are representative of three experiments performed once.
Finally, in terms of selectivity, all the monovalent nanobodies tested were shown to be inactive against the related chemokine receptor CXCR1 (Supplemental Fig. 1 shows the results for 127D1 and 163E3, as representatives from each class).
Pharmacologic Characterization of Bivalent and Biparatopic Nanobodies.
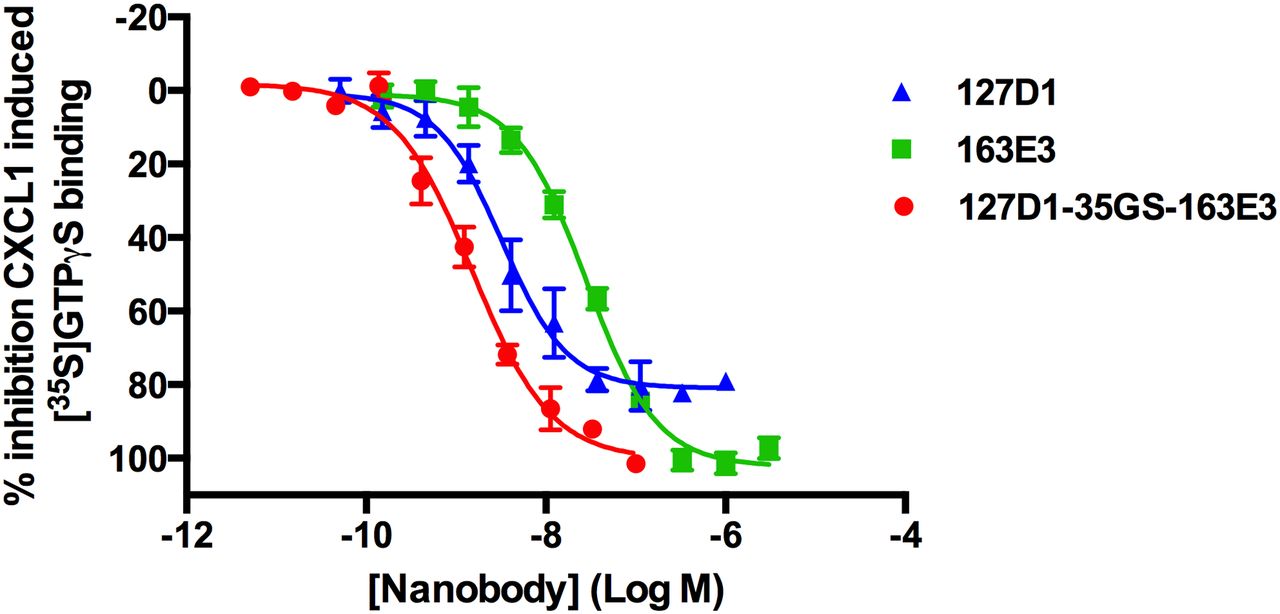
Next, we engineered a series of bivalent but also biparatopic nanobody constructs using the monovalent building blocks described previously, linked together with either 9GS or 35GS linkers. A number of trends were observed from an initial characterization, which compared 6 bivalent and 15 biparatopic nanobodies and their ability to inhibit labeled CXCL1 binding to CHO-CXCR2 cells (Supplemental Table 1). First, a combination of two identical or different class 2 (non 1–19 peptide binding) nanobodies did not show a strong increase in potency over their monovalent counterparts, as exemplified by 163E3-35GS-163E3 in Table 2, where at best a fourfold increase in potency was observed for the inhibition of CXCL1-stimulated [35S]GTPγS binding to CHO-CXCR2 cell membranes. Second, a combination of two identical class 1 (1–19 peptide binding) nanobodies showed significantly increased potency over their monovalent counterpart, as exemplified by 2B2-9GS-2B2 in Table 2, with a 134-fold increase in potency in the whole blood neutrophil shape change assay. However, this bivalent construct was still unable to fully inhibit CXCL1-stimulated [35S]GTPγS binding to CHO-CXCR2 cell membranes; in whole blood neutrophil shape change, at high concentrations of CXCL1 (20 × EC50), it was completely inactive (data not shown). Finally, biparatopic nanobodies that were composed of one class 1 nanobody and one class 2 nanobody (e.g., 127D1-35GS-163E3; Fig. 4 and Table 2) showed the greatest increases in potency when compared with their class 2 monovalent counterparts, and increased efficacy when compared with their class 1 monovalent counterparts, although the gains in potency were not as large in the labeled CXCL1-binding assay and CXCL1-stimulated [35S]GTPγS binding assay (Table 2) as compared with all the other assay formats. As with the monovalent nanobodies, the bivalent and biparatopic nanobodies could inhibit CXCL8-stimulated [35S]GTPγS binding with similar potency and efficacy as for CXCL1 (Table 2) and were completely inactive at the CXCR1 receptor (Supplemental Fig. 1).

Effect of generating biparatopic nanobody construct on potency in CXCL1-stimulated [35S]GTPγS binding assay using CHO-CXCR2 cell membranes. Data are presented as the mean ± S.E.M. of three separate experiments.
To further characterize the biparatopic nanobody 127D1-35GS-163E3, we performed a Schild analysis, and the data are shown in Fig. 5A. Parallel rightward shifts of the CXCL1 concentration effect curve were observed, indicative of a competitive antagonist, and the Schild plot (Fig. 5C) yielded a straight line with a mean slope of 0.60 ± 0.01 and a mean pKB of −9.97 ± 0.03. If the slope of the line is equal to 1, as is the case for monovalent 163E3 (Fig. 2C), then this could be indicative of a competitive antagonist. However, for the biparatopic nanobody, the slope is significantly less than 1, suggesting that the mechanism of inhibition of the formatted nanobody may not be identical to a competitive antagonist. Finally, we performed a Schild analysis on equimolar mixtures of monovalent components 127D1 and 163E3, which were added to the assay at the same time (Fig. 5B). The resulting Schild plot yielded a mean slope of 0.80 ± 0.06 and a mean pKB of −8.95 ± 0.38 (Fig. 5C). This pKB value is a log order of magnitude lower in potency than the biparatopic nanobody, suggesting that when linked, the binding of one monomer forces the second tethered one to stay close to its corresponding binding site. This forced proximity favors the binding and rebinding (once dissociated) of each nanobody to their discreet epitopes and hence leads to the increased potency/affinity that we observe with the biparatopic constructs (Vauquelin and Charlton, 2013).
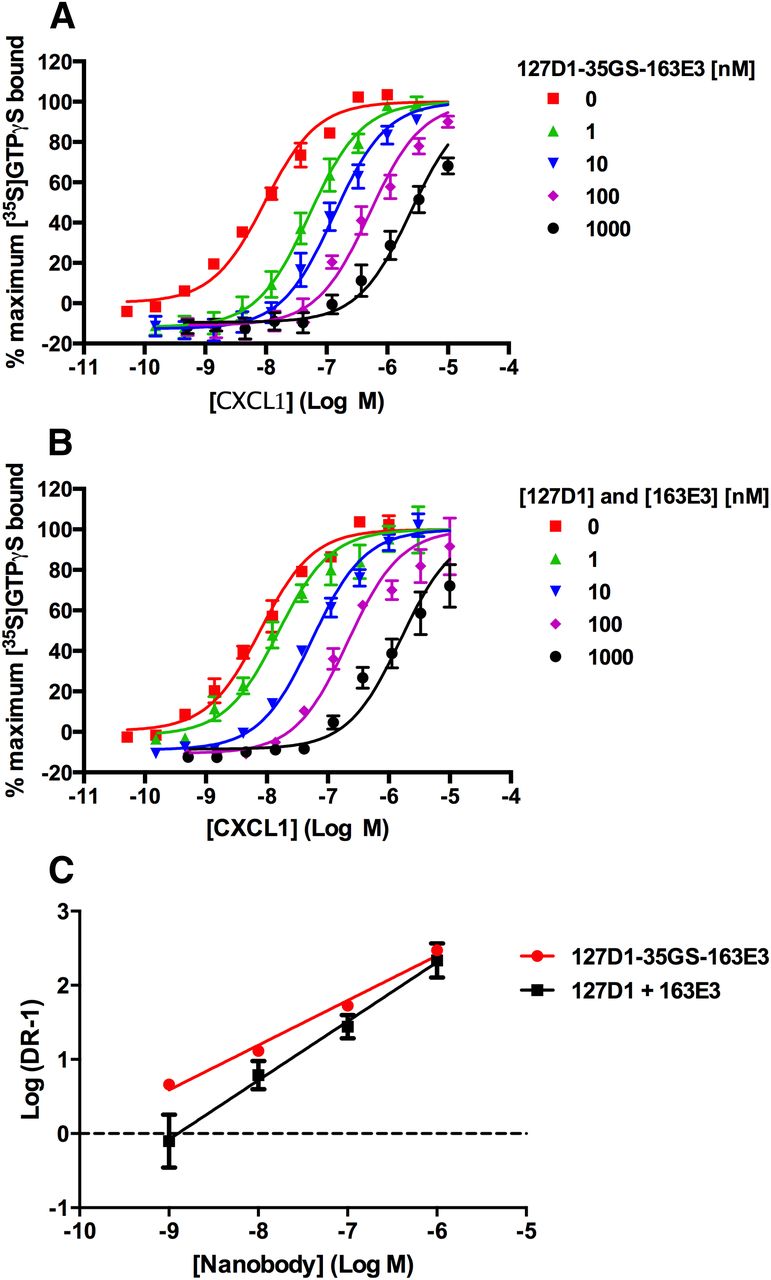
Schild experiments performed using CXCL1-stimulated [35S]GTPγS binding assay with CHO-CXCR2 cell membranes in the presence of a range of concentrations of (A) 127D1-35GS-163E3 (B) 127D1 and 163E3 monovalent nanobodies added together. (C) Schild plots derived from data shown in (A) and (B). Data are presented as the mean ± S.E.M. of three separate experiments.
Finally, the biparatopic nanobody showed inverse agonist effects on basal [35S]GTPγS binding to CHO-CXCR2 membranes (Fig. 6) and produced a mean pIC50 value of 9.17 ± 0.10, which is in line with the potency derived in the presence of the agonist. In addition, Fig. 6 shows that the monovalent nanobodies 127D1 and 163E3 also exhibited inverse agonist effects on basal [35S]GTPγS binding to CHO-CXCR2 membranes, with mean pIC50 values of 8.48 ± 0.61 and 7.89 ± 0.28, respectively. These potency values are in line with those generated in the presence of the agonist, and the percentage decrease of basal [35S]GTPγS binding is less with 127D1 as compared with 163E3, correlating with the reduced efficacy we observed with the class 1 nanobodies.

Inhibition of basal [35S]GTPγS binding to CHO-CXCR2 cell membranes using monovalent and biparatopic nanobodies. Data are presented as the mean ± S.E.M. of three separate experiments.
Epitope Mapping of Nanobody Binding Sites.
To define the binding sites of 127D1 and 163E3 nanobodies, we applied the shotgun mutagenesis technology platform from Integral Molecular (Philadelphia, PA), in addition to generating both single-point mutations and a chimeric CXCR2/CCR9 receptor. The shotgun mutagenesis analysis indicated that 127D1 and 163E3 bind to distinct, nonoverlapping binding sites on the CXCR2 receptor as shown in Fig. 7. As expected, the class 1 nanobody 127D1 mapped to the N-terminal region of the receptor, specifically to residues F11, F14, and W15; when mutated to alanine residues, these resulted in less than 30% binding of 127D1, when compared with wild-type CXCR2 (Supplemental Table 2). The close proximity of the critical residues (F11, F14, and W15) suggests that the epitope is linear in nature, hence the ability of this class of nanobodies to bind to the CXCR2 1–19 peptide. The CXCR2 receptor with the single-point mutation W15A in the N-terminus was expressed on the surface of CHO Flp-In cells, and fluorescence image analysis showed that the binding of 127D1 was lost in the presence of this mutation (Fig. 8, Fig. 9B, and Table 3) while binding of 163E3 was retained, thus confirming the shotgun mutagenesis data.

Epitope mapping of CXCR2 nanobodies: snake plot representation of human CXCR2 with highlighted amino acids involved in the binding of CXCL1, CXCL8, and nanobodies 127D1 and 163E3 [blue: Katancik et al., 2000; yellow: 127D1; and red: 163E3]. In addition, residues are highlighted that affect the epitope of 163E3 but are believed not to directly contact the nanobody (green). Residues shown in yellow, red, and green were identified after shotgun mutagenesis (Integral Molecular) and when mutated to alanine resulted in diminished binding (≤30% compared with wild-type CXCR2) of the respective nanobody.

Nanobody binding to mutated CXCR2 receptors expressed in CHO cells (blue fluorescent nuclear counterstaining with Hoechst dye) and measured using Alexa647-conjugated anti-His antibody, as shown by red fluorescent cell membrane staining.
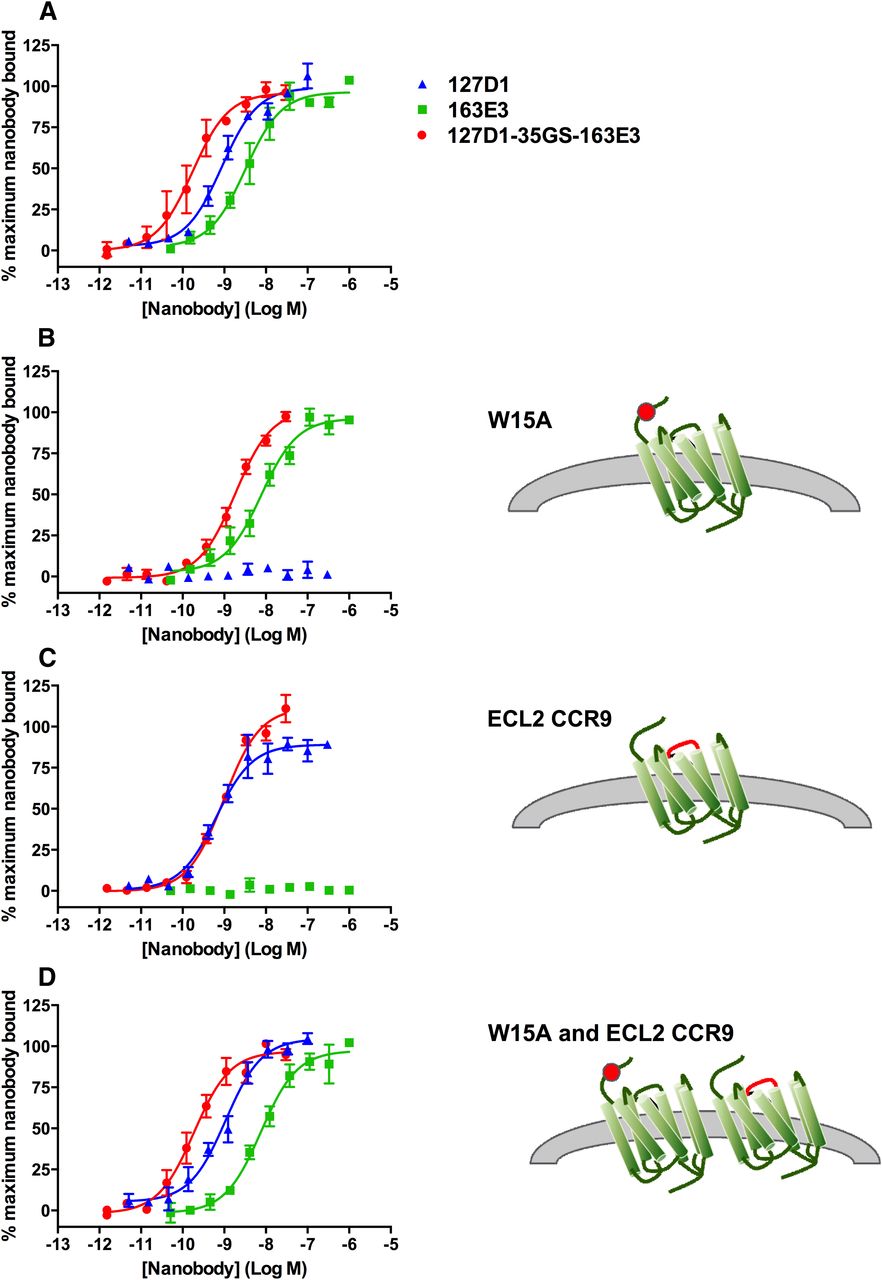
Effect of CXCR2 receptor mutations on affinity of monovalent and biparatopic nanobodies measured using Alexa647-conjugated anti-His antibody on (A) CHO-CXCR2 wild-type cells, (B) CHO-W15A cells, (C) CHO-CXCR2-CCR9-ECL2 cells, and (D) CHO-CXCR2-W15A:CXCR2-CCR9-ECL2 cells. Data are presented as the mean ± S.E.M. of three separate experiments.
Nanobody affinity measured using Alexa647-conjugated anti-His antibody on wild-type and mutated CXCR2 receptor CHO cell lines
In contrast to the linear epitope for the class 1 nanobody, the class 2 nanobody 163E3 appears to bind to a conformationally complex epitope formed primarily by extracellular loops 1 and 3 (ECL1 and ECL3). When mutated to alanine, residues W112, F114, G115, D274, I282, T285, and D293 all exhibited loss of 163E3 binding (≤30% binding when compared with wild-type CXCR2) (Supplemental Table 2). The epitope also appears to be highly sensitive to the conformational state of the receptor, as many mutations located in the transmembrane (TM) domains (primarily TM5 and TM6, data shown in Supplemental Table 3); cysteine residues in the extracellular loops (Supplemental Table 2) appear to affect the binding of this nanobody, as shown in Fig. 7. The single-point mutations W112A (ECL1) and D274A (ECL3), that form part of the epitope for the class 2 nanobodies, were both successfully expressed in CHO-Flp-In cells. Both single residue substitutions, although they did not completely inhibit the binding of 163E3, produced >10-fold loss in affinity compared with CXCR2 wild-type (data not shown) with no effect on the affinity of 127D1, confirming the shotgun mutagenesis data. Interestingly, CXCL1 stimulated [35S]GTPγS binding was completely abolished in cells expressing these single-point mutations, suggesting that these residues play a critical role in CXCR2 receptor activation.
As the class 2 nanobodies appear to use a more complex epitope, compared with those of class 1, a more significant epitope disruption may be required to completely inhibit 163E3 binding. This was demonstrated by generation of a CXCR2-CCR9 chimeric receptor, whereby ECL2 of the CXCR2 receptor was replaced with the equivalent region from the CCR9 receptor. The CCR9 receptor was chosen as it showed the least sequence homology to the CXCR2 receptor in a comparison of chemokine receptor family members. This ECL2 replacement fully inhibited the binding of all class 2 monovalent nanobodies. As shown in Fig. 8, Fig. 9C, and Table 3, determination of nanobody binding by fluorescence image analysis demonstrated that ECL2 replacement completely inhibited binding of 163E3, while retaining binding of 127D1.
Biparatopic Nanobody Binding to Cells Coexpressing Mutated CXCR2 Receptors.
To gain an understanding of the binding of the biparatopic nanobody at a molecular level, we determined the apparent affinity of the biparatopic nanobody for both the CXCR2-W15A mutant and the CXCR2-CCR9-ECL2 chimera. We predicted that due to the total loss of binding of one of its constituent building blocks, the affinity of the biparatopic nanobody would match that of the building block that was still capable of binding to the mutated receptor. In the case of the CXCR2-CCR9-ECL2 chimera, this is exactly what was observed (Fig. 9C and Table 3). In the case of the CXCR2-W15A mutant, the data show that although the biparatopic nanobody shows approximately a 10-fold loss in affinity with this mutation compared with CXCR2 wild-type, it exhibits a fivefold increased affinity compared with monovalent 163E3.
One possible explanation could be that the binding of the 163E3 moiety of the biparatopic nanobody brings the 127D1 moiety within close enough proximity to the N-terminus of the CXCR2-W15A receptor and as such allows some binding, although with much reduced affinity, to the other key residues within its epitope (F11 and/or F14). The CXCR2-CCR9-ECL2 chimeric receptor was successfully combined with the W15A point mutant in one cell line, and Fig. 8 shows binding curves of both monovalent and biparatopic nanobodies to this cell line. Figure 9D and Table 3 show that for this cell line the affinities of both monovalent nanobodies and biparatopic nanobody are comparable to the affinities observed with the CHO-CXCR2 wild-type cells. Most importantly, the statistics confirm that the biparatopic nanobody binds equally well to both CXCR2 wild-type and CXCR2-W15A:CXCR2-CCR9-ECL2 cells.
Given the reduced affinity of the biparatopic nanobody for the individual CXCR2-W15A and CXCR2-CCR9-ECL2 cell lines, the most likely explanation for reinstatement of its affinity on the combined cell line is binding across two different mutant CXCR2 receptors. Essentially, these data indicate that the class 1 nanobody moiety 127D1 of the biparatopic construct is binding to the wild-type N-terminal region of the CXCR2-CCR9-ECL2 receptor and that the class 2 nanobody 163E3 moiety is binding to the wild-type ECL2 region of the CXCR2-W15A receptor.
Discussion
In this study we describe the generation and characterization of highly potent, selective, and efficacious nanobodies against the chemokine receptor CXCR2. Interestingly, these nanobodies exhibit a very different mechanism of inhibition as compared with small-molecule antagonists of CXCR2, which have been shown to bind to an intracellular binding site and display an allosteric mechanism of action (Bradley et al., 2009; Salchow et al., 2010).
A broad panel of CXCR2 nanobodies was identified, all of which fell into two distinct classes, based on whether they bound to a 1–19 amino acid peptide derived from the first 19 residues of the CXCR2 receptor. Nanobodies that were able to bind the peptide were termed class 1; these had higher potencies than the other class of nanobodies, but they were unable to fully inhibit CXCL1 binding and function, particularly at high agonist concentrations. The class 2 nanobodies did not bind the 1–19 peptide and were of lower potency compared with class 1. However, they were able to fully inhibit CXCL1 binding and function and appeared to be competitive in Schild analysis experiments. Both classes of nanobody displayed inverse agonism, class 1 to a lesser extent, but this suggests they can both inhibit the active conformation of the receptor. In addition, it was demonstrated that nanobodies from different classes did not compete with each other for binding to CXCR2.
Shotgun mutagenesis studies confirmed that these two classes of nanobodies bind to distinct, nonoverlapping regions on the CXCR2 receptor (Fig. 7). The epitope of the class 1 nanobodies was formed of three key residues within the N-terminus of the receptor, and the epitope of the class 2 nanobodies was a more conformationally complex epitope formed primarily by ECL1 and ECL3, although ECL2 was also demonstrated to be important in maintaining epitope conformation. A study by Katancik et al. (2000) identified amino acid residues of the CXCR2 receptor that are critical for the binding of CXCL1 and CXCL8 (also shown in Fig. 7). Four key amino acids were identified in the N-terminus of the receptor, in very close proximity to the class 1 nanobody epitope, and three key amino acid residues in ECL1, which are positioned either side of key residues which form part of the class 2 nanobody epitope. These data suggest that the nanobodies inhibit key interactions within the binding sites for CXCL1 and CXCL8 at the CXCR2 receptor. These data also suggest that the binding sites of CXCL1 and CXCL8 span two distinct regions of the receptor, which indicates that it might be possible to inhibit only one binding site while potentially retaining agonist binding at the other site. It is therefore possible that the class 1 nanobodies can bind to the N-terminus of the CXCR2 receptor, while either CXCL1 (or CXCL8) is simultaneously bound to the ECL1 region of the receptor, resulting in “partial” competition of this class of nanobodies with the chemokines. Partial competitive inhibition has previously been proposed for a number of antagonists of class B GPCRs and has been described in terms of the Charnière effect (Hoare, 2007). More recently it has been shown that this mode of inhibition can result in incomplete inhibition curves, as we observed in this study for the class 1 nanobodies, which could be misinterpreted as an allosteric effect (Vauquelin et al., 2015).
The two classes of nanobody also behaved very differently when bivalent constructs were generated; for the class 1 nanobodies, this resulted in large increases in potency compared with the monovalent molecules, suggesting simultaneous binding of the linked monomers to epitopes that are close enough together to allow this to occur (model 1 as described by Vauquelin and Charlton (2013)). In contrast, the class 2 nanobodies did not show any strong increase in potency as bivalent constructs, suggesting that the two building blocks do not bind simultaneously as the epitopes are too far apart (model 2, as described by Vauquelin and Charlton, 2013). Receptor dimerization, specifically the concept of asymmetric activation of GPCR dimers (Damian et al., 2006), could also be an explanation for the data we have observed with the bivalent nanobodies. Asymmetric activation proposes that there is one active conformation and one inactive conformation of each monomer within the dimer after binding of the agonist. Our data suggest that the epitope for the class 2 nanobodies is more complex than for the class 1 nanobodies and also that it is more sensitive to changes in conformation. Applying asymmetric activation to the binding of a bivalent class 2 nanobody to CXCR2 would mean that the binding of one of the nanobodies to a CXCR2 monomer within the dimer could produce an allosteric conformational change that inactivates the other CXCR2 receptor and thus prevents the simultaneous binding of the second linked nanobody.
The generation of the biparatopic nanobody formed by linking together a building block from each class resulted in the most desirable pharmacologic profile, combining high potency and efficacy in one molecule. When discussing binding modes of the bivalent nanobodies identified in this study, the assumption was that to see gains in potency, simultaneous binding of the linked nanobodies to two epitopes on two separate receptors had to occur. As the epitopes for the two classes of nanobodies are distinct and nonoverlapping, the individual building blocks (in a biparatopic format) could bind simultaneously within one receptor, or across two separate receptors. Competition assays with the two different classes of monovalent nanobodies demonstrated that they do not compete with each other. To investigate whether a biparatopic nanobody could also bind across two separate receptors, we generated a cell line that expressed two CXCR2 receptor variants, one with a mutation in the N-terminal (class 1) nanobody epitope and a chimeric replacement of a key region of the class 2 nanobody epitope. Each of these mutated receptors could only bind one of the two building blocks comprising the biparatopic nanobody. As anticipated, the affinity of the biparatopic nanobody for this cell line was similar to that for wild-type CXCR2, revealing that the biparatopic nanobody can bind across two CXCR2 receptors. There is evidence that the CXCR2 receptor can form dimers (Trettel et al., 2003) as well as CXCL8 (Horcher et al., 1998) and more recently CXCL1 (Ravindran et al., 2013). Although we cannot definitively rule out that overexpression of our mutant receptors in a recombinant system has “forced” an otherwise unnatural receptor dimerization or increased receptor colocalization to occur, the fact that we observe significant gains in potency with the biparatopic and bivalent class 1 formats in the neutrophil assays suggests that the receptors are also sufficiently colocalized/dimerized in an endogenous system. Overall, our data suggest that it may be possible for the biparatopic nanobodies to bind within one CXCR2 receptor or across two receptors.
Comparisons can be made between our data and that produced for nanobodies binding the CXCR4 or CXCR7 chemokine receptors (Jähnichen et al., 2010; Maussang et al., 2013). N-terminal CXCR7 nanobodies partially inhibited [125I]CXCL12 binding, suggesting that within the chemokine receptor family at least, inhibiting binding of the agonist to the N-terminus is not sufficient to fully inhibit receptor function. In addition, when two identical non–N-terminal CXCR7 nanobodies were linked, there was no more than a threefold increase in affinity, whereas with a biparatopic CXCR7 nanobody composed of building blocks that bound to distinct nonoverlapping regions of the receptor, larger increases in potency were observed. Jähnichen et al. (2010) also observed an increase in affinity and potency with an anti-CXCR4 biparatopic format, the major difference being that it was composed of two nanobodies that compete with each other and bind to different but overlapping epitopes in ECL2. The inverse agonism that we observe with both nanobody classes is unique when compared with CXCR4, in which inverse agonism was only observed with the biparatopic format (Jähnichen et al., 2010). In addition, we do appear to have observed much greater gains in potency as a result of generating the biparatopic format for CXCR2, when compared with both CXCR4 and CXCR7, and this may be due to the epitopes of the different nanobody classes and/or differences in CXCR2 receptor activation. Finally, it was postulated for the anti-CXCR4 biparatopic nanobody that it must be binding to two CXCR4 molecules in close proximity, given that the epitopes for the monovalent nanobodies were distinct but overlapping (Jähnichen et al., 2010). However, we believe that the data we have generated are the first time that this has been demonstrated experimentally for such a biparatopic nanobody specific for a G protein–coupled receptor, whose epitopes are distinct. Finally, the potential for both intramolecular and intermolecular avid binding with the biparatopic nanobody opens up the possibility for the inhibition of 1) monomeric CXCR2, 2) homodimeric CXCR2, and 3) heterodimeric CXCR2. In other words, whatever the relevant signaling conformation(s) of the receptor is/are, the biparatopic nanobody should be able to effectively bind and inhibit any/all of these possibilities.
Acknowledgments
The authors thank Catelijne Stortelers for her help with the DNA immunizations, Peter Hunt for his help in generating the CXCR2-CCR9-ECL2 chimera and for the useful discussions on nanobody binding mechanisms, Lindsay Fawcett for providing molecular biology tools for the generation of mutated CXCR2 receptors, and Cheryl Paes, Rachel Fong, and Edgar Davidson from Integral Molecular for their help with generating Fig. 7.
Authorship Contributions
Participated in research design: Bradley, Brown, Charlton, Cromie, Dombrecht, Grot, Kent, Laeremans, Manini, Steffensen, Van Heeke, Vlerick, Willis.
Conducted experiments: Bradley, De Taeye, Grot, Laeremans, Manini, Roobrouck, Steffensen, Van den Heede, Vlerick, Willis.
Contributed new reagents or analytic tools: Bradley, De Taeye, Van den Heede, Grot, Kent.
Performed data analysis: Bradley, Brown, Charlton, De Taeye, Dombrecht, Grot, Laeremans, Manini, Roobrouck, Steffensen, Van den Heede, Van Heeke, Vlerick, Willis.
Wrote or contributed to the writing of the manuscript: Bradley, Brown, Charlton, Cromie, Dombrecht, Kent, Steffensen, Van Heeke.
Footnotes
- Received August 27, 2014.
- Accepted December 2, 2014.
M.E.B. and B.D. contributed equally to this work.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- CHO
- Chinese hamster ovary
- ECL
- extracellular loop
- FACS
- fluorescence-activated cell sorter
- FBS
- fetal bovine serum
- GPCR
- G protein–coupled receptor
- HCAb
- heavy chain-only antibody
- PBS
- phosphate-buffered saline
- Sch527123
- 2-hydroxy-N,N-dimethyl-3-{2-[[(R)-1-(5-methyl-furan-2-yl)-propyl]amino]-3,4-dioxo-cyclobut-1-enylamino}-benzamide
- TM
- transmembrane
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}