


Abstract
It is established that long-chain free fatty acids including ω-3 fatty acids mediate an array of biologic responses through members of the free fatty acid (FFA) receptor family, which includes FFA4. However, the signaling mechanisms and modes of regulation of this receptor class remain unclear. Here, we employed mass spectrometry to determine that phosphorylation of mouse (m)FFAR4 occurs at five serine and threonine residues clustered in two separable regions of the C-terminal tail, designated cluster 1 (Thr347, Thr349, and Ser350) and cluster 2 (Ser357 and Ser361). Mutation of these phosphoacceptor sites to alanine completely prevented phosphorylation of mFFA4 but did not limit receptor coupling to extracellular signal regulated protein kinase 1 and 2 (ERK1/2) activation. Rather, an inhibitor of Gq/11 proteins completely prevented receptor signaling to ERK1/2. By contrast, the recruitment of arrestin 3, receptor internalization, and activation of Akt were regulated by mFFA4 phosphorylation. The analysis of mFFA4 phosphorylation-dependent signaling was extended further by selective mutations of the phosphoacceptor sites. Mutations within cluster 2 did not affect agonist activation of Akt but instead significantly compromised receptor internalization and arrestin 3 recruitment. Distinctly, mutation of the phosphoacceptor sites within cluster 1 had no effect on receptor internalization and had a less extensive effect on arrestin 3 recruitment but significantly uncoupled the receptor from Akt activation. These unique observations define differential effects on signaling mediated by phosphorylation at distinct locations. This hallmark feature supports the possibility that the signaling outcome of mFFA4 activation can be determined by the pattern of phosphorylation (phosphorylation barcode) at the C terminus of the receptor.
Introduction
Rather than acting simply as sources of energy, fatty acids derived from the diet have recently been appreciated to also act as signaling molecules that play integrative roles in dietary homeostasis (Dranse et al., 2013; Hara et al., 2014; Offermanns, 2014). Key contributions of such fatty acids to metabolic and inflammatory control, as well as to dysregulation of processes that are associated with obesity and metabolic syndrome (Oh et al., 2014; Watterson et al., 2014; Sekiguchi et al., 2015), are produced via specific interactions with G protein–coupled receptors (GPCRs) present at the surface of cells within tissues (including the gut, pancreas, and adipose tissue) that control metabolite distribution, flux, and storage (Oh et al., 2014; Watterson et al., 2014; Sekiguchi et al., 2015). Two GPCRs, free fatty acid receptor (FFA) 1 (also known as GPR40) and FFA4 (also known as GPR120), are known to respond selectively to longer-chain free fatty acids (Milligan et al., 2015). FFA1 is highly expressed by pancreatic β cells (Mancini and Poitout, 2013) and plays a key role in stimulating release of insulin in a glucose concentration–dependent manner (Mancini and Poitout, 2013). This receptor has attracted considerable attention as a means to control glucose homeostasis and thus to serve as a potential therapeutic target for the treatment of type 2 diabetes (Mancini and Poitout, 2013). Indeed, the FFA1 agonist fasiglifam entered phase III clinical trials; although it was clearly able to regulate glycemia and to produce clinically relevant lowering of hemoglobin A1c levels in patients with type 2 diabetes (Mancini and Poitout, 2013, 2015; Kaku et al., 2015), it was withdrawn from further trials because of concerns of possible liver toxicity. Although FFA4 is expressed in the pancreas (Stone et al., 2014; Suckow et al., 2014), where it may play roles in the regulation of glucagon production (Suckow et al., 2014), it is also expressed in various enteroendocrine cells (Parker et al., 2009; Iwasaki et al., 2015; Liu et al., 2015), adipocytes (Oh et al., 2010, 2014; Liu et al., 2015) and macrophages (Oh et al., 2010, 2014; Im, 2015; Liu et al., 2015). Potential combinations of effects on the release of incretins and/or satiety-regulating hormones in the gut, differentiation of and uptake of glucose by adipocytes, and control of the release of inflammatory mediators by macrophages have suggested that FFA4 might also be a useful therapeutic target to modulate insulin resistance and glucose homeostasis (Ichimura et al., 2014; Oh et al., 2014; Liu et al., 2015; Milligan et al., 2015). A nonsynonymous polymorphic variant of FFA4 was reported to be linked to obesity in humans (Ichimura et al., 2012); however, this variant does not appear to be linked to risk of type 2 diabetes or variation of fasting insulin levels (Bonnefond et al., 2015). However, genetic elimination of FFA4 in mice has been noted to result in obesity, glucose intolerance, and fatty liver with decreased adipocyte differentiation when such animals are fed a high-fat diet (Ichimura et al., 2012).
Murine models of diabetes and obesity make important contributions to preclinical predictions and decisions regarding possible therapeutic approaches. Therefore, understanding details of the responsiveness, function, and regulation of murine (m)FFA4 in response to both endogenously generated and synthetic ligands (Oh et al., 2014) and how these may vary compared with the human receptor is vital. Herein, we address these issues by exploring both G protein–dependent and G protein–independent signals produced by mFFA4, defining sites of regulatory phosphorylation on the receptor, and generating and employing phosphorylation site–specific antibodies that can define the intrinsic activity state of the receptor in situ. In this way, we describe here the signaling responses that are regulated by mFFA4 phosphorylation within two clusters of phosphoacceptor sites at the C-terminal tail of the receptor.
Materials and Methods
Materials.
Unless otherwise stated, all biochemicals and reagents were from Sigma-Aldrich Co. (Dorset, UK). Tissue culture reagents and buffers were from Life Technologies Inc. (Paisley, UK). Primers were purchased from Thermo Fisher Scientific (Ulm, Germany). TUG-891 [3-(4-((4-fluoro-4′-methyl-[1,1′-biphenyl]-2-yl)methoxy)phenyl)-propanoic acid] was synthesized as described previously (Shimpukade et al., 2012). Compound 39 was synthesized according to Sparks et al. (2014). This compound is also known as AH 7614 (4-methyl-N-9H-xanthen-9-yl-benzenesulfonamide) and can be purchased from Tocris (Bristol, UK). The Gq/G11 inhibitor YM-254890 [(1R)-1-{(3S,6S,9S,12S,18R,21S,22R)-21-acetamido-18-benzyl-3-[(1R)-1-methoxyethyl]-4,9,10,12,16,22-hexamethyl-15-methylene-2,5,8,11,14,17,-20-heptaoxo-1,19-dioxa-4,7,10,13,16-pentaazacyclodocosan-6-yl}-2-methylpropyl rel-(2S,3R)-2-acetamido-3-hydroxy-4-methylpentanoate] was a kind gift of Astellas Pharma Inc. (Osaka, Japan). Cellular signaling assay kits [pAkt-Ser473, phosphorylated extracellular signal regulated protein kinase 1 and 2 (pERK1/2)] were obtained from Cisbio Bioassays (Codolet, France). Antibodies (pAkt-Ser473, pERK1/2, and total ERK1/2) were obtained from Santa Cruz Biotechnology Inc. (Heidelberg, Germany). Anti–green fluorescent protein (GFP) mouse monoclonal antibodies and anti-GFP rabbit polyclonal antibodies were purchased from Abcam (Cambridge, UK). Protease and phosphatase inhibitor cocktails were from Roche Applied Science (Burgess Hill, UK). [32P]-orthophosphate (5 mCi, 185 MBq) was from PerkinElmer (Waltham, MA). Enhanced chemiluminescence (ECL) developing reagent was from Millipore (Watford, UK). Protein A–sepharose beads, ECL, and X-ray films were obtained from GE Healthcare (Amersham, Buckinghamshire, UK). Xograph compact ×2 film developer and developing solutions were obtained from Xograph Healthcare (Gloucestershire, UK).
Plasmids and Mutagenesis.
To generate an HA epitope–tagged form of mFFA4, the receptor coding sequence was amplified by PCR that incorporated the HA tag sequence at the C-terminal end followed by a stop codon. The mFFA4 receptor was also fused at its C terminus to enhanced yellow fluorescent protein (eYFP) and to an N-terminal FLAG epitope in pcDNA5/FRT/TO. This construct was used as a template to generate various mutants (see the Results) using the QuikChange II method (Stratagene, Berkshire, UK). The identity of all new constructs generated was verified by nucleotide sequencing. Bovine arrestin 3 was fused to Renilla luciferase as previously described (Hudson et al., 2014; MacKenzie et al., 2014).
Cell Lines.
Flp-In T-REx 293 cells able to express constructs of interest from the tetracycline/doxycycline T-REx–inducible locus were generated by cotransfecting FLAG-mFFA4-eYFP or one of the mutants (FLAG-mFFA4-TDTS-AAA-eYFP, FLAG-mFFA4-ADAA-SSS-eYFP, or FLAG-mFFA4-ADAA-AAA-eYFP) with the pOG44 plasmid into parental Flp-In T-REx 293 cells (Life Technologies Inc.). After transfection, 200 μg/ml hygromycin B was added to the culture medium, allowing for polyclonal selection of cells. Chinese hamster ovary (CHO) cells, which stably and constitutively expressed the non–C-terminally tagged C-terminal HA or eYFP epitope–tagged mFFA4 or mFFA4 containing mutations within the C-terminal tail, were generated using the Flp-In system. CHO Flp-In cells (Life Technologies Inc.) were cotransfected with pcDNA5/FRT containing mFFA4 and pOG44, transfected cells were selected with 400 µg/ml hygromycin B, and expression of mFFA4 was confirmed by immunoblotting with anti-HA or anti-GFP antibody.
[32P]-Orthophosphate Labeling and mFFA4 Immunoprecipitation.
CHO Flp-In cells expressing mFFA4 were seeded at 200,000 cells/well in six-well plates and grown for 48 hours. They were then serum starved overnight at 37°C. Cells were washed three times in 1 ml phosphate-free Krebs/HEPES buffer (10 mM HEPES, 118 mM NaCl, 4.69 mM KCl, 1.18 mM MgSO4.7H2O, 1.3 mM CaCl2, 25.0 mM NaHCO3, and 11.7 mM glucose, pH 7.4) and then incubated with 100 μCi/ml [32P]-orthophosphate for 1.5 hours at 37°C. Cells were treated with ligands for an appropriate time at 37°C. The reactions were terminated by rapid aspiration of the buffer followed by addition of 1 ml ice-cold radioimmunoprecipitation buffer [10 mM Tris, 2 mM EDTA, 20 mM glycerol-2-phosphate, 160 mM NaCl, 1% (v/v) Nonidet-P40, and 0.5% (w/v) sodium deoxycholate, pH 7.4] for 30 minutes on ice. Cell lysates were cleared by centrifugation (20,000 × g for 10 minutes) and receptors were immunoprecipitated from precleared lysates using 1 μg/sample of the appropriate antibody for 1 hour or overnight at 4°C. Immunocomplexes were isolated on protein A–sepharose beads and the beads were washed three times with ice-cold buffer (10 mM Tris, 2 mM EDTA, and 20 mM glycerol-2-phosphate, pH 7.4). Immunocomplexes were resuspended in 2× SDS-PAGE sample buffer (125 mM Tris, 200 mM dithiothreitol, 4% SDS, 20% glycerol, and 0.05% bromophenol blue, pH 6.8) and placed in a 60°C water bath for 3–5 minutes. Receptor proteins were resolved on 10% SDS-PAGE gels (200 v; 1 hour, 20 minutes) and electroblotted onto nitrocellulose membranes using the semidry transfer method and Tris-glycine transfer buffer (25 mM Tris, 190 mM glycine, and 20% methanol). Receptor phosphorylation was detected on X-ray films and developed using Xograph film developer. The films were scanned and bands were quantified using AlphaImager software (Alpha Innotech, San Leandro, CA). Parallel Western blotting was performed on the immunoprecipitated samples to check for loading consistency.
mFFA4 Purification and Mass Spectrometry.
Cells were stimulated with 10 µM TUG-891 for 5 minutes at 37°C and then harvested using phosphate-buffered saline (PBS)/1 mM EDTA (pH 7.4). Membranes were prepared and receptors were solubilized in PBS/1% NP-40 supplemented with protease and phosphatase inhibitors for 4 hours at 4°C. Samples were centrifuged (20,000 × g, 20 minutes) and the receptors in the supernatant were immunoprecipitated using anti-HA antibody coupled to agarose beads. The purified receptors were resolved on 8% SDS-PAGE gel and the gel was stained with colloidal Coomassie blue to reveal receptor band(s). The bands were excised and cut into 1- to 2-mm squares and then incubated with 1 μg sequencing grade trypsin (Promega, Southampton, UK) in 50 mM ammonium bicarbonate overnight at 37°C. The supernatant was transferred to fresh tubes and the gel pieces were washed twice with 0.1% trifluoroacetic acid dissolved in 50% acetonitrile. The supernatants were pooled and subjected to liquid chromatography–tandem mass spectrometry (MS/MS) analysis.
Liquid chromatography–MS/MS was carried out using an LTQ Orbitrap mass spectrometer (Thermo Fisher Scientific, Rockford, IL). The pooled supernatants containing receptor peptides were loaded at a high flow rate onto a reverse-phase trapping column (0.3mm i.d. × 1 mm), containing 5 µm C18 300-Å Acclaim PepMap media (Dionex, Hempstead, UK) and eluted through a reverse-phase capillary column (75 µm i.d. × 150 mm) containing Symmetry C18 100-Å media (Waters, Elsetree, UK) that was self-packed using a high-pressure packing device (Proxeon Biosystems, Odense, Denmark). The resulting spectra were searched against the UniProtKB/SwissProt database using MASCOT (Matrix Science Ltd., London, UK) software with peptide tolerance set to 5 ppm and the MS/MS tolerance set to 0.6 Da. Fixed modifications were set as carbamidomethyl cysteine with variable modifications of phosphoserine, phosphothreonine, phosphotyrosine, and oxidized methionine. The enzyme was set to trypsin/P and up to two missed cleavages were allowed. Peptides with a MASCOT score greater than 20 and where the probability that the observed match was a random event was < 0.05 were included in the analysis. The spectra of peptides reported as being phosphorylated were interrogated manually to confirm the precise sites of phosphorylation.
Generation of Phosphorylation Site–Specific FFA4 Antisera.
Phosphorylation-specific antisera were raised against two peptides: IFTDTS(P)VRRNDLNDLS and GAIFT(P)DTS(P)VRRND, which correspond to amino acids 343–357 of mFFA4 in which Ser350 was phosphorylated in the first peptide and Thr347 and Ser350 were phosphorylated in the second peptide. The 87-day program, which included four immunizations, was performed by Eurogentec (Leige Science Park, Seraing, Belgium). The resulting antiserum was purified against the immunizing peptide.
Antibody Characterization Using Phosphatase Treatment.
To characterize potential phosphospecific antibodies, phosphatase treatment experiments were performed. Cells seeded at 200,000 cells/well in six-well plates were grown for 48 hours at 37°C and then serum starved overnight at 37°C. Cells were stimulated with an agonist and the receptors were purified by immunoprecipitation as described above. The immunoprecipitated receptors were washed three times with 150 μl calf intestinal alkaline phosphatase (CIAP) buffer supplemented with protease inhibitors and 0.2% octyl glucoside. The beads were resuspended in 50 μl CIAP buffer in the presence of 40 U CIAP and incubated overnight at 37°C. Positive controls that consist of immunoprecipitated receptors incubated in CIAP buffer were included. The CIAP buffer was aspirated and the beads were resuspended in SDS-PAGE sample buffer. Samples were run on 8% SDS-PAGE and analyzed by Western blotting.
Immunocytochemistry.
CHO cells expressing FLAG-mFFA4-eYFP–tagged receptors were seeded onto 20-mm glass coverslips for 48 hours prior to experimentation. Cells were serum starved overnight at 37°C and then treated with vehicle, the FFA4 antagonist compound 39, or agonist for the indicated time. Cells were fixed in PBS containing 4% paraformaldehyde for 30 minutes at room temperature. Phosphorylation-specific antibody pThr347/pSer350 was used at 0.52 ng/ml followed by Alexa Fluor 546 goat anti-rabbit secondary antibody at 1:1000 dilution. Data were acquired using an LSM510 laser-scanning confocal microscope (Karl Zeiss, Oberkochen, Germany).
mFFA4–Arrestin 3 Interaction Assay.
Arrestin 3 recruitment to mFFA4 was assessed using a bioluminescence resonance energy transfer (BRET) assay (Jenkins et al., 2010; Butcher et al., 2014; Hudson et al., 2014; MacKenzie et al., 2014). Briefly, human embryonic kidney 293T (HEK293T) cells were cotransfected with arrestin 3-Renilla luciferase and eYFP-tagged receptor plasmids in a 1:4 ratio using polyethyleneimine. After 24-hour incubation, cells were subcultured into poly(d-lysine)–coated white 96-well microplates and incubated for a further 24 hours prior to the assay. Cells were washed and incubated in Hanks’ balanced salt solution for 30 minutes prior to conducting the assay. The luciferase substrate coelenterazine h (2.5 μM) (Nanolight Tech, Pinetop, CA) was added to the cells and incubated for 10 minutes at 37°C before test compounds were added. After a further 5-minute incubation at 37°C, luminescent emissions at 535 and 475 nm were measured using a PHERAstar FS (BMG Labtech, Aylesbury, UK). BRET signals were represented as the 535/475 ratio and for all mutant forms of mFFA4; the BRET ratios obtained were expressed as a percentage of the maximum ratio obtained for TUG-891 at the wild-type receptor. Kinetic experiments of arrestin 3 recruitment were carried out on transiently transfected HEK293T cells by incubating the cells at 37°C in the PHERAstar FS after addition of 5 μM coelenterazine h, with BRET measurements taken at 6-second intervals. In these experiments, test compounds were added using PHERAstar FS injectors allowing for continuous measurement during and immediately after compound addition.
Visualization of mFFA4 Internalization.
Flp-In T-REx 293 cells expressing the different mutant constructs were cultured on poly(d-lysine)–coated glass coverslips and cultured for 24 hours before treatment with doxycycline (100 ng/ml) to induce receptor expression. Live cells were then imaged using a Zeiss VivaTome spinning disk confocal microscopy system (Karl Zeiss). Images were taken before the addition of ligand and every 5 minutes after ligand addition for a total of 60 minutes.
Cell Surface Enzyme-Linked Immunosorbent Assay.
Flp-In T-REx 293 cells expressing the different constructs were cultured on poly(d-lysine)–coated flat-bottom 96-well plates for 24 hours before treatment with doxycycline (100 ng/ml) and were treated with ligand for the indicated times. Total receptor surface expression was measured on paraformaldehyde-fixed cells. These were subsequently incubated in PBS containing 5% bovine serum albumin to block nonspecific binding sites (30 minutes at room temperature), followed by incubation with an anti-FLAG monoclonal primary antibody (30 minutes at room temperature), and finally with an anti-mouse horseradish peroxidase–conjugated secondary antibody (30 minutes at room temperature). Cells were washed three times with PBS before measuring Hoechst fluorescence using a PolarStar Omega plate reader (BMG Labtech, Offenburg, Germany). After washing a final time with PBS and incubating with 3,3′,5,5′-tetramethylbenzidine horseradish peroxidase substrate in the dark at room temperature, the absorbance at 620 nm was measured on a PolarStar Omega plate reader. To calculate surface expression, the 620-nm absorbance was corrected for cell number based on Hoechst fluorescence.
Akt(Ser473) Phosphorylation Assay.
mFFA4 signaling to phospho-Akt Ser473 (also called protein kinase B) was detected by using the HTRF Phospho-Akt (Ser473) kit (Cisbio Biassays, Codolet, France). In brief, cells were seeded in 96-well plates at 20,000 cells/well and grown for 48 hours before being serum starved overnight at 37°C. Cells were stimulated with an agonist for the indicated times and lysed with 50 µl lysis buffer for 1 hour at room temperature. Lysates [16 µl] were transferred to 384-well plates and 4 µl premixed d2/cryptate antibodies [1:1 (v/v)] were added to each lysate. The reaction mixtures were incubated for 2 hours at room temperature and the plate was read on the ClarioStar plate reader (BMG Labtech). For inhibition experiments, cells were treated with an inhibitor (Gαq/11 inhibitor/pertussis toxin) for the indicated time before the addition of the agonist. Confirmatory assays were performed using Western blotting with total and phospho-Akt (Ser473) antibody purchased from Santa Cruz Biotechnology Inc. (Heidelberg, Germany).
ERK1/2 Phosphorylation Assay.
Receptor-mediated ERK1/2 phosphorylation was determined by using the Cisbio HTRF phospho-ERK (Thr202/Tyr204) cellular assay kit in a protocol identical to the phospho-Akt assay above.
Immunoblotting.
Nitrocellulose membranes containing resolved receptor proteins were incubated in blocking solution [20 mM Tris, 150 mM NaCl, 0.1% Tris-buffered salt plus Tween 20 (TBST), and 5% nonfat dried milk, pH 7.5] for 1 hour at room temperature or overnight at 4°C. Membranes were probed with the appropriate primary antibody (diluted in blocking solution) for 1 hour at room temperature. Membranes were washed three times for 15 minutes each in TBST and then incubated with a secondary antibody (diluted in blocking solution) for 1 hour at room temperature. After three 15-minute washes in TBST, the membranes were dried by blotting the edge onto a piece of tissue paper. The membranes were placed on glass plates and ECL developing reagent was added for 5 minutes. Immunoreactivity was captured on ECL films and developed using a Xograph film developer. The films were scanned and bands were quantified using AlphaImager software (Alpha Innotech, San Leandro, CA).
Data Analysis.
All data presented represent means ± S.E.M. of at least three independent experiments. Data analysis and curve fitting was carried out using the GraphPad Prism software package (GraphPad Software Inc., La Jolla, CA). Concentration-response data were fitted to three-parameter sigmoidal concentration-response curves. Statistical analyses were carried out using standard t tests, one-way analysis of variance with Tukey’s post hoc analysis, or two-way analysis of variance combined with Bonferroni post hoc analysis as appropriate.
Results
Murine FFA4 Becomes Phosphorylated in an Agonist-Dependent Fashion.
To investigate whether the mFFA4 receptor becomes phosphorylated in an agonist-dependent manner, we performed [32P]-orthophosphate labeling of CHO Flp-In cells stably expressing a form of mFFA4 that incorporated a C-terminal HA-epitope tag (mFFA4-HA). Subsequent to treatment of the cells with vehicle, an ω-3 fatty acid agonist [α-linolenic acid (αLA), 100 μM], or the synthetic FFA4 agonist TUG-891 [10 μM, Fig. 1], samples were immunoprecipitated with anti-HA antibody, resolved by SDS-PAGE, and exposed to X-ray film. Under basal conditions, [32P] was incorporated predominantly into a polypeptide with molecular mass of approximately 45 kDa (Fig. 1A), which correlated with the molecular mass of the mFFA4 construct as determined in anti-HA immunoblots (Fig. 1B). The extent of incorporation of the radiolabel was significantly increased in samples that had been exposed to either αLA or TUG-891 (Fig. 1C). Global incorporation of [32P] in response to treatment with TUG-891 was rapid, peaking within 3 minutes (Fig. 1D). Although it showed a tendency to decline, TUG-891–induced phosphorylation remained significantly elevated over vehicle treatment of at least 30 minutes (Figs. 1, D–F). These data indicate that mFFA4 is phosphorylated in the basal state and undergoes a rapid and sustained increase in phosphorylation after agonist stimulation.

Murine FFA4 becomes phosphorylated in an agonist-dependent and sustained manner. (A–F) Flp-In CHO cells stably expressing mFFA4-HA were labeled with [32P]-orthophosphate and treated for 5 minutes (A–C) or for varying times (D–F) with vehicle (−) or the indicated concentrations of αLA or TUG-891. (A and D) After anti-HA immunoprecipitation, samples were resolved by SDS-PAGE and subjected to autoradiography. (C and F) Such autoradiograms were quantified by densitometry. (G) The structure of TUG-891 is shown. Data are given as means ± S.E.M. **P < 0.01; ***P < 0.001 (significantly greater than vehicle-treated cells). (B and E) Similar levels of the receptor construct in each sample was demonstrated in anti-HA immunoblots. mono, monoclonal; WB, Western blot.
Agonist-Mediated Phosphorylation Occurs within Two Clusters of Residues within the Intracellular C-Terminal Tail of mFFA4.
To identify specific amino acids that became phosphorylated in response to TUG-891, we performed mass spectrometry. Phosphorylation of wild-type mFFA4-HA was detected at multiple residues in these studies (Fig. 2). We obtained excellent coverage of the intracellular domains of mFFA4 with identification of tryptic peptides containing more than 70% of the serine and threonine residues contained within the intracellular loops and C-terminal tail. All of the phosphorylation sites identified were within the C-terminal tail and appeared in two spatially resolved clusters. Cluster 1 contained phosphorylation sites on residues Thr347, Thr349, and Ser350 and cluster 2 contained sites on Ser357 and Ser361. Hence, five of six serine/threonine residues in the C-terminal tail were phosphorylated. Despite identifying peptides containing the one remaining serine in this region, there was no evidence from mass spectrometry that this residue, Ser360, was phosphorylated.

Mass spectrometry analysis of sites of agonist-promoted phosphorylation in mFFA4. CHO Flp-In cells stably expressing C-terminally HA-tagged mFFA4 and treated with TUG-891 (10 μM, 5 minutes) were used to immunoprecipitate and then digest the receptor for analysis using mass spectrometry. (A–E) Representative mass spectra and associated fragmentation tables are shown that cover the five phosphorylated residues identified in various experiments: Thr347, Thr349, Ser350, Ser357, and Ser361. (F) The cartoon shows mFFA4 with the sequence from the conserved NPXXY motif (NPILY in this receptor) at the bottom of transmembrane domain VII to the C-terminal tail, with amino acids denoted by the “one-letter” code. Amino acids that were identified as being phosphorylated are shown in bold type. (G) A summary of the overall data set is provided.
Antisera Generation and Characterization.
To further characterize the phosphorylation status of mFFA4 and its regulation, we generated antisera predicted to specifically identify phosphorylation of both Thr347 and Ser350 of the receptor, or only of Ser350. These residues were selected on the basis that phosphorylation at these amino acids was readily observed in the mass spectrometry studies and phosphopeptides containing these sites were predicted to be highly immunogenic. After anti-GFP/eYFP immunoprecipitation of lysates from Flp-In CHO cells expressing a form of mFFA4 incorporating an N-terminal FLAG-epitope tag and C-terminal yellow fluorescent protein (eYFP) (FLAG-mFFA4-eYFP) and that that had been treated with TUG-891, immunoblotting with either pThr347/pSer350 or pSer350 antibodies detected an approximately 80-kDa polypeptide that potentially reflected the appropriately phosphorylated form of the receptor (Fig. 3, A and B). Confirmation that the antibodies specifically identified phosphorylated forms of FLAG-mFFA4-eYFP was provided by pretreatment of the samples with CIAP, to remove phosphate groups, prior to resolution by SDS-PAGE in which neither pT347/pS350 nor pS350 antisera were able to identify the FLAG-mFFA4-eYFP protein (Fig. 3, A and B). Immunoblots using an anti-GFP antiserum demonstrated the presence of similar levels of receptor protein in all samples (Fig. 3C). Importantly, even without the immunoenrichment provided by anti-GFP/eYFP immunoprecipitation, the pThr347/pSer350 and pSer350 antisera were both able to detect phosphorylated FLAG-mFFA4-eYFP in lysates of cells expressing the receptor construct and, as anticipated, recognition was enhanced substantially after treatment of the cells with TUG-891 (Fig. 3, D–F).
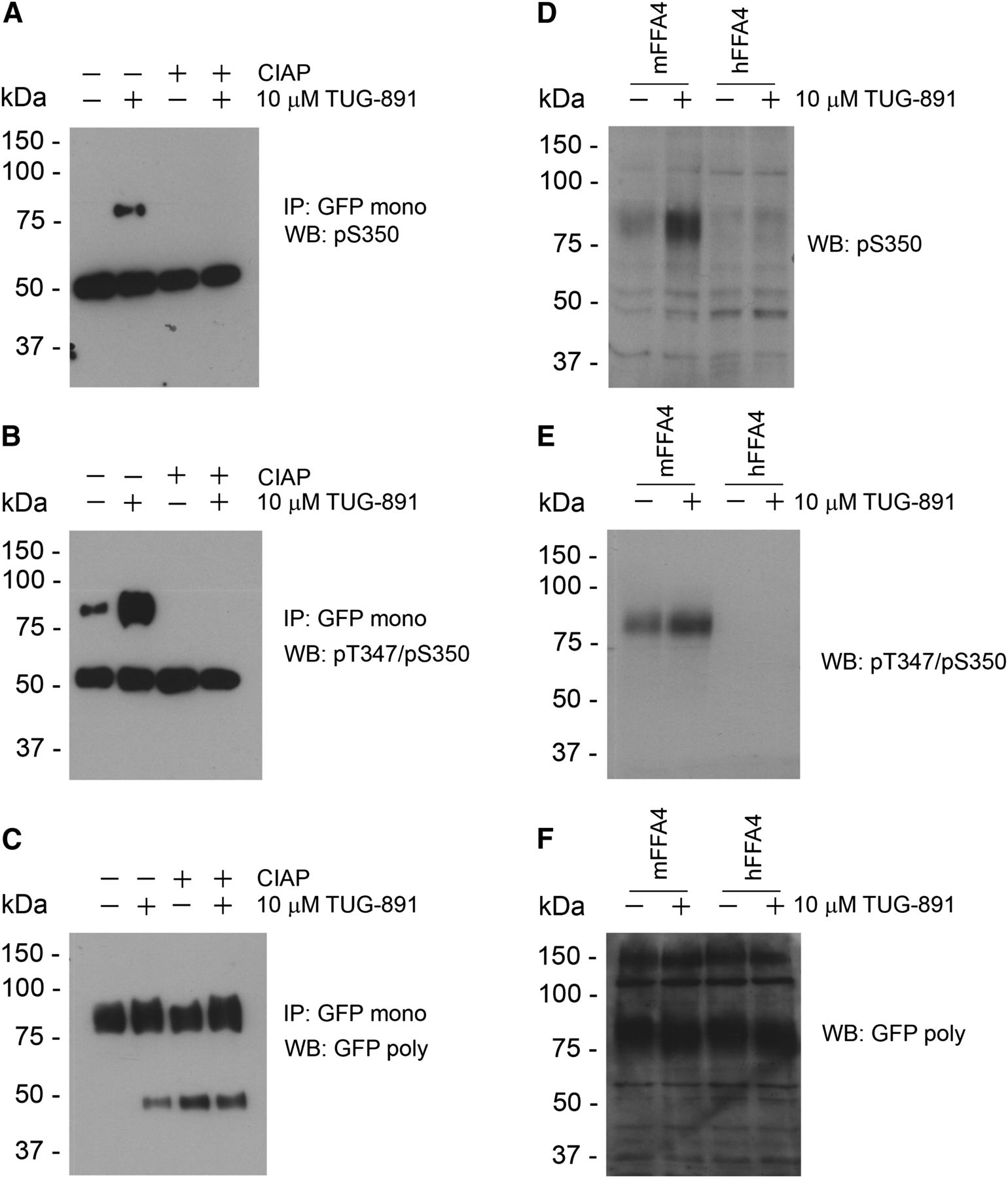
Generation and characterization of phosphorylation state–specific mFFA4 antibodies. (A, B, D, and E) Antisera were generated against peptides incorporating either only phospho-Ser350 (A and D) or both phospho-Thr347 and phospho-Ser350 (B and E). Affinity purified antibodies were used to assess the phosphorylation status of FLAG-mFFA4-eYFP after treatment with vehicle or TUG-891 (10 µM, 5 minutes). (C and F) Parallel immunoblots with an anti-GFP antiserum defined equal loading and levels of receptor expression in each sample. (A and B) Confirmation that the phospho-Thr347/Ser350 and phospho-Ser350 antibodies were indeed phosphorylation site specific was produced by pretreating samples prior to resolution on SDS-PAGE with CIAP. (D–F) The phosphospecific antibodies displayed marked species specificity. Although human FFA4 also becomes phosphorylated in response to addition of TUG-891 and we have previously described phosphospecific antibodies able to recognize this protein (Butcher et al., 2014), neither of the mFFA4 phosphospecific antibodies was able to identify the phosphorylated human receptor (D and E). Immunoblots with anti-GFP/eYFP confirmed that the human receptor was expressed at similar levels (F). IP, immunoprecipitation; mono, monoclonal; poly, polyclonal; WB, Western blot.
Furthermore, the pThr347/pSer350 antibody was able to detect mFFA4 receptor phosphorylation in the presence and absence of C-terminal tail eYFP tag (Supplemental Fig. 1). This suggests that at least for these two sites, the presence of eYFP tag at the C-terminal tail does not interfere with receptor phosphorylation.
Interestingly, although mice and humans share a high degree of sequence similarity within the C-terminal tail, they are not identical. In particular, the epitope recognized by the antibodies in the mouse and human FFA4 receptors differ by two amino acid residues (mouse: GAIFTDTSVRRNDLS; and human: GAILTDTSVKRNDLS). Neither the mFFA4 pThr347/pSer350 nor mFFA4 pSer350 antibodies were able to identify human FLAG-FFA4-eYFP that had been phosphorylated by treatment with TUG-891 (Fig. 3, D–F). The phosphospecific antibodies therefore displayed exquisite species selectivity, as previously also noted for equivalent phosphospecific antibodies to human FFA4 (Butcher et al., 2014). Phosphorylation of mFFA4 in response to TUG-891 as detected by the pThr347/pSer350 antibodies was concentration dependent (Fig. 4A). Moreover, this effect of TUG-891 was blocked by coaddition of the FFA4 antagonist, compound 39 (Sparks et al., 2014) (Fig. 4, B and D), and this was also true for identification using the mFFA4 pSer350 antiserum (Fig. 4B). In addition to phosphorylation of mFFA4 by the synthetic agonist, activation of mFFA4 with either of the ω-3 fatty acids αLA or docosahexaenoic acid also resulted in phosphorylation of the receptor as detected by the pThr347/pSer350 antiserum in a time-dependent and sustained fashion (Fig. 4C).

Identification of agonist regulation of mFFA4 phosphorylation status using phosphorylation site–specific antibodies. Flp-In T-REx 293 cells were induced to express FLAG-mFFA4-eYFP by addition of doxycycline. (A) Lysates of cells exposed to the indicated concentrations of TUG-891 for 5 minutes were resolved by SDS-PAGE and phosphorylated receptors detected by immunoblotting with the phospho-Thr347/Ser350 antibodies. Experiments akin to (A) were performed using combinations of the FFA4 agonist TUG-891 and the FFA4 antagonist compound 39 (Sparks et al., 2014). (B) Samples were subsequently immunoblotted with the phospho-Thr347/Ser350 (left) or the phospho-Ser350 (right) antibodies. Cells as in (A) were exposed to the indicated concentrations of the fatty acid ligands docosahexaenoic acid or αLA or the synthetic agonist TUG-891. (C) The phosphorylated receptor was detected using phospho-Thr347/Ser350 antibodies. (D) The structure of compound 39 is shown. Comp, compound; dox, doxycycline.
The pThr347/pSer350 antibodies were also capable of identifying TUG-891–activated mFFA4 in immunocytochemical studies. In Flp-In CHO cells expressing FLAG-mFFA4-eYFP, these antibodies illuminated the plasma membrane of agonist-treated cells and merging of images of the antibody staining with those of the location of the eYFP tag showed co-incidence (Fig. 5A). pThr347/pSer350 antibody staining was substantially less intense in vehicle-treated cells (Fig. 5B) and cell surface staining was completely abolished in cells that had been pretreated with the antagonist compound 39 (Fig. 5C). These data, coupled with a lack of significant antibody staining of nontransfected cells (Fig. 5D), indicated the specificity of the antibody recognition of the phosphorylated form(s) of the receptor (Fig. 5).
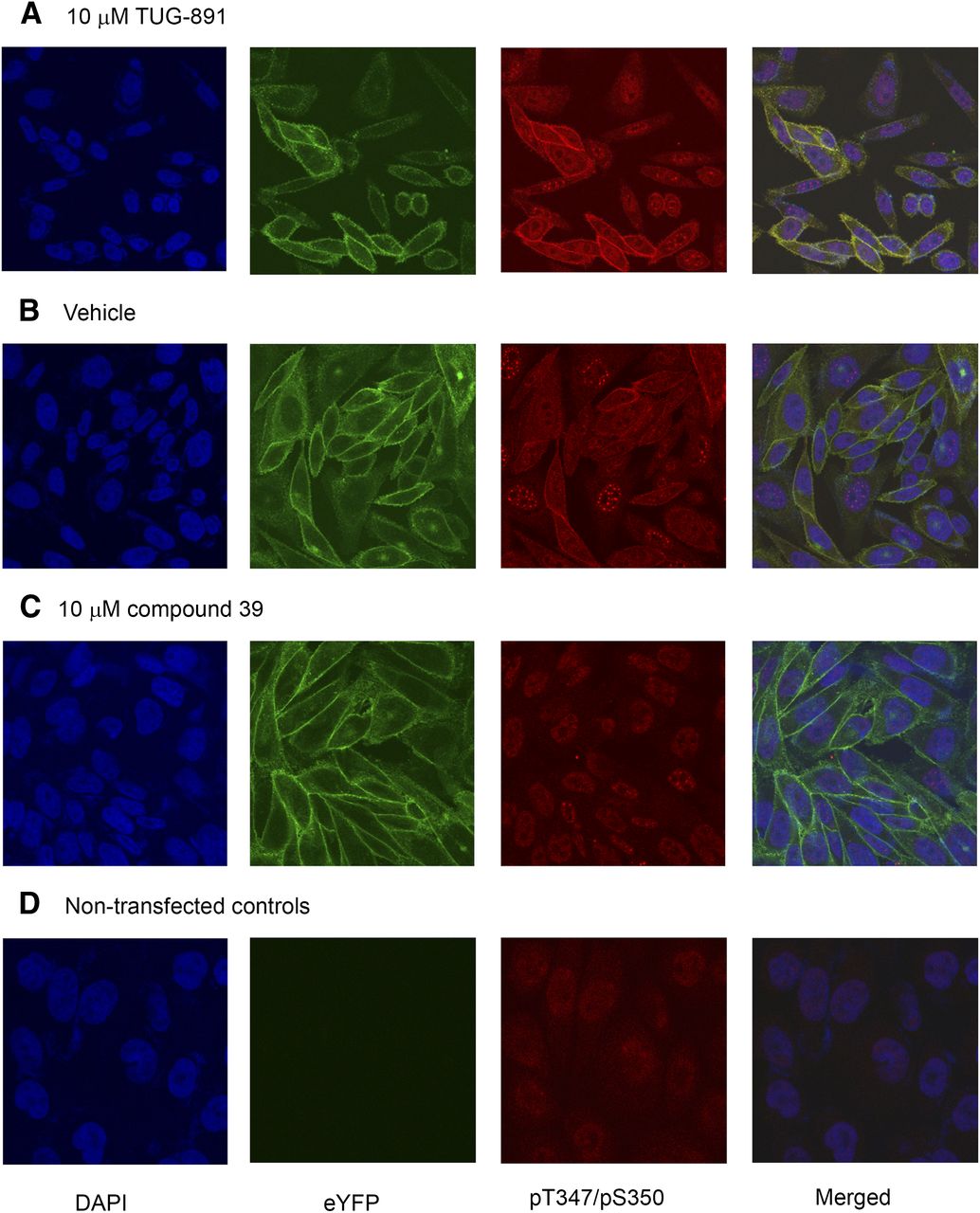
Phosphorylated mFFA4 is identified in immunocytochemical studies. (A–D) Flp-In CHO cells transfected to express FLAG-mFFA4-eYFP (A–C) or parental cells (D) were grown on coverslips. Those expressing FLAG-mFFA4-eYFP were treated with TUG-891 (10 μM, 10 minutes) (A), vehicle (B), or compound 39 (10 μM, 60 minutes) (C). After fixation, cells were treated with the phospho-Thr347/Ser350 antibodies and with DAPI to identify cell nuclei. Blue, DAPI; green, eYFP; and red, phospho-Thr347/Ser350 antibodies. The merged images (right panels) illustrate colocalization of eYFP-tagged receptor and the phosphorylation site–specific antibodies (A and B). Note the enhanced staining with the phosphoreceptor-specific antibodies after treatment with TUG-891 (A versus B) and the reduction in basal antibody staining after treatment with compound 39 (C versus B). DAPI, 4′,6-diamidino-2-phenylindole.
Generation of Phosphorylation-Deficient mFFA4 Variants.
To define whether residues identified as being phosphorylated in the mass spectrometry studies played important roles in potential interactions between mFFA4 and arrestin 3, we generated a series of site-directed mutants of the receptor. [32P]-orthophosphate labeling experiments were performed in Flp-In CHO cells stably expressing either the wild-type FLAG-mFFA4-eYFP construct or receptor variants that had been mutated to convert various serine and/or threonine residues to alanine (Fig. 6A). Consistent with the data from mFFA4-HA (Fig. 1), the wild-type construct, which now migrated as an approximately 75-kDa polypeptide due to the eYFP tag compared with the HA-tagged receptor in Fig. 1, showed basal levels of phosphorylation in [32P]-orthophosphate labeling experiments that significantly increased with agonist (TUG-891) stimulation (Fig. 6B). The two clusters of serine/threonine phosphorylation sites identified by mass spectrometry (cluster 1: amino acids 347–350; and cluster 2: amino acids 357–361) were altered as either two separate groups or the two sets of mutations were combined to eliminate all of the potential sites of phosphorylation (Fig. 6A). The variant in which serine and threonine residues within cluster 1 (347TDTS350) were replaced with alanine (designated mFFA4-ADAA-SSS) was substantially less phosphorylated upon addition of TUG-891 (Fig. 6B). Similarly, the variant (designated mFFA4-TDTS-AAA), in which the serine residues within cluster 2 (Ser357, Ser360, and Ser361) were replaced with alanine, also showed substantially lower incorporation of [32P] after addition of TUG-891 (Fig. 6B). Combination of these two groups of alterations produced a form of the receptor (mFFA4-ADAA-AAA) in which both basal and agonist-induced phosphorylation was absent (Fig. 6B). This was despite this form of the receptor being expressed as well as the wild type and the other two mutants (Fig. 6C). The level of agonist-mediated phosphorylation of mFFA4-ADAA-SSS was similar to that of mFFA4-TDTS-AAA (Fig. 6D), suggesting that both clusters of Ser/Thr residues contribute to a similar extent to the phosphorylation capacity of mFFA4 in response to TUG-891. These phosphoacceptor variants also confirmed the specificity of the pT347/pS350 antiserum. Immunoblots performed on lysates from Flp-In T-REx 293 cells stably expressing eYFP-tagged versions of each of the three mutants showed a lack of recognition of both mFFA4-ADAA-AAA and mFFA4-ADAA-SSS, either with or without treatment with TUG-891 (Fig. 6E). By contrast, mFFFA4-TDTS-AAA was identified by the pT347/pS350 antiserum in an agonist-dependent manner (Fig. 6E). Identification of mFFFA4-TDTS-AAA was reduced, however, compared with the wild-type receptor, despite maintaining the specific target epitope. This may reflect that the peptide antigen sequence contains other residues of mFFA4 that may help define the quaternary structure of this region.

Mutagenesis studies define two clusters of amino acids that become phosphorylated in response to TUG-891. The C-terminal sequence of mFFA4 is shown using the amino acid one-letter code. Aliphatic hydroxy amino acids (serine and threonine) in this region that are potential targets for modification by phosphorylation are in bold. (A) Designation of variants in which clusters of these residues were mutated to alanine is shown, and these changes are noted in the sequence. Wild-type FLAG-mFFA4-eYFP and each of the indicated mutants were expressed stably in Flp-In CHO cells. (B) [32P]-orthophosphate labeling and incorporation of this into the forms of the receptor with or without treatment with TUG-891 for 5 minutes was performed as in Fig. 1. (C) Parallel immunoblotting studies using anti-GFP/eYFP indicated that each form of the receptor was expressed to similar levels. Quantification of the extent of incorporation of [32P] into the receptor was performed as in Fig. 1D. *P < 0.05 (different from the wild type). (E and F) Cells expressing mFFA4-eYFP, mFFA4-TDTS-AAA, mFFA4-ADAA-SSS, or mFFA4-ADAA-AAA were treated for 5 minutes with TUG-891 and immunoblots performed on cells lysates using the phospho-Thr347/Ser350 (E) or anti-FLAG (F) antisera. poly, polyclonal; WB, Western blot; WT, wild type.
Interactions between mFFA4 and Arrestin 3.
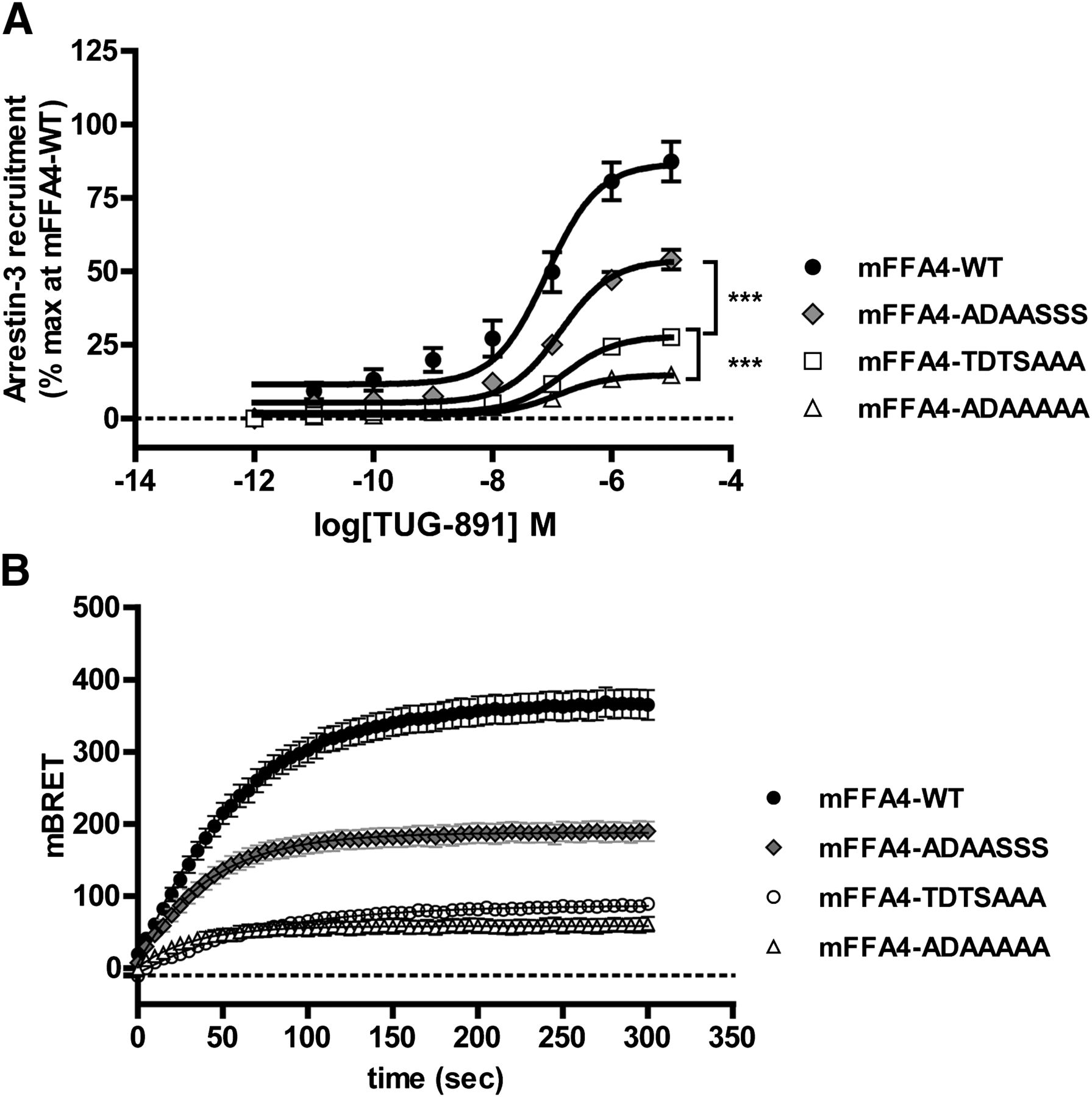
Agonist-induced interactions between the mutant forms of mFFA4 and arrestin 3 (also sometimes designated β-arrestin 2) were then explored in a series of BRET studies performed in HEK293T cells coexpressing a form of mFFA4-eYFP and arrestin 3 tagged with Renilla luciferase. In these studies, stimulation of wild-type FLAG-mFFA4-eYFP with TUG-891 produced a large, concentration-dependent (mean pEC50 = 7.00 ± 0.07, n = 22) increase in the BRET signal (Fig. 7A). Removal of all of the phosphorylation sites contained in clusters 1 and 2 at the C-terminal tail of mFFA4 (mFFA4-ADAA-AAA) resulted in a substantial decrease in agonist-mediated interaction with arrestin 3 (Fig. 7). In these experiments, the maximal BRET signal obtained with mFFA4-ADAA-AAA was only 19.7% ± 1.2% (n = 10) of that of the wild-type receptor (Fig. 7). Linked with the fact that mFFA4-ADAA-AAA showed no evidence of receptor phosphorylation (Fig. 6), this supports the notion that agonist-induced recruitment of arrestin 3 to mFFA4 is largely dependent on receptor phosphorylation. We next tested the relative contributions of each of the cluster 1 and cluster 2 phosphorylation sites to the recruitment of arrestin 3. Conversion of serine residues within cluster 2 to alanine (mFFA4-TDTS-AAA) had a significant effect on the interaction of arrestin 3, reducing the maximal BRET signal to 31.9% ± 2.3% (n = 7) of that observed for the wild-type receptor (Fig. 7A; Table 1). In the case of cluster 1 (mFFA4-ADAA-SSS), mutating the serine and threonine residues to alanine also resulted in a reduction in arrestin 3 recruitment (Emax = 54.3% ± 2.1%, of that observed for the wild-type receptor, n = 12) but this was not to the same extent (P < 0.001) as that seen for the cluster 2 mutant (Fig. 7A; Table 1). Equivalent conclusions as to the contribution of these two clusters to arrestin 3 recruitment were drawn from kinetic BRET experiments carried out by treating the cells with a single maximal concentration of TUG-891 (10 µM) and recording the receptor–arrestin 3 interaction over time (Fig. 7B). Thus, it would appear that although cluster 1 and cluster 2 provide equal contribution to the overall phosphorylation of mFFA4 in response to agonist activation (Fig. 6), cluster 2 has a larger effect on the recruitment of arrestin 3.

Elimination of C-terminal hydroxyl amino acids from mFFA4 compromises agonist-induced interaction with arrestin 3. BRET studies were performed in HEK293T cells transfected to coexpress arrestin 3–Renilla luciferase and each of wild-type FLAG-mFFA-eYFP, mFFA4-ADAA-SSS, mFFA4-TDTS-AAA, or mFFA4-ADAA-AAA. (A) After addition of the indicated concentrations of TUG-891 for 5 minutes, interaction was recorded. Although the alterations to the receptor sequence did not affect the potency of TUG-891, each of the three variants produced a smaller effect than the wild type. Differences between the individual mutants in terms of maximal signal were also significant as noted (***P < 0.001). (B) Similar studies were performed, but after interactions were followed over time addition of TUG-891 (10 μM) . Representative traces are shown. WT, wild type.
Concentration response and kinetic profiles of TUG-891 at the mFFA4-WT and mutant receptors in an arrestin 3 BRET assay
Data represent the mean ± S.E.M. of at least five independent experiments.
Relationship between Receptor Phosphorylation, Arrestin 3 Recruitment, and mFFA4 Internalization.
Interactions with arrestins are frequently essential for agonist-induced receptor internalization (Marchese et al., 2008; Luttrell and Gesty-Palmer, 2010). To test the relationship between receptor phosphorylation, arrestin 3 recruitment and receptor internalization, both imaging of the cellular location of the eYFP tag (Fig. 8A) and enzyme-linked immunosorbent assay studies that measured removal of the N-terminal FLAG-epitope tag from the surface of cells (Fig. 8B) were used to define the extent of internalization of the FLAG-mFFA4-eYFP receptor constructs after exposure to TUG-891. Consistent with the canonical pathway, in which receptor phosphorylation promotes receptor–arrestin interaction (which in turn mediates receptor internalization), the variant mFFA4-ADAA-AAA (which contains alanines in place of all of the serine/threonine residues in clusters 1 and 2 and interacted poorly with arrestin 3) showed a large reduction in TUG-891–mediated internalization (Figs. 8). Conversion to alanines of the cluster 2 phosphorylation sites (mFFA4-TDTS-AAA) also inhibited agonist-mediated internalization and to the same extent as mutation of all of the phosphoacceptor sites. By contrast, removal of cluster 1 phosphorylation sites (mFFA4-ADAA-SSS) had no effect on TUG-891–induced receptor internalization (Fig. 8). Hence, as observed for arrestin 3 recruitment (Fig. 7), phosphorylation of residues within cluster 2 has a larger effect on receptor internalization than phosphorylation within cluster 1.

Elimination of C-terminal hydroxyl amino acids from mFFA4 compromises agonist-induced internalization of the receptor. N-terminally FLAG– and C-terminally e-YFP–tagged forms of wild-type mFFA, mFFA4-TDTS-AAA, mFFA4-ADAA-SSS, or mFFA4-ADAA-AAA were expressed stably in Flp-In T-REx 293 cells. After doxycycline-induced expression of the receptor constructs for 24 hours, cells were challenged with vehicle (0 minutes) or 10 μM TUG-891 (60 minutes). (A) Cells were imaged to identify the location of eYFP. Representative images of groups of cells are shown. (B) Cell surface enzyme-linked immunosorbent assays were performed using an anti-FLAG antibody to detect cell surface receptor on nonpermeabilized cells treated as in (A) with vehicle (0 minutes) or 10 μM TUG-891 (60 minutes). Data are given as means ± S.E.M. of triplicate data points from a single experiment, representative of four. Internalization of mFFA4-ADAA-SSS in response to the agonist was unaffected compared with wild-type mFFA4, whereas internalization of both mFFA4-TDTS-AAA and mFFA4-ADAA-AAA was substantially reduced (***P < 0.001). WT, wild type.
Relationship between mFFA4 Phosphorylation and Coupling to ERK1/2 and Protein Kinase B/Akt.
Agonist activation of FFA4 can result in downstream regulation of a number of signaling pathways. Although generally viewed as a receptor that couples selectively to Gq/G11 family G proteins (Butcher et al., 2014; Milligan et al., 2015), there are also reports of key functions of the receptor that reflect engagement with pertussis toxin–sensitive Gi-family proteins (Engelstoft et al., 2013). In CHO cells stably expressing FLAG-mFFA4-eYFP, addition of TUG-891 resulted in rapid phosphorylation of ERK1/2 mitogen-activated protein kinases. This effect was blocked completely by the selective Gq/G11 inhibitor YM-254890 (Takasaki et al., 2004; Canals et al., 2006; Nishimura et al., 2010) (Fig. 9A), indicating that mFFA4 coupling to ERK1/2 was transduced via Gq/G11 proteins. By contrast, mFFA4 activation of protein kinase B/Akt was independent of Gq/G11 proteins since this response was entirely unaffected by YM-254890 pretreatment (Figs. 9, B and C). This was the case whether Akt phosphorylation was measured via an HTRF-based assay (Fig. 9B) or in immunoblots using a pAkt Ser473 antibody (Fig. 9C). Therefore, we next assessed whether TUG-891 produced phosphorylation of protein kinase B/Akt via a Gi-family G protein–mediated cascade. However, pretreatment of cells with pertussis toxin to cause ADP-ribosylation of Gi-family G proteins also did not affect this pathway and again this was the outcome whether using HTRF- or immunoblot-based assays (Fig. 9, D and E, respectively).

Signaling of mFFA4 to pAkt Ser473 is G protein independent. (A–E) Flp-In CHO cells expressing FLAG-mFFA4-eYFP were used to assess the potential contribution of Gq/G11 (A–C) or Gi-family G proteins (D and E) to TUG-891–mediated regulation of pERK1/2 (A) or pAkt Ser473 (B–E) phosphorylation in either HTRF (A, B, and D) or immunoblotting (C and E) studies. In certain studies, cells were pretreated with the Gq/G11 inhibitor YM-254890 (A–C) or the Gi inhibitor pertussis toxin (D and E). Effects of TUG-891 with and without inhibitors were assessed. Although YM-254890 fully blocked the effect of TUG-891 in pERK1/2 assays (A), it had no effect on pAkt Ser473 (B and C); pertussis toxin also did not modify TUG-891 regulation of this signal end point (D and E) (**P < 0.01). n.s., not significantly different; PTX, pertussis toxin; WB, Western blot.
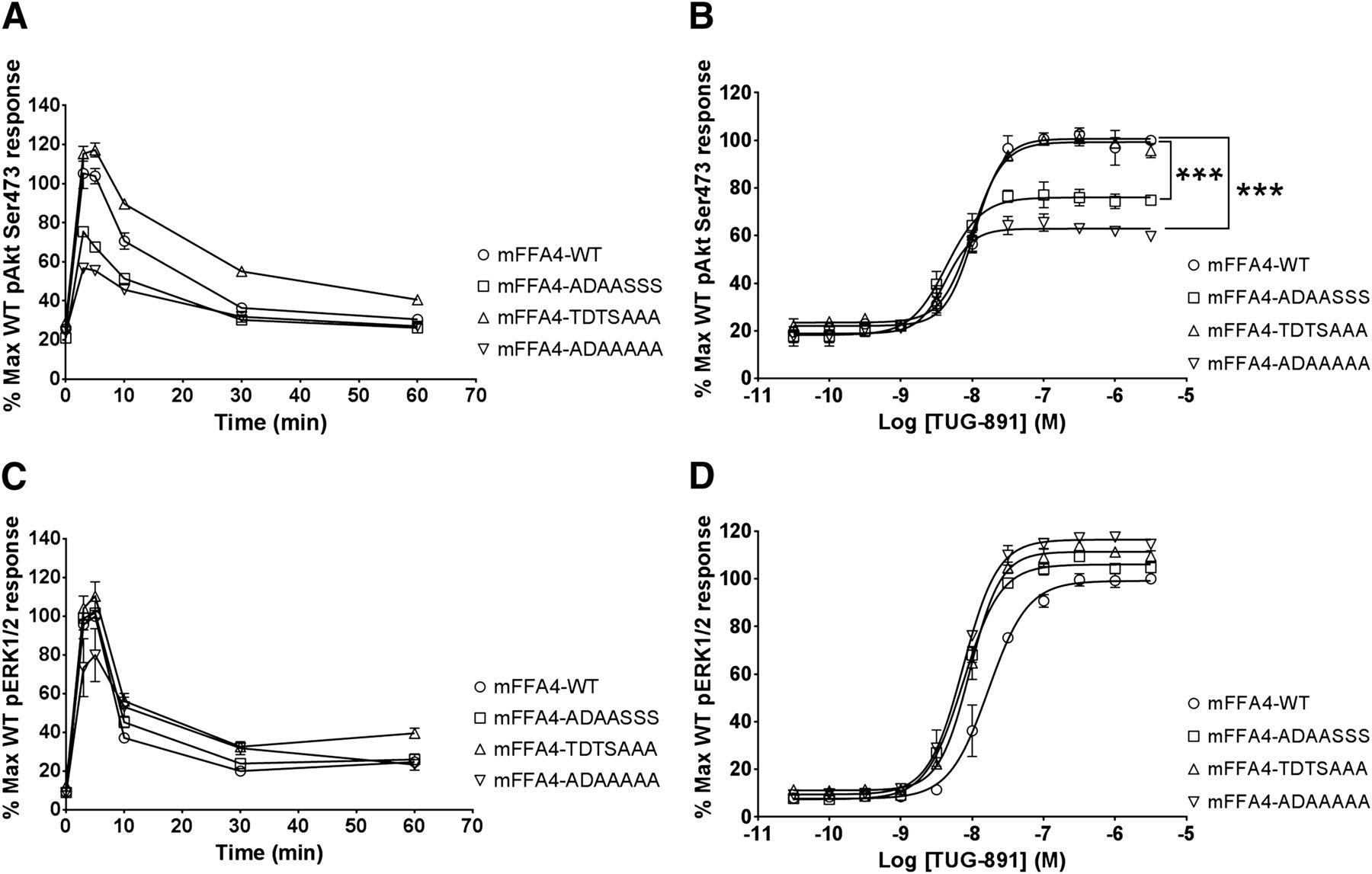
It was possible that receptor phosphorylation-dependent pathways, rather than a canonical G protein–mediated pathway, might be responsible for coupling the mFFA4 to Akt signaling. The rapidly induced peak of protein kinase B/Akt Ser473 phosphorylation was substantially lower for the combined cluster 1 and 2 mutant mFFA4-ADAA-AAA both in time-course experiments and in concentration-response studies employing TUG-891 (Fig. 10, A and B) compared with the wild type, indicating that receptor phosphorylation-dependent mechanisms do play a role in Akt signaling. Importantly, a similar reduction in peak Akt Ser473 phosphorylation was observed for the cluster 1 mutant, mFFA4-ADAA-SSS (Fig. 10, A and B; Table 2). Notably, by contrast, the peak Akt Ser473 phosphorylation response for the cluster 2 mutant mFFA4-TDTS-AAA was equivalent to that of the wild-type receptor (Fig. 10, A and B; Table 2). Thus, mFFA4 phosphorylation appears to play a role in coupling the receptor to Akt activation and amino acids within cluster 1 (Thr347, Thr349, and Ser350) are the primary phosphorylation targets involved. By contrast, removal of serines and threonines in cluster 1, cluster 2, or both groups did not uncouple mFFA4 signaling to ERK1/2 (Fig. 10, C and D). This demonstrates that receptor phosphorylation, arrestin 3 recruitment, and receptor internalization, all of which are disrupted in these mutants, have no role in coupling mFFA4 to the ERK1/2 pathway.

Signaling of mFFA4 to pAkt Ser473 is dependent on the capacity for and location of phosphorylation within the receptor. (A–D) pAkt Ser473 (A and B) or pERK1/2 (C and D) was assessed in HTRF assays in Flp-In CHO cells expressing mFFA4-eYFP (circles), mFFA4-TDTS-AAA-eYFP (triangles), FLAG mFFA4-ADAA-SSS-eYFP (squares), or FLAG mFFA4-ADAA-AAA-eYFP (inverted triangles). Effects of TUG-891 (10 μM) for varying times (A and C) or of varying concentrations of the agonist for 5 minutes (B and D) were assessed.
Potency (pEC50) and relative efficacy (Emax) of TUG-891 at the mFFA4-WT and mutant receptors in pERK1/2 and pAkt (Ser473) assays
Values represent the mean ± S.E.M. from three independent experiments performed in triplicate.
Discussion
Many GPCRs fashion cell type–specific signals despite being activated by the same ligand(s). Although this is poorly explored, these differences may reflect the relative prevalence of distinct G protein–mediated and noncanonical pathways that a receptor can couple to in different cell types. Importantly, such variation expands the diversity and utility of endogenous ligands and their receptors (Thompson et al., 2014) beyond the currently fashionable view of “biased” ligands that favor engagement with one signaling pathway over another in a single cell type (Violin et al., 2014; Luttrell et al., 2015). FFA4 is such a pleotropic GPCR (Milligan et al., 2015), able to generate a range of physiologic endpoints in different cell types by engaging distinct signaling pathways. For example, although FFA4 mediates elevation of Ca2+ via Gq/G11 G proteins and hence promotes the release of incretin hormones, such as glucagon-like peptide-1, from enteroendocrine cells (Hirasawa et al., 2005), this receptor is also reported to regulate release of satiety-promoting hormones from gut cells via activation of pertussis toxin–sensitive Gi-family G proteins (Engelstoft et al., 2013). Moreover, a number of anti-inflammatory effects of ω-3 fatty acids produced via macrophages are mediated by FFA4, reportedly via engagement with non–G protein–dependent pathways involving recruitment of arrestin 3 and subsequent downstream interactions between transforming growth factor kinase protein binding protein and transforming growth factor kinase protein or transforming growth factor-beta (TGF-beta)-activated kinase 1 (Oh et al., 2010, 2014). Although there may be opportunities to identify agonist ligands at FFA4 that display marked bias in stabilizing conformations of the receptor that allow differential engagement with Gq/G11 versus Gi G proteins or other pathways, current synthetic ligands of FFA4 are, in essence, carboxylic acid–containing, fatty acid–like molecules (Milligan et al., 2015). Therefore, there is currently insufficient chemical diversity to hope to engage distinct signaling pathways with high selectivity compared with the endogenous ligands and to test this hypothesis directly.
In the absence of such ligands, our approach has been to determine the sites of phosphorylation and, by mutating these sites, generate a mutant receptor that is not phosphorylated and is therefore uncoupled from phosphorylation-dependent processes such as receptor interaction with arrestins and receptor internalization. We previously described these receptor mutants as G protein biased because they predominately coupled to heterotrimeric G proteins over noncanonical pathways such as those mediated by arrestins (Kong et al., 2010). Here, we employed mass spectrometry to determine that mFFA4 shows agonist-regulated phosphorylation within two specific clusters of serine/threonine residues (cluster 1 and cluster 2) located within the C-terminal tail. Mutation of the observed phosphoacceptor amino acids to alanine resulted in a receptor that was fully resistant to agonist-induced phosphorylation. Interestingly, this receptor mutant coupled normally to the ERK1/2 pathway, indicating that mFFA4 coupling to ERK1/2 was phosphorylation independent. Moreover, by employing an inhibitor of Gq/G11 proteins, we were able to define that mFFA4 coupling to ERK was dependent on G protein signaling. This is in contrast with mFFA4 recruitment of arrestin 3, mFFA4 internalization, and Akt activation, which were all regulated by mFFA4 phosphorylation. Thus, we were able to deconvolute the mFFA4 signaling responses that are downstream of G protein coupling and mFFA4 phosphorylation.
One of the primary functions of GPCR phosphorylation is to promote the interaction of the receptor with members of the arrestin-adaptor protein family (arrestins 1–4) (Reiter and Lefkowitz, 2006). Although the extent to which phosphorylation increases the affinity of the activated receptor for an arrestin depends on the receptor subtype (see Tobin, 2008), we show here that agonist-mediated recruitment of arrestin 3 to mFFA4 was almost totally dependent on phosphorylation of residues at the C-terminal tail of the receptor. In this respect, mFFA4 behaves in a very similar manner to human FFA4 (Butcher et al., 2014). Also consistent with human FFA4, the phosphoacceptor sites on mFFA4 appeared in two clusters: cluster 1 (Thr347, Thr349, and Ser350) and cluster 2 (Ser357 and Ser361). Based on incorporation of [32P] orthophosphate, each cluster contributed approximately equally to the overall phosphorylation of the receptor in response to agonist; however, phosphorylation at cluster 2 had a larger effect on arrestin 3 recruitment. These data point toward site-specific roles for phosphorylation of mFFA4 and argue against the concept that phosphorylation at multiple sites at the C-terminal tail simply adds bulk negative charge that drives arrestin interaction, as has been suggested might be the case for rhodopsin (Ohguro et al., 1993).
Our study extended the analysis of the site-specific role of phosphorylation by consideration of other phosphorylation-dependent processes. This included receptor internalization that similarly was affected more by removal of the phosphorylation sites in cluster 2 than cluster 1. This correlates with reduced interaction with arrestin 3 seen in the cluster 2 mutant and is consistent with the known role of arrestins in driving receptor internalization (Shenoy and Lefkowitz, 2011). However, the relationship between receptor phosphorylation and the activation of Akt was more complex. Here, the peak Akt response was significantly reduced by a mutant mFFA4 in which all of the phosphoacceptor sites were removed, indicating an important regulatory role of phosphorylation in this response. Selective removal of cluster 2 phosphoacceptor sites did not, however, significantly affect the Akt response. By contrast, removal of phosphoacceptor sites within cluster 1 had as large an effect on the Akt response as removal of all of the phosphoacceptor sites in mFFA4. Therefore, these data further support the hypothesis that different sites of phosphorylation at the C terminus of mFFA4 have different signaling outcomes.
Because the cluster 1 mutant, despite showing poor interaction with arrestin 3, still activated Akt fully, it might be concluded that arrestin 3 plays only a minor role in the activation of this pathway by mFFA4. However, neither blockade of Gq/G11 or Gi–G protein family interactions with the receptor affected this signal. Moreover, as a downstream measure of mFFA4 activation, it would be expected that regulation of Akt would show a significant degree of amplification and, therefore, substantially reduced interaction with arrestin 3 is compatible with maintenance of a level of signal generation. This is a common feature in “knockdown” studies in which a nonlinear relationship between the degree of protein reduction and effect is often seen and is to be expected. Thus, our data can be interpreted to indicate that phosphorylation at cluster 2 regulates Akt signaling in an arrestin-dependent manner. Moreover, a number of other receptors have also been shown to activate the Akt pathway in an arrestin-dependent manner (Cianfrocca et al., 2010; Kendall et al., 2014). Given the possibility that mFFA coupling to Akt is phosphorylation and arrestin 3 dependent, it is possible that phosphorylation at either cluster 1 or 2 induces distinct and differential conformations of arrestin 3 and that these mediate different signaling outcomes from the arrestin. Assessing this hypothesis will be an important challenge for the future. In the case of Akt activation, it may be that phosphorylation within cluster 2 induces an arrestin conformation that drives Akt activation. Removal of phosphoacceptor sites in cluster 2 will thereby prevent arrestin from adopting a conformation that mediates Akt signaling. We and others have suggested that the multisite phosphorylation of GPCRs might represent a barcode that determines receptor signaling outcomes and that different signaling outcomes can be achieved by differential regulation of the phosphorylation barcode (Tobin et al., 2008; Butcher et al., 2011; Nobles et al., 2011). It is possible that different phosphorylation barcodes mediate different arrestin conformations and that this results in different arrestin-dependent signaling (Nobles et al., 2011). Although it is now clear that arrestin adopts an active conformation on interaction with receptors (Zhuo et al., 2014) and that a recent crystal structure of arrestin in complex with a phosphorylated C-terminal tail peptide of a GPCR provides atomic-level detail of the active arrestin conformation (Shukla et al., 2013), it is not yet clear whether differential phosphorylation can result in different active conformational states. Our data do, however, support the hypothesis that differential receptor phosphorylation can result in different signaling outcomes. In the case of mFFA4, it would appear that phosphorylation within cluster 1 can preferentially effect arrestin interaction and receptor internalization, whereas phosphorylation within cluster 2 has less of an effect on arrestin recruitment and internalization but preferentially regulates Akt signaling.
Our studies clearly predict that identification or design of novel mFFA4 ligands that drive differential phosphorylation at cluster 1 and cluster 2 would show differential coupling to phosphorylation-dependent processes that include arrestin recruitment, receptor internalization, and Akt signaling. In this way, an mFFA4 ligand that can modulate the site-specific phosphorylation of residues within cluster 1 and 2 would have a highly refined signaling profile and, as a consequence, might activate a restricted number of physiologic responses compared with a ligand that activated nonselectively both phosphorylation-dependent and phosphorylation-independent signaling. This may be very important in the development of therapeutic compounds that target FFA4 (e.g., to regulate glycemia in type 2 diabetes) (Oh et al., 2014; Watterson et al., 2014; Milligan et al., 2015; Sekiguchi et al., 2015), in which it is desirable to activate those pathways that mediate therapeutically beneficial outcomes over pathways that lead to adverse/toxic responses. Thus, the study presented here might provide the framework to develop mFFA4 biased ligands that specifically promote signaling with improved therapeutic potential.
Acknowledgments
The authors thank Dr. John D. Pediani (University of Glasgow) for assistance with imaging experiments. The authors also thank Dr. Sharad C. Mistry and Dr. Andrew R. Bottrill for providing assistance in performing mass spectrometry and phosphorylation sites identification.
Authorship Contributions
Participated in research design: Ulven, Miller, Tobin, Milligan.
Conducted experiments: Prihandoko, Alvarez-Curto, Hudson, Butcher.
Contributed new reagents or analytic tools: Ulven, Tobin.
Performed data analysis: Prihandoko, Alvarez-Curto, Hudson, Butcher.
Wrote or contributed to writing of manuscript: Prihandoko, Alvarez-Curto, Tobin, Milligan.
Footnotes
- Received October 12, 2015.
- Accepted February 11, 2016.
R.P. and E.A.-C. contributed equally to this work.
This research was supported by the Research Councils UK Biotechnology and Biosciences Research Council [Grants BB/K019864/1 and BB/K019856/1] and the Research Councils UK Medical Research Council Toxicology Unit [5TR60].
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- AH 7614
- 4-methyl-N-9H-xanthen-9-yl-benzenesulfonamide
- αLA
- α-linolenic acid
- BRET
- bioluminescence resonance energy transfer
- CHO
- Chinese hamster ovary
- CIAP
- calf intestinal alkaline phosphatase
- ECL
- enhanced chemiluminescence
- ERK1/2
- extracellular signal regulated protein kinase 1 and 2
- eYFP
- enhanced yellow fluorescent protein
- FFA
- free fatty acid receptor
- GFP
- green fluorescent protein
- GPCR
- G protein–coupled receptor
- HEK293T
- human embryonic kidney 293T
- MS/MS
- tandem mass spectrometry
- PBS
- phosphate-buffered saline
- pERK1/2
- phosphorylated ERK1/2
- TBST
- Tris-buffered salt plus Tween 20
- TUG-891
- 3-(4-((4-fluoro-4′-methyl-[1,1′-biphenyl]-2-yl)methoxy)phenyl)-propanoic acid
- YM-254890
- (1R)-1-{(3S,6S,9S,12S,18R,21S,22R)-21-acetamido-18-benzyl-3-[(1R)-1-methoxyethyl]-4,9,10,12,16,22-hexamethyl-15-methylene-2,5,8,11,14,17,-20-heptaoxo-1,19-dioxa-4,7,10,13,16-pentaazacyclodocosan-6-yl}-2-methylpropyl rel-(2S,3R)-2-acetamido-3-hydroxy-4-methylpentanoate
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}