


Visual Overview

Abstract
The nicotinic acetylcholine receptor (nAChR) belongs to a superfamily of pentameric ligand-gated ion channels involved in many physiologic and pathologic processes. Among nAChRs, receptors comprising the α7 subunit are unique because of their high Ca2+ permeability and fast desensitization. nAChR agonists elicit a transient ion flux response that is further sustained by the release of calcium from intracellular sources. Owing to the dual ionotropic/metabotropic nature of α7 receptors, signaling pathways are activated. The α7 subunit is highly expressed in the nervous system, mostly in regions implicated in cognition and memory and has therefore attracted attention as a novel drug target. Additionally, its dysfunction is associated with several neuropsychiatric and neurologic disorders, such as schizophrenia and Alzheimer’s disease. α7 is also expressed in non-neuronal cells, particularly immune cells, where it plays a role in immunity, inflammation, and neuroprotection. Thus, α7 potentiation has emerged as a therapeutic strategy for several neurologic and inflammatory disorders. With unique activation properties, the receptor is a sensitive drug target carrying different potential binding sites for chemical modulators, particularly agonists and positive allosteric modulators. Although macroscopic and single-channel recordings have provided significant information about the underlying molecular mechanisms and binding sites of modulatory compounds, we know just the tip of the iceberg. Further concerted efforts are necessary to effectively exploit α7 as a drug target for each pathologic situation. In this article, we focus mainly on the molecular basis of activation and drug modulation of α7, key pillars for rational drug design.
Introduction
Nicotine has been a key molecule for the advancement of pharmacology since the beginning of the 20th century, when Langley (1905), through fundamental experiments, concluded that muscle contraction was mediated by a “receptive substance” present on the muscle. The muscle nicotinic acetylcholine receptor (nAChR) was thus a pillar in the discovery of neurotransmitter receptors (Langley, 1905). Still, it was not until 1970 that the first neurotransmitter receptor, nAChR, was identified (Changeux et al., 1970; Miledi and Potter, 1971). With the later advent of the molecular biology revolution in the 1980s, the nAChR family was first identified and an extended family of homologous pentameric receptors was revealed (Patrick et al., 1983; Le Novère and Changeux, 1995). This class of receptors was first known as Cys-loop receptors because all family subunits contain a conserved pair of disulfide-bonded cysteines separated by 13 residues. The discovery of orthologs in prokaryotes (Tasneem et al., 2005), which lack the double cysteines, has extended the Cys-loop family to the superfamily of pentameric ligand-gated ion channels (pLGIC).
In vertebrates, the pLGIC superfamily includes cationic channels, nAChRs and serotonin 5-HT3 receptors, and anionic channels activated by GABA or glycine (Le Novère and Changeux, 2001; Lester et al., 2004; Sine and Engel, 2006; Bartos et al., 2009). Their vital role in converting chemical recognition into an electrical impulse makes these receptors prime loci for learning, memory, and disease processes, as well as targets for clinically relevant drugs.
The nAChR is widely distributed throughout the animal kingdom, from nematodes to humans (Le Novère and Changeux, 1995). nAChRs are expressed in many regions of the central and peripheral nervous system, in addition to non-neuronal tissues. The muscle nAChR plays a major role in neuromuscular transmission and is the target of muscle relaxants (Sine, 2012), whereas nAChRs in the brain represent a broad heterogeneous family of ubiquitously expressed receptors. nAChR responses to endogenous acetylcholine (Ach) and choline and exogenous nicotine are involved in a number of physiologic processes and pharmacological effects (Dani and Bertrand, 2007; Albuquerque et al., 2009; Hurst et al., 2013).
The homopentameric α7, one of the most abundant nAChRs in the nervous system, is also expressed in many non-neuronal cells. Its unique activation properties, high calcium permeability, ionotropic/metabotropic dual action, ubiquitous distribution, and involvement in a range of neurologic, psychiatric, and inflammatory disorders have made α7 an important emerging drug target; the participation of α7 in pathologic conditions and the therapeutic potential of α7 ligands has been well documented (see for example Taly and Changeux, 2008; Wallace and Porter, 2011; Lendvai et al., 2013; Wallace and Bertrand, 2013; Uteshev, 2014; Dineley et al., 2015). In this article, we focus on the unique properties of activation and drug modulation of α7 and its relationship with disease and therapy.
nAChR Structure and Function
nAChR subunits are classified as two types, α and non-α, with the α-type containing a disulfide bridge in the agonist binding site. Five muscular (α1, β, γ, ε, and δ) and eleven neuronal (α2–α7, α9, α10, and β2–β4) nAChR subunits have been identified in the mammalian genome (ligand-gated ion channel database, http://www.ebi.ac.uk/compneur-srv/LGICdb/cys-loop.php).
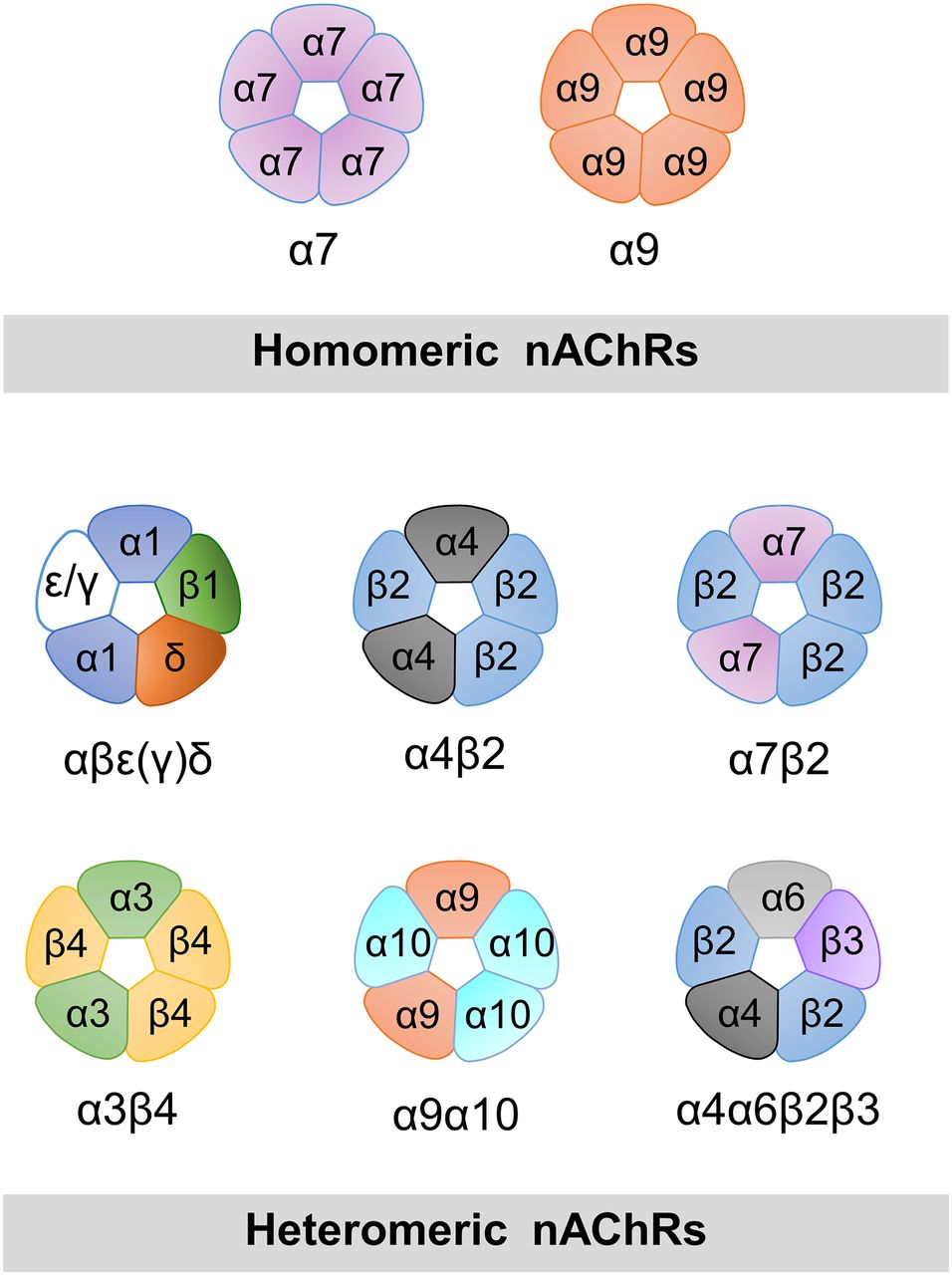
nAChRs are assembled from five identical (α7 or α9) or different subunits (at least two α-type subunits), and can form a variety of different heteromeric receptors with a broad spectrum of pharmacological and kinetic properties (Fig. 1). The resolution of the three-dimensional structures of pLGICs has been the subject of intense efforts over the last decade (Brejc et al., 2001; Dellisanti et al., 2007; Hilf and Dutzler, 2008, 2009; Bocquet et al., 2009; Hibbs and Gouaux, 2011; Corringer et al., 2012; Hassaine et al., 2014; Miller and Aricescu, 2014; Sauguet et al., 2014; Cecchini and Changeux, 2015). However, no high-resolution structure of the full length α7 has been reported to date; an extracellular domain of α7/AChBP chimera (Li et al., 2011; Nemecz and Taylor, 2011) and a nuclear magnetic resonance (NMR) structure of α7 transmembrane domain have been described (Bondarenko et al., 2014).
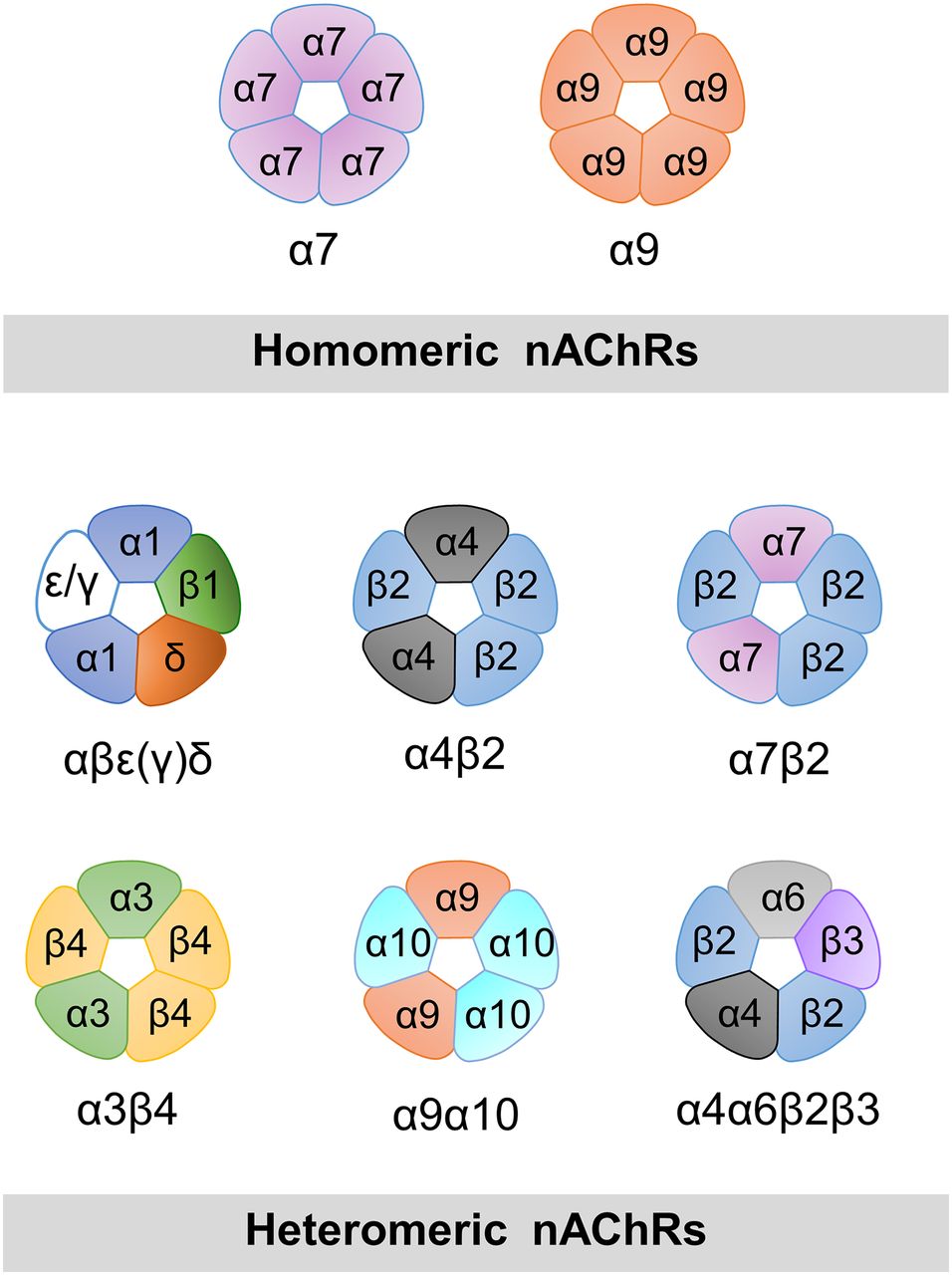
Models of pentameric arrangements of homomeric and some heteromeric nAChRs. α7 and α9 are the only α-type subunits capable of forming functional homomeric receptors, which contain five identical binding sites. In α7, occupancy of only one site is required for activation. Examples of possible combinations of α and non-α subunits in heteromeric arrangements are shown. An α-type subunit is required for the principal face of the binding site. The muscle nAChR contains two functional binding sites at α/δ and α/ε(γ) interfaces. Some subunits can assemble with different stoichiometries, such as α4 and β2. In addition to the arrangement shown, (α4)3(β2)2 receptors are also functional.
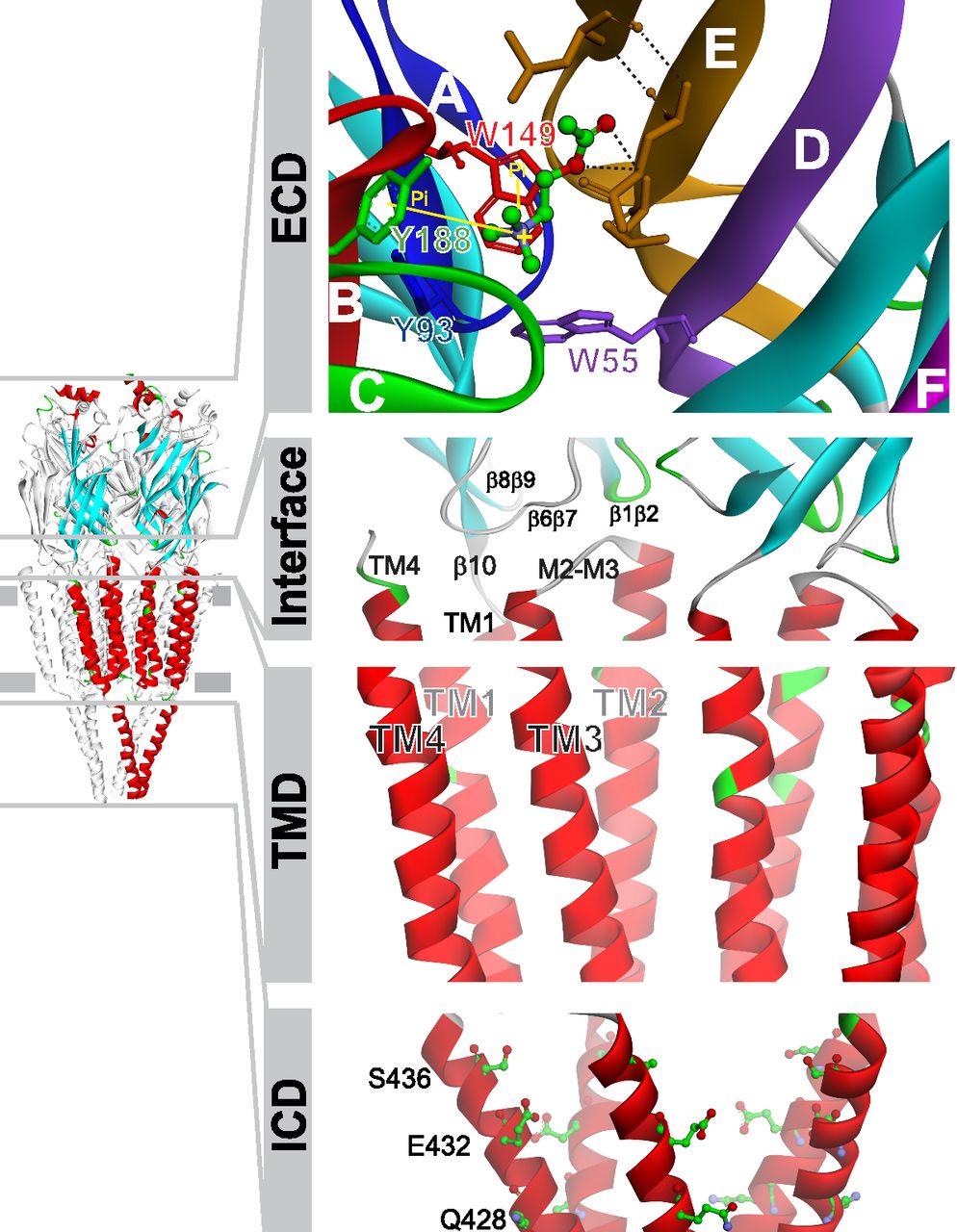
All pLGICs share a conserved organization with five subunits symmetrically arranged around a central ion pore (Fig. 2). Functional domains include the extracellular domain (ECD), which carries the agonist binding sites at subunit interfaces; the transmembrane domain (TMD), which contains the ion pore and the gate; and the intracellular domain (ICD), which contains determinants of channel conductance and sites for regulation and intracellular signaling (Paulo et al., 2009; Jones et al., 2010; King et al., 2015) (Fig. 2). The interface between the ECD and TMD, also referred to as the coupling region, is important for coupling agonist binding to channel opening (Bouzat et al., 2004; Lee and Sine, 2005; Castillo et al., 2006; Bartos et al., 2009), as well as for determining open channel lifetime and rate of desensitization (Bouzat et al., 2008; Yan et al., 2015) (Fig. 2).

Structural model of pLGICs. The ECD is folded into a highly conserved immunoglobulin-like β-sandwich. The agonist binding site is found in a cavity at an interface between two adjacent subunits (Sine, 2012). The principal or positive face is provided by an α-type subunit and includes three loops that span β strands (named as Loops A–C) that harbor predominantly aromatic residues essential for binding and gating. The adjacent subunit, which forms the complementary or minus face, contributes with residues clustered in segments called Loops D–F (Brejc et al., 2001; Sine, 2012). ACh docked into the binding site is shown (α7/AChBP chimera, pdb code 3SQ6). Key aromatic residues from the principal face are Tyr188, Trp149, and Tyr93, and from the minus face, Trp55. The transmembrane domain (TMD) is composed of four transmembrane-spanning helices (TM1–TM4). The TM2 forms the walls of the ion pore, which contains the gate (ring of leucines at 9′ position) and determinants for selectivity. The outer ring of fifteen α-helices (TM1, TM3, and TM4) shields the inner TM2 ring from the lipids (reviewed in Althoff et al., 2014). The interface between the ECD and TMD, also named as coupling region, includes the conserved Cys-loop (β6β7 loop), β1β2 and β8β9 loops from the ECD, and the M2–M3 linker from the TMD. The long intracellular region (ICD) between TM3 and TM4 is highly variable and particularly short in prokaryotic pLGICs. It is thought to be associated with cytoskeletal proteins and involved in channel modulation in all pLGICs (Kabbani et al., 2013). As shown in the figure, in α7, this region contains determinants of channel conductance (Andersen et al., 2011, 2013) and mediates several intracellular signals (Paulo et al., 2009; Jones et al., 2010).
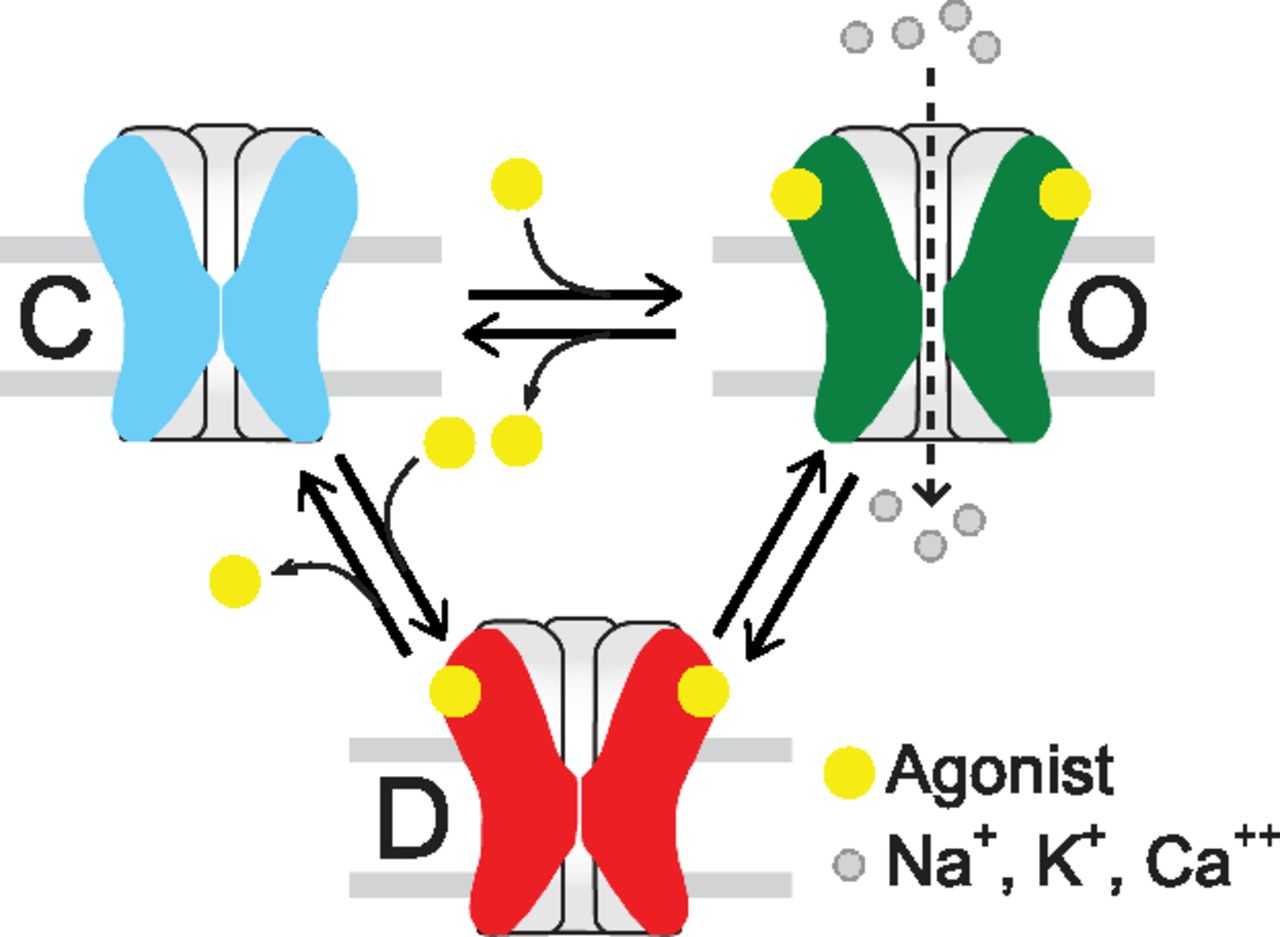
The possible structural events that translate neurotransmitter binding at the ECD into opening of the transmembrane ion channel 60 Å away is an issue of intense research that has been discussed in recent reviews (Corringer et al., 2012; Althoff et al., 2014; Sauguet et al., 2014; Cecchini and Changeux, 2015). On the basis of the Monod-Wyman-Changeux model (Monod et al., 1965), the functional response of a pLGIC can be interpreted as a selection from a few global and discrete conformations elicited by the binding of agonist: closed, open, and desensitized, the latter showing high agonist affinity at the same time being impermeable to ions (Zhang et al., 2013) (Fig. 3). Intermediate states between closed and open or open and desensitized states have been proposed for nAChRs and several pLGICs (Lape et al., 2008; Corradi et al., 2009; Mukhtasimova et al., 2009; Cecchini and Changeux, 2015). Therefore, the number of states in the main conformations and the rate of transition between states determine receptor kinetics, and this tunes each receptor to its physiologic role. In turn, drugs, by binding to different states, can differently modulate receptor function.
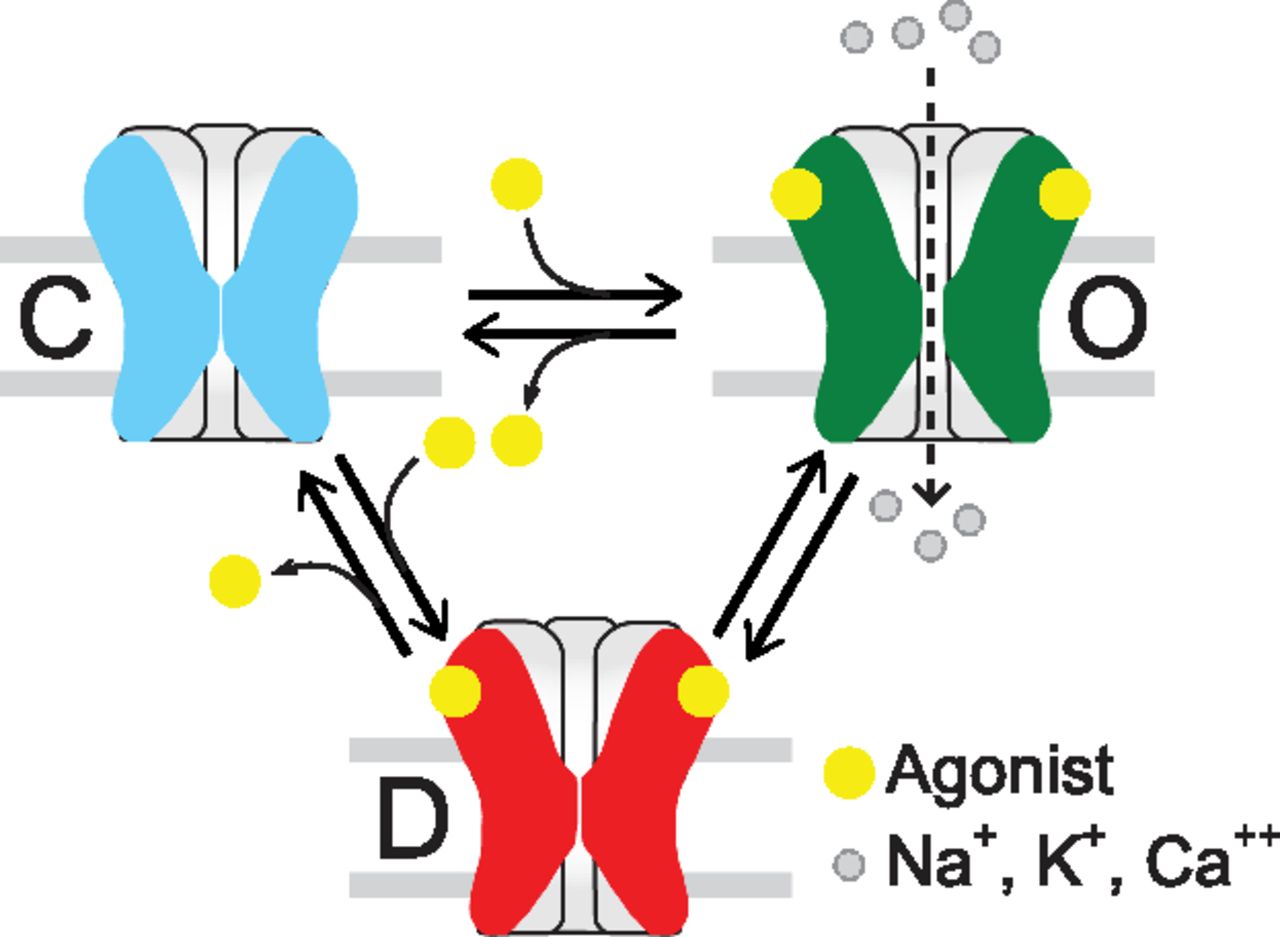
Minimal model of nAChR activation. pLGICs have three main classes of conformational states: Closed (C), open (O), and desensitized (D). Intermediate states have also been detected.
α7 in the Nervous System in Healthy and Disease States
α7 is one of the most abundant nAChRs in the central nervous system. It is particularly expressed in regions implicated in cognitive function and memory, such as hippocampus, cortex, and several subcortical limbic regions (see Lendvai et al., 2013). It is also expressed on non-neuronal cells, including astrocytes, microglia, oligodendrocyte precursor cells, and endothelial cells, where it plays a role in immunity, inflammation, and neuroprotection (Shytle et al., 2004; Shen and Yakel, 2012; Dineley et al., 2015).
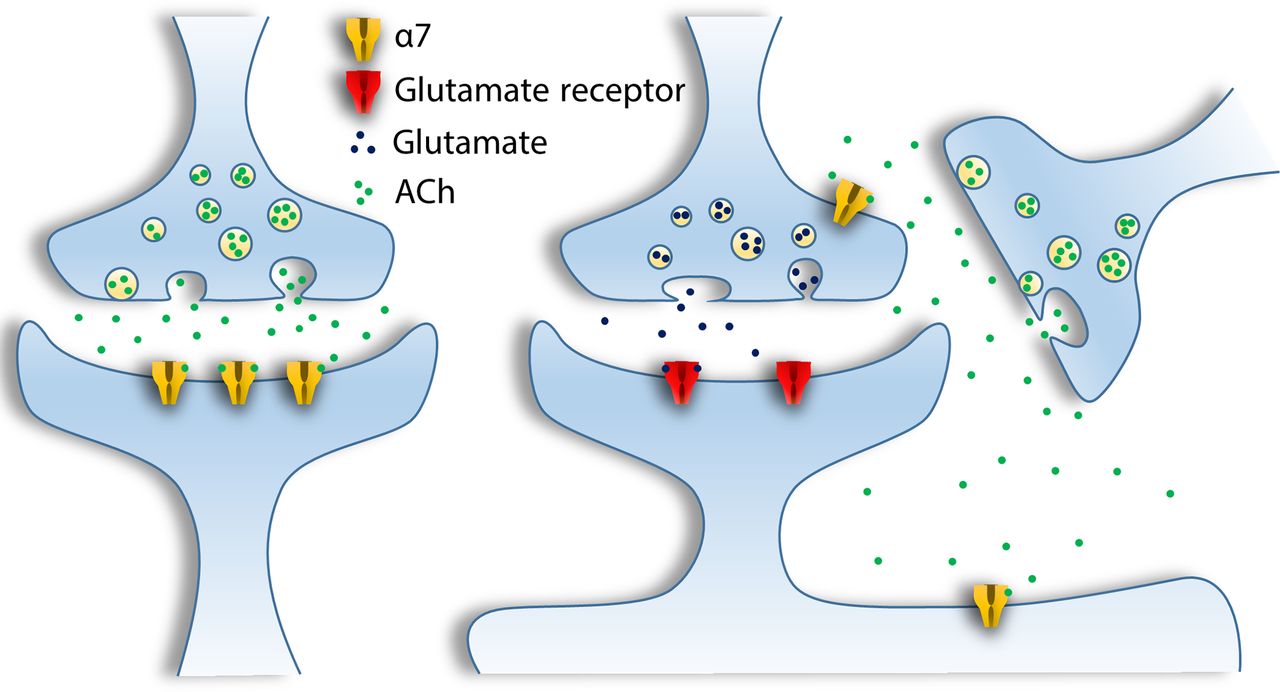
In neurons, α7 receptors localize presynaptically on GABAergic and glutamatergic terminals in the hippocampus and other regions to facilitate release of neurotransmitters. Postsynaptically, α7 receptors mediate fast synaptic transmission, and in perisynaptic locations they modulate other inputs to neurons and activate a variety of signaling pathways through volume transmission (Gotti and Clementi, 2004; Jones and Wonnacott, 2004; Dani and Bertrand, 2007; Dickinson et al., 2008; Albuquerque et al., 2009; Sinkus et al., 2015) (Fig. 4).

Neurotransmission mediated by α7 in the mammalian brain. α7 receptors can be postsynaptic, presynaptic (with a role in regulation of neurotransmitter release), or perisynaptic when they are involved in volume transmission.
α7 contributes to cognitive functioning, sensory processing information, attention, working memory, and reward pathways, and a large body of evidence shows that enhancing α7 activity improves attention, cognitive performance, and neuronal resistance to injury (reviewed in Uteshev, 2014). Significant reduction of α7 in the brain, particularly in the hippocampus, has been reported in Alzheimer disease (Guan et al., 2000; Kadir et al., 2006) and schizophrenic patients (Schaaf, 2014; Dineley et al., 2015). The α7 gene, CHRNA7 on chromosome 15, is genetically linked to multiple disorders with cognitive deficits, including schizophrenia, intellectual disability, bipolar disorder, autism spectrum disorders, attention deficit hyperactivity disorder, epilepsy, Alzheimer disease, and sensory processing deficit (Sinkus et al., 2009, 2015; Schaaf, 2014; Dineley et al., 2015; Deutsch et al., 2016). A partial duplication of CHRNA7 resulting in the chimeric gene CHRFAM7A, which is present only in humans, has been associated with schizophrenia (Sinkus et al., 2009; Neri et al., 2012). Its gene product, dupα7, lacks part of the binding site and may act as a negative modulator of α7 (Wang et al., 2014).
Despite its homomeric character, α7 can assemble with other subunits to form heteromeric receptors. In particular, α7β2 heteromeric receptors have been detected in several brain areas (Liu et al., 2009, 2012; Moretti et al., 2014; Thomsen et al., 2015; Zoli et al., 2015). Interestingly, α7β2 are highly sensitive to blockade by Aβ1–42, suggesting that they may play a unique role in the neuropathology of Alzheimer disease (Liu et al., 2009). Additionally, these heteromeric receptors exhibit high sensitivity to volatile anesthetics, and therefore could be targets for anesthetic action (Mowrey et al., 2013). Thus, α7β2 nAChR may represent a novel molecular target requiring selective α7β2 ligands.
Extraneuronal α7 and Its Pleiotropic Roles
α7 is present in various non-neuronal tissues, such as glia (Sharma and Vijayaraghavan, 2001), blood cells (Kawashima and Fujii, 2004; De Rosa et al., 2005; Báez-Pagán et al., 2015), keratinocytes (Maus et al., 1998), epithelial cells and fibroblasts (Zia et al., 1997), endothelial cells (Macklin et al., 1998), cells of the digestive system and lung cells (reviewed in Wessler and Kirkpatrick, 2008), spermatogonia, spermatocytes, and seminiferous tubular and Sertoli cells (Schirmer et al., 2011). The functional role of α7 in these cells is being intensively investigated and has been associated with differentiation, migration, adhesion, cell contact, apoptosis, and angiogenesis processes (Ni et al., 2010; Egea et al., 2015; Zdanowski et al., 2015).
In particular, α7, present in all types of immune cells, including lymphocytes (T and B cells), dendritic cells and macrophages (reviewed in Egea et al., 2015), has attracted considerable attention as an important drug target for inflammation. α7 is an important player in the “cholinergic anti-inflammatory pathway,” which is a link between vagal efferent fibers and innate immune system (Martelli et al., 2014). α7 modulates intracellular signal pathways [Janus kinase 2 (JAK2)/signal transducer and activator of transcription (STAT3) and PI3K/Akt] in immune cells, which results in potent anti-inflammatory effects through cytokine production inhibition and overexpression of heme oxygenase 1 (Báez-Pagán et al., 2015; Egea et al., 2015) (Fig. 5). A brain cholinergic pathway also exists that regulates microglial activation through α7 (Shytle et al., 2004). This pathway is crucial for neuroprotection (Park et al., 2007) and is probably important in Parkinson disease (Stuckenholz et al., 2013), oxygen and glucose deprivation (Parada et al., 2013), and global ischemia (Guan et al., 2015).

Dual ionotropic/metabotropic nature of α7: Intracellular pathway signaling mediated by α7 activation. α7 allows transient flux of Na+, K+, and Ca2+. The transient increase of calcium may also lead to a sustained calcium response by a calcium-induced calcium release (CICR) mechanism through IP3 receptors. As a metabotropic receptor, α7 mediates intracellular signals by binding to Gα and Gβγ proteins and several other partners (Kabbani et al., 2013; King et al., 2015). In immune cells, α7 has been shown to be involved in several intracellular pathways; although not yet fully deciphered mechanistically, these pathways lead to potent anti-inflammatory effects. For example, α7 has been shown to activate JAK2/STAT3 in some immune cells, which leads to blockade of nuclear factor (NF)-κB nuclear translocation and inhibition of NF-κB, resulting in inhibition of proinflammatory cytokine production. Also, α7 has been shown to activate the PI3K/Akt pathway that promotes Nrf-2 translocation to the nucleus and overexpression of heme oxygenase 1 (HO-1), resulting in potent anti-inflammatory effects (see de Jonge and Ulloa, 2007; Báez-Pagán et al., 2015; Egea et al., 2015). DAG, diacylglycerol; GPCR, G protein-coupled receptor; PKC, protein kinase C; ROS, reactive oxygen species.
Therefore, α7 nAChR is emerging as an important drug target for the modulation of inflammation in different pathologic contexts, including sepsis, ischemia/reperfusion, rheumatoid arthritis, and pancreatitis.
α7 Pharmacology and Ion Selectivity
Hallmark features of α7 receptors include high Ca2+ permeability, relatively low sensitivity to ACh, full activation by choline, high-affinity for α-bungarotoxin (α-BTX), relatively low affinity for nicotine, and fast desensitization that occurs on the submillisecond time scale.
Dose-response curves show EC50 values of ∼100–200 μM for ACh [Hill coefficient (nH) ∼1] (Andersen et al., 2013), 0.4–1.6 mM for choline, and 18–91 μM for nicotine (Wonnacott and Barik, 2007). Several α7-specific agonists have been synthetized, including PNU-282987 (EC50 128 nM), AR-R17779 (EC50 ∼10–20 μM), compound A (EC50 ∼14 nM to 0.95 μM), and partial agonists GTS-21 (EC50 ∼6–26 μM), and SSR180711 (EC50 ∼1–4 μM). Selective competitive antagonists are α-BTX (IC50 ∼1–100 nM), which has been widely used to detect α7 in tissues, and methyllycaconitine (MLA, IC50 10–200 nM) (Wonnacott and Barik, 2007).
α7 allows flux of Na+ and K+ and is highly permeable to Ca2+. The PCa2+/PNa+ ratio is ∼10–20, which is greater than that of other nAChRs and similar to N-methyl-d-aspartate receptors (Séguéla et al., 1993; Albuquerque et al., 1997). The high Ca2+ permeability underlies most of α7 functions: facilitation of neurotransmitter release, depolarization of postsynaptic cells, and initiation of many cellular processes through its action as a second messenger (Gotti and Clementi, 2004). The transient increase in intracellular Ca2+ is converted into a sustained, wide-ranging phenomenon by calcium release from intracellular stores through a calcium-induced calcium release mechanism, a process involving IP3 and ryanodine receptors (Tsuneki et al., 2000; Dajas-Bailador et al., 2002; Guerra-Álvarez et al., 2015) (Fig. 5).
The concept of α7 as a dual metabotropic/ionotropic receptor is attracting increasing attention (Kabbani et al., 2013). α7 binds both Gα and Gβγ proteins through the M3–M4 loop and enables a downstream calcium signaling response that can persist beyond the expected time course of channel activation (Fig. 5) (Kabbani et al., 2013; King et al., 2015). Moreover, in lymphocyte T cells, mobilization of Ca2+ through the α7 channel is not necessarily required for the nicotine-induced release of Ca2+ from the internal stores (Razani-Boroujerdi et al., 2007). Channel-independent signal transduction has been related to the role of α7 in inflammation (de Jonge and Ulloa, 2007; Thomsen and Mikkelsen, 2012) and in neurite growth (Nordman and Kabbani, 2012; Kabbani et al., 2013).
α7 is not only permeable to Ca2+ but is also modulated by the ion; Ca2+ has been shown to regulate agonist efficacy and cooperativity (Bonfante-Cabarcas et al., 1996; Albuquerque et al., 1997). As in other pLGICs (Zimmermann et al., 2012), the divalent modulatory sites may be located at the ECD.
α7 Channel Kinetics
Heterologous expression of α7 in oocytes and mammalian cells combined with electrophysiological experiments has provided information about the receptor’s unique activation properties. The surface expression of recombinant α7 requires coexpression of the chaperone RIC-3 (Williams et al., 2005), a transmembrane protein that is required for efficient receptor folding, assembly, and functional expression (Castillo et al., 2005; Millar, 2008). Recently, another chaperone, NACHO, has been identified. NACHO is a transmembrane protein of neuronal endoplasmic reticulum that mediates assembly of α7 by promoting protein folding, maturation through the Golgi complex, and expression at the cell surface (Gu et al., 2016).
Typical α7 receptor-mediated currents decay rapidly in the presence of agonist as a result of desensitization (Fig. 6A). Given the fast kinetics, the temporal resolution of agonist exchange and most recording systems limit the accurate estimation of the true desensitization rate (Zhou et al., 1998; Lovinger et al., 2002; Bouzat et al., 2008), which may partially account for the great variability of desensitization rates found in the literature. Outside-out patches rapidly perfused with ACh, which allows for a more accurate determination of the desensitization rate, show current decay time constants of ∼0.4 milliseconds (Bouzat et al., 2008). Owing to their fast decay, α7 peak responses occur in advance of complete solution exchange. Therefore, more accurate EC50 values are obtained if the net charge, which represents the time integration of all channel activation, rather than the peak current, is used for the analysis of the concentration-effect relationship (Papke and Porter Papke, 2002) (Fig. 6A).
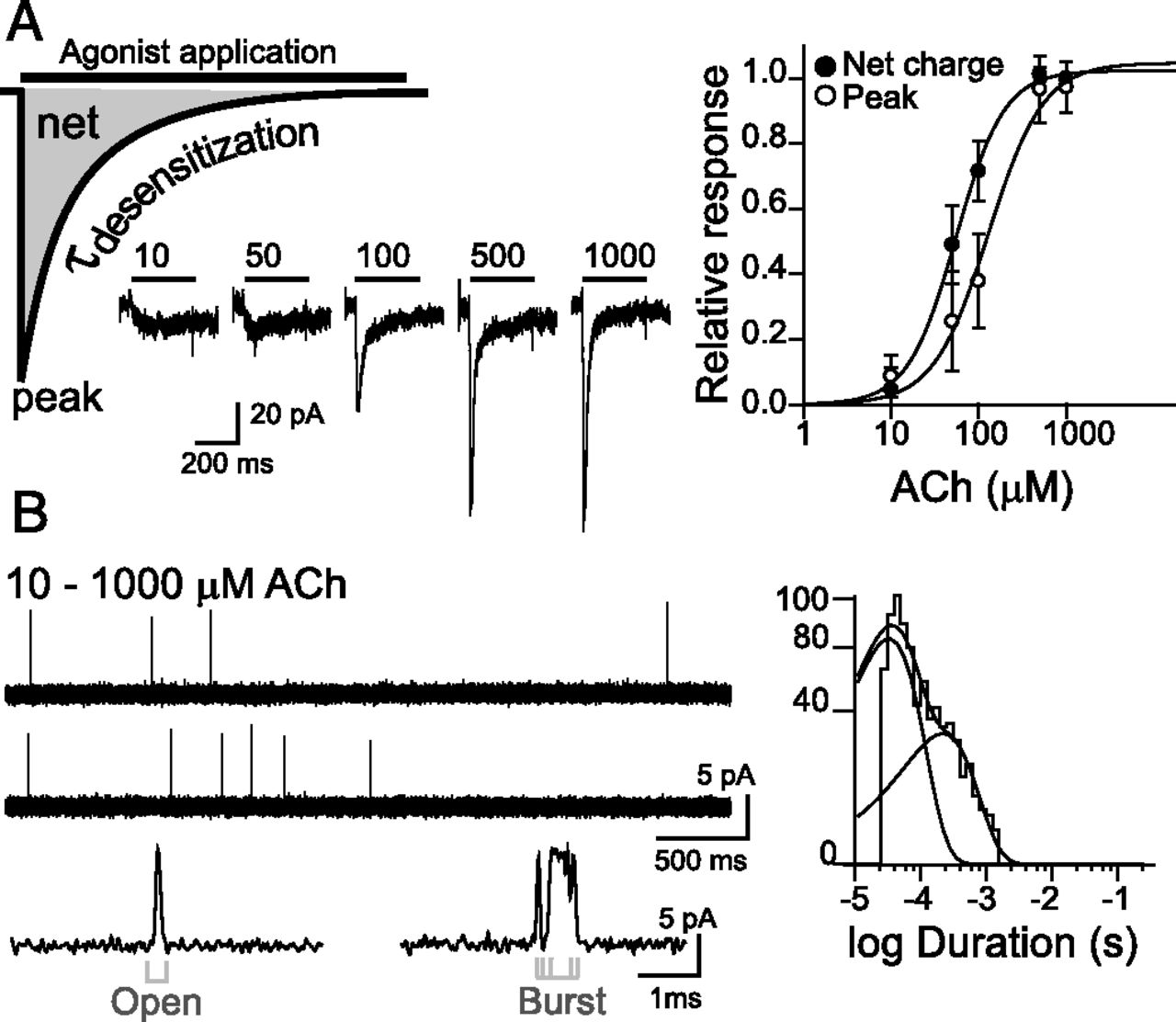
α7 single-channel and macroscopic currents. (A) Typical whole-cell currents elicited by different ACh concentrations (from 10 to 1000 μM) from cells transfected with human α7. At left, a schematic representation of a macroscopic current with the measured parameters. Both the maximal current (peak) and the net charge (net) can be used to construct dose-response curves as shown in the figure. (B) Single-channel activity from cell-attached patches of cells expressing human α7 appears as very brief (∼0.1–0.3 milliseconds) and isolated openings or less often as short bursts. Channel openings are shown as upward deflections. Membrane potential, –70 mV; filter, 9 kHz. At right, a typical open duration histogram (Bouzat et al., 2008).
At the single-channel level, channel activity appears as isolated brief pulses (0.1–0.3 milliseconds) flanked by long closed periods and, less often, as two or three brief pulses in quick succession (bursts) (Fig. 6B). Thus, α7 has a very low open probability. Single-channel openings exhibit a broad distribution of current amplitudes, probably owing to limited time resolution of the brief openings, with a maximum of ∼10 pA at –70 mV (Mike et al., 2000; Bouzat et al., 2008; Andersen et al., 2013; daCosta and Sine, 2013; Yan et al., 2015).
Although there is no general consensus for an α7 kinetic model, an interesting aspect is that the temporal pattern of single ACh-activated currents is similar at 10 μM or 1 mM ACh (Bouzat et al., 2008) (Fig. 6B). This lack of concentration-dependence combined with the fact that most receptor activation episodes consist of a single opening with a duration similar to the desensitization time constant suggests that desensitization is the predominant pathway for channel closing, a unique feature among nAChRs. Control of open-channel lifetime through desensitization has potential consequences for inter-response latency at a synapse where the neurotransmitter pulse is transient. Recovery from desensitization depends on agonist concentration and exposure duration, since different desensitized states may exist. Desensitized α7 receptors expressed in human embryonic kidney cells recover with a time constant of ∼1 second (Bouzat et al., 2008), whereas 15–30 seconds are required for full recovery in the hippocampus (Frazier et al., 1998). Thus, after α7 brief response, a latency of several seconds is required to generate another response of full amplitude. The fast desensitization and brief open duration may avoid cell toxicity caused by increased intracellular Ca2+ owing to α7 overstimulation.
Another intriguing aspect of α7 activation is the relationship between ACh occupancy and activation. We set out to determine how many of the five identical agonist binding sites are required to activate α7 by developing a strategy that utilizes coexpression of an inactivated binding-site subunit and a reporter amplitude subunit. This allows for the determination of the number of ACh-occupied sites from the amplitude of each individual single-channel opening (Rayes et al., 2009; Andersen et al., 2011, 2013). The results revealed that ACh occupancy of only one of five α7 binding sites is necessary for activation, and that open-channel lifetime of a single-occupied receptor is indistinguishable from that of receptors containing five intact binding sites (Andersen et al., 2013). Also, occupancy of a single site by the antagonist methylcaconitine (Palma et al., 1996) or α-BTX (daCosta et al., 2015) will inhibit α7 function.
Whole-cell experiments have revealed the physiologic consequence of having more sites than those required for activation; saturation of receptors in cells expressing a wild-type and a binding-site mutant subunit (α7Y188T) is achieved at higher ACh concentrations when the proportion of receptors with fewer functional binding sites increases (Andersen et al., 2013; Williams et al., 2011a). This suggests that α7 is a highly sensitive sensor of ACh and is therefore adapted to function with submaximal occupancy of its sites, a property appropriate for volume transmission.
The unique activation properties of α7 also suggest that any slight change in the energy barriers between active, closed, and/or desensitized states may have a deep impact on receptor function, which makes these receptors very sensitive drug targets.
α7 Modulation as a Therapeutic Strategy
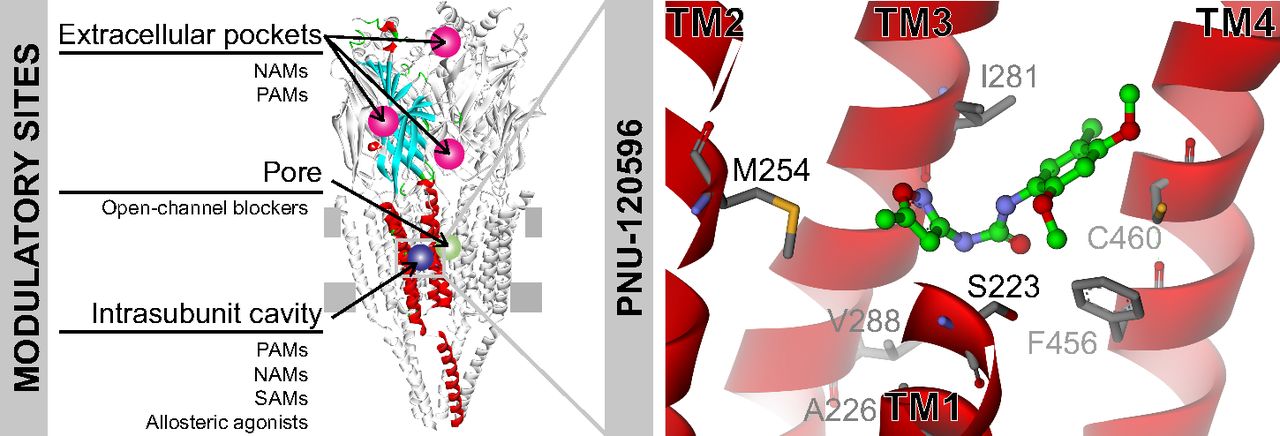
In addition to agonists and antagonists that bind to the orthosteric site, a large number of compounds modulate α7 function by binding to allosteric sites. These compounds may act as: 1) positive allosteric modulators (PAMs) that potentiate currents only in the presence of the agonist; 2) allosteric agonists that activate receptors from nonorthosteric sites; 3) negative allosteric modulators (NAMs), which either act as open-channel blockers by binding to the pore or inhibit activation allosterically; and 4) silent allosteric modulators (SAMs) that have no effect on orthosteric agonist responses but block allosteric modulation (Fig. 7).

Allosteric modulatory sites in α7. Structural model of the α7 receptor viewed from the side representing several potential sites for allosteric modulators. At the TMD, the intrasubunit transmembrane cavity is a site for a great variety of compounds that may elicit different pharmacological effects (potentiation (PAMs), inhibition (NAMs), no effect (SAMs), or activation (allosteric agonists)). PNU-120596 docked into this cavity is shown. Open-channel blockers inhibit response by transiently blocking the flux of ions through the pore. At the ECD, different potential sites for inhibitors and potentiators have been proposed (Ludwig et al., 2010; Spurny et al., 2015).
Since stimulation of α7 improves attention, cognitive performance, and neuronal resistance to injury in addition to eliciting robust analgesic and anti-inflammatory effects, α7 potentiation has emerged as a potential therapeutic strategy, and the search for novel potentiators is an active research field. The potential therapeutic use of several α7 partial agonists and PAMs on animals and humans has been documented in several recent reviews (e.g., Wallace and Porter, 2011; Thomsen and Mikkelsen, 2012; Lendvai et al., 2013; Dineley et al., 2015) (Table 1). Still, no drug has reached phase III clinical stage.
α7 modulators with potential clinical applications.
Compared with agonists, PAMs are promising therapeutic tools because they: 1) better maintain the temporal spatial characteristics of endogenous activation; 2) show higher selectivity, since the orthosteric site is more conserved among nAChRs than allosteric sites (Yang et al., 2012); 3) allow greater diversity in structure and final effects; 4) reduce tolerance attributable to α7 desensitization; and 5) act as neuronal protectors (Kalappa et al., 2013; Sun et al., 2013; Uteshev, 2014). In particular, because neuronal damage elevates choline levels near the site of injury, the presence of a PAM may not require coapplication of an agonist for the mediation of a local neuroprotective effect (Uteshev, 2014). Therefore, it is increasingly accepted that targeting allosteric sites can provide novel medications with greater structural diversity and specificity.
PAMs have been classified on the basis of their macroscopic effects as type I or type II (Fig. 8). Type I PAMs mainly enhance agonist-induced peak currents without significantly affecting current decay and do not reactivate desensitized receptors, whereas type II PAMs delay desensitization and reactivate desensitized receptors (Bertrand and Gopalakrishnan, 2007; Arias and Bouzat, 2010; Williams et al., 2011b). The ratio of the changes in net charge/peak current induced by type I PAMs is close to one, whereas it is higher than one in the presence of type II PAMs (Andersen et al., 2016; Williams et al., 2011c).
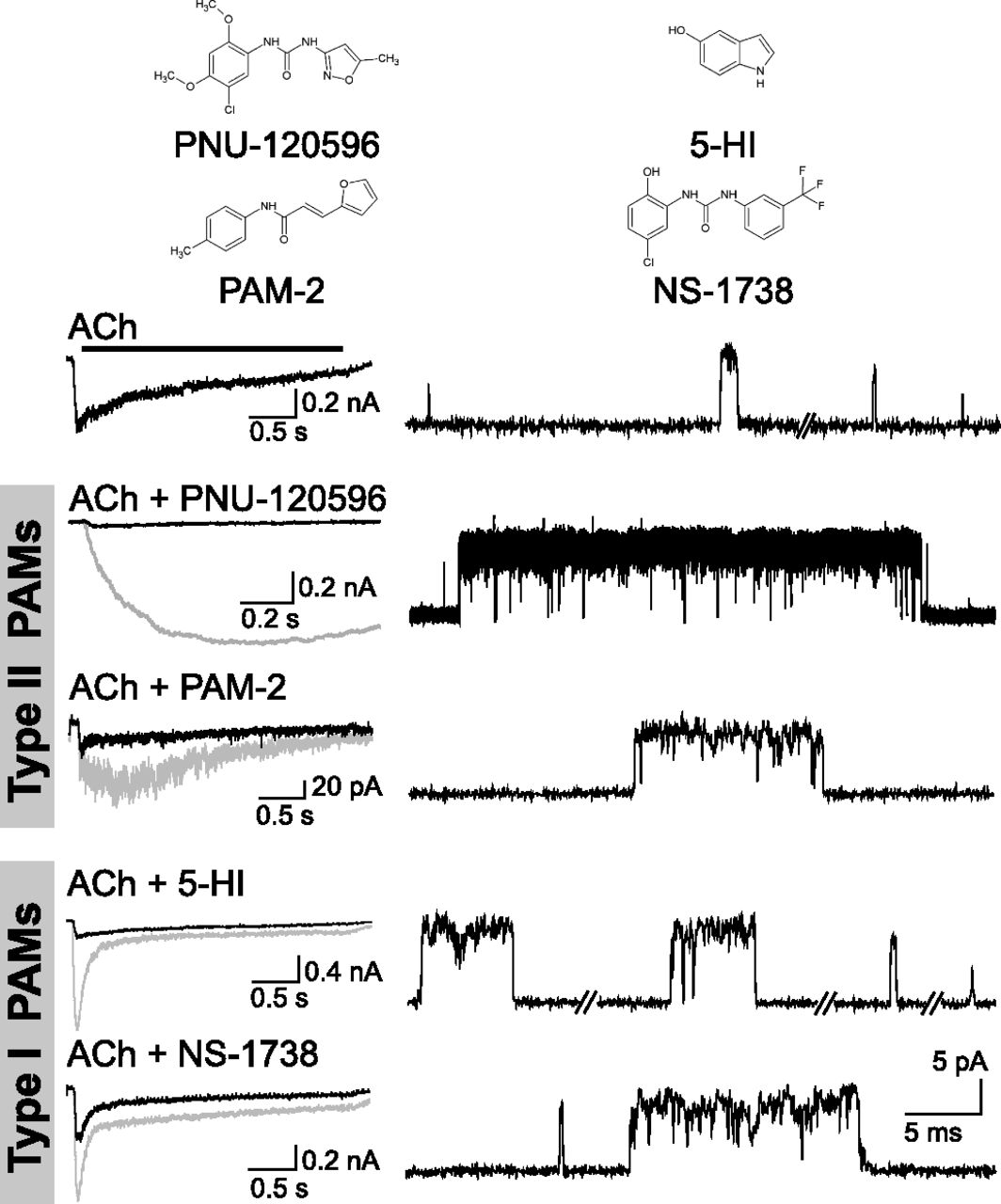
Macroscopic and single-channel current profiles of human α7 in the presence of typical type I and type II PAMs. Macroscopic current profiles have been used to classify PAMs into type II (i.e., PNU-120596 and PAM-2), which increase the decay time constant, or type I, which only increase the peak current (5-HI and NS-1738). Left: Macroscopic currents elicited by ACh in the absence (black) and presence of the specified PAM (gray traces). Right: Traces of 50–100 μM ACh-activated single α7 channels in the absence or in the presence of 1 μM PNU-120596, 5 μM PAM-2, 2 mM 5-HI, 10 μM NS-1738. Membrane potential: –70 mV. Channel openings are shown as upward deflections. All PAMs enhance open channel lifetime and elicit activation episodes formed by successive opening events.
Single-channel recordings provide an invaluable tool for understanding the foundations of these macroscopic effects. In the presence of either type I or type II PAMs, ACh-activated α7 channels show prolonged open durations and appear in longer activation episodes, revealing that both PAM types affect activation kinetics (Andersen et al., 2016) (Fig. 8). The most efficacious PAM to date is PNU-120596, a type II PAM (Hurst et al., 2005). This compound elicits significantly prolonged openings that appear grouped in bursts, which in turn coalesce into long activation periods of several seconds, referred to as clusters (daCosta et al., 2011, 2015; Williams et al., 2011b; Pałczyńska et al., 2012; Andersen et al., 2016). However, the single-channel profile in the presence of a weaker type II PAM, PAM-2 (3-furan-2-yl-N-p-tolyl-acrylamide) (Arias et al., 2011), more closely resembles that of type I 5-HI or NS-1738 PAMs than that of type II PNU-120596 (Fig. 8). Thus, when analyzed at the molecular level, potentiation is more complex than initially believed. Moreover, PAMs showing macroscopic intermediate type I/II properties were proposed (Dunlop et al., 2009; Malysz et al., 2009; Dinklo et al., 2011; Sahdeo et al., 2014; Chatzidaki and Millar, 2015). Therefore, the classification of type I and type II appears to be an oversimplification resulting mainly from macroscopic observations, and a more thorough classification may be required.
It is a generally accepted statement that type II PAMs may increase the energetic barrier for desensitization, which allows successive opening/closing events (daCosta et al., 2011) and/or reversal of some forms of agonist-induced desensitization (Williams et al., 2011b). Also, it has been proposed that PNU-120596 binds predominantly to a fast desensitized state and induces a set of conformations in which the opening of the pore is energetically more favorable (Szabo et al., 2014). On the other hand, only the decrease in the energetic barrier for opening has been proposed as the underlying mechanism for enhancement of peak currents by type I PAMs (Williams et al., 2011b; Hurst et al., 2013). Such a decrease might explain the appearance of bursts of openings owing to rapid reopening of the closed channel. However, it would only explain the increase in open-channel duration if reopening were so fast that the associated brief closings could not be detected, thus making openings appear longer. Alternatively, the increase in open duration could be the result of either slight changes in desensitization that are not detectable from whole-cell macroscopic currents or to the induction of different open states. Thus, there seems to be more than one mechanism by which PAMs prolong open-channel lifetime and activation episodes.
α7 PAM potentiation is particularly dependent on temperature. For PNU-120596, such dependence is revealed by decreased potentiated macroscopic currents (Dunlop et al., 2009; Williams et al., 2012) and the absence of long clusters (Andersen et al., 2016) at physiologic temperature compared with room temperature. Thus, for better extrapolation to the in vivo situation, in vitro studies should be carried out at physiologic temperatures.
α7 Allosteric Binding Sites
Computational studies (Dey and Chen, 2011), electrophysiological studies from mutant or chimeric receptors (Bertrand et al., 2008; Young et al., 2008; Collins et al., 2011; daCosta and Sine, 2013), crystallographic studies, (Spurny et al., 2015), and NMR studies (Bondarenko et al., 2014) have suggested the existence of several allosteric binding sites, some of which are common to other pLGICs (Sauguet et al., 2014).
The Intrasubunit Transmembrane Cavity.
In silico and electrophysiological studies show that several PAMs, including LY-2087101, PNU-120596, and TQS bind to an intrasubunit transmembrane cavity (Young et al., 2008) (Fig. 7). An α7 receptor with mutations at five residues lining this cavity was not potentiated by PNU-120596, PAM-2, or type I PAM NS-1738, indicating common structural determinants for their potentiation (daCosta et al., 2011; Andersen et al., 2016). The macrocyclic lactone ivermectin (a type I PAM) also appears to bind in close proximity to this intrasubunit site (Collins and Millar, 2010), although it binds at an intersubunit transmembrane site in the glutamate-activated Cys-loop receptor (Hibbs and Gouaux, 2011).
This cavity is also the site for allosteric agonists that mediate α7 activation in the absence of an orthosteric agonist (Gill et al., 2011, 2012, 2013; Pałczyńska et al., 2012). Single-channel activity of α7 in the presence of allosteric agonists resembles that of the receptor in the presence of ACh and a PAM. Both are characterized by long bursts instead of isolated brief ACh-elicited openings, indicating activation with significantly reduced desensitization (Pałczyńska et al., 2012).
Moreover, α7-selective allosteric modulators showing subtle structural changes and displaying distinct pharmacological effects (typical of type I PAMs, type II PAMs, NAMs, SAMs, and allosteric agonists) may bind to this common site (Gill et al., 2013; Gill-Thind et al., 2015). Thus, the intrasubunit transmembrane cavity appears to be a promiscuous binding site with a strategic location for allosteric modulation. Furthermore, this cavity may be a common modulatory site within the pLGIC superfamily from which a large variety of different compounds that bind to and mediate a great spectrum of allosteric effects (Corradi et al., 2011; Nury et al., 2011; Jayakar et al., 2013; Sauguet et al., 2014).
Extracellular Allosteric Binding Sites.
The putative binding site of galantamine, an α7 potentiator, is located at the outer surface of the ECD in the vicinity of the ACh site (Hansen and Taylor, 2007; Ludwig et al., 2010). Three different allosteric sites in the ECD of the α7/AChBP chimera were also identified by X-ray crystallography (Spurny et al., 2015; Fig. 7). Although all the allosteric binders behaved on human α7 as negative allosteric modulators, it was proposed that their chemical modification could lead to a change in functional activity.
Binding to potential ECD sites has been proposed for the type I PAMs 5-HI and NS-1738, although other reports support a TMD location (Placzek et al., 2004; Bertrand et al., 2008; Hu and Lovinger, 2008; Gronlien et al., 2010; Collins et al., 2011; Andersen et al., 2016). A virtual screening revealed that some PAMs that bind to the TMD, such as PNU-120596 and TQS, also dock into potential allosteric sites at the ECD (Dey and Chen, 2011). Therefore, for any given PAM, multiple binding sites or domains may be involved in the conformational changes associated with potentiation, which could account for these controversial results. Also, until the location of an allosteric binding site is unequivocally defined, it is advisable to refer to structural determinants of potentiation instead of a binding site.
Despite the large body of experimental evidence supporting α7 potentiation as a promising therapeutic strategy, there are still many unsolved challenges: 1) Potentiation by exogenous agonists may inhibit α7 response owing to desensitization. Thus, PAMs might have therapeutic benefits in situations where stronger agonist responses are desirable. 2) Given the presence of other nAChRs and homologous receptors, high PAM selectivity is required. However, PAMs targeting multiple receptors might show better efficacy (Iturriaga-Vásquez et al., 2015; Möller-Acuña et al., 2015). 3) Excessive receptor activation, particularly with efficacious nondesensitizing PAMs, might lead to cytotoxicity, which is an issue of concern and controversy (Ng et al., 2007; Liu et al., 2009; Williams et al., 2012; Guerra-Álvarez et al., 2015; Uteshev, 2016). 4) The ubiquitous distribution of α7 and its interplay with different signal pathways could make the cell response to a given PAM variable among cell types or conditions. Given the broad spectrum of effects and molecular mechanisms of PAMs, it is probable that each patient or pathologic situation could require a unique PAM.
Concluding Remarks
α7 has emerged as an important drug target for improving cognition and memory in several neuropsychiatric disorders, and as a target for inflammatory processes. α7 is unique owing to its high calcium permeability and fast desensitization, and it behaves as a ACh-sensitive sensor harboring a built-in filtering mechanism against excessive stimulation. Transient calcium responses are further sustained by the release of calcium from intracellular sources, and several signaling pathways are also activated because α7 has a dual ionotropic/metabotropic nature. Its ubiquitous location and pleiotropic effects make α7 an interesting but complex drug target. A better understanding of the molecular basis underlying allosteric modulation and its wide spectrum of effects, as well as the availability of high resolution structures of α7, will help in the rational design of therapeutics for the receptor.
Acknowledgments
ASPET thanks Dr. Katie Strong for copyediting of this article.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Bouzat, Corradi.
Footnotes
- Received March 7, 2016.
- Accepted May 5, 2016.
This work was supported by grants from Universidad Nacional del Sur (UNS), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), FONCYT, and the Bill and Melinda Gates Foundation to C.B.
Abbreviations
- ACh
- acetylcholine
- α-BTX
- α-bungarotoxin
- ECD
- extracellular domain
- JAK
- Janus kinase
- LY-2087101
- (2-amino-5-keto)thiazole), [2-[(4-fluorophenyl)amino]-4-methyl-5-thiazolyl]-3-thienyl-methanone
- nAChR
- nicotinic acetylcholine receptor
- NAM
- negative allosteric modulators
- NS-1738
- 1-(5-chloro-2-hydroxyphenyl)-3-(2-chloro-5-trifluoromethylphenyl)urea
- PAM
- positive allosteric modulator
- pLGIC
- pentameric ligand-gated ion channels
- PNU-120596
- 1-(5-chloro-2,4-dimethoxyphenyl)-3-(5-methylisoxazol-3-yl)urea
- SAM
- silent allosteric modulator
- STAT
- signal transducer and activator of transcription
- TMD
- transmembrane domain
- TQS
- 4-(naphthalen-1-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Visual Overview
- Abstract
- Introduction
- nAChR Structure and Function
- α7 in the Nervous System in Healthy and Disease States
- Extraneuronal α7 and Its Pleiotropic Roles
- α7 Pharmacology and Ion Selectivity
- α7 Channel Kinetics
- α7 Modulation as a Therapeutic Strategy
- α7 Allosteric Binding Sites
- Concluding Remarks
- Acknowledgments
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters