


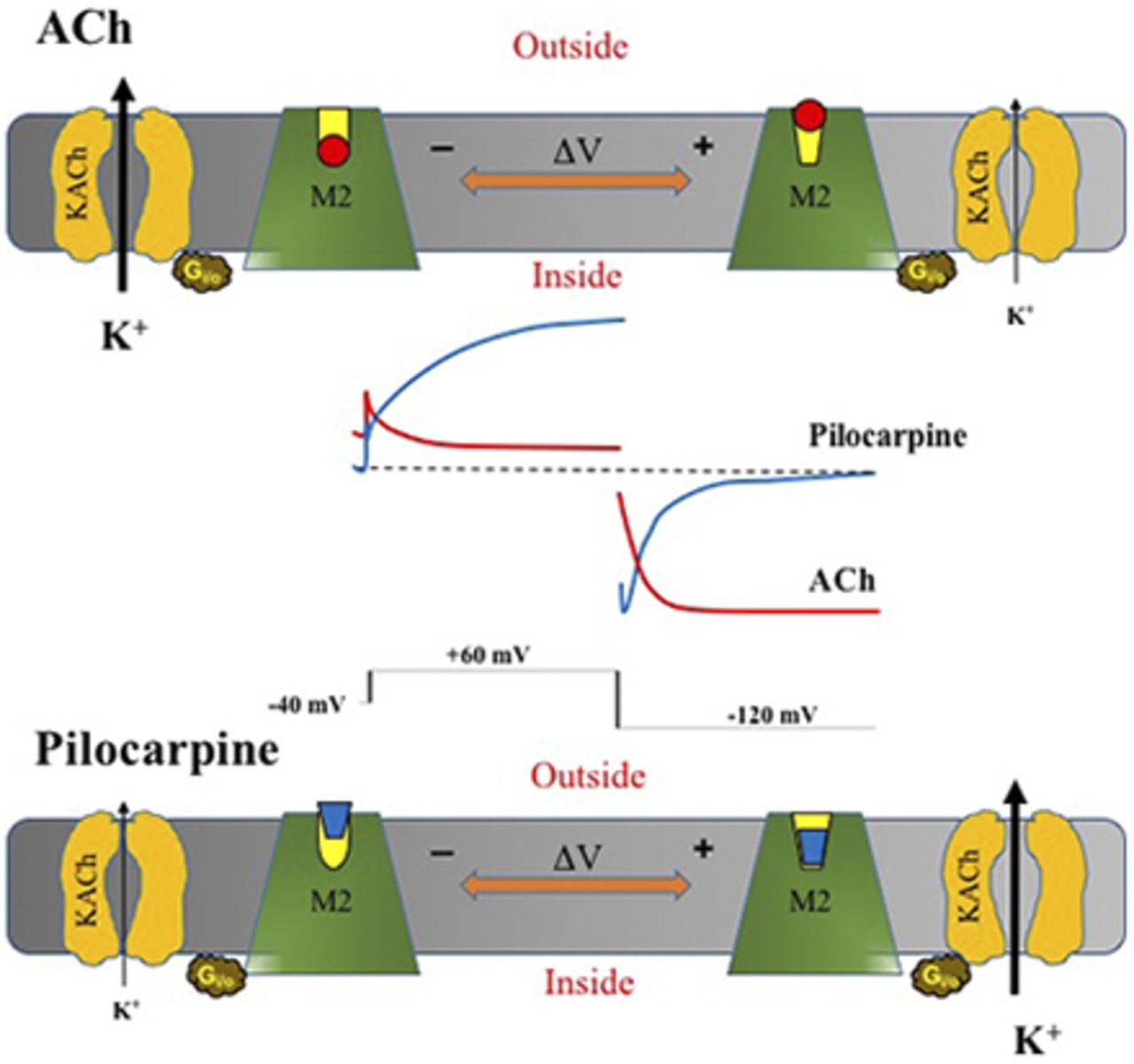
Visual Overview

Abstract
Potassium (K+) channels are crucial for determining the shape, duration, and frequency of action-potential firing in excitable cells. Broadly speaking, K+ channels can be classified based on whether their macroscopic current outwardly or inwardly rectifies, whereby rectification refers to a change in conductance with voltage. Outwardly rectifying K+ channels conduct greater current at depolarized membrane potentials, whereas inward rectifier channels conduct greater current at hyperpolarized membrane potentials. Under most circumstances, outward currents through inwardly rectifying K+ channels are reduced at more depolarized potentials. However, the acetylcholine-gated K+ channel (KACh) conducts current that inwardly rectifies when activated by some ligands (such as acetylcholine), and yet conducts current that outwardly rectifies when activated by other ligands (for example, pilocarpine and choline). The perplexing and paradoxical behavior of KACh channels is due to the intrinsic voltage sensitivity of the receptor that activates KACh channels, the M2 muscarinic receptor (M2R). Emerging evidence reveals that the affinity of M2R for distinct ligands varies in a voltage-dependent and ligand-specific manner. These intrinsic receptor properties determine whether current conducted by KACh channels inwardly or outwardly rectifies. This review summarizes the most recent concepts regarding the intrinsic voltage sensitivity of muscarinic receptors and the consequences of this intriguing behavior on cardiac physiology and pharmacology of KACh channels.
Introduction
Potassium (K+) channels are cell membrane proteins involved in multiple physiologic processes, and their modulation entails important functional implications at the cellular and systemic level. K+ channels are present in almost all cell types and constitute the most diverse group of ion channels in living organisms (Gonzalez et al., 2012; Jan and Jan, 2012). In excitable cells, their function determines the shape, duration, and frequency of action-potential firing (Hille, 2001). More than 40 genes encoding pore-forming α subunits are expressed in cardiac cells, where voltage-gated (Kv) and inward rectifier (Kir) K+ channels comprise the two major types of K+ channels (Nerbonne and Kass, 2005). Broadly speaking, K+ channels can be classified based on whether their macroscopic current outwardly or inwardly rectifies, whereby rectification refers to a change in conductance with voltage (Nichols and Lopatin, 1997). Outwardly rectifying Kv channels conduct greater current at depolarized membrane potentials, whereas inward rectifier channels conduct greater current at hyperpolarized membrane potentials (Fig. 1, A–C). Delayed rectifier K+ channels are outwardly rectifying Kv channels that contribute to the latter phases of cardiac action-potential repolarization. By contrast, the outward current of Kir channels typically decreases at more depolarized potential (Fig. 1C), and thus these channels principally contribute to the resting membrane potential (Yang and Nerbonne, 2016). Intriguingly, the acetylcholine-gated inward rectifying K+ channel (KACh) conducts current that inwardly rectifies when activated by some ligands (such as acetylcholine) and yet conducts current that outwardly rectifies when activated by other ligands, such as choline (Fig. 1, D–F) (Navarro-Polanco et al., 2013). The perplexing and paradoxical behavior of KACh channels is due to the intrinsic voltage sensitivity of the receptor that activates KACh channels and serves as the focus of this review.

Schematics of typical currents from Kir and Kv channels. Representation of characteristic current families from Kir (A), Kv (B), and KACh channels activated by ACh (D) or choline (E) obtained when using the indicated voltage step protocol. The dashed lines indicate the zero current level. Diagrams of the I-V relationships depicted in (A) and (B) (C) and in (D) and (E) (F).
The Molecular Basis for Inward Rectification of Kir Channels
The initial cloning of outward and inward rectifier K+ channels revealed fundamental structural differences that underlie their unique functional properties (Papazian et al., 1987; Kubo et al., 1993). K+ channels are composed of four identical (homotetramers) or different (heterotetramers) pore-forming α subunits that are often associated with accessory β subunits. The outward rectifier Kv subunits are composed of six membrane-spanning domains whereby domains S1–S4 function as the voltage-sensing domain, whereas domains S5–S6 form the ion conduction pore (Papazian et al., 1987). The outward rectifier Kv channels are closed (deactivated) at negative membrane potentials and progressively activate with membrane depolarization, resulting in the outwardly rectifying current-voltage relationship depicted in Fig. 1. By contrast, the inward rectifier Kir subunits contain two membrane-spanning domains that correspond generally to the S5–S6 pore-forming domains of Kv channels (Papazian et al., 1987; Tao et al., 2009; Hansen et al., 2011). Unlike Kv channels, Kir channels conduct current at negative membrane potentials, with progressively less current conducted at depolarized potentials, resulting in the inwardly rectifying current-voltage relationship illustrated in Fig. 1. Currently, 16 Kir subunit genes have been cloned that correspond to seven subfamilies (Kir1.x to Kir7.x) (see Hibino et al., 2010; Swale et al., 2014), and these subfamilies exhibit variable mechanisms of regulation, as well as variable degrees of inward rectification. Multiple lines of evidence demonstrate that inward rectification is due to a voltage-dependent blockade of Kir channels by intracellular charged particles, such as polyamines and Mg2+ (Nichols and Lopatin, 1997; Baronas and Kurata, 2014) (Fig. 2). Thus, unlike the intrinsic voltage-dependent activation of Kv channels, Kir channel activity is regulated extrinsically by voltage-dependent blocking particles that reduce conductance at depolarized potentials.

Simplified illustration of the prototypical parasympathetic signaling pathway in cardiac cells. The binding of ACh to M2R induces dissociation of the βγ-complex (from Gi/o proteins) to directly interact with the KACh channel and then decreases cardiac excitability.
The Muscarinic G-Protein–Coupled Inward Rectifier KACh Channel
KACh channels are localized in cardiac cells and represent a subclass of inward rectifier channels that are associated with muscarinic G-protein–coupled receptors (GPCRs) when activated by neurotransmitter acetylcholine (ACh) (Hibino et al., 2010). KACh channels are heterotetramers composed of two homologous G-protein–gated inwardly rectifying K+ channel subunits, Kir3.1 and Kir3.4 (Corey et al., 1998), and these channels play a crucial role in heart-rate regulation (Wickman et al., 1998; Mesirca et al., 2013) and susceptibility to atrial arrhythmias (Kovoor et al., 2001). Postganglionic parasympathetic (vagal) stimulation releases ACh, which binds to the M2 muscarinic receptor (M2R) to activate trimeric Gi/o proteins (Breitwieser and Szabo, 1985; Pfaffinger et al., 1985; Kurachi et al., 1986). The trimeric protein dissociates with the βγ subunit binding to the Kir subunits to increase KACh channel activity (Logothetis et al., 1987; Ito et al., 1992) (Fig. 2), resulting in membrane hyperpolarization, shortening of action-potential duration (APD), and a reduction in heart-rate and atrioventricular conduction (Dhein et al., 2001). Although five subtypes of muscarinic cholinergic receptors have been identified, from M1 to M5 (Wess, 1996; Caulfield and Birdsall, 1998), M2R is the predominant muscarinic subtype in the mammalian heart (Brodde and Michel, 1999; Dhein et al., 2001; Krejcí and Tuček, 2002; Kitazawa et al., 2009), and is preferentially expressed in the sinoatrial node, working atrial myocardium, and atrioventricular node. Even more, the physiologic role of decreasing the cardiac cell excitability is determined by M2R (Stengel et al., 2000; Wess, 2004; Kitazawa et al., 2009), and the only current activated by this receptor is the ACh-activated K+ current (IKACh) (Breitwieser and Szabo, 1985; Pfaffinger et al., 1985; Kurachi et al., 1986) since the hyperpolarization-activated current (If) and the L-type calcium current (ICaL) are both inhibited.
An Outward Rectifier K+ Current Activated by Muscarinic Ligands
The properties of the IKACh conducted by KACh channels have been extensively studied since the initial description of this cardiac current in the late 1970s (Noma and Trautwein, 1978). Similar to ACh, other muscarinic agonists activate IKACh, with its characteristic inwardly rectifying current-voltage relationship (Watanabe et al., 1996; Yamada, 2002). Unexpectedly, Fermini and Nattel (1994) reported that choline, a compound widely used for replacing extracellular sodium, activated both a background current and a time- and voltage-dependent outwardly rectifying current similar to a delayed rectifier K+ current (Fig. 1, E and F). Interestingly, the delayed rectifier-like current was inhibited by the muscarinic antagonist atropine, suggesting that the current was activated by a muscarinic receptor (Fermini and Nattel, 1994). Also incidentally, Navarro-Polanco and Sanchez-Chapula (1997) discovered that the K+ channel blocker 4-aminopyridine (4-AP) paradoxically increased both a background current and a time- and voltage-dependent outward current in feline atrial cells, similar to that evoked by choline in canine atrial cells (Fermini and Nattel, 1994). The 4-AP–induced conductance was antagonized by atropine and mediated through a pertussin toxin–sensitive Gi/o protein, suggesting that M2R or the M4 muscarinic receptor was involved in activating the current. Single-channel recordings confirmed that ACh and 4-AP both activated the same channel: the KACh channel (Navarro-Polanco and Sanchez-Chapula, 1997).
Subsequently, other groups also reported that various compounds elicited delayed rectifier-like K+ currents in atrial myocytes that were mediated by muscarinic receptor activation. Based on their sensitivity to muscarinic antagonists and to pretreatment with pertussis toxin, it was suggested that the current induced by 4-AP was mediated by M4 muscarinic receptor (Shi et al., 1999a, 2003). The current evoked by choline, tetramethylammonium, and the muscarinic agonist pilocarpine (Pilo) was proposed to be mediated by the M3 muscarinic receptor, and the current was named IKM3 (Shi et al., 1999a,b, 2003; Wang et al., 1999, 2004). This group postulated that IKM3 modulates cardiac electrical activity (Shi et al., 1999b; Wang et al., 1999) and plays a role in the pathophysiology of atrial fibrillation (Yeh et al., 2007) and cytoprotection against myocardial injuries (Yang et al., 2005). Despite the prediction of a novel channel, the molecular correlates of the K+ channel subunits that underlie IKM3 remained elusive (Yang and Nerbonne, 2016). The puzzling ability of some muscarinic agents to activate an inwardly rectifying current while others activate outwardly rectifying current remained unexplained until more recent studies demonstrated that M2R is intrinsically voltage-sensitive, and that the affinity of the receptor for various ligands is dependent upon membrane potential.
Muscarinic Receptors Possess Intrinsic Voltage Sensitivity
Voltage-gated ion channels are prototypical membrane proteins that possess an inherent voltage sensitivity by virtue of charged amino acid residues that comprise the voltage sensor. The observation that some GPCRs are voltage-sensitive (Marty and Tan, 1989; Cohen-Armon and Sokolovsky, 1991; Itoh et al., 1992; Mahaut-Smith et al., 1999; Ong et al., 2001) was quite surprising given that these proteins lack an obvious voltage-sensing domain. Using heterologously expressed muscarinic receptors and Kir3.1/Kir3.2 channels (which are neuronal molecular correlates, homologous to cardiac KACh channels), Ben-Chaim et al. (2003) found that membrane depolarization decreased the potency of ACh for the M2R as measured by a reduction in channel activation. Binding experiments directly confirmed a reduced affinity of M2R toward ACh, which explained the reduced potency at depolarized potentials. Activation of the signaling pathway downstream of M2R, by overexpressing Gβγ or intracellular injection of guanosine 5′-3-O-(thio)triphosphate (GTPyS), caused no additional voltage dependence, supporting the notion that M2R is the signaling element with intrinsic voltage sensitivity (Ben-Chaim et al., 2003). Subsequently, the ability of muscarinic receptors to sense changes in membrane potential was confirmed by recording gating currents in M1 muscarinic receptors and M2R (Fig. 4A) (Ben-Chaim et al., 2006). Gating currents result from the movement of gating charges across the membrane electrical field and represent voltage-dependent conformational changes in the receptor in response to changes in membrane potential, representing the most important evidence about the voltage sensitivity of these muscarinic receptors. Likewise, other GPCRs have been noted to display intrinsic voltage dependence (Dekel et al., 2012; Rinne et al., 2013, 2015).
Subsequently, our group confirmed the ACh voltage-dependent modulation of M2R-IKACh in cardiac cells (Navarro-Polanco et al., 2011). Additionally, we discovered that M2R displays opposite voltage sensitivity in a ligand-specific manner—that is, whereas membrane depolarization decreases ACh potency and affinity, depolarization increases the potency of Pilo (Fig. 3A) by enhancing, in part, the affinity of this muscarinic agonist for M2R. In addition, we observed that ACh and Pilo induce marked differences in the magnitude and kinetics of M2R gating currents (Fig. 4, B and C), suggesting that these agonists induce distinct conformational changes within the receptor (Navarro-Polanco et al., 2011). Moreover, mutations in the ligand-binding site alter the voltage-dependent M2R charge movement, implying that a clear interaction between the binding site and the (still unidentified) voltage sensor exists. Therefore, we postulated that variations in membrane potential directly induce conformational changes in the ligand-binding site that facilitate the binding of some agonists and hinder the binding of other agonists (Navarro-Polanco et al., 2011). This proposal is supported by findings in D2 dopaminergic (Sahlholm et al., 2008, 2011), α2A adrenergic (Rinne et al., 2013), and Gq-coupled muscarinic receptors, M1 muscarinic receptor, M3 muscarinic receptor, and M5 muscarinic receptor (Rinne et al., 2015). Agonist-specific voltage sensitivity was termed to denote the relationship between specific agonists and voltage (Sahlholm et al., 2008, 2011; Navarro-Polanco et al., 2011; Moreno-Galindo et al., 2016).

Opposite voltage-dependent behavior of IKACh. (A) Diagram of the concentration-response relationships for the ACh and Pilo activation of IKACh at −100 mV (red line) versus +50 mV (black line). Arrows represent the change in the current level induced by voltage. (B) Schema of superimposed IKACh induced by depolarizing and hyperpolarizing voltage steps in the presence of ACh (red line) and Pilo (black line). The dashed line indicates the zero current level.

Opposite effects of ACh and Pilo on M2R charge movement. (A) M2R gating currents assessed by the cut-open oocyte voltage-clamp technique. (B) Effect of ACh (red) and Pilo (blue) on control (black) M2R gating currents at a voltage step of +60 mV. (C) Diagram of M2R charge-voltage relationships obtained in the absence and presence of ACh and Pilo. Ctrl, control.
The Mechanism for Pharmacological Conversion of an Inward Rectifier into an Apparent Delayed Rectifier K+ Channel
Our recent knowledge of the intrinsic voltage dependence of muscarinic receptors sheds new light on the perplexing behavior of Pilo-, choline-, and 4-AP–activated currents. Our data reveal that not only is the M2R voltage sensitive, but the voltage sensitivity varies in a ligand-specific manner. For example, depolarization induces a conformational change in the M2R agonist binding pocket that decreases the affinity for ACh and increases the affinity and efficacy for Pilo and choline. As a result of decreased ACh affinity, IKACh current magnitude decreases with increasing depolarization (and current increases with hyperpolarization), resulting in current that inwardly rectifies. By contrast, the Pilo- and choline-induced current magnitude increases with depolarization as a consequence of increased ligand affinity and efficacy (and current decreases with hyperpolarization). The time-dependent increase in current with Pilo and choline at depolarized potentials is reminiscent of the time-dependent increase in a voltage-sensitive delayed rectifier channel and thus has created confusion that a novel channel underlies Pilo- and choline-activated currents (Shi et al., 1999a,b, 2003; Wang et al., 1999, 2004).
Subsequent pharmacological and electrophysiological characterization of the Pilo- and choline-activated delayed rectifier-like K+ currents supports the notion that these ligands activate KACh channels. The currents evoked by Pilo and choline were completely abolished by tertiapin-Q (Moreno-Galindo et al., 2011; Rodriguez-Martinez et al., 2011; Navarro-Polanco et al., 2013), a potent and specific cardiac KACh channel blocker (Drici et al., 2000; Kitamura et al., 2000). Moreover, the single-channel properties of choline-activated channels are identical to KACh channels, with the exception that the open probability is increased at depolarized potentials (Navarro-Polanco et al., 2013). Furthermore, a Markov model of M2R-IKACh that incorporates agonist-selective, voltage-dependent parameters recapitulates the general features of the Pilo- and choline-induced currents, as well as those of ACh (Moreno-Galindo et al., 2011; Navarro-Polanco et al., 2013).
Taken together, the emerging evidence supports the concept that voltage-dependent changes in agonist affinity (and efficacy for Pilo and choline) occur as a consequence of voltage-dependent conformational changes in M2R and determine whether the evoked current behaves like an inwardly rectifying or outwardly rectifying current. But what explains the time-dependent nature of the outwardly rectifying current? It turns out that IKACh also displays a time-dependent activation, a process called “relaxation gating.” Relaxation gating, originally described in the late 1970s (Noma and Trautwein, 1978), refers to a slow decrease in current magnitude with membrane depolarization (Ishii et al., 2001). Our data suggest that relaxation gating represents a voltage-dependent change in agonist affinity as a consequence of conformational changes in M2R (Moreno-Galindo et al., 2011). Because the voltage sensitivity is also ligand-specific, the time-dependent activation of KACh channels by Pilo represents “opposite” relaxation gating. Thus, relaxation gating is mediated by the intrinsic voltage sensitivity of the muscarinic receptor.
The Effect of Voltage on the Affinity and Efficacy of the M2R-Agonist Interaction
For any given biologic system or signaling pathway, the actions of ligands at receptors (measured as potency in concentration-response relationships) depend on two fundamental properties: affinity and efficacy. Affinity refers to the strength by which the ligand binds to its receptor. Efficacy denotes the ability of a ligand to affect the receptor and its downstream associated signaling systems to produce a cellular response (Kenakin, 2006). Based on experimental data, it is clear that membrane potential modifies the affinity of agonists for their receptors, i.e., voltage-induced conformational changes within the M2R modify the strength with which specific agonists bind to the receptor (Ben-Chaim et al., 2003, 2006; Navarro-Polanco et al., 2011). In addition, membrane potential also modulates the efficacy of the signaling pathway for partial muscarinic agonists. For example, the partial agonist choline activates ∼50% of available IKACh at −100 mV, and yet activates ∼98% of available IKACh at +50 mV, as compared with the full agonist ACh (Navarro-Polanco et al., 2013). That is, choline behaves as a partial agonist at negative membrane potentials, but exerts a full agonist effect at positive potentials. These observations add a new paradigm to the classic concept that a given ligand, assessed in the same biologic system, functions as an antagonist, partial agonist, or full agonist. Rather, ligand function may be dynamically modulated by the membrane voltage (Gurung et al., 2008), mainly in excitable cells that express voltage-sensitive GPCRs.
The ability of M2R and other voltage-sensitive GPCRs to respond differentially to both voltage and ligands adds additional complex dimensions to the regulation of downstream signaling pathways in excitable cells. Moreover, the voltage-dependent modulation offers unique opportunities for drug development. One can envision the tailoring of compounds to exploit voltage-dependent changes in affinity or efficacy to exert distinct effects at negative versus positive membrane potentials.
Physiologic Implications of M2R Voltage Sensitivity for Parasympathetic Regulation of the Heart
The observation that M2Rs are intrinsically voltage-sensitive has profound implications for cellular signaling in excitable tissues, such as the heart. Over the past 30 years, the role of IKACh in mediating the purely chronotropic effects of low (or “physiologic”) ACh concentrations has been debated. Low concentrations of ACh (e.g., 50 nM) and weak vagal stimulation reduce spontaneous pacemaker rate without altering APD or increasing the maximal diastolic potential (Shibata et al., 1985). DiFrancesco et al. (1989) proposed that weak vagal stimulation primarily inhibited the hyperpolarization-activated current (If) with little to no contribution from IKACh. However, subsequent studies have corroborated an important role for IKACh in mediating the chronotropic effects of weak vagal stimulation and externally applied low ACh concentrations (Boyett et al., 1995; Lyashkov et al., 2009; Han and Bolter, 2011) and confirmed that basal vagal tone activates IKACh to slow heart rate (Wickman et al., 1998). Our experimental data provide a mechanistic basis to explain the participation of IKACh in the modest chronotropic effects induced by resting vagal tone. As a result of conformational changes in the M2R, the affinity for ACh varies throughout the cardiac action potential such that low ACh concentrations preferentially activate IKACh during diastolic membrane voltages to slow the spontaneous firing rate without appreciably altering APD.
Although speculative, alterations in the voltage sensitivity of M2R could theoretically contribute to cardiovascular disease states associated with changes in vagal tone. For example, parasympathetic induction and maintenance of atrial arrhythmias is a well described phenomenon, first reported in 1930 (Andrus and Carter, 1930). Thus, mutations or polymorphisms in M2R that shift the voltage sensitivity in the hyperpolarized direction might explain a subset of patients with vagally mediated atrial fibrillation, who would present with markedly shortened atrial APD in the setting of normal (basal) ACh concentrations. Alternatively, positive shifts in M2R voltage sensitivity would decrease vagal modulation of heart rate, thereby favoring sympathetic stimulation that might cause elevated basal heart rate, as seen in the inappropriate sinus tachycardia syndrome. In fact, a recent study confirmed an exaggerated sympathetic tone and decreased parasympathetic tone in patients with the inappropriate sinus tachycardia syndrome (Nwazue et al., 2014). Although speculative, the hypothesis that altered M2R voltage sensitivity might impact clinical disease provides a novel mechanistic foundation to study disorders such as atrial fibrillation and inappropriate sinus tachycardia.
Conclusions
The discovery that M2Rs are intrinsically voltage-sensitive and that the voltage-sensitive properties of the receptor vary in a ligand-specific manner allowed for a novel mechanistic understanding of muscarinic-activated currents and their physiologic consequences. The concept that the affinity for ACh varies dynamically throughout the cardiac action potential helps to explain, in part, the greater contribution of IKACh in slowing the firing rate of pacemaker cells under basal vagal tone without appreciably shortening APD. The observation that depolarization decreases the affinity of M2R for some ligands, while increasing affinity for others, explains how KACh channels behave like an inward rectifier when activated by some ligands and an apparent delayed rectifier for other ligands. Finally, the ability of membrane voltage to convert ligand effects from partial to full agonism allows for the creation of compounds that selectively alter the diastolic or systolic components of the cardiac cycle, either to preferentially slow heart rate without affecting repolarization or vice versa.
Acknowledgments
ASPET thanks Dr. Katie Strong for copyediting of this article.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Moreno-Galindo, Sanchez-Chapula, Tristani-Firouzi, Navarro-Polanco.
Footnotes
- Received April 27, 2016.
- Accepted May 27, 2016.
This study was supported by SEP-CONACYT, México [Grants CB-2011-01-167109 to E.G.M.-G., CB-2013-01-220546 to J.A.S.-C., and CB-2013-01-220742 to R.A.N.-P.]
Abbreviations
- ACh
- acetylcholine
- APD
- action-potential duration
- 4-AP
- 4-aminopyridine
- GPCR
- G-protein–coupled receptors
- ICaL
- L-type calcium current
- If
- hyperpolarization-activated current
- IKACh
- ACh-activated K+ current
- KACh
- acetylcholine-gated K+ channel
- Kir
- inward rectifier K+
- Kv
- voltage-gated K+
- M2R
- M2 muscarinic receptor
- Pilo
- pilocarpine
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Visual Overview
- Abstract
- Introduction
- The Molecular Basis for Inward Rectification of Kir Channels
- The Muscarinic G-Protein–Coupled Inward Rectifier KACh Channel
- An Outward Rectifier K+ Current Activated by Muscarinic Ligands
- Muscarinic Receptors Possess Intrinsic Voltage Sensitivity
- The Mechanism for Pharmacological Conversion of an Inward Rectifier into an Apparent Delayed Rectifier K+ Channel
- The Effect of Voltage on the Affinity and Efficacy of the M2R-Agonist Interaction
- Physiologic Implications of M2R Voltage Sensitivity for Parasympathetic Regulation of the Heart
- Conclusions
- Acknowledgments
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters