


Abstract
Understanding the agonist-receptor interactions in the neuropeptide Y (NPY)/peptide YY (PYY) signaling system is fundamental for the design of novel modulators of appetite regulation. We report here the results of a multidisciplinary approach to elucidate the binding mode of the native peptide agonist PYY to the human Y2 receptor, based on computational modeling, peptide chemistry and in vitro pharmacological analyses. The preserved binding orientation proposed for full-length PYY and five analogs, truncated at the amino terminus, explains our pharmacological results where truncations of the N-terminal proline helix showed little effect on peptide affinity. This was followed by receptor mutagenesis to investigate the roles of several receptor positions suggested by the modeling. As a complement, PYY-(3-36) analogs were synthesized with modifications at different positions in the common PYY/NPY C-terminal fragment (32TRQRY36-amide). The results were assessed and interpreted by molecular dynamics and Free Energy Perturbation (FEP) simulations of selected mutants, providing a detailed map of the interactions of the PYY/NPY C-terminal fragment with the transmembrane cavity of the Y2 receptor. The amidated C-terminus would be stabilized by polar interactions with Gln2886.55 and Tyr2195.39, while Gln1303.32 contributes to interactions with Q34 in the peptide and T32 is close to the tip of TM7 in the receptor. This leaves the core, α-helix of the peptide exposed to make potential interactions with the extracellular loops. This model agrees with most experimental data available for the Y2 system and can be used as a basis for optimization of Y2 receptor agonists.
Introduction
The human neuropeptide Y (NPY) signaling system consists of three peptides (NPY, peptide YY (PYY) and pancreatic polypeptide (PP)) and four receptors (Y1, Y2, Y4 and Y5) (Michel et al., 1998). Cancer and obesity are among the many pathophysiological functions regulated by this neural and endocrine system (Zhang et al., 2011). In particular, food intake is stimulated by NPY through activation of the Y1 and Y5 receptors (Fekete et al., 2002; Lecklin et al., 2002; Mashiko et al., 2009), or inhibited through activation of receptors Y4 (by PP) (Sato et al., 2009) or Y2 by PYY-(3-36) (Degen et al., 2005; Neary and Batterham, 2009; Pedersen et al., 2010), an endogenous truncated version of PYY. Consequently, many efforts have been made to develop drugs for regulating this system to treat obesity, either with Y1/Y5 antagonists or Y2/Y4 agonists (Brothers and Wahlestedt, 2010; Walther et al., 2011; Yulyaningsih et al., 2011). Of particular interest is the pathway regulated by the Y2 receptor which is located in the arcuate nucleus of the basal hypothalamus. Here, stimulation by PYY-(3-36), arriving via the blood from the gastrointestinal tract, reduces release of the appetite-stimulating peptides NPY and AGRP in the paraventricular nucleus (Schneeberger et al., 2014). In the search for therapeutically useful agonists to regulate this pathway, several peptides and peptidomimetics have been generated by modification of the common C-terminus of NPY/PYY peptides, which shed light on the structural determinants of high affinity and selectivity (Kirby et al., 1997; Balasubramaniam et al., 2000; Pedersen et al., 2009; Albertsen et al., 2013; Ehrlich et al., 2013; Pedragosa-Badia et al., 2013; Nishizawa et al., 2017).
The crystallization of G-protein-coupled receptor (GPCR) structures has blossomed in the last decade (Katritch et al., 2013). Still, the majority of receptors have to be investigated through computational modeling, a process that benefits greatly from site-directed mutagenesis studies to characterize receptor-ligand binding modes (Kristiansen, 2004; Salon et al., 2011; Zhukov et al., 2011). In this field, our group has recently contributed with a computational protocol, based on free energy perturbation (FEP), to estimate the effects of single point mutations on ligand binding affinity with high accuracy and precision (Boukharta et al., 2014; Keränen et al., 2014, 2015; Nøhr et al., 2017; Vasile et al., 2017). The protocol was first developed and validated within the scope of the present project, in particular in the case of antagonist binding to the NPY/PYY receptor Y1 (Boukharta et al., 2014), and later applied with success to understand both agonist and antagonist binding to the A2A adenosine receptor (Keränen et al., 2014, 2015) and more recently to the deorphanization of the GPR139 orphan receptor (Nøhr et al., 2017).
To date, there is no crystal structure of any member of the NPY-family receptor family. Instead, several homology models of the human Y1 and Y2 receptors have been proposed on the basis of the available experimental structures of rhodopsin-like GPCRs and assessed by mutagenesis and pharmacological studies (Sautel et al., 1996; Kannoa et al., 2001; Sjödin et al., 2006; Merten et al., 2007; Åkerberg et al., 2010; Fällmar et al., 2011; Xu et al., 2013; Boukharta et al., 2014; Pedragosa-Badia et al., 2014; Kaiser et al., 2015). Our most recent hY2 model was built on the basis of the neurotensin receptor 1 (NTS1) active-like crystal structure (White et al., 2012), and verified with site-directed mutagenesis and binding studies. We showed the utility of this model to elucidate the binding mode of the C-terminal coil of the agonist peptides, 32TRQRY36-NH2, in the transmembrane (TM) regions of the receptor (Xu et al., 2013). Later, Beck-Sickinger and co-workers reported a different modeling approach, complemented by mutagenesis, functional profiling and NMR data (Kaiser et al., 2015). The two studies are in agreement about the binding mode of the C-terminal coil of the PYY/NPY peptides, although presenting different interaction partners for the C-terminal amidated tyrosine residue (Y36-CONH2).
In the present work, we refine and extend our Y2-agonist model to the complex with the full PYY peptide, which is used as a basis to design and characterize new receptor mutants and peptide analogs exploring the binding of the conserved PYY/NPY C-terminal fragment. The binding and functional assays were complemented with FEP simulations, and the positions investigated also assessed by evolutionary comparisons of receptor sequences. Based on these results, we propose a detailed binding model of native agonist peptides to the human Y2 receptor that should facilitate design of subtype-selective ligands.
Materials and Methods
Computational Modeling
Generation of the Y2 Receptor Model in Complex with PYY and Analogs.
Our previously reported computational model of wild-type human Y2 receptor (WT-hY2) in complex with NPY (Xu et al., 2013) was used as a starting point in this work. Briefly, the structure of the hY2 receptor was obtained by homology modeling using the crystal structure of rat neurotensin receptor 1 (rNTS1, PDB code 4GRV) as a template. This template presents the advantages of belonging to the same β-branch of class-A GPCRs as the NPY receptors, and since it was crystallized with a peptidic agonist it is referred to as an active-like conformation (White et al., 2012). The quality of the model was assessed with Procheck (Laskowski et al., 1993), confirming the good stereochemical quality (85% residues in the most favored regions, close to the 89% value obtained for the template, rNTS1). The docking of the NPY peptide in our homology model was done in two stages: the amidated C-terminal pentapeptide (32TRQRY36-NH2, note the residue numbering on the peptide in superscript along this manuscript) was initially docked with the Induced Fit Docking protocol in GLIDE (Sherman et al., 2006) and the resulting complex was equilibrated in the membrane with our PyMemDyn protocol, for full atom molecular dynamics (MD) simulations of GPCRs (Gutiérrez-de-Terán et al., 2013). The resulting pose of the C-terminal fragment was then used as anchoring point to manually dock the full NPY peptide, starting from its NMR determined structure (Monks et al., 1996), by molecular superposition and energy minimization with the Schrödinger suite (Schrödinger LCC, New York, NY). We here employed this same software to generate a new complex of the Y2-PYY structure, given the high sequence and structural homology between the two peptides (see Supplemental Fig. 1). The truncated analogs of the PYY at the amino tail were generated from this initial Y2-PYY complex by simply removing the corresponding residues.
Molecular Dynamics (MD) Simulations.
The generated peptide-receptor complexes were inserted in the membrane and equilibrated under periodic boundary conditions (PBC) using the PyMemDyn protocol (Gutiérrez-de-Terán et al., 2013). Shortly, the structure is automatically embedded in a hexagonal prism-shaped box of pre-equilibrated membrane of POPC (1-palmitoyl-2-oleoyl phosphatidylcholine) lipids, with the TM bundle aligned to its vertical axis. This box is then soaked with bulk water and energy minimized with GROMACS 4.6 (Hess et al., 2008), using the OPLS-AA force field (Kaminski et al., 2001) for both the receptor and the peptide, combined with the Berger parameters for the lipids (Berger et al., 1997). The same setup is used for a 2.5 nanosecond MD equilibration, where initial restraints on protein and ligand atoms are gradually released from 1000 to 200 kJ/molÅ2, followed by a 2.5 nanosecond run in which the 200 kJ/molÅ2 positional restraint is applied only in the C-alpha trace of the protein as described in detail in reference (Gutiérrez-de-Terán et al., 2013). For each of the complexes with PYY and truncated analogs, five unrestrained replica simulations of 100 nanosecond each were then produced, under the following MD conditions: isobaric NPT ensemble using a Nose-Hoover thermostat (Nosé and Klein, 1983) with a target temperature of 310 K. Electrostatic interactions beyond a cutoff of 12 Å were estimated with the particle mesh Ewald method. Analyses of these MD runs were conducted with several GROMACS utilities, VMD (Humphrey et al., 1996) and MDTraj (McGibbon et al., 2015). Molecular superimpositions, trajectory visualizations, and molecular images were obtained with PyMOL. For the MD simulations of each peptide in water, a cubic box was generated to solvate the peptide (12 Å minimum distance to the boundary) and subjected to a short equilibration, consisting on 0.1 nanosecond in the NVT ensemble followed by 0.2 nanosecond in NPT (constant pressure) conditions. Thereafter, five unrestrained replica simulations of 10 nanosecond followed, under the same conditions as for the complexes.
Free Energy Perturbation Calculations.
The model of the WT-hY2 in complex with PYY (holo form) and the corresponding reference (apo form, obtained by deleting the ligand and resolvating the originated cavity) were retrieved from the previous MD equilibration under PBC. Each system was transferred to a spherical boundary system for free energy perturbation (FEP) calculations, performed with the software Q (Marelius et al., 1998) using the same force field parameters described for the PBC simulations. A sphere with radius 25 Å, centered on the alpha-carbon of Q34 of PYY in the complex, was created. A 10 kcal/molÅ2 positional restraint was applied on solute atoms within the outer shell of the sphere (i.e., 22–25 Å from the center), while solvent atoms were subjected to polarization and radial restrains, using the surface constrained all-atom solvent (SCAAS) (King and Warshel, 1989; Marelius et al., 1998) model to mimic the properties of bulk water at the sphere surface. Solvent bond and angles were constrained using the SHAKE algorithm (Ryckaert et al., 1977). Atoms lying outside the simulation sphere were tightly constrained to their initial positions with a 200 kJ/molÅ2 force constant, and excluded from the calculation of non-bonded interactions. Long range electrostatics interactions beyond a 10 Å cut-off were treated with the local reaction field method (Lee and Warshel, 1992), except for the atoms of the sidechains involved in the FEP transformations, to which no cut-off was applied. Ionizable residues near the boundary of the sphere were neutralized, to avoid artifacts due to missing dielectric screening (Åqvist, 1996), while all titratable residues outside the sphere were neutralized.
The spherical systems were equilibrated with an initial heating phase of 0.11 nanosecond, the temperature slowly raised up from 1 to 310 K while the positional restraints initially applied to the solute atoms (25 kcal/molÅ2) were released, followed by 0.5 nanosecond of unrestrained equilibration (for mutations involving charged residues the equilibration time was doubled) at the final constant temperature of 310 K (bath coupling of 0.1 femtosecond) with a time step of 1 femtosecond. MD sampling under the same conditions followed, accounting for the sampling for FEP calculations. Data collection consisted of 10 replica simulations, each starting with different random velocities, for each simulated state (i.e., apo and ligand-bound receptor). Each independent simulation lasted for 4–7 nanoseconds (depending on the mutation), leading to a total sampling time of 2 (states) × 10 (replicates) × [4–7] (nanosecond per replica per state) ≈100 nanosecond per simulated mutation. The SEM was reported for each case based on the statistical analysis of the 10 replicate simulations. Our FEP protocol for amino acid mutations has been described elsewhere (Boukharta et al., 2014; Keränen et al., 2014, 2015). Briefly, a given mutation of any residue to alanine was divided in several smaller subperturbations to allow for a smoother transition between the end-states. Three steps were introduced for groups of atoms (charge groups) starting with the group with the highest topological distance (number of atoms) from the protein backbone: 1) removal of partial charges per charge group, 2) introduction of a soft core van der Waals potential and 3) full annihilation of the involved atom(s). The last step included the introduction of the Cβ hydrogen atom of the alanine residue. For non-alanine mutations, initial models of mutant receptors were created with PyMol, using the most probable rotamer of the mutated residue, and a double thermodynamic cycle was joined (i.e., WT -> Ala and mut -> Ala), meaning that in these cases double simulation time was needed (i.e., ∼200 nanosecond per mutation) and that the associated SEM is expected to go above the 1 kcal/mol value typical of Ala mutations (Boukharta et al., 2014; Keränen et al., 2014, 2015). For mutations of charged residues, a counter ion (chloride for mutation of Glu, potassium for Arg mutations) was placed in the bulk solvent, partially restrained and simultaneously charged, to maintain the total charge of the sphere constant along the FEP transformation. In these cases, the heating and the equilibration times were doubled and the sidechain of the mutated residue was constrained to its initial position with 5 kcal/molÅ2 during the equilibration phase to increase the convergence of the simulations. To compare with the experimental mutant binding data, we converted the experimental Ki values into ΔΔG using:

Mutagenesis
The primers for the mutagenesis were designed using web-based software PrimerX (http://www.bioinformatics.org/primerx/) and synthesized by Eurofins Genomics (Ebersberg, Germany) and purified with high pure salt free method. The QuikChange II site-directed mutagenesis kit (Agilent Tachnologies, Santa Clara, CA,) or AccuPrime Pfx SuperMix (Thermo Fisher Scientific, Waltham, MA) was used for introducing mutations to the human Y2 receptor (WT-hY2, UniProt P49146) coding region inserted in a pcDNA-DEST47 vector (Thermo Fisher Scientific) according to manufacturer’s protocols. The generated PCR product was transformed into One Shot Top10 E.coli cells (Thermo Fisher Scientific). The Y2 receptor mutations were confirmed by sequencing of the whole Y2 coding region (Eurofins). The plasmids containing different mutations were purified with Pulink plasmid purification kit (Thermo Fisher Scientific) eluted in pure water and stored at −20°C for future transfections.
Membrane Preparation
HEK293 cells were grown on 55 cm2 Nunclon cell culture dishes (Thermo Fisher Scientific) to 90%–95% confluency and transfected with Y2 wild type or mutant plasmids. After 72 hours, plates were washed in ice cold phosphate-buffered saline, and scraped mechanically from the plates. The cells were then transferred to tubes and centrifuged for 5 minute at 1000g. Pellets were resuspended in ice cold binding buffer (25 mM HEPES, 2.5 mM CaCl2, 1.0 mM MgCl2 and 0.2% Bacitracin, pH 7.4) used for binding assays described below and then homogenized for 30 seconds using an Ultra-Turrax homogenizer at medium speed. The homogenate was centrifuged at 3500g for 5 minute, the supernatant was discarded and fresh cold buffer was added. Homogenization of the pellet was repeated three times. The final pellet was resuspended in binding buffer to 1 ml and stored at −80°C.
Synthesis of PYY and Its Analogs
All chemicals were of analytical grade or higher. Triisopropylsilane (TIPS), N,N´-diisopropylcarbodiimide (DIC), acetic anhydride, thioanisole, collidine and formic acid (FA) (≥98%) were from Sigma–Aldrich, Chemie GmBH (Steinheim, Germany). Acetonitrile (ACN), dichloromethane (DCM) (LiChrosolve), trifluoroacetic acid (TFA), and diethyl ether were purchased from Merck KGaA (Darmstadt, Germany). Water came from a MilliQ equipment (Advantage A10) from Millipore (Molsheim, France). 2-Chlorotrityl chloride polystyrene, Rink-amide or PAL polystyrene resins were purchased from Merck KGaA. Standard Fmoc amino acids, resins and coupling reagents, 1-hydroxybenzotriale (HOBt) and OxymaPure Novabiochem were from Merck KGaA or Protein Technologies (Tucson, AZ). N-Methylpyrrolidone (NMP) dimethylformamide (DMF) and piperidine were from Biosolve (Dieuze, France).
Peptide analogs (see Supplemental Fig. 2) were prepared using a Prelude or Prelude X peptide synthesizer (Protein Technologies) using a Fmoc-chemistry protocol for solid phase peptide synthesis and either Fmoc-Rink-amide or Fmoc-PAL polystyrene resins. Each coupling used 6–8 eq. Fmoc-amino acid-OH and OxymaPure in DMF (both 6–8 eq.) and activated by DIC and collidine also both 6–8 eq. The amino acid solutions were all 0.3 M containing also 0.3 M OxymaPure. The DIC and collidine solutions were both 3 M and added as 1/10 volume compared with the volume of Fmoc-amino acid solutions. For deprotection of Fmoc, 25% piperidine in NMP or DMF was used for 4 + 4 minute (or 2 + 2 minute using heating 55°C on Prelude X). Coupling time was set to 60 minute (or 30 minute using heating 45–55°C on Prelude X) while double coupling was performed for Fmoc-Arg(Pbf)-OH and a total coupling time of 1 hour (or 20 + 40 minute using heating 55°C on Prelude X).
Synthesis of PYY-(3-36) methylamide was done using the linker (3-formylindolyl)acetamidomethyl polystyrene (Merck Millipore) from Epstep (Estep et al., 1998). Coupling of Fmoc-Tyr(Otbu)-OH onto this linker was done using 8 eq, 8 eq DIC and 8 eq OxymaPure in DMF and heating at 55°C for 2 hours. After coupling the resin was capped using 10 eq acetic anhydride in DMF for 30 minute.
Synthesis of PYY-(3-36)-ol (the C-terminal as primary alcohol) was done by coupling 5 eq of Fmoc-(O-tert-butyl)-tyrosinol in DCM containing 3% DPEA to 2-chlorotrityl chloride polystyrene resin for 5 hours at room temperature. Coupling of residues 1-35 for both of the above analogs was done by standard SPPS Fmoc-chemistry.
Peptides were cleaved and deprotected with TFA/TIPS/thioanisole or TFA/TIPS/H2O (95:3:2) for 1–3 hour and precipitated with diethyl ether. After washing with diethyl ether four to five times through a 0.45 μm filter or by centrifugation, the peptides were dried. The peptides were purified by preparative RP-HPLC using a linear gradient: 15%–40% ACN with 0.1% TFA over 40 minute on a SymmetryPrep C18 19 × 300 mm, 7 μm column (Waters Corporation, Milford, MA) eluting at 20 ml/min. The purity of peptides was determined by analytical RP-UPLC on a Waters Acquity UPLC System with a Waters BEH column C18 using 0.05% TFA in H2O (solvent A1) and 0.05% TFA in ACN (solvent B1). Molecular weights were determined using matrix-assisted laser desorption and ionization time-of-flight mass spectroscopy, recorded on a Microflex or Autoflex (Bruker Daltonics, Bremen,Germany). A matrix of α-cyano-4-hydroxy cinnamic acid was used. Alternatively, characterization was performed by UPLC–MS on a setup consisting of a Waters Acquity UPLC system connected to a LCT Premier XE mass spectrometer from Micromass, or by HPLC–MS on an Agilent 1200 series HPLC connected to an Agilent 6230 time-of-flight (TOF) system using 0.02% TFA in MilliQ (solvent A2) and (0.02% TFA in ACN (solvent B2). The purified analogs were dissolved in 80% DMSO with 20% H2O as stock solutions to a final concentration of 200 μM as quantified using a chemiluminescent nitrogen detector and RP-UPLC.
Binding Assays
125I-porcine PYY (125I-pPYY) was used as radioligand with a specific activity of 2200 Ci/mmol (PerkinElmer, Waltham, MA). Two different binding methods were used in this study: a filtration method and a scintillation proximity assay (SPA). The same binding buffer was used for both binding methods: 25 mM HEPES, 2.5 mM CaCl2, 1.0 mM MgCl2 and 0.2% Bacitracin, pH 7.4. Both the saturation and competition experiments were performed in U-bottom flexible 96-well plates (PerkinElmer) in a final volume of 100 or 200 μl and incubated for 3 hours at room temperature. All binding studies were performed in duplicate and repeated independently at least three times. The saturation experiments were carried out using serial dilutions of radioligand, 125I-pPYY, whereas the competition experiments were performed with serial dilutions of the competing ligand and a constant concentration of radioligand, 50 pM. In both types of binding experiments, non-specific binding was defined in the presence of 100 nM hPYY.
For the filtration method, the incubation was terminated by filtration with ice cold 50 mM Tris (pH 7.4) at 4°C through Filtermat A GF/C filters (PerkinElmer) pre-soaked in 0.3% (v/v) polyethyleneimine (Sigma-Aldrich) using a Tomtec cell harvester. The filters were dried at 50°C, then covered with MeltiLex scintillator sheets (PerkinElmer). For the SPA based assay, 0.5 mg/well wheat germ agglutinin SPA beads (PerkinElmer) was used and the assays were terminated by centrifugation at 500g for 10 minutes. Light emission from scintillation beads or filters was measured using a Wallac 1450 Microbeta counter (PerkinElmer). Each assay was optimized by adding a suitable amount of receptors, and in each case the dilution factor was determined empirically by performing test binding assays where we checked that less than 10% of the radioligand was bound.
Inositol Phosphate Accumulation Assay
For the mutants with low expression level or too low binding affinity, a functional signal transduction assay was performed. The assay was performed according to previous descriptions (Xu et al., 2013). Briefly, each receptor construct was transfected into HEK293 cells using Lipofectamine 2000 (Thermo Fisher Scientific), together with a chimeric G protein construct that changes the signaling pathway from Gi to Gq (Kostenis, 2002). Myo2-3H-inositol (PerkinElmer) at 3 μCi/ml was added the day after transfection. On the third day, the cells were detached from plates after incubation with phosphate-buffered saline/EDTA mixture (0.2 g/l) and resuspended in assay buffer with 10 mM LiCl, 20 mM Hepes, 137 mM NaCl, 5 mM KCl, 0.44 mM KH2PO4, 4.2 mM NaHCO3, 1.2 mM MgCl2, 1 mM CaCl2, and 10 mM glucose. After 10 minute pre-incubation, the cells were stimulated with serial dilution of peptides for 25 minute at 37°C. The cells were lysed for 60 minute at 4°C with an equal volume of 0.8 M perchloric acid, and then neutralized with KOH/KHCO3 solution. Ion exchange chromatography on AG1-X8 resin, formate form (Bio-Rad, Hercules, CA) was used to collect the generated 3H-inositol phosphates. The resin was washed with buffer containing 5 mM Na2B4O7 and 60 mM NH4-formate and eluted with buffer containing 1 M NH4-formate and 0.1 M formic acid. Then the samples were mixed with OptiPhase HiSafe (PerkinElmer) and the 3H radioactivity was measured with a Tri-Carb 2910TR liquid scintillation counter (PerkinElmer). The assays were performed in triplicate for each concentration and repeated at least three times.
Data Analysis
The pharmacology assay results, Kd from saturation assay, Ki from competition assay and EC50 values from functional assay, were calculated with the GraphPad Prism 5.0 software using non-linear regression curve fitting function. The data were presented as geometric mean with 95% confidence interval. Kd values were determined with either filtration or SPA methods, and used to calculate the Ki values from the assays. The statistical analyses of pKi, and pEC50 were performed using one-way ANOVA with Dunnett or Tukey post hoc tests.
Results
MD Simulations and Binding Affinities of PYY-(3-36) and Truncated Analogs.
The affinities of full-length PYY and its four truncated analogs [PYY-(3-36), PYY-(18-36), PYY-(19-36) and PYY-(22-36)] for the wild-type human Y2 receptor (WT-hY2) are summarized in Table 1. To facilitate the analysis, the relative fold in affinity is depicted taking as a reference the Ki value of the endogenous agonist PYY-(3-36). Full-length PYY binds with about sevenfold higher affinity than PYY-(3-36), with both of these native agonists displaying affinities in the subnanomolar range (Table 1). Further truncations up to position 19 showed modest (less than fivefold) reduction in affinity. Removal of the first 21 amino acids had a more dramatic effect with 25-fold affinity loss as compared with PYY-(3-36).
Binding affinities of PYY and its truncated analogs for WT-hY2
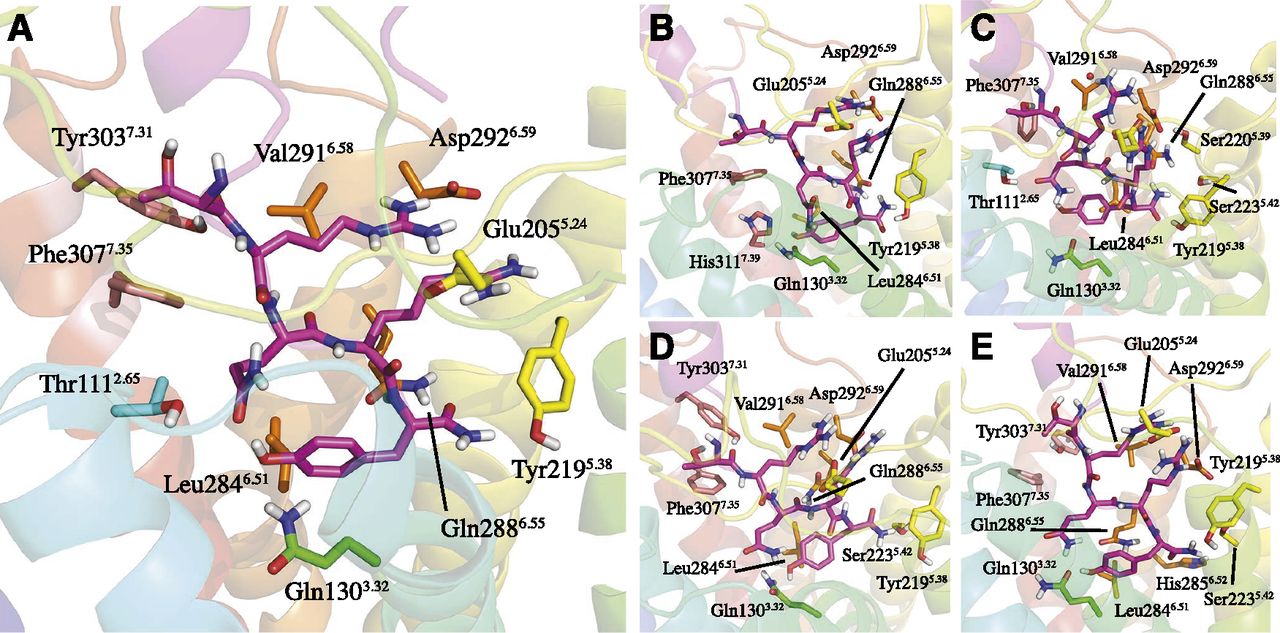
To characterize the peptide-receptor interactions at the structural level, we used our previously published structure of the Y2-NPY complex as a starting point (Xu et al., 2013). According to this model, the interaction network for the amidated C-terminal pentapeptide fragment (32TRQRY36-NH2) is defined within the TM cavity, which was later recognized by an independent study that measured receptor potency, not affinity (Kaiser et al., 2015). The characteristic secondary structure common to the NPY/PYY/PP peptides consists of a α-helix (positions 13–31), which protrudes out in the extracellular loop (EL) region, and is followed by a proline-rich helix (positions 1–12) that interacts through several H-bonds with the α-helix domain. Subsequently, we built an initial model of the Y2-PYY complex by replacing the varying amino acids between NPY and PYY peptides, and used this as a basis to build the complexes of Y2 with the four N-terminal truncated analogs of PYY described above. Each complex was subjected to five MD simulations of 100 nanosecond each, where we analyzed the stability of the complex and characterized the main interactions of the common C-terminal fragment. These interactions are well preserved and show high stability, with the residues involved in the peptide – receptor interactions being essentially the same in all five cases (see Fig. 1; Supplemental Table 1). The slight differences in the particular peptide-receptor interactions between some peptides are not statistically significant and can thus be related to the dynamic nature of peptide-receptor binding.

The binding mode of the amidated C-terminus of PYY and four truncated analogs in human Y2: (A) WT-PYY; (B) PYY-(3-36); (C) PYY-(18-36); (D) PYY-(19-36); (E) PYY-(22-36). The interacting residues were selected on the basis of hydrogen bonds occurring at least 40% of the time in each replicate of the MD simulations (Supplemental Table 1) or Cα- Cα distances under 9 Å.
However, we noted that the folding of the secondary structure elements described above (α-helix and proline helix domains) is maintained stable only in the case of PYY and PYY-(3-36). In contrast, for the shorter peptides (PYY-(18-36), PYY-(19-36) and PYY-(22-36)) misfolding of the helical part of the peptides is consistently observed (see Supplemental Fig. 3). To determine if this was due to the particular environment of the modeled receptor binding site, analogous MD simulations of all the five peptides were performed in water. The same trend was observed in solution, i.e., PYY and PYY-(3-36) maintain the typical interactions between the proline-rich helix and the central α-helix, whereas for the shorter peptides lacking the proline-rich helix and part of the α-helix, the remaining fragment of the α-helix is less stable (see Supplemental Fig. 4). The progressive loss in the stability of the secondary structural elements of the truncated peptides might be related to the moderate to a high decrease in binding affinity for the Y2 receptor described above (Table 1).
Rational Design and Binding Affinities of hY2 Receptor Mutants and PYY-(3-36) Analogs.
As presented above, the interactions of the 32TRQRY36-NH2 C-terminal fragment with the receptor are quite preserved, and the statistics on the H-bond interactions involved in this binding mode are summarized in Supplemental Table 1. Since this fragment is common to all truncated peptides and also involves a conserved region between the three peptides of the NPY family, we focused on a further characterization of the interaction pattern here proposed by comparison with available mutagenesis data, and rational design of new mutagenesis experiments, as summarized in Fig. 2. The effects on PYY-(3-36) affinity of previously investigated mutants for some of these positions are noted as a fold change in binding affinity as compared with the WT-hY2 receptor values, while the new positions selected for mutagenesis studies are indicated. Table 2 shows the effect on PYY-(3-36) affinity (or potency, in a few indicated cases) for each of the mutants generated. To further characterize some of the binding motifs proposed, we complemented the receptor mutagenesis studies with the generation of a number of PYY-(3-36) derivatives, which include mutation of certain peptide positions to natural amino acids, plus chemical modifications of the amidated C-terminus (Fig. 3; Supplemental Table 2).

Proposed binding mode of 32TRQRY36-NH2 to the hY2 receptor, with the receptor residues participating ligand binding and studied by site-directed mutagenesis denoted in sticks and labeled in bold. The effect in fold change in agonist affinity (or potency, indicated by a) is indicated for mutations reported in references Merten et al. (2007), Xu et al. (2013), where (nb) indicates no detectable binding. Residues selected for mutagenesis in this study are indicated in red, and the corresponding results reported in Table 2.
Binding affinities or potencies of PYY-(3-36) for WT-hY2 and mutants

Affinity values (pKi = –Log Conc (M), Y axis) of PYY-(3-36) analogs for WT-hY2 and its mutants. The upper panels depict data for peptide modifications on position T32 (T32A, T32F) and Q34 (Q34L). Lower panels refer to modifications on the amidated C-terminus, replaced by three different chemical groups (NHCH3, COOH, CH2OH). Within each panel, affinity for a given mutant (indicated by three-letter amino acid code) is given for PYY-(3-36) or the corresponding peptide modification (indicated in parenthesis). Error bars indicate SEM. The statistical significance of pairwise comparison is indicated with asterisks following the code: *P < 0.05; **P < 0.01; ***P < 0.001.
The first group of residues selected for mutagenesis was predicted to surround T32 in PYY (and NPY), namely Tyr3037.31 and Phe3077.35 (the numbering of receptor residues is followed by the Ballesteros-Weinstein topological position commonly used for GPCRs, in superscript) (Ballesteros and Weinstein, 1995). Tyr3037.31Ala has a negligible effect on binding affinity, while Phe3077.35Ala importantly decreases the affinity of PYY-(3-36) (45-fold as compared with the WT-hY2). One of the differences of our model as compared with that recently reported by Kaiser et al. is an interaction proposed between Tyr1102.64 and T32 in the latter (Kaiser et al., 2015). Therefore, we further evaluated the effect of the Tyr1102.64Ala mutation on peptide binding in our present model, which was found to have only a minor effect (Table 2). The peptide modification T32A affected binding to the WT and mutagenized receptors, as shown in Fig. 3 and Supplemental Table 2. The affinity for WT-hY2 is considerably reduced (53-fold), an effect that is mostly recovered when this peptide analog binds to the Tyr1102.64Ala receptor mutant and, to a lesser extent, to the Tyr3037.31Ala mutant. Conversely, the same (T32A)-PYY-(3-36) peptide shows essentially the same affinity for the Phe3077.35Ala receptor mutant than for the wild-type. Substitution by the bulkier Phe (T32F) in the peptide has a milder effect on the affinity both for the WT receptor and for the Tyr3037.31Ala and Phe3077.35Ala mutants, while showing slightly less affinity for the Tyr1102.64Ala receptor than the (T32A)-PYY-(3-36) peptide analog (Fig. 3; Supplemental Table 2).
Continuing along the peptide sequence we find a pattern of interactions for R33 and R35 that was already identified in our previous model (Xu et al., 2013). These two arginines form salt bridges with Asp2926.59 and Glu2055.24 (in EL2), with additional contacts between R35 and Gln2886.55. This interaction pattern is supported by previous mutagenesis of Asp2926.59 to Glu, Asn, or Ala (Merten et al., 2007), and of Glu2055.24 to Ala (Merten et al., 2007) or Arg (Xu et al., 2013), all of which reduce agonist binding or potency. We have further complemented these mutational data with a substitution by a flexible but apolar sidechain, i.e., Glu2055.24Leu, which had a similar effect on agonist binding (Table 2). To further assess the conformation of the EL2 region in our model, we also tested the effect of alanine mutations of Arg1874.64 and Glu1884.65, both of which are in the distal part of EL2 (close to TM4) and predicted to be in the vicinity of Glu2055.24. The moderately negative impact on binding affinity when removing the positively charged Arg is in agreement with its possible role in stabilizing the negative charge of Glu2055.24. In the same trend is the slight gain in affinity when removing the negative charge of Glu1884.65. Although not forming a stable interaction in our MD simulations, the initial model also suggested a role for Ser2205.39 in an H-bond with this arginine, which would indeed be supported by the moderate decrease in binding affinity for the corresponding Ala mutation (see Table 2).
The other position in contact with R35 is Gln2886.55. The previously reported conserved mutation Gln2886.55Asn did not reduce affinity (Xu et al., 2013). However, the fact that this position is also predicted to interact with the amidated C-terminal residue motivated us to explore this position by introducing less conservative replacements (see below). The peptide sidechain of glutamine Q34, located between the two arginines, shows hydrogen bonds with Gln1303.32 and Thr1112.65 in our model. We investigated the latter side chain, and the results indicated that the corresponding alanine mutation considerably reduces the affinity (33-fold, Table 2), while Thr1112.65Leu had a negligible effect suggesting an unspecific, non-polar interaction for the native Thr (Table 2). However, removing the polarity of the peptide sidechain (Q34L mutant, Fig. 3; Supplemental Table 2) results in a moderate loss of affinity for WT-hY2, only slightly recovered when combined with the Thr1112.65Leu receptor mutant. The polar interaction of Q34 with Gln1303.32, on the other side, is supported by the decrease in affinity reported for its corresponding His and Glu mutants ((Xu et al., 2013), see Fig. 2). To further test the role of residues in TM3 in ligand binding, we evaluated the effect of breaking the predicted proline kink provided by Pro1273.29, which would be important to create the binding cavity needed for Y36 and Q34 (see Fig. 2). The results support this model, as Ala and especially Leu mutants have a great impact on ligand potencies determined in functional assays (Table 2).
The amidated C-terminal residue of all agonist peptides is Y36. The phenol sidechain of this residue shows in our model van der Waals and H-bond interactions with Leu2846.51 and Gln2886.55, respectively, while the -NH2 of the amidated C-terminus can form H-bonds with Gln2886.55, Ser2235.42, Tyr2195.38 and His2856.52 (Supplemental Table 1). Previously reported alanine mutants of Leu2846.51 and Tyr2195.38 showed a moderate effect on agonist binding ((Xu et al., 2013) and Fig. 2). While the Tyr2195.38Leu mutant also maintained this trend ((Xu et al., 2013) see Fig. 2), we found that in the case of Leu2846.51Tyr the binding affinity is recovered, and even increased as compared with WT-hY2. Mutations of Ser2235.42Ala, Ser2235.42Leu and His2856.52Phe showed a negligible effect on binding affinity, thus not supporting the predicted H-bonds with the peptide. Finally, the binding site of Y36 is optimally delimited by Cys1032.57 in TM2, a model that is in agreement with the big decrease in ligand potency observed after the introduction of a bulky side chain in the Cys1032.57Phe mutant (Table 2).
As for the role of the conserved Gln2886.55, the previously reported negligible effect of a conservative Asn mutation ((Xu et al., 2013) and Fig. 2) was here complemented with mutations to Ala, Leu, and Glu, all of which showed negligible to small effects in agonist binding (Table 2), whereas the introduction of a bulkier sidechain with a positive charge in the Gln2886.55Arg showed a dramatic 79-fold reduction in the affinity of PYY-(3-36) (see Table 2). Finally, we complemented the mutagenesis exploration with other residues that in our model would be in contact with the α-helical part of the peptide. Given the minor or negligible effects on ligand binding and the difficulty in interpreting the anticipated indirect effects from mutations in this area in the absence of a crystal structure, we summarize these results in Supplemental Table 3.
The intriguing role of the amidated C-terminus in ligand binding was here further explored with the characterization of three peptide analogs that involved modifications of the amide group with a carboxylate (introducing a negative charge), a primary alcohol (removal of the carbonyl group) or methylamide (adding a methyl to amid nitrogen), all of which showed a dramatic loss of affinity (Fig. 3; Supplemental Table 2). The methylamide analog was further tested against mutated versions of the receptor that in principle should allow the accommodation of the bulkier methyl-amide, i.e., Tyr2195.38Ala and Gln2886.55Ala: the affinity was not regained, but was decreased even further. In an attempt to explore polar contacts with a positively charged Arg, double mutants of PYY-(3-36)-CH2OH-Gln2886.55Arg and PYY-(3-36)-COOH-Gln2886.55Arg were also tested but showed the same negative trend.
In Silico Evaluation of Site-Directed Mutagenesis with Free Energy Calculations.
In a final iteration, we went back to our structural model of the Y2-PYY complex to interpret the results of the mutagenesis and functional experiments. Most of the interactions were initially proposed based on similar structural models by us (Xu et al., 2013) and others (Kaiser et al., 2015) and showed good agreement with the results of the specifically designed mutations. The interaction pattern of the amidated C-terminus in our models remained more variable (see Supplemental Table 1), which is in contrast with the tight interaction of this group with Gln1303.32 proposed by the Beck-Sickinger group (Kaiser et al., 2015). We therefore computed the effect of selected point mutations on the binding free energy of the common C-terminal fragment of agonist peptides. In particular, mutations of the positions proposed to bind the amidated C-terminus, namely Gln1303.32, Gln2886.55 and Tyr2195.38, were simulated with our recently proposed MD/FEP protocol (Boukharta et al., 2014; Keränen et al., 2015) and the results compared with the experimental values extracted from this work and from our previous study (Xu et al., 2013). As shown in Fig. 4 and Supplemental Table 4, the calculated relative binding free energies are in good agreement with the experimental values, with mean absolute error of 1.3 kcal/mol and an average SEM of 1.47 kcal/mol (which drops to 1.16 kcal/mol if we exclude the unconverged Gln2886.55Glu data point, see below).

Calculated and experimental relative binding free energies (ΔΔG, kilocalories per moles) for PYY-(3-36) to nine selected hY2 mutants compared with WT-hY2. Black bars indicate  (values from Table 2 and (*) from Xu et al., 2013), gray bars represent
(values from Table 2 and (*) from Xu et al., 2013), gray bars represent  (values from Supplemental Table 4). The SEM is shown with error bars (calculated from 10 independent replica simulations for the FEP binding free energies, or from three independent experiments for the experimental data).
(values from Supplemental Table 4). The SEM is shown with error bars (calculated from 10 independent replica simulations for the FEP binding free energies, or from three independent experiments for the experimental data).
Removal of the polar side chain of glutamine in the Gln2886.55Ala mutant is found in our model to have a very small impact on peptide affinity, thereby providing a structural as well as an energetic explanation for the experimental negligible effect of this mutation. We observed an insertion of a water molecule in the Ala mutant allowing retention of the interactions with Y36, partially counterbalancing the effect of the mutation. This water-mediated interaction is not stable, which is reflected in the slight over-prediction of the experimental data. Notably, the small to negligible effect in our experimental studies for mutations to sidechains that maintain the bulkiness (Leu) or the polarity (Asn) of the WT Gln, is perfectly reproduced in the FEP simulations. The same applies for the mutation to the negatively charged Glu, though in this case the simulations did not properly converge as indicated by the high associated error (Supplementary Table 4), due to the different ways of reorganization of the binding site to accommodate this negative charge. These effects are in stark contrast with the dramatic reduction of peptide affinity observed upon mutation to a longer and positively charged sidechain of Arg, which is reproduced (although overpredicted) in our model. The model indeed provides a structural interpretation of this effect, as the Arg mutation perturbs dramatically the interactions with the neighboring positively charged residues in the peptide (R33 and R35).
As outlined above, the amino group of the C-terminal amide would mostly interact with Tyr2195.38 in our model, which makes the most important difference with previous suggestions of an amide-amide interaction with the sidechain of Gln1303.32 (Kaiser et al., 2015). In our model, this residue would be involved in interactions with Q34 and the peptide backbone. Notably, the FEP simulations based on our 3D-structure of the Y2-PYY complex perfectly explain the experimental effect of mutations of these two sidechains. Replacement of Gln1303.32 with a negatively charged Glu or a neutral histidine is detrimental for peptide binding in both cases. A similar effect is observed and correctly reproduced when removing the sidechain of Tyr2195.38 (mutation to Ala), an effect that is less pronounced both experimentally and in our calculations with the introduction of a bulky, hydrophobic chain (Leu). As shown in Fig. 4, the calculated relative binding free energies are in good agreement with the experimental drop in affinity for all nine mutants, a remarkable result considering that the random probability of such a qualitative agreement is as low as (1/2)9 = 0.002. Moreover, the scatter plot for the remaining eight mutations (calculated vs. experimental free energy shifts, Supplemental Fig. 5) shows a good quantitative correlation, with a Pearson correlation coefficient of 0.63, which goes up to 0.85 (statistical significance for the correlation of P < 0.005) if we leave out the aforementioned overpredicted data point in our model Asn2886.55Arg. Thus, our computational modeling provides further support for the network of interactions proposed herein for the 32TRQRY36-NH2 peptide fragment, including the hydrogen bonding of the carboxyterminal amide with Tyr2195.38 while Q34 makes contacts with Gln1303.32.
Alignment of NPY Receptors and RFamide Receptors.
It has been proposed that the NPY family receptors and the RFamide receptors for NPFF and PRLH, as well as QRFP (Elphick and Mirabeau, 2014; Larhammar et al., 2014; Yun et al., 2014), share a common ancestor from which they arose by gene duplications followed by sequence divergence. The conservation of the carboxy terminus of the peptides, with RYamide for the NPY family and RFamide for the others, correlates with a critical role of the carboxy terminus for receptor interactions for all of these receptors (Beck-Sickinger et al., 1994; Mauduit et al., 1998; Roland et al., 1999; Boyle et al., 2005; Le Marec et al., 2011; Findeisen et al., 2012). This should be reflected by a corresponding conservation of receptor residues that are critical for binding of the carboxy terminus of the respective peptides. Therefore, alignment of NPY receptor subtypes with RFamide receptors across vertebrates has been a constant frame of reference throughout this work. The receptor positions that we have investigated in human Y2 are marked in the alignment with other human NPY receptor subtypes and human RFamide receptors shown in Fig. 5. We have also made alignments with all of these receptors across vertebrates. The Y2 receptor residues proposed to interact with 32TRQRY36-NH2 indeed show high conservation among the RY/RFamide receptors. The following positions are almost completely conserved among the NPY-family receptors in human and many other vertebrates: Thr1112.65 (Val in Y5), Gln1303.32, Glu2055.24 (Asp in human Y1), Tyr2195.38, Leu2846.51, Asp2926.59, Phe3077.35, and His3117.39. Among these, positions 2.65 and 7.35 and 6.55 differ in all RFamide receptors in comparison with the NPY-family receptors, (Fig. 5) which may explain the relative tolerance of mutations at these positions in the Y2 receptor.

Alignment of human NPY family receptors and RF amide receptors. The Y2 transmembrane regions have been marked with frames based on the transmembrane region prediction of GPCRDB. The residues selected for mutagenesis were marked with light gray, and the most conserved position in each helix (TM.50, according to Ballesteros-Weinstein) is highlighted in dark gray.
Discussion
In this study, we used a combination of experimental, evolutionary and theoretical approaches to provide a detailed map of interactions between the C-terminus of NPY/PYY and the human Y2 receptor. Taking as a starting point our recently reported computational model of the Y2-agonist (NPY) complex (Xu et al., 2013), we initially investigated through MD simulations the interactions of Y2 with the peptide PYY and four truncated analogs on the N-terminus. Due to the intrinsic difficulty in the modeling of the EL region of GPCRs (Cavasotto and Palomba, 2015), the most speculative part of the model corresponds to the loop regions of the receptor and their potential interactions with the N-terminal fragment of the natural agonist. On the other side, we found that the binding mode of the common amidated C-terminus was almost identical for PYY and the four truncated analogs, in particular around the 32TRQRY36-NH2 motif, while truncations of the proline helix mainly affected the folding of the peptides. This would explain the gradual, smooth affinity loss of truncations up to residue 18, while PYY-(22-36) exhibited a larger reduction of affinity, in agreement with earlier studies on NPY truncations (Kaiser and Kézdy, 1983; Fuhlendorff et al., 1990). Our simulations indicate that the interactions between the α-helix and the proline-helix region are critical to maintain the PP-fold, characteristic of all Y-receptor agonist peptides (Glover et al., 1984; Bjørnholm et al., 1993; Nygaard et al., 2006). PYY-(3-36) also maintains this fold, as revealed in NMR studies (Nygaard et al., 2006), but more severe truncations of the peptide lead to misfolding, in line with the experimental data available (Minakata and Iwashita, 1990; Hu et al., 1994). Taken together, our binding affinity data and the subsequent MD analysis support the current model for Y2 agonism, where the activation trigger is located in the amidated C-terminus fragment. The function of the remaining peptide might be related to the kinetics of peptide binding, as earlier indicated for NPY (Fuhlendorff et al., 1990). While intriguing, this point should be investigated with further kinetic experiments of the binding of different peptides.
The pattern of interactions derived from our MD analysis for the 32TRQRY36-NH2 motif with hY2 was essentially the same as proposed in our earlier study (Xu et al., 2013). While many of these interactions were further supported by existing mutagenesis data, either previously reported or designed and tested in that study (Fig. 2), we herein investigated novel potential interactions with receptor mutagenesis and peptide modifications. Position T32 in the peptide is predicted close to Phe3077.35, which was confirmed by the experimental data: while the alanine mutant of this residue had a moderate impact on affinity of the peptide, this was partially recovered for the (T32A)PYY-(3-36) peptide analog, which otherwise showed lower affinity for the WT-Y2 receptor. Conversely, the corresponding introduction of an aromatic sidechain in the peptide, i.e., (T32F)PYY-(3-36) analog, was better tolerated by the WT-Y2 receptor suggesting a π-stacking interaction in the peptide mutant. The proximity of position Phe3077.35 with residue T32 in the peptide is in contrast with the previous suggestion of an interaction of T32 with Tyr1102.64, a possibility that we could rule out based on the minor effect of peptide binding of the corresponding Tyr1102.64Ala mutant.
The pair of arginines in the C-terminal tail of the peptide (R33 and R35) are involved in salt-bridge interactions with Glu2055.24 and Asp2926.59, as independently proposed in the most recent models of agonist-Y2 receptor binding (Xu et al., 2013; Kaiser et al., 2015). The glutamine residue in between this arginine pair (Q34) protrudes in the opposite direction, facing receptor residues Gln1303.32 and Thr1112.65. This interaction pattern is supported by our previous and current site-directed mutagenesis data. Mutations of Thr1112.65 were here combined with the Q34L peptide analog, the results suggesting a smooth interaction between the two positions. Thus, residue Gln1303.32 appears to be the main anchoring point for Q34, in line with the high sensibility to peptide binding of conservative mutations (His, Glu) of this position. (Xu et al., 2013). We found further evidence for this interpretation in our FEP calculations of the Gln1303.32Glu and Gln1303.32His mutants. This is in contrast with the alternative role proposed for this residue in the model from Beck-Sickinger and co-workers, where this residue is the anchoring point of the amidated C-terminus (Kaiser et al., 2015). In addition, Gln1303.32 might have a structural role by participating in a network of interhelical interactions with Thr1072.61 and His3117.39, supported by the lack of receptor expression in the corresponding Ala and Leu mutants (Xu et al., 2013). Our model, on the other hand, suggests a direct interaction between Y36 and Leu2846.51. This is in line with our previous data that showed an increased binding affinity for the Leu2846.51Tyr mutant, indicative of a possible π-stacking of the aromatic rings. This would indeed leave the amidated C-terminal group exposed to more diffuse polar interactions with Gln2886.55 and Tyr2195.38. This possibility was thoroughly investigated here by receptor mutants, combined with peptide modifications on the C-terminal amide group, and the results interpreted with FEP simulations of the generated mutants. For mutations at these two positions, our FEP calculations consistently reproduce the small to moderate effect observed when removing the sidechain (Gln2886.55Ala, Tyr2195.38Ala), replacing it with an apolar side chain (Leu mutants) or even the small effects of more conservative mutants like Gln2886.55Asn/Glu. On the other hand, the introduction of a flexible and positively charged arginine in Gln2886.55 was not tolerated by our model, in clear agreement with the experimental data on that mutant. Additional verification of this polar network was given by the conspicuous loss of binding affinity observed for the peptide modifications of the amidated C-terminus. To further test the binding mode proposed, we designed three mutants that would be expected to hinder the peptide to go into the binding pocket: Pro1273.29Ala, Pro1273.29Leu and Cys1032.57Phe. All three of these exhibited too low affinities to be measured, precisely, but potencies could be determined accurately. This confirmed our hypothesis that Pro3.29 is key to maintain the integrity of the binding pocket in the upper side of the TM2-TM3 region, where Cys1032.57Phe could act as an obstacle at the Y36 binding pocket.
We repeatedly found that single-amino acid replacements had a greater impact in the peptides than in the receptors. The reason for this is probably that the receptor has multiple points of interaction with each residue in the peptide. A given peptide residue is usually surrounded by, and interacts with, many receptor residues. Each receptor residue would thereby contribute relatively little to the interactions with the peptide. For the peptide, on the other hand, the number of residues that interact with the receptor is more limited, meaning that each one makes a greater contribution to receptor binding.
Another important aspect is that most of the identified residues that are involved in the pentapeptide interactions are highly conserved among NPY family receptor subtypes and also the more distantly related RFamide receptors in human and other vertebrates. This suggests that the binding modes of these receptors share structural similarities, reflecting their common evolutionary ancestry. Furthermore, the binding pockets formed by these conserved residues in the NPY-family receptors are likely to explain the conservation of the C-terminus of the NPY family peptides. The positions that differ between peptide-binding receptors for peptides ending with RFamide, rather than the RYamide of PYY and NPY, are likely to contribute to peptide-receptor preferences. This information may be used for synthesis of Y2-selective agonists. It is important to take into consideration that some of the positions in the peptides and receptors may be subjected to negative selection pressures to avoid cross-binding to other members of these families.
Our study presents a unique combination of computational and in vitro pharmacological analyses, focused on the effect on ligand binding of both receptor mutagenesis and peptide modifications. This broad approach allowed us to deduce a more detailed model for the interactions of the native PYY agonist with the human Y2 receptor. By also including evolutionary comparisons of the critical positions, we have identified several positions that are likely to be important for PYY binding to Y2 and are also likely to be involved in PYY binding to the other NPY-family receptor subtypes. The interaction pattern described between 32TRQRY36-NH2 and hY2 may guide the optimization of Y2-selective agonists, with potential applications in the control of appetite.
Authorship Contributions
Participated in research design: Xu, Østergaard, Wulff, Gutiérrez-de-Terán, Larhammar.
Conducted experiments: Xu, Østergaard, Paulsson, Pruner.
Performed calculations: Vasile.
Performed data analysis: Xu, Vasile, Åqvist, Gutiérrez-de-Terán, Larhammar.
Wrote or contributed to the writing of the manuscript: Xu, Vasile, Østergaard, Paulsson, Åqvist, Wulff, Gutiérrez-de-Terán, Larhammar.
Footnotes
- Received September 12, 2017.
- Accepted January 19, 2018.
B.X. and S.V. contributed equally to this study as shared first authors. H.G.-T. and D.L. A.M. and H.G.T. contributed equally to this study as shared corresponding authors.
This work was supported by a postdoctoral fellowship for B.X. from Novo Nordisk A/S, DK-2880 Bagsvaerd, Denmark, the Swedish Research Council (Department of Cell and Molecular Biology, Grant 521-2014-2118) and the Swedish Strategic Research Program eSSENCE. The computations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC).
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- ACN
- acetonitrile
- DCM
- dichloromethane
- DIC
- N,N´-diisopropylcarbodiimide
- DMF
- N,N-dimethylformamide
- FA
- formic acid
- FEP
- free energy perturbation
- Fmoc
- fluorenylmethyloxycarbonyl
- HOBt
- 1-hydroxybenzotriazole
- MD
- molecular dynamics
- NMP
- N-methylpyrrolidone
- NPFF
- neuropeptide FF
- NPY
- neuropeptide Y (for tyrosine)
- PP
- pancreatic polypeptide
- PRLH
- prolactin releasing hormone
- PYY
- peptide YY (for tyrosine-tyrosine)
- QRFP
- pyroglutamylated RFamide peptide
- PBC
- periodic boundary condition
- RP-HPLC
- reverse phase high-performance liquid chromatography
- RP-UPLC
- reverse phase ultra-performance liquid chromatography
- SEM
- standard error of the mean
- SPA
- scintillation proximity assay
- TFA
- trifluoracetic acid
- TIPS
- triisopropylsilane
- WT
- wildtype.
- Copyright © 2018 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}