


Abstract
Chimeric D1/D2 receptors were constructed to identify structural determinants of drug affinity and efficacy. We previously reported that chimeras that had D1 receptor transmembrane domain VII together with amino-terminal sequence from the D2 receptor were nonfunctional. D2/D1 chimeras were constructed that contained D2 receptor sequence at the amino- and carboxyl-terminal ends and D1 receptor sequence in the intervening region. Chimeric receptors with D2 sequence from transmembrane domain 7 to the carboxyl terminus together with D2 receptor sequence from the amino terminus through transmembrane helix 4 (D2[1–4,7]) and 5 (D2[1–5,7]) bound [3H]spiperone with high affinity, consistent with the hypothesis that D2 receptor transmembrane domain I or II is incompatible with D1 receptor transmembrane domain VII. D2[1–4,7] and D2[1–5,7] had affinities similar to D1 and D2 receptors for most nonselective dopamine antagonists and had affinities for most of the selective antagonists that were intermediate between those of the parent receptors. D2[1–4,7] and D2[1–5,7]mediated dopamine receptor agonist-induced stimulation and inhibition, respectively, of cAMP accumulation. The more efficient coupling of D2[1–5,7] to inhibition of cAMP accumulation, compared with the coupling of D2[5–7] and D2[3–7], supports the view that multiple D2 receptor cytoplasmic domains acting in concert are necessary for receptor activation of Gi. In contrast, D2[1–4,7], which contains only one cytoplasmic loop (the third) from the D1 receptor, is capable of activating Gs. D2[1–4,7]exhibited several characteristics of a constitutively active receptor, including enhanced basal (unliganded) stimulation of cAMP accumulation, high affinity for agonists even in the presence of GTP, and blunted agonist-stimulated cAMP accumulation. A number of dopamine receptor antagonists were inverse agonists at D2[1–4,7], inhibiting basal cAMP accumulation. Some of these drugs were also inverse agonists at the D1 receptor. Interestingly, several antagonists also potentiated forskolin-stimulated cAMP accumulation via D2[1–5,7] and via the D2 receptor, which could reflect inverse agonist inhibition of native constitutive activity of this receptor.
The dopamine receptor family comprises D1-like (D1 and D5) and D2-like (D2L, D2S, D3, and D4) receptors (1). The D1-like dopamine receptors have a shorter IC3 and a longer carboxyl terminus than the D2-like receptors. D1-like receptors have high affinity for benzazepine ligands, such as SCH23390, whereas D2-like receptors have high affinity for benzamide and butyrophenone ligands, such as sulpiride and spiperone. D1-like dopamine receptors couple to the G protein Gs, stimulating adenylate cyclase, whereas the D2-like receptors couple to Gi or Go, inhibiting adenylate cyclase. D1- and D2-like receptors both regulate phosphoinositide turnover, and D2-like dopamine receptors also modulate arachidonic acid release, Na+/H+ exchange, K+ currents, and Ca2+currents (2).
One approach to identification of the structural features of G protein-coupled receptors that determine function is to construct mutant and chimeric receptors. Mutagenesis of single amino acids in dopamine receptors has identified a number of residues in the putative transmembrane helices that are involved in the binding of ligands and agonist activation of D1 and D2 receptors (3-8). Although point mutations often can identify residues that are critical for specific functions, some receptor properties are likely to be attributable to multiple, contiguous amino acid residues that cannot be identified by point mutations.
Our analysis of chimeric D1/D2 receptors has suggested that structural determinants of selective potency and efficacy can differ, with TMVI of the D2 dopamine receptor implicated in selective efficacy of some agonists and TMVII of D1 and D2 receptors involved in the selective potency of some ligands. We demonstrated that there are determinants of receptor down-regulation in TMV and IC3 and confirmed the importance of IC3 in stimulation of adenylate cyclase by D1 receptors, although inhibition of adenylate cyclase via D2 receptors seems to require both IC2 and IC3 (9, 10).
It often is observed that the function of certain chimeric receptors is dramatically impaired due to incompatibilities between adjacent transmembrane regions (11). Such chimeras can be used to identify intramolecular interactions in the parent receptors (12, 13). When we constructed chimeric receptors containing the D2receptor TMI and TMII and D1 receptor TMVII, the chimeras were nonfunctional (9). Because three-dimensional models of G protein-coupled receptors and mutagenesis data suggest that TMI, TMII, and TMVII are adjacent to one another and that interactions between pairs of amino acids in these adjacent helices may stabilize the receptor structure (12-15), we hypothesized that incompatibilities between D1 TMVII and D2 TMI and TMII produced the nonfunctional state of these chimeric receptors. We now report that replacement of the D1 TMVII in the nonfunctional chimeras with D2 TMVII restored function. Interestingly, one of the chimeric receptors exhibited several characteristics of a constitutively active receptor. Constitutive activity results from mutations that increase the probability of spontaneous isomerization of the receptor to its active conformation, which increases the affinity of the receptor for agonists and enhances “basal,” or unliganded, stimulation of effectors by the receptor (16).
Experimental Procedures
Materials.
[3H]Spiperone (80 Ci/mmol) was purchased from Amersham (Arlington Heights, IL). [3H]SCH23390 (70 Ci/mmol) and [3H]cAMP (30 Ci/mmol) were purchased from Dupont-New England Nuclear (Boston, MA). SCH23390, SKF-38393, spiperone, chloro-APB (SKF 82958), 6-chloro-PB, quinpirole (LY17155), apomorphine, bromocriptine, lisuride, and forskolin were purchased from Research Biochemicals (Natick, MA). DHX (Dr. Richard Mailman, University of North Carolina, Chapel Hill, NC), epidepride (NCQ 219; Dr. Tomas de Paulis, Vanderbilt University, Nashville, TN), fenoldopam (SKF82526; Dr. Richard Wilcox, University of Texas at Austin), and pergolide (Eli Lilly, Indianapolis, IN)) were generous gifts. Dopamine (3-hydroxytyramine), 3-isobutyl-1-methylxanthine, and most other reagents were purchased from Sigma Chemical (St. Louis, MO).
Construction of chimeric receptor cDNAs.
Eight chimeric cDNAs (previously referred to as CH1–8; new nomenclature depicted in Fig. 1, A and B) were constructed bytrans-polymerase chain reaction and cloned intoHindIII and EcoRI sites of pcDNA-1. The construction, expression in C6 glioma cells, and characterization of these receptors were described previously (9). The D1 TMVII and cytoplasmic tail was removed from D2[1–2] (CH5), D2[1–4] (CH6), and D2[1–5] (CH7) by digestion withClaI and EcoRI and replaced with a fragment containing D2 TMVII and cytoplasmic tail from D2[7] (CH4), creating D2[1–2,7], D2[1–4,7], and D2[1–5,7] (Fig. 1C).

Structure of the chimeric dopamine D2/D1 receptors. D1 sequence shows putative TM helices (open rectangles) and intracellular and extracellular hydrophilic regions (thin lines). D2 sequence shows putative TM helices (filled rectangles) and hydrophilic regions (thick lines).
Expression of recombinant receptors.
D1, D2L, and chimeric cDNAs (D2[1–2,7], D2[1–4,7], and D2[1–5,7]) were stably expressed in HEK 293 cells by electroporation. HEK 293 cells (2.2 × 107/ml) were resuspended with the appropriate cDNA (15 μg) and pBabe Puro (2 μg), to confer resistance to puromycin (17), in Dulbecco’s modified Eagle’s medium supplemented with 10% BCS and 5 mm N,N-bis-(2-hydroxyethyl)-2-aminoethanesulfonic acid in a total volume of 400 μl. With a 0.4-cm cuvette gap, the electroporator settings were 0.17 kV and 950 μF, yielding time constants between 40 and 50 msec. The cells were split into four 10-cm-diameter tissue culture plates and grown in Dulbecco’s modified Eagle’s medium supplemented with 5% fetal bovine serum, 5% BCS, penicillin G (50 units/ml), and streptomycin (50 μg/ml) in a humidified incubator at 37° in the presence of 10% CO2. After 48 hr, the medium was replaced with growth medium containing puromycin (2 μg/ml). Puromycin-resistant colonies were transferred to duplicate wells and tested for binding of D1- and D2-selective radioligands.
Radioligand binding assay.
Confluent plates of cells were lysed by replacing the medium with ice-cold hypotonic buffer (1 mm Na+ HEPES, pH 7.4, 2 mm EDTA). After swelling for 10–15 min, the cells were scraped off the plate and centrifuged at 24,000 × gfor 20 min. The crude membrane fraction was resuspended in Tris-buffered saline with a Polytron homogenizer (Brinkmann Instruments, Westbury, NY) at setting 6 for 10 sec and used for radioligand binding assays. For determinations ofKi values, the affinity of D2[1–4,7], D2[1–5,7], and D2 receptors for ligands was assessed by inhibition of the binding of [3H]spiperone, whereas the affinity of the D1 receptor for ligands was quantified by inhibition of the binding of [3H]SCH23390. Aliquots of the membrane preparation (5–100 μg of protein) were added to duplicate assay tubes containing (final concentrations): 50 mmTris·HCl, pH 7.4, with 155 mm NaCl (Tris-buffered saline), 0.001% bovine serum albumin, radioligand, and appropriate drugs. (+)-Butaclamol (2 μm for wild-type or 20 μm for chimeras) was used to define nonspecific binding. Incubations for binding studies were carried out at 30° for 1 hr and terminated by filtration through glass-fiber filters on a 96-well Tomtec cell harvester. The filters were dried before the addition of BetaPlate scintillation fluid, and radioactivity on the filters was determined with a Wallac (Gaithersburg, MD) 1205 BetaPlate scintillation counter.
For competition binding studies in which displacement of radioligand binding by agonists was assessed, the crude membrane fraction was resuspended in Tris-buffered saline containing 4 mmMgCl2 before addition to assay tubes containing Tris-buffered saline and (final concentrations) 4 mmMgCl2, 1 mm EDTA, 200 μm GTP, 0.0025% ascorbic acid, 0.001% bovine serum albumin, radioligand, and appropriate drug concentrations. For competition binding studies in which dopamine displacement of binding in the absence and presence of GTP (200 μm) was assessed, the crude membrane fraction was added to assay tubes containing 20 mm HEPES, pH 7.5, with 6 mmMgCl2, 1 mm EDTA, 1 mmEGTA, 1 mm dithiothreitol, 0.0025% ascorbic acid, and 0.001% bovine serum albumin, together with radioligand and dopamine concentrations as indicated. Incubations were carried out and filtered as detailed above.
cAMP accumulation assay.
Cells were plated at a density of ∼100,000 cells/well in 48-well tissue culture clusters. After 2–4 days, when the cells were confluent, the plates were used for adenylate cyclase stimulation or inhibition experiments. The rates of division of the clones were similar, so that each had 300,000–400,000 cells/well at the time of assay. The cAMP accumulation assay of adenylate cyclase activity was carried out essentially as described previously (18). Cells were rinsed on ice two times with 200 μl of assay buffer (Earle’s balanced salt solution, containing 0.02% ascorbic acid, 2% BCS, and 500 μm 3-isobutyl-1-methylxanthine) for 5 min followed by the addition of assay buffer with or without forskolin (10 μm final concentration) and appropriate drugs. Incubations were carried out at 37° for 10 min, except for agonist stimulation and inverse agonist inhibition of D2[1–4,7], which was carried out at 40°, a temperature that produced more consistent results. The assays were terminated by decanting the buffer, placing the plates on ice, and lysing the cells with 3% trichloroacetic acid. The plates were centrifuged at 1000 × g for 15 min and stored at 4° for ≥1 hr before quantification of cAMP.
Quantification of cAMP.
cAMP was quantified using a competitive binding assay (19) as described previously (18). Samples of the cell lysate from each well (5–20 μl) were added to duplicate assay tubes. [3H]cAMP (∼1 pmol) in cAMP assay buffer buffer (100 mm Tris·HCl, pH, 7.4, 100 mm NaCl, 5 mm EDTA) was added to each tube, followed by cAMP-binding protein (100 μg of crude bovine adrenal extract in cAMP assay buffer) for a final volume of 500 μl. The reaction tubes were incubated on ice for 2–5 hr. The contents of the tubes were harvested by filtration (Whatman GF/C filters or Wallac Filter Mat A) using a 96-well Tomtec cell harvester. Filters were dried, and BetaPlate scintillation fluid was added to each sample. Radioactivity on the filters was determined using a Wallac BetaPlate scintillation counter. The cAMP concentration in each sample was estimated from a standard curve ranging from 0.1 to 100 pmol of cAMP.
Data analysis.
Saturation isotherms, radioligand displacement curves, and dose-response curves for cAMP accumulation were analyzed by nonlinear regression using the program GraphPAD Prism (GraphPAD Software, San Diego, CA).Ki values are geometric mean values from three or more independent experiments ± the asymmetrical standard error. For dopamine, the goodness of fit for one- and two-site analyses was compared using an F test. At a value ofp < 0.05 for improvement of the fit assuming two classes of binding sites, data were analyzed in terms of two classes of binding sites. Other statistical comparisons were made using Student’s paired t test (two-tailed) as indicated. The change in free energy of binding of a drug was calculated using the equation ΔG° = −RTln(1/Ki ) where ΔG° is the change in free energy, R is the gas constant, T is temperature in °K, and Ki is the apparent affinity of the drug.
Results
To test the hypothesis that incompatible interactions between D1 TMVII and D2 TMI and TMII resulted in nonfunctional chimeric receptors, we modified D2[1–2], D2[1–4], and D2[1–5] (Fig. 1), so both the amino- and carboxy-terminal ends were from the D2 receptor, with D1 receptor sequence in the intervening region. The chimeric and wild-type D1 and D2 receptors were stably expressed in HEK 293 cells, and the binding of two radioligands was assessed. The chimeric receptors D2[1–4,7] and D2[1–5,7] bound [3H]spiperone, a D2-selective ligand, with high affinity. Saturation analysis of radioligand binding to these receptors determined that the density of D2[1–4,7] was 730 ± 70 fmol/mg of membrane protein, with aKD value for [3H]spiperone of 0.60 ± 0.04 nm (11 experiments), the density of D2[1–5,7] was 300 ± 30 fmol/mg, with aKD value of 2.00 ± 0.33 nm (13 experiments), and the density of D2 receptors was 480 ± 30 fmol/mg, with aKD value of 0.071 ± 0.018 nm (9 experiments). Although we were able to detect specific binding of [3H]spiperone and [3H]clozapine to D2[1–2,7] and specific binding of [3H]SCH23390 to D2[1–2,7], D2[1–4,7], and D2[1–5,7], the binding was of such low affinity that accurate estimates ofKD andB max values could not be obtained.
Three clonal lines of D1 receptor-expressing HEK 293 cells were used for these experiments. The density of D1 receptors on the clones used to assess stimulation of adenylate cyclase was 2700 ± 490 fmol/mg (9 experiments) and 1100 ± 90 fmol/mg (4 experiments) of membrane protein.
Characterization of antagonist and agonist binding.
For catecholamine receptors, structural determinants of ligand binding seem to be primarily in the conserved α helical regions (20). Therefore, we analyzed the chimeras in terms of the number of transmembrane helices that were contributed by D1 or D2 receptors. As has been done previously (9,21), we calculated the change in the free energy of binding of a ligand (ΔG°) at each chimeric receptor. If each transmembrane region contributes an equal amount, or one seventh, of the difference between the affinity of a given ligand for the D1 and D2 receptors, then a plot of ΔG° at each chimeric and wild-type receptor versus the number of transmembrane regions that are from the D2 receptor should be linear, with the values for the chimeric receptors falling on a line drawn between the wild-type receptors. The 95% confidence interval of the mean ΔG° was calculated for each drug. If the line drawn between ΔG° for the drug at the wild-type receptors did not intersect the 95% confidence interval for the ΔG° at a given chimera, then the affinity of the chimera for the drug was considered to be significantly different from the value predicted by the null hypothesis that each transmembrane region contributes equally to the ΔG° of binding. A value of ΔG° for a chimeric receptor significantly above or below the line could indicate that a particular transmembrane region contributes more or less, respectively, than the average contribution of the other transmembrane regions to the selectivity of the ligand being tested.
Three of the antagonists that were tested, (+)-butaclamol, clozapine, and cis-flupenthixol, were relatively nonselective for D1 and D2 dopamine receptors. (+)-Butaclamol had similar affinities for all four receptors (D1, D2, D2[1–4,7], and D2[1–5,7]) (Table1 and Fig.2A), but the affinity ofcis-flupenthixol for D2[1–5,7] was significantly below the line drawn between wild-type receptors. The affinity of D2[1–4,7] for clozapine however, was significantly greater than the affinity of either wild-type receptor for the drug (Table 1)
Affinity of D1, D2, and chimeric dopamine receptors for antagonists
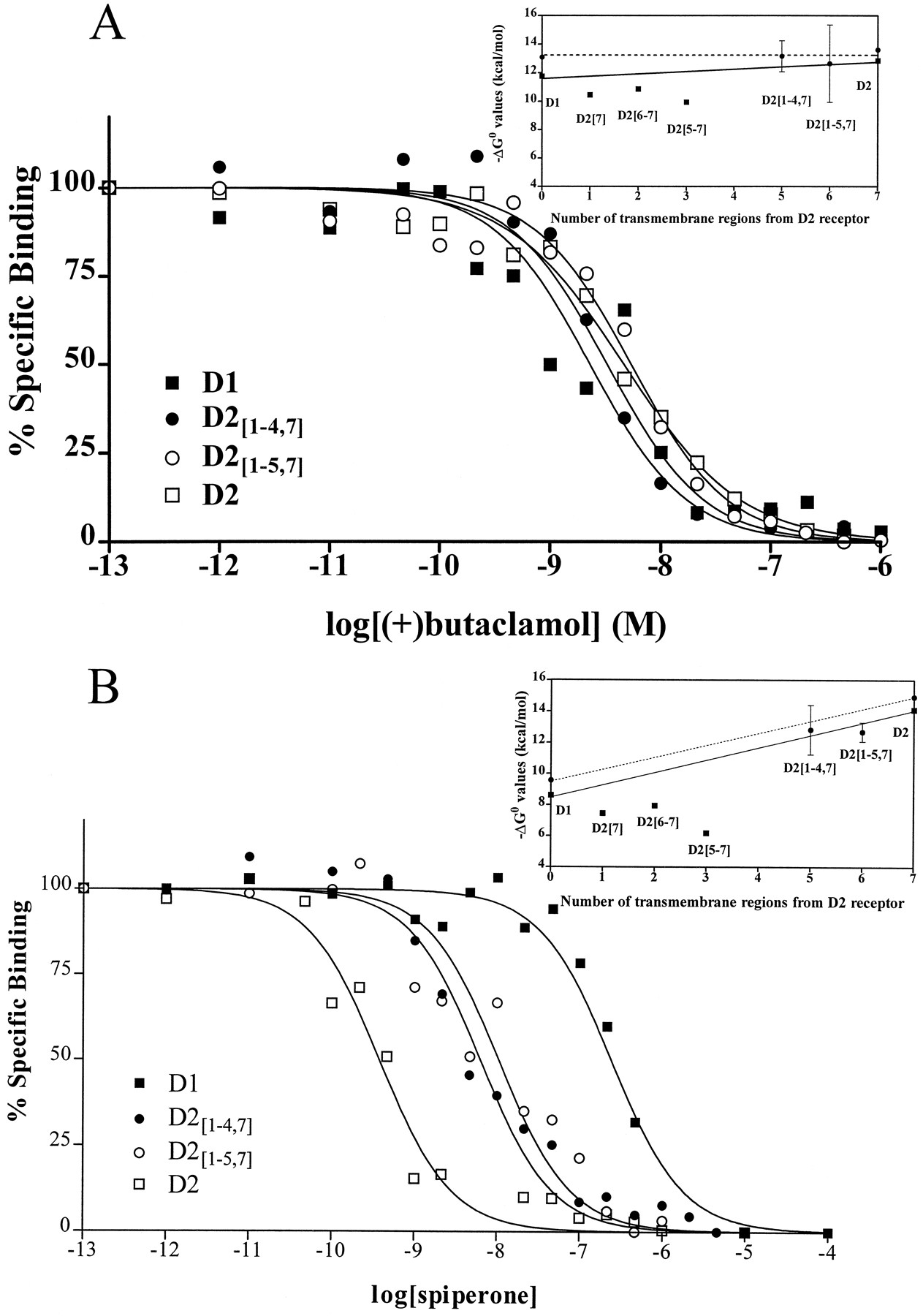
Binding of antagonists. Data are shown from one of three or more independent experiments in which inhibition of radioligand binding was determined for the indicated drugs. Data are plotted as a percentage of the specific binding in the absence of inhibitor versus the logarithm of the concentration of (A) (+)-butaclamol or (B) spiperone. Insets, Averaged data from all experiments are expressed as the free energy change of binding (in Kcal/mol) plotted versus the number of transmembrane regions from the D2 receptor. Error bars, 95% confidence intervals. ▪, Data from our previous report using chimeric and wild-type receptors expressed in C6 glioma cells (9). Solid line, line drawn between D1 and D2 receptor ΔG° values from our previous report. •, Data from the current study using receptors expressed in HEK 293 cells.Dotted line, line drawn between D1 and D2 ΔG° values from the current study.
Although the mean ΔG° values for the binding of the D2-selective antagonists epidepride, haloperidol, spiperone, and sulpiride were slightly below the line drawn between values for each drug at D1 and D2 dopamine receptors, only for the binding of spiperone and epidepride to D2[1–5,7] did the line not intersect the 95% confidence interval of the mean (Table 1and Fig. 2B). There was no difference in the affinity of D2[1–4,7] and D2[1–5,7] for these D2-selective ligands.
The affinity was assessed of D1-selective benzazepines SCH23390, SKF38393, and chloro-PB for D2[1–4,7] and D2[1–5,7]. The mean ΔG° values were not significantly different from the line drawn between D1 and D2 receptors (Table2 and Fig.3A). Also, the affinity of SCH23390 and SKF38393 for D2[1–5,7] and D2 receptors was not significantly different. For another D1-selective benzazepine, chloro-APB, the affinity of D2[1–5,7] was significantly less than predicted, whereas the potency of chloro-APB for D2[1–4,7] was higher than would be predicted by the line drawn between D1 and D2 receptors (Fig. 3B).
Affinity of D1, D2, and chimeric dopamine receptors for agonists

Binding of benzazepine. Data are shown from one of three or more independent experiments in which inhibition of radioligand binding was determined for the indicated drugs. Data are plotted as a percentage of the specific binding in the absence of inhibitor versus the logarithm of the concentration of (A) SCH23390 or (B) chloro-APB in the presence of GTP. Insets, averaged data from all experiments are expressed as free energy change of binding (in Kcal/mol) plotted versus the number of transmembrane regions from the D2 receptor. Error bars, 95% confidence intervals. ▪, Data from our previous report using chimeric and wild-type receptors expressed in C6 glioma cells (9). Solid line, line drawn between D1and D2 receptor ΔG° values from our previous report. •, Data from the current study. Dotted line, linedrawn between D1 and D2 ΔG° values from the current study.
For most agonists, ΔG° values for D2[1–5,7] were not significantly different from those predicted by the line drawn between D1and D2 receptors. However, D2[1–4,7] receptors had significantly higher affinity for many dopamine agonists than did D1and D2 receptors (Table 2). The affinity of D2[1–4,7] receptors was higher than either wild-type dopamine receptor for 6,7-ADTN, apomorphine, NPA, DHX, dopamine, lisuride, and pergolide. For every agonist tested, except the partial agonist SKF38393, ΔG° values for D2[1–4,7] were above the line drawn between D1 and D2 receptors (Table2 and Fig. 4, A and B), indicating a higher-than-expected affinity of D2[1–4,7] for most agonists. Because one characteristic of constitutively active receptors is high affinity for agonists (16), these results suggested that D2[1–4,7] is constitutively active.

Binding of agonists. Data are shown from one of three or more independent experiments in which inhibition of radioligand binding was determined for the indicated drugs. Data are plotted as a percentage of the specific binding in the absence of inhibitor versus the logarithm of the concentration of (A) NPA or (B) quinpirole in the presence of GTP. Insets, Averaged data from all experiments are expressed as the free energy change of binding (in Kcal/mol) plotted versus the number of transmembrane regions from the D2 receptor. Error bars, 95% confidence intervals. ▪, Data our previous report using chimeric and wild-type receptors expressed in C6 glioma cells (9). Solid line, the line drawn between D1 and D2receptor ΔG° values from our previous report. •, Data from the current study. Dotted line, line drawn between D1 and D2 ΔG° values from the current study.
To assess the hypothesis that D2[1–4,7] was constitutively active, we examined agonist binding in the presence and absence of GTP. When a receptor is constitutively active, the equilibrium between active and inactive receptor conformations shifts spontaneously toward the active conformation, resulting in high affinity binding of agonists even in the presence of GTP (16). We were able to detect both high (KH ) and low (KL ) affinity states in the absence of GTP for D2, D1, D2[1–4,7], and D2[1–5,7] receptors (Table3 and Fig.5). However, for D2[1–4,7], >75% of the receptors were in a high affinity state, whereas for D2, D1, and D2[1–5,7], <45% of the receptors were in a high affinity state (43%, 30%, and 33%, respectively). In several assays in the absence of GTP, the goodness of fit of the binding data for D1 and D2[1–4,7] receptors was not significantly improved by assuming the presence of two classes of binding sites. For the D1 receptor, theKi value for dopamine when only one class of binding sites was detected was close to theKL value when the best fit assumed two classes of binding sites. On the other hand, when only one class of binding sites could be detected for dopamine at D2[1–4,7], theKi value was similar to the value forKH (Table 3). Furthermore, theKi value for dopamine at D2[1–4,7] was unchanged by the addition of GTP, whereas for D1, D2, and D2[1–5,7] receptors, only the low affinity state KL value could be detected in the presence of GTP.
Affinity of D1, D2, and chimeric dopamine receptors for dopamine in the presence and absence of GTP

GTP sensitivity of the binding of dopamine. Representative data are shown from one of three or more independent experiments in which inhibition of radioligand binding to D2[1–4,7] and D2[1–5,7] was determined for dopamine. Data are plotted as the percentage of the specific binding versus the logarithm of the concentration of dopamine. Inset, Averaged data from all experiments are expressed as the free energy change of binding (in Kcal/mol) plotted versus the number of transmembrane regions from the D2 receptor. ▪, ΔG° values for K H. □, ΔG° values for K L in the absence of GTP. •, ΔG° values in the presence of GTP. Solid line, line drawn between D1 and D2receptor ΔG° values in the presence of GTP. Dotted line, line drawn between high affinity states. Dashed line, line drawn between low affinity states of the D1 and D2 receptor ΔG° values in the absence of GTP.
Stimulation of adenylate cyclase via D1 and D2[1–4,7] receptors.
Consistent with the hypothesis that D2[1–4,7] was constitutively active, cells that expressed D2[1–4,7] receptors had basal cAMP levels that were significantly elevated compared with nontransfected HEK 293 cells and compared with cells expressing D1, D2[1–5,7], and D2 receptors (Table4). Interestingly, basal cAMP accumulation in nontransfected HEK 293 cells was significantly lower than in cells expressing D1 receptors (Table 4), suggestive of a low level of unliganded activity by D1 receptors expressed in HEK 293 cells. Assays incubated at either 37° or 40° resulted in similar levels of basal cAMP accumulation (Table 4). Because agonist-stimulated accumulation of cAMP was more consistent at 40° for cells expressing D2[1–4,7] receptors, those assays were carried out at 40°.
Basal levels of cAMP in nontransfected cells or cells expressing D1, D2, and chimeric dopamine receptors
Most dopamine receptor agonists stimulated cAMP accumulation via D2[1–4,7] and D1receptors (Fig. 6, top). Apomorphine, NPA, chloro-APB, SKF38393, DHX, lisuride, pergolide, and bromocriptine were as efficacious as dopamine via D2[1–4,7]. Fenoldopam and chloro-PB did not stimulate cAMP accumulation via D2[1–4,7], although both drugs are agonists at D1 receptors. Interestingly, the D2 receptor agonist quinpirole stimulated adenylate cyclase via D2[1–4,7], although quinpirole lacks efficacy at D1receptors. We were unable to detect stimulation of cAMP accumulation by dopamine via D2[1–2,7].

Modulation of cAMP accumulation via D1and D2[1–4,7] receptors. Data shown are the average ± standard error from three or more independent experiments in which (top) stimulation by agonists (1 μm, except 10 μm quinpirole) or (bottom) inhibition by antagonists (1 μm) of cAMP accumulation via D1 and D2[1–4,7] receptors was assessed. Data are expressed as pmol/well. Assays were performed at 37°, except for agonist stimulation and antagonist inhibition of D2[1–4,7], which was at 40°. In other experiments, no stimulation of cAMP accumulation via the D1 receptor was observed in the presence of 100 μm quinpirole (data not shown). ∗, p < 0.05 compared with basal, Student’s t test for paired means. ∗∗,p < 0.05 compared with D1 basal, Student’s t test for paired means.
Stimulation of adenylate cyclase via D1 receptors resulted in cAMP levels of >450 pmol/well for many of the agonists tested, whereas agonist-mediated stimulation of adenylate cyclase via D2[1–4,7] receptors was blunted, resulting in maximal cAMP levels that were only ∼150 pmol/well (Fig. 6,top). To determine whether the reduced ability to stimulate cAMP accumulation was due to coupling of the D2cytoplasmic domains of D2[1–4,7] to Gi/o, we treated cells with pertussis toxin (25 ng/ml for 18–22 hr). Treatment with pertussis toxin elevated cAMP accumulation stimulated by 1 μm dopamine from 137 ± 10 to 327 ± 43 pmol/well (three experiments). The cAMP accumulation stimulated by 10 μm forskolin in these cells was not altered by treatment with pertussis toxin (data not shown), indicating that the treatment did not nonspecifically alter the responsiveness of adenylate cyclase.
Inverse agonism at D2[1–4,7] and D1receptors.
Another characteristic of constitutively active receptors is the ability of inverse agonists to antagonize basal second messenger generation (22). A number of nonselective antagonists and D2-selective antagonists seemed to be inverse agonists at D2[1–4,7] receptors. (+)-Butaclamol, clozapine, epidepride, cis-flupenthixol, haloperidol, and spiperone decreased basal cAMP levels in the absence of agonist by ∼70% [Fig. 6, bottom; data for (+)-butaclamol, epidepride, and cis-flupenthixol not shown]. Inhibition of basal cAMP accumulation by clozapine was not altered by pretreatment with pertussis toxin (data not shown). Although not statistically significant, there was a trend for nonselective antagonists, such as clozapine and haloperidol, to decrease basal cAMP formation in cells expressing D1 receptors. The D1-selective antagonist, SCH23390, did not inhibit basal levels of cAMP formation at either D1 or D2[1–4,7]receptors. There was a tendency for SCH23390 to increase D1 receptor-mediated cAMP formation, consistent with its reported weak partial agonist activity at D1 receptors (23).
In other experiments, inhibition was assessed of forskolin-stimulated cAMP accumulation by antagonists. Spiperone and clozapine both inhibited forskolin-stimulated cAMP accumulation via D2[1–4,7] in a dose-dependent manner, with EC50 values of 1.9 ± 0.2 and 24 ± 10 nm, respectively (Fig. 7A). To confirm that some antagonists are inverse agonists at the wild-type D1 receptor, we determined that clozapine,cis-flupenthixol, and haloperidol inhibited forskolin-stimulated activity via the D1 receptor with EC50 values of 180 ± 24, 61 ± 5, and 540 ± 200 nm, respectively (Fig. 7B).

Inhibition of forskolin-stimulated cAMP accumulation by inverse agonists. Data shown are representative of four independent experiments in which inverse agonist inhibition of cAMP accumulation in the presence of 100 nm forskolin was assessed in cells expressing (A) D2[1–4,7] or (B) D1 receptors. Data are plotted as a percentage of forskolin-stimulated cAMP accumulation in the absence of inverse agonist versus the logarithm of the concentration of inverse agonist.
Agonist modulation of D2 and D2[1–5,7]receptor-mediated cAMP accumulation.
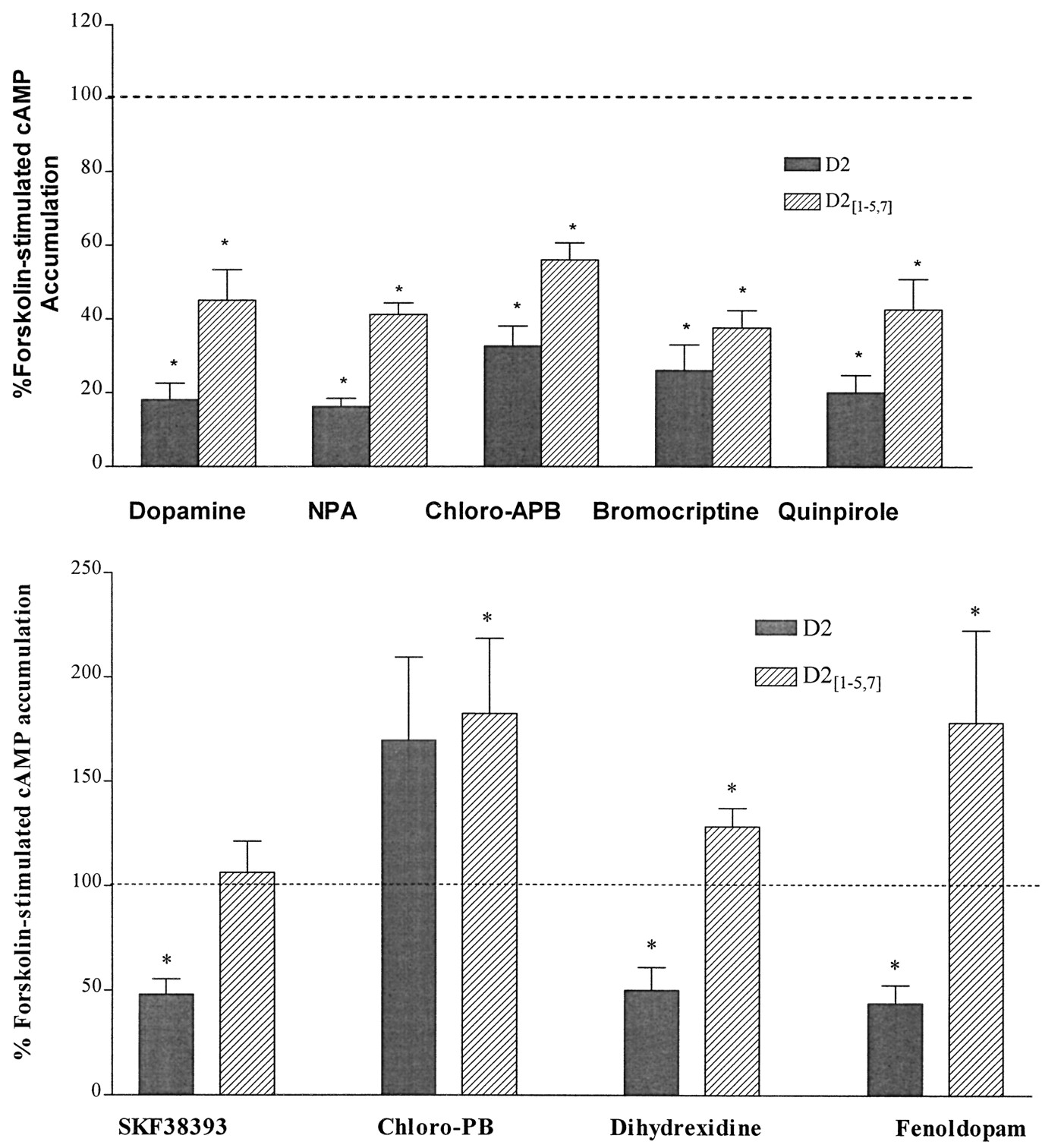
Inhibition of adenylate cyclase activity was assessed in cells expressing wild-type and chimeric receptors. The nonselective agonists dopamine, NPA, and apomorphine inhibited ∼80% of forskolin-stimulated adenylate cyclase activity via the D2 receptor and 50–60% of activity via D2[1–5,7] (Fig.8, top; data for apomorphine not shown). Similarly, the D2-selective agonists bromocriptine, lisuride, pergolide, and quinpirole inhibited adenylate cyclase activity via the D2 receptor by 70–75% and via D2[1–5,7] by 40–50% (Fig. 8,top; data for pergolide and lisuride not shown). However, only one of the D1-selective agonists tested, chloro-APB, inhibited adenylate cyclase activity via D2[1–5,7] (60%; Fig. 8, top). At this chimera, other D1-selective agonists (SKF38393, 6-chloro-PB, DHX, and fenoldopam) either had no effect or potentiated forskolin stimulation of cAMP (Fig. 8, bottom). SKF38393, DHX, and fenoldopam inhibited adenylate cyclase via D2 receptors by ∼50%, whereas 6-chloro-PB did not inhibit adenylate cyclase. Although 6-chloro-PB was an agonist at D1 receptors, it seemed to be an antagonist or an inverse agonist at D2, D2[1–4,7], and D2[1–5,7] receptors

Modulation of cAMP accumulation via D2and D2[1–5,7] receptors. The data shown are the average ± standard error of four or more independent experiments in which (top) inhibition or (bottom) potentiation of forskolin-stimulated (10 μm) adenylate cyclase activity by D1 receptor agonists (1 μm) was assessed. Data are expressed as a percentage of forskolin-stimulated cAMP accumulation in the absence of receptor agonist. ∗, p < 0.05 compared with control, Student’s t test for paired means.
Inverse agonism at D2[1–5,7] and D2receptors.
We also evaluated the ability of antagonists to act as inverse agonists at D2 receptors. The presence of (+)-butaclamol, epidepride, cis-flupenthixol, or haloperidol more than doubled forskolin-stimulated cAMP accumulation in cells expressing D2 receptors (Fig. 8; data for (+)-butaclamol not shown). Clozapine, spiperone, and SCH23390 produced a more modest potentiation of forskolin-stimulated cAMP accumulation, ∼40% over control values. Because the binding profile of D2[1–5,7] was similar to D2 receptors for most antagonists, we also evaluated the effect of antagonists on forskolin-stimulated cAMP accumulation in cells expressing D2[1–5,7]receptors. Epidepride, cis-flupenthixol, and haloperidol significantly increased forskolin-stimulated cAMP accumulation in cells expressing D2[1–5,7] by 40–50% (Fig.9). Antagonists had no effect on forskolin-stimulated cAMP accumulation in untransfected HEK 293 cells (data not shown).

Potentiation of forskolin-stimulated adenylate cyclase activity by antagonists. Data shown are the average ± standard error of four or more independent experiments in which potentiation of forskolin-stimulated (10 μm) adenylate cyclase activity by antagonists (1 μm) was assessed. Data are expressed as a percentage of forskolin-stimulated cAMP accumulation in the absence of antagonist. ∗, p < 0.05 compared with control, Student’s t test for paired means.
Discussion
We described previously the construction of eight chimeric receptors. Four D1/D2chimeras were functional (Ref. 9; Fig. 1A), whereas reciprocal D2/D1 chimeras (Fig. 1B) were not. We hypothesized that the chimeric receptors were nonfunctional due to incompatible interactions between D1 TMVII and D2 TMI and TMII, regions that other mutagenesis and modeling data have suggested to be adjacent (12-14). Specifically, amino acid residues in TMII of the serotonin 5-hydroxytryptamine2A, gonadotropin-releasing hormone, and adrenergic receptors and TMI of the muscarinic m2 and m5 receptors interact with amino acid residues in TMVII (12, 14, 15, 24). For example, mutation of Asn87 in TMII to aspartic acid in the GnRH receptor resulted in a nonfunctional receptor with no detectable binding of ligands, whereas addition of the reciprocal mutation of Asp318 in TMVII to asparagine restored function to the mutant receptor (15). Also, mutation of Asn312 in β2-adrenergic receptors to the amino acid residue found in the homologous domain of the α2-adrenergic receptor (phenylalanine) resulted in a nonfunctional protein, whereas replacement of TMI and TMII of the β2-adrenergic receptor mutant with α2-adrenergic receptor sequence restored function to the mutant receptor (12). These findings suggest that the low affinity of D2[5–7], D2[6–7], and D2[7] for a number of ligands (9) and the lack of detectable antagonist binding to D2[1–2], D2[1–4], D2[1–5], and D2[1–6]are due to structural distortions of the chimeric receptors that are not observed when TMI, TMII, and TMVII are from the same receptor. The affinity of D2[1–4,7] and D2[1–5,7] for most antagonists was not significantly different from that predicted by the null hypothesis that all transmembrane regions contribute equally to ligand binding.
These experiments highlight the difficulty of distinguishing effects on ligand binding that are due to interactions of specific receptor domains with the ligands from effects that are due to disruption of helix packing or other nonspecific perturbations of structure. The latter is apparently the explanation for the lack of ligand binding to D2[1–2], D2[1–4], D2[1–5], and D2[1–6], and perhaps also the low affinity of D2[5–7], D2[6–7], and D2[7] for some ligands, but it is difficult to make firm conclusions regarding other effects, such as the high affinity of D2[1–4,7] and D2[1–5,7] for clozapine or the low affinity of D2[1–2,7] for the radioligands tested. One finding that was consistent for all the chimeras tested in these experiments and previous work (9) is that TMVI of the D1 receptor apparently contributes little to the selective binding of most benzazepine ligands.
These difficulties of interpretation also apply to functional studies, in which it may be difficult to distinguish results due to the loss or addition of domains that interact specifically with G proteins from nonspecific structural effects. Still, the results presented here and in our previous work (9), such as the ability of D2[1–4,7] and D2[6–7]and the inability of D2[1–5,7] and D2[5–7] to stimulate adenylate cyclase, demonstrate consistently that the IC3 of D1receptors is necessary and sufficient for coupling to Gs. Furthermore, the lack of inhibition of isoproterenol-stimulated cAMP accumulation by D2[5–7] and the modest inhibition by D2[3–7] indicated that both IC2 and IC3 from the D2 receptor are required for inhibition of adenylate cyclase (9). Dopamine activation of D2[3–7] receptors inhibits cAMP accumulation by only ∼20%, but dopamine was able to cause ∼60% inhibition via the D2[1–5,7] receptor. These results suggest that the first cytoplasmic loop (IC1) of the D2receptor is also important for inhibition of adenylate cyclase activity via the D2 receptor. If all three cytoplasmic loops are involved in coupling to G proteins that inhibit adenylate cyclase, it might be predicted that D2[1–4,7]would activate both Gi/o and Gs. Consistent with this, pertussis toxin treatment enhanced stimulation of adenylate cyclase by D2[1–4,7].
The extended (allosteric) ternary complex model of receptor activation of G proteins proposes that receptors spontaneously isomerize between inactive (R) and active (R*) conformations, with agonists having higher affinity for and stabilizing or inducing the active conformation (16). This model predicts that most receptors will have some ability to activate G proteins in the absence of agonist, with the extent of unliganded receptor activity depending on the density of the receptor and the constant (J) that describes the equilibrium between the active and inactive conformations of that receptor. For example, the D1 and D5 receptors both exhibit a receptor density-dependent ability to stimulate adenylate cyclase activity in the absence of agonist, but the slope of the line that describes the relationship between receptor density and basal activity is much steeper for the D5receptor than for the D1 receptor, indicating greater unliganded activity of the former (23). Constitutive activation of a receptor describes any manipulation that tends to increase the formation of R* in the absence of agonist. In the current study, basal levels of cAMP accumulation were 4-fold higher in HEK cells expressing D2[1–4,7] than in HEK cells expressing the wildtype D1 receptor. Basal cAMP accumulation was in turn higher in HEK cells expressing the D1receptor than in untransfected cells or cells expressing the D2 receptor. In these experiments, the level of expression of D2[1–4,7] was lower than that of the wild-type D1 receptor, indicating that the enhanced activity was due to mutation-enhanced formation of R* rather than to a greater density of receptors. In addition to the enhanced basal levels of cAMP in cells expressing the D2[1–4,7] receptor, other characteristics of constitutively active G protein-coupled receptors were observed, including increased affinity for agonists, attenuation of agonist-induced second messenger signaling, and the ability of inverse agonists to block basal second messenger generation (16, 22).
Although the affinity of most agonists for D2[1–5,7] was approximately what would be expected if all transmembrane domains contribute equally to ligand binding, the affinity of most agonists for D2[1–4,7] was much higher than would be predicted. The high affinity of the receptor for dopamine was resistant to addition of GTP, indicating that it did not result from coupling to G proteins.
Maximal agonist stimulation of adenylate cyclase via D2[1–4,7] receptors was only ∼30% of the maximal stimulation via the D1 receptor. Some of the reduced responsiveness was apparently due to coupling of D2[1–4,7] to both Gi/oand Gs because pertussis toxin treatment more than doubled the stimulation of cAMP accumulation by dopamine to 72% of stimulation via the D1 receptor. In addition, stimulation may have been attenuated because even in the absence of agonist, the chimeric receptor was desensitized, decreasing the ability of the chimera to respond to agonist stimulation. Constitutive engagement of cellular desensitization mechanisms has been demonstrated for constitutively active mutants of the β2-adrenergic receptor (25) and the α2-adrenergic receptor (26). On the other hand, because D2[1–4,7] is a mutant receptor, stimulation might have been attenuated because the receptor is missing some structural elements required for full agonist efficacy or is globally perturbed in a way that modestly interferes with coupling to Gs. Agonist-mediated cAMP accumulation via D2[6–7] and D2[7]receptors was also attenuated compared with the D1 receptor (9).
Every D1 or D2 receptor antagonist tested at the D2[1–4,7] receptor, with the exception of SCH23390, inhibited basal cAMP accumulation. In addition, clozapine and spiperone both caused dose-dependent decreases in forskolin-stimulated cAMP accumulation, with EC50 values similar to their affinities at D2[1–4,7]. Although inverse agonism at a chimeric receptor does not necessarily mean that the drugs are also inverse agonists at the wild-type D1 receptor, we determined that three presumed dopamine receptor antagonists (clozapine, haloperidol, and cis-flupenthixol) dose-dependently inhibited forskolin-stimulated cAMP accumulation in HEK cells expressing the D1 receptor. Unliganded activity of D1 receptors and the inhibition of this activity by dopamine receptor antagonists confirms the results of Tiberi and Caron (23).
A mutant D1 receptor (L286A) with enhanced unliganded stimulation of adenylate cyclase activity was recently described (8), but the characteristics of this receptor differ in several respects from the constitutively active chimeric receptor, D2[1–4,7]. In particular, the affinity of L268A (1.2 μm) for dopamine was similar to the affinity of the wild-type D1 receptor (1.9 μm). The sensitivity to GTP of high affinity agonist binding, and the ability of antagonists to inhibit basal cAMP accumulation via L268A were not assessed.
It has been proposed that the structural instability of a mutant β2-adrenergic receptor confers on the receptor its constitutive activity because it is able to isomerize more readily between the R and R* conformations (27). The constitutive activity of D2[1–4,7] may reflect enhanced formation of R* due to a global effect on helix packing, as proposed for a chimeric m2/m5 muscarinic receptor that constitutively activates Gq (28). Because the IC3 (29) and peptides derived from the IC3 of G protein-coupled receptors (30-33) have an intrinsic ability to activate G proteins, we propose that this intrinsic activity is typically constrained by intramolecular interactions among the transmembrane helices. Disruption of helix packing in D2[1–4,7] may mimic the agonist-induced relaxation of the receptor that permits functional coupling to G proteins (34).
We also observed evidence for unliganded activity of the wild-type D2 receptor. All the antagonists that we tested enhanced forskolin-stimulated cAMP accumulation in HEK 293 cells expressing the wild-type D2 receptor but not in untransfected cells, suggesting that the D2receptor constitutively inhibits adenylate cyclase. The potentiation of cAMP accumulation at these receptors likely reflects inverse agonist inhibition of the constitutively active receptors, consistent with the haloperidol-induced enhancement of prolactin release from GH4C1 cells expressing D2 receptors (35). It is noteworthy that the density of receptors on the cells used in the current study (480 fmol/mg) is similar to the density of D2receptors in brain regions in which the receptors are expressed most abundantly (36).
In summary, restoration of function in D2/D1 chimeric receptors that have D2 receptor sequence in TMI, TMII, and TMVII is consistent with a model in which interactions among these helices maintain receptor function. Our data support the view that multiple D2 receptor cytoplasmic domains acting in concert are necessary for receptor activation of Gi, whereas a chimeric receptor with only one cytoplasmic domain from the D1 receptor, IC3, was capable of activating Gs. Furthermore, this chimeric receptor constitutively activated adenylate cyclase, had high affinity for agonists, and revealed the inverse agonism of a number of antagonists. The wild-type D2 receptor may constitutively inhibit adenylate cyclase activity, as indicated by antagonist-induced potentiation of cAMP accumulation.
Acknowledgments
We thank Drs. Aaron Janowsky and Val Watts for careful reading of the manuscript.
Footnotes
- Received April 7, 1997.
- Accepted August 29, 1997.
-
Send reprint requests to: Dr. Kim Neve, Research Service (151LL), VA Medical Center, 3710 S.W. U.S. Veterans Hospital Road, Portland, OR 97201. E-mail: nevek{at}teleport.com
-
This work was supported by the Veterans Affairs Merit Review and Research Career Scientist Programs.
Abbreviations
- ΔG°
- change in free energy
- 6
- 7-ADTN, (±)-2-amino-6,7-dihydroxy-1,2,3,4-tetrahydronaphthalene
- BCS
- bovine calf serum
- DHX
- dihydrexidine
- Gi
- G protein that inhibits adenylate cyclase
- Gs
- G protein that stimulates adenylate cyclase
- ICX
- intracellular loop, where Xis the loop number
- KH
- high affinity binding state
- KL
- low affinity binding state
- NPA
- N-propylnorapomorphine
- TM
- transmembrane domain
- HEK
- human embryonic kidney
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}