


Abstract
Hypoglycemic sulfonylureas (e.g., glibenclamide, glipizide, and tolbutamide) exert their stimulatory effect on excitatory cells by closure of ATP-sensitive potassium (KATP) channels. These channels are heteromultimers composed with a 4:4 stoichiometry of an inwardly rectifying K+ channel (KIR) subunit 6.x plus a sulfonylurea receptor (SUR). SUR1/KIR6.2 reconstitutes the neuronal/pancreatic β-cell channel, whereas SUR2A/KIR6.2 and SUR2B/KIR6.1 (or KIR6.2) are proposed to reconstitute the cardiac and the vascular smooth muscle-type KATP channels, respectively. SUR2A and SUR2B are splice variants of a single gene differing only in their C-terminal 42 amino acids. Affinities of sulfonylureas for rat SUR2A, rat or human SUR2B, and a SUR2 chimera containing the C-terminal 42 amino acids of SUR1 did not differ significantly, implying that the C terminus does not form part of the binding pocket. Consistent with these findings, reconstituted SUR2A/KIR6.2 and SUR2B/KIR6.2 channels revealed similar sensitivities for glibenclamide and tolbutamide. Dissociation constants of sulfonylureas for SUR2A and SUR2B were 10- to 400-fold higher than for SUR1, however, amazingly the benzoic acid derivative meglitinide did not show lower affinity for SUR2 isoforms. Potencies of glibenclamide, glipizide, tolbutamide, and meglitinide to inhibit activity of SUR1/KIR6.2 and SUR2B/KIR6.2 channels were 3- to 6-fold higher than binding affinities of these drugs with concentration-inhibition relations being significantly steeper (Hill coefficients 1.23–1.32) than binding curves (Hill coefficients 0.93–1.06). The data establish that the C terminus of SURs does not affect sulfonylurea affinity and sensitivity. We conclude that occupation of one of the four SUR sites per channel complex is sufficient to induce KATP channel closure.
Hypoglycemic sulfonylureas (e.g., glibenclamide, glipizide, and tolbutamide) are widely used in the therapy of noninsulin-dependent diabetes mellitus. These drugs exert their stimulatory effect on insulin secretion by interaction with a high-affinity sulfonylurea receptor in the plasma membrane of pancreatic β cells (SUR1). Occupation of this receptor induces closure of the ATP-sensitive potassium (KATP) channel of these cells thereby depolarizing the plasma membrane and initiating the events finally leading to exocytosis of insulin (Ashcroft and Rorsman, 1991; Edwards and Weston, 1993; Aguilar-Bryan et al., 1998). Recent progress resulted in cloning of KATP channels and elucidation of their subunit composition (Aguilar-Bryan et al., 1995, 1998; Inagaki et al., 1995, 1996; Isomoto et al., 1996; Clement et al., 1997; Yamada et al., 1997). These channels are assembled with a tetradimeric stoichiometry, (SUR/Kir6.x)4, from two structurally distinct subunits, the regulatory SUR plus a pore-forming inwardly rectifying K+ channel (KIR) subunit 6.1 or 6.2. Three isoforms of SURs have been cloned, SUR1 and two splice products of a single gene, SUR2A and SUR2B, differing only in their C-terminal 42 to 45 amino acids (Isomoto et al., 1996; Chutkow et al., 1996; Aguilar-Bryan et al., 1998). SUR1/KIR6.2 has been proposed to reconstitute the neuronal/pancreatic β-cell (Inagaki et al., 1995), SUR2A/KIR6.2 the cardiac (Inagaki et al., 1996;Okuyama et al., 1998), and SUR2B/KIR6.1 (or KIR6.2) the vascular smooth muscle-type KATP channels (Isomoto et al., 1996; Yamada et al., 1997; Schwanstecher et al., 1998).
Still, important questions of sulfonylurea action have not been addressed. SURs have been shown to represent the receptors for potassium channel openers (KCOs) and the C terminus to be critical for binding of these drugs (Schwanstecher et al., 1998). However, the role of the C terminus in sulfonylurea binding and action has yet not been defined. Although it seems clear that sulfonylureas exert their effect by interaction with the SUR subunit, it is unknown how many of the four subunits per complex have to be occupied to induce channel closure.
In this report, we establish that the C terminus of SURs does not affect sulfonylurea affinity and sensitivity. The data indicate that potencies of sulfonylureas to close recombinant SUR1/KIR6.2 or SUR2B/KIR6.2 channels are significantly higher than affinities of the drugs. These findings and steeper concentration inhibition curves are explained by a model in which channel closure is induced by occupation of one of the four SUR sites per channel complex.
Experimental Procedures
Materials.
[3H]P1075 (specific activity 116 Ci/mmol) was purchased from Amersham Pharmacia Biotech (Freiburg, Germany). [3H]glibenclamide (specific activity 51 Ci/mmol) was obtained from NEN (Dreieich, Germany). All other chemicals and drugs were obtained from the sources described elsewhere (Schwanstecher et al., 1992a, 1994). Stock solutions of all drugs were prepared in KOH (20–50 mM) or dimethyl sulfoxide with a final solvent concentration in the media below 1%.
Molecular Biology.
SUR2/ct1 and SUR2/ctB were constructed as described (Schwanstecher et al., 1998), substituting the complementary DNA coding for the C-terminal 42 amino acids of rat SUR2A with the corresponding sequences from hamster SUR1 or human SUR2B. Rat SUR2B was obtained from SUR2/ctB by replacing asparagine in position 1538 against asparatic acid. Hamster SUR1 and rat KIR6.2 were fused tail-head through a six glycine linker (SUR1∼KIR6.2; Clement et al., 1997). Hamster SUR1 1540X (Table 1) was constructed by substitution of the cDNA triplet coding for histidine in position 1541 by a stop codon. The resulting products were subcloned into the pECE vector and sequenced to verify chimeric constructs or point mutations and PCR fidelity before transfection.
Sulfonylurea affinities of SUR isoforms in membranes or intact cells transiently expressed with or without inward rectifier subunit
Binding Assays.
Transfections and membrane preparations were performed as described (Schwanstecher et al., 1992a, 1998). Briefly, COS-7 cells cultured in Dulbecco’s modified Eagle’s medium (DMEM) HG (10 mM glucose), supplemented with 10% fetal calf serum, were plated at a density of 5 × 105cells per dish (94 mm) and allowed to attach overnight. Two hundred micrograms of pECE-SUR with or without pSV-mouse KIR6.2 or pECE-rat KIR6.1 complementary DNA were used to transfect ten plates. For transfection the cells were incubated 4 h in a Tris-buffered salt solution containing DNA (5–10 μg/ml) plus DEAE-dextran (1 mg/ml), 2 min in HEPES-buffered salt solution plus dimethyl sulfoxide (10%), and 4 h in DMEM-HG plus chloroquine (100 μM). Cells were then returned to DMEM-HG plus 10% fetal calf serum and were used 60 to 72 h post-transfection to assay binding or prepare membranes as described (Schwanstecher et al., 1992a). To assay binding to the intact cells, they were washed twice, resuspended (final concentration 2–4 · 105 cells/ml) and incubated in “extracellular solution” (final concentrations in mM: 140 NaCl, 5.6 KCl, 1.2 MgCl2, 2.6 CaCl2, 5 Tris, pH 7.4) containing [3H]P1075 (final concentration 3 nM, nonspecific binding defined by 100 μM pinacidil) or [3H]glibenclamide (final concentration 3 or 10 nM and nonspecific binding defined by 100 nM or 30 μM glibenclamide for SUR1 or SUR2B, respectively) and displacing drugs as indicated in Table 1. Incubations were carried out for 2 h at room temperature and were terminated by rapid filtration through Whatman GF/B filters. To measure binding to membranes from COS-cells or pancreatic islets the resuspended fraction (final protein concentration 5–50 μg/ml) was incubated in Tris-buffer (50 mM, pH 7.4) containing either [3H]glibenclamide (final concentration 0.3 nM, nonspecific binding defined by 100 nM glibenclamide) or [3H]P1075 (final concentration 3 nM, nonspecific binding defined by 100 μM pinacidil) and other additions as shown in the figures and tables. The free Mg2+concentration was kept close to 0.7 mM. ADP (0.3 mM) was added to the incubations with SUR1 to warrant identical nucleotide concentrations in binding and patch-clamp experiments (Fig.1, Tables 1 and2). ATP (0.1 mM) was added to incubation media for SUR2 isoforms to enable [3H]P1075 binding (Schwanstecher et al., 1998). Incubations were carried out for 1 h at room temperature and were terminated by rapid filtration through Whatman GF/B filters. Membranes from pancreatic islets of obese-hyperglycemic mice were prepared as described (Schwanstecher et al., 1991).

Binding affinities for SUR1 and potencies of sulfonylureas and meglitinide to inhibit SUR1/KIR6.2 channels. A, [3H]glibenclamide (0.3 nM) displacement assays were done with membranes from COS-7 cells expressing wild-type hamster SUR1. All incubations were performed in Tris-buffer (50 mM, pH 7.4) containing 0.3 mM ADP, 0.7 mM free Mg2+, and displacing drugs as indicated. The IC50 values (half-maximally inhibitory concentrations) and Hill coefficients are: 1.02 ± 0.09 nM, 0.94 (glibenclamide, ●); 24 ± 4 nM, 0.94 (glipizide, ○); 9.8 ± 1.0 μM, 0.93 (meglitinide, ▪); 41 ± 2 μM, 0.98 (tolbutamide, ■). B, glipizide-induced inhibition of hamster SUR1/KIR6.2 channels transiently expressed in COS-7 cells. Representative current recorded from an inside-out patch at −50 mV. Inward currents are shown as downward deflections. Free Mg2+ was maintained at 0.7 mM in all solutions. The patch was exposed to nucleotides and glipizide as indicated by the lines above the record. ADP was added to enhance maximal drug-induced inhibition (see Experimental Procedures). C, potencies of sulfonylureas and meglitinide to inhibit recombinant hamster SUR1/KIR6.2 channels. Channel inhibition was recorded in inside-out patches as shown in part B. Results are expressed as percentage of channel activity in control solution before and after application of test substances. The EC50 values (half-maximally effective concentrations) and Hill coefficients are: 0.13 ± 0.06 nM, 1.23 (glibenclamide, ●); 3.8 ± 1.2 nM, 1.26 (glipizide, ○); 1.2 ± 0.3 μM, 1.26 (meglitinide, ▪); 4.9 ± 1.6 μM, 1.30 (tolbutamide, ■). Results shown are mean ± S.E.M. (n = 4–8).
Sulfonylurea affinities and potencies in recombinant and native KATP channels
Electrophysiology.
Transfections were performed as described above with the following modification. COS cells were plated at a density of 8 × 104 cells per dish (35 mm). Twenty micrograms of pECE-hamster SUR1, rat SUR2A, or rat SUR2B complementary DNA and 20 μg of pSV-mouse KIR6.2 complementary DNA were mixed and used to transfect six 35-mm plates. Experiments in the inside-out configuration of the patch-clamp technique were performed at room temperature as described previously (Schwanstecher et al., 1994). Membrane patches were clamped at −50 mV. The intracellular bath solution contained (mM) 140 KCl, 2 CaCl2, 0.7 free Mg2+, 10 EGTA, 5 HEPES (pH 7.3) and the pipette solution 146 KCl, 2.6 CaCl2, 1.2 MgCl2 and 10 HEPES (pH 7.4). ADP (0.3 mM) enhances maximal sulfonylurea-induced inhibition of SUR1/KIR6.2 but not of SUR2A/KIR6.2-channels (Gribble et al., 1998). It was added to the bath solution of SUR1/KIR6.2 registrations to expedite registration of concentration-response curves. For construction of these curves patches were chosen with little “run-down” over the measuring period and drug effects were corrected for this constitutive loss of channel activity by use of linear interpolation. Artifacts due to incomplete drug wash-out or slow reversibility were excluded by making sure that cumulative experiments with stepwise increase or decrease of the drug concentration yielded identical EC50 values and slope factors. Channel activity (A) was defined as the product of the number of functional channels (N) and the probability of the channels being in the open state (p). (A) was calculated by dividing the mean current (I) by the single-channel current amplitude (i). Density of KATP channels per patch ranged from 15 to 50. Varying channel densities did not affect EC50values or Hill coefficients.
Data.
Data analysis (including calculation ofK Ds from IC50 values) and statistics were performed as described (Schwanstecher et al., 1992a, 1994). Results shown are mean ± S.E.M. (n= 3–16). Theoretical channel activity (A) in the presence of a given concentration of test drug (c) was calculated as follows: (i) (1 − b)4 (one site model) (ii) (1 − b)4 + 4 b · (1 − b)3 (two-site model) (iii) 1 − [b4 + 4 b3 · (1 − b)] (three-site model) (iv) 1 − b4(four-site model) with b = probability of drug binding at c assuming K D and Hill coefficient = 1 (see legend to Fig. 3D).

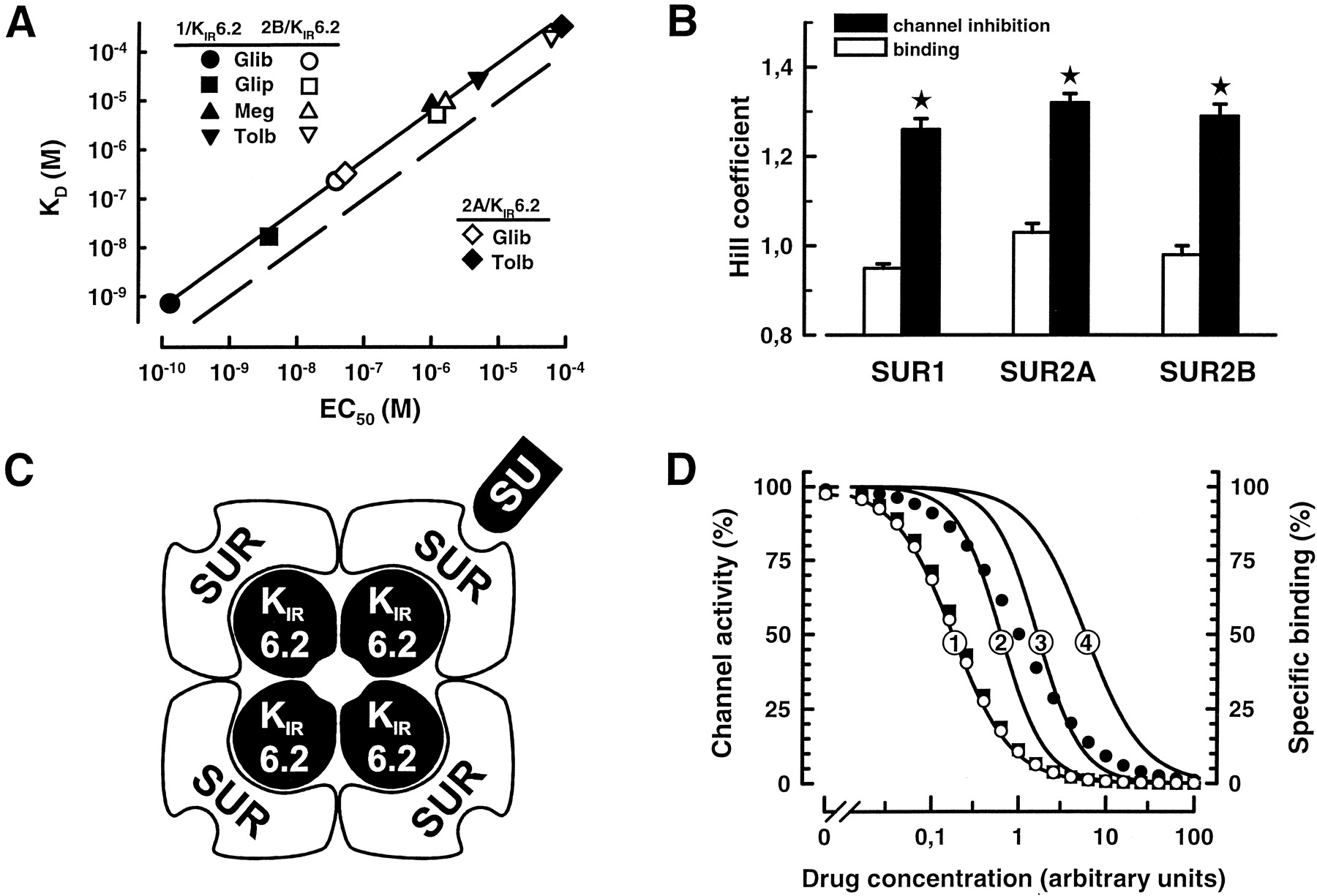
Stoichiometry of sulfonylurea action. A, potencies of sulfonylureas are higher than their affinities.K D and EC50 values were taken from Fig. 1, 2, and Table 1 (1 = hamster SUR1; 2A = rat SUR2A; 2B = rat SUR2B; Glib = glibenclamide, Glip = glipizide, Meg = meglitinide, Tolb = tolbutamide). The dashed line represents the relation expected for K D= EC50. B, slope factors for drug action are higher than those for drug binding. Hill coefficients for drug binding to SUR isoforms (hamster SUR1; rat SUR2A and B) or drug-induced inhibition of channels reconstituted with KIR6.2 were taken from Fig. 1,2, and Table 1. Results shown are mean ± S.E.M. (n = 6–18). *p < .01 for comparison with the corresponding Hill coefficient for drug binding. C, tetradimeric architecture of KATP channels (Clement et al., 1997). D, sulfonylurea-induced KATP channel closure is probably due to occupation of a single site. Theoretical concentration-inhibition curves were constructed assuming: 1) noncooperative binding (Hill coefficient = 1) withK D = 1 (the corresponding curve is represented by closed circles, ●, right ordinate) and 2) channel inhibition induced by occupation of one (1), two (2), three (3), or four (4) binding sites per channel (no matter which; seeExperimental Procedures).K D/EC50 ratios and Hill coefficients resulting from these models are: 1) 5.75 and 1.27, 2) 1.62 and 1.71, 3) 0.62 and 1.71, and 4) 0.17 and 1.27. Arithmetic means ofK D/EC50 ratios and Hill coefficients of channel inhibition for the four drugs tested were 5.42 ± 0.33 and 1.26 ± 0.02 (hamster SUR1/KIR6.2, Fig. 1, Table 1; the corresponding curve is represented by open circles, ○) or 4.93 ± 0.67 and 1.29 ± 0.02 (rat SUR2B/KIR6.2, Fig. 2, Table 2; the corresponding curve is represented by closed squares, ▪).
Results
SUR1/KIR6.2 Channels.
Competition binding experiments were performed to characterize the properties of the high-affinity sulfonylurea binding site on hamster SUR1. Unlabeled glibenclamide, glipizide, meglitinide, and tolbutamide induced complete monophasic inhibition curves with Hill coefficients close to 1 (0.93–0.98) yielding dissociation constants (K Ds) of 0.72 nM, 17 nM, 6.9 μM, and 29 μM (Fig. 1A; Table 1).
To assess the functional relevance of the sulfonylurea site on SUR1 the ability of the drugs to close reconstituted SUR1/KIR6.2 channels was tested. All of the drugs strongly inhibited KATP channel activity in inside-out patches transiently coexpressing SUR1 and KIR6.2 (Fig. 1, B and C). EC50 values (glibenclamide, 0.13 nM; glipizide, 3.8 nM; meglitinide, 1.2 μM; tolbutamide, 4.9 μM) were in the same rank order (glibenclamide < glipizide < meglitinide < tolbutamide) but 4.5- to 6-fold lower thanK D values for binding to SUR1 (Figs. 1, A and C, and 3A). Inhibition was reversible for all drugs tested with recovery of channel activity being particularly slow after application of glipizide (Fig. 1B) or glibenclamide. Hill coefficients for drug-induced channel closure were significantly higher than one ranging between 1.23 and 1.30 (Figs. 1C and 3B).
SUR2B/KIR6.2 Channels.
The affinity of [3H]glibenclamide for rat SUR2B was too weak to allow direct detection of binding to membrane fractions by use of the filtration assay. Therefore, binding of sulfonylureas and meglitinide to SUR2B was measured indirectly through the allosteric inhibition of high-affinity P1075 binding (Hambrock et al., 1998; Schwanstecher et al., 1998). Similarly to SUR1 all drugs induced complete monophasic displacement with Hill coefficients close to one (0.93–1.02) yielding, however, apparent dissociation constants (glibenclamide, 0.25 μM; glipizide, 6.1 μM; meglitinide, 9.2 μM; tolbutamide, 260 μM; Fig.2A, Table 1), which were up to 400-fold higher than K Ds for binding to hamster SUR1. Interestingly, however, meglitinide did not show markedly lower affinity for binding to SUR2B (K D = 9.2 μM; Fig. 2A, Table 1) than SUR1 (K D = 6.9 μM; Fig. 1A, Table 1).

Binding affinities for SUR2B and potencies of sulfonylureas and meglitinide to inhibit SUR2B/KIR6.2 or SUR2A/KIR6.2 channels. A, [3H]P1075 (3 nM) displacement assays were done with membranes from COS-7 cells expressing wild-type rat SUR2B. All incubations contained 100 μM MgATP (to enable P1075 binding, see Experimental Procedures), 0.7 mM free Mg2+, and displacing drugs as indicated. The IC50 values (half-maximally inhibitory concentrations) and Hill coefficients are: 0.33 ± 0.04 μM, 0.99 (glibenclamide, ●); 7.9 ± 0.4 μM, 0.99 (glipizide, ○); 12 ± 1.7 μM, 0.93 (meglitinide, ▪); 340 ± 20 μM, 1.02 (tolbutamide, ■). B, glibenclamide-induced inhibition of rat SUR2B/KIR6.2 channels transiently expressed in COS-7 cells. Representative current recorded from an inside-out patch at −50 mV. Inward currents are shown as downward deflections. Free Mg2+ was maintained at 0.7 mM in all solutions. The patch was exposed to glibenclamide and ATP as indicated by the lines above the record. C, potencies of sulfonylureas and meglitinide to inhibit recombinant rat SUR2B/KIR6.2 or rat SUR2A/KIR6.2-channels. Channel inhibition was recorded in inside-out patches as shown in part B. Results are expressed as a percentage of maximal drug-induced inhibition of channel activity (60–80% of activity in control solution before and after application of test substances). The EC50 values (half-maximally effective concentrations) and Hill coefficients are: 42 ± 14 nM, 1.27 (glibenclamide, SUR2B, ●); 1.2 ± 0.4 μM, 1.32 (glipizide, SUR2B, ○); 1.6 ± 0.7 μM, 1.26 (meglitinide, SUR2B, ▪); 88 ± 21 μM, 1.29 (tolbutamide, SUR2B, ■); 45 ± 12 nM, 1.35 (glibenclamide, SUR2A, Δ); 85 ± 18 μM, 1.30 (tolbutamide, SUR2A, ▴). Results shown are mean ± S.E.M. (n = 3–8).
All drugs rapidly and reversibly inhibited activity of transiently expressed SUR2B/KIR6.2 channels with EC50 values (glibenclamide, 42 nM; glipizide, 1.2 μM; meglitinide, 1.6 μM; tolbutamide, 88 μM; Fig. 2, B and C), which, similarly to SUR1/KIR6.2 channels, were significantly lower than K D values for binding to SUR2B (3–6 fold; Figs. 2, A and C, and 3A). Hill coefficients for the concentration-inhibition curves ranged between 1.26 and 1.32 (Figs. 2C and 3B).
Role of C Terminus in Sulfonylurea Binding.
To analyze the importance of the SUR C terminus for sulfonylurea binding we assessed the affinities of glibenclamide, glipizide, meglitinide, and tolbutamide for rat SUR2A and a chimera containing the rat SUR2 “backbone” and the 42 C-terminal residues of hamster SUR1 (SUR2/ct1). The dissociation constants of these two isoforms did not differ significantly from those of rat or human SUR2B (Table 1). Consistent with these findings potencies of glibenclamide or tolbutamide to close SUR2A/KIR6.2 channels (EC50 = 45 nM or 85 μM, respectively; Fig. 2C) were similar to those observed for SUR2B/KIR6.2 channels (Fig. 2C).
Discussion
This study provides a new insight into the mechanism of sulfonylurea-induced closure of KATP channels strongly supporting the idea that interaction with only one of four receptor sites per channel is sufficient for the drugs to exert their effect. This conclusion is based on two findings: 1) potencies of sulfonylureas to inhibit activity of SUR1/KIR6.2, SUR2A/KIR6.2 or SUR2B/KIR6.2 channels were significantly (3.0- to 6.4-fold) higher than binding affinities (Fig. 3A) and 2) for all drugs tested, Hill coefficients for channel inhibition were notably higher than one (1.23–1.35; Fig. 3B).
The leftward shift of potencies versus affinities was neither induced by differences in the composition of the media in binding and patch-clamp experiments nor by loss of associated proteins (e.g., cytoskeletal elements) in the membrane preparation or coexpression with KIR6.2. This was shown by controls indicating that substitution of Tris-buffer with intracellular solution, assay of affinities in intact cells, and coexpression with or fusion to KIR subunits do not alterK Ds (Table 1). The conclusion that our data obtained in membranes and inside-out patches reflect the properties of the physiologic channel complexes is reinforced by close correlation with results from native tissues (Table 2).
Slope factors (Hill coefficients) for binding of sulfonylureas to SUR1, SUR2A, and SUR2B were entirely close to one, pointing to homogenous populations of noncooperative binding sites (Figs. 1A, 2A, 3B; Tables 1and 2). Thus, slope factors higher than one in channel-inhibition curves can not be explained by positive cooperativity of drug binding but strongly suggest positive functional interaction of the sites. High concentrations of sulfonylureas have been shown to directly act on a low-affinity site residing on KIR6.2 thereby closing the channel (Gribble et al., 1998). However, significant effects via this site were ruled out by choosing drug concentrations too low to affect KIR6.2 directly.
The most likely explanation for the apparent discrepancy between drug binding and action results from the subunit architecture of KATP channels. These channels require four SUR molecules to be active (Fig. 3C; Clement et al., 1997; Shyng and Nichols, 1997). Although stoichiometry of sulfonylurea binding to SUR1 is not yet clear, the displacement studies suggest one binding site per molecule and accordingly four sites per channel. Any combination of these sites might mediate channel closure, and we have constructed some of the resulting theoretical concentration-inhibition curves (Fig. 3D). One of these models, assuming inhibition from binding to just one and any of the four sites, almost precisely describes our findings (Fig.3D). EC50 values for inhibition of SUR1/KIR6.2 or SUR2B/KIR6.2 channels were 5.42 ± 0.33- or 4.93 ± 0.67-fold lower than the corresponding dissociation constants with Hill coefficients of 1.26 ± 0.02 or 1.29 ± 0.02, respectively. The one-site model predicts aK D/EC50 ratio of 5.75 and a slope factor of 1.27 (Fig. 3D). When using the inside-out configuration of the patch-clamp technique we expect a reduced concentration of lipophilic drugs at the cytoplasmic side of membrane patches due to diffusion into the pipette solution. Thus EC50 values are probably slightly overestimated, which would provide a plausible explanation for the small difference between measuredK D/EC50 ratios (5.42 or 4.93, see above) and the theoretical value (5.75). We conclude that binding of sulfonylureas to any of the four sites per channel is sufficient to induce closure of KATP channels.
KATP channel activity could not be suppressed completely by sulfonylurea concentrations 10- to 80-fold higher than EC50 values (Figs. 1C and 2C). This finding is consistent with previous reports for native pancreatic β-cells and SUR1/KIR6.2 or SUR2A/KIR6.2 channels (Schwanstecher et al., 1994; Gribble et al., 1997,1998). The reason for the failure to suppress channel openings entirely is unknown. However, presumably probability that sulfonylurea binding results in channel closure is lower than one. Consistent with that idea, the maximal amount of suppressable channel activity was found to depend on the SUR subtype, being higher for SUR1/KIR6.2 (85–95%, Fig. 1C) than SUR2(A or B)/KIR6.2 channels (60–80%, Fig. 2C).
KD values for binding to hamster SUR1 correspond well to those in membranes from mouse pancreatic islets (Table 2) strongly supporting the conclusion that SUR1 represents the SUR of pancreatic β cells. Affinity of rat SUR2A and SUR2B was too low to allow direct detection of [3H]glibenclamide binding to membranes and thus interaction with SUR2 isoforms was measured indirectly via negative allosteric coupling of the receptor sites for sulfonylureas and KCOs (Hambrock et al., 1998; Schwanstecher et al., 1998). Displacement of low-affinity [3H]glibenclamide binding to intact COS-cells transiently expressing SUR2B yieldedKDs that did not differ significantly from those obtained by use of the [3H]P1075 assay either in membranes or intact cells (Table 1). These data validate use of allosteric P1075 displacement to measure sulfonylurea affinities of SUR2 isoforms.
[3H]P1075 displacement gave regular curves for all drugs tested with Hill coefficients near one proposing binding to the same noncooperative site (Fig. 2A; Table 1). Sulfonylureas had the same rank order of affinities found for SUR1 (glibenclamide > glipizide > tolbutamide) with, however, significantly higherK Ds. Identical rank orders (Table 1, Fig.3A), negative allosteric coupling to the KCO site (SUR1, Schwanstecher et al., 1998; SUR2A, Table 1; SUR2B, Hambrock et al., 1998, Fig. 2A and Table 1), and similar EC50/K D ratios (Fig.3A) indicate a high degree of similarity within binding sites, suggesting that small sequence differences might be responsible for either high or low sulfonylurea affinity. Amazingly, the benzoic acid derivative meglitinide did not show markedly lower affinity for SUR2 isoforms and thus this structure could represent a basis for the development of SUR2-specific drugs.
Affinities and potencies were strictly correlated (Fig. 3A) indicating that the sulfonylurea binding sites detected on SUR1 or SUR2B represent the functionally relevant receptor sites. Interestingly, affinities for human SUR1 or SUR2B did not differ significantly from those for the corresponding hamster or rat isoforms (Table 1) supporting the hypothesis that conservation of the receptor sites might be important for regulation by endogenous ligands (Heron et al., 1998).
Affinities of sulfonylureas for SUR2A did not differ significantly from those for SUR2B and a SUR2 construct containing the C terminus of SUR1 (SUR2/ct1; Table 1). We conclude that the C-terminal 42 amino acids are not essential for sulfonylurea binding and thus most probably are not involved in formation of the binding pocket. Consistently, deletion of the C-terminal 42 amino acids (ha SUR1 1540X) does not affect sulfonylurea affinity of hamster SUR1 (Table 1). The data predict identical sulfonylurea sensitivities of SUR2A/KIR6.2 and SUR2B/KIR6.2 channels and, according to that idea, similar potencies were observed for glibenclamide (42 or 45 nM, respectively; Fig. 2C), meglitinide (1.6 or 0.5 μM; Fig. 2C, Gribble et al., 1998), and tolbutamide (88 or 85 μM; Fig. 2C). The conclusion that the two channel subtypes don’t differ in sulfonylurea sensitivity also conforms with published data for native cardiac and vascular KATP channels (Belles et al., 1987; Venkatesh et al., 1991; Findlay, 1992; Xu and Lee, 1994; Quayle et al., 1995).
Recently, sulfonylurea potencies have been reported that were significantly weaker than sensitivities determined in this study (EC50 for inhibition of SUR1/KIR6.2 by glibenclamide = 4 nM; EC50 for inhibition of SUR2A/KIR6.2 by tolbutamide = 1.7 mM;Gribble et al., 1998), suggesting that drug action might be underestimated using the Xenopus expression system.
Our data present new insight into molecular pharmacology of KATP channels establishing that the C terminus of SURs does not affect sulfonylurea affinity and sensitivity. We conclude that occupation of one of the four SUR sites per channel complex is sufficient to induce KATP channel closure.
Acknowledgments
We thank Dr. Joseph Bryan (Baylor College of Medicine, Houston, TX) for providing us with human SUR1, human SUR2B, the chimeric SUR2/ct1 construct, and the SUR1∼KIR6.2 fusion, and H. Fürstenberg, U. Herbort, G. Müller, C. Rattunde, and S. Warmbold for excellent technical assistance.
Footnotes
- Received January 13, 1999.
- Accepted March 24, 1999.
-
Send reprint requests to: Dr. M. Schwanstecher, Institut für Pharmakologie und Toxikologie, Universität Braunschweig, Mendelssohnstraβe 1, 38106 Braunschweig, Germany. E-mail: M.Schwanstecher{at}tu-bs.de
-
This work was supported by grants from the Deutsche Forschungsgemeinschaft (M.S. and C.S.).
Abbreviations
- KATP channel
- ATP-sensitive potassium channel
- SUR
- sulfonylurea receptor
- KIR
- inwardly rectifying K+ channel
- KCO
- potassium channel opener
- DMEM
- Dulbecco’s modified Eagle’s medium
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}