


Abstract
The mechanism by which receptors activate G proteins is unclear because a connection between the receptor and the nucleotide binding site has not been established. To investigate this mechanism, we evaluated the roles in receptor interaction of three potential receptor contact sites in αs: the α2/β4, α3/β5, and α4/β6 loops. Substitutions of αi2 homologs for αsresidues in the α2/β4 loop and alanine substitutions of residues in the α4/β6 loop do not affect activation by the β2-adrenergic receptor. However, replacement of five αs residues in the α3/β5 loop region with the homologous αi2 residues decreases receptor-mediated activation of αs and increases the affinity of Gs for this receptor. The substitutions do not alter guanine nucleotide binding or hydrolysis, or activation by aluminum fluoride, indicating that the effects on receptor interaction are not due to a destabilization of the guanine-nucleotide bound state. In a model of the receptor-G protein complex, the α3/β5 loop maps near the second and third intracellular loops of the receptor. The effects of the α3/β5 substitutions suggest that the wild-type residues may be receptor contact sites that are optimized to ensure the reversibility of receptor-G protein interactions. Furthermore, the α3/β5 region corresponds to an exchange factor contact site in both EF-Tu and Ras, suggesting that the mechanisms by which seven-transmembrane receptors and exchange factors catalyze nucleotide exchange may share common elements.
Heterotrimeric G proteins transmit signals from cell surface receptors to intracellular effector proteins that modulate a wide variety of physiological processes (Neer, 1995). The α- and βγ-subunits of G proteins are associated in the inactive GDP-bound form. Receptors activate G proteins by catalyzing replacement of GDP by GTP on the α-subunit. Binding of GTP leads to dissociation of the receptor from α · GTP and βγ, each of which can transmit signals to effectors.
Our understanding of the mechanism of receptor-mediated activation of G proteins is incomplete. The predicted membrane-facing side of the heterotrimer includes the amino and carboxyl termini of the α-subunit and places the nucleotide binding site too far away to contact the receptor (Fig. 1). Therefore, receptors are thought to stimulate nucleotide exchange through currently undefined conformational changes transmitted from the sites of receptor binding to the nucleotide binding pocket (Bohm et al., 1997; Bourne, 1997).

Model of the receptor-G protein complex. The G protein heterotrimer is an αt/αi1 chimera complexed with βtγt (Lambright et al., 1996). The receptor model is the MII rhodopsin model (Pogozheva et al., 1997). Positioning of the heterotrimer relative to the receptor is based on the model proposed by Bourne (1997). The GTPase domain of the α-subunit is light blue. The helical domain of the α-subunit is pink. The GDP is yellow. Switch I is dark blue, switch II is green, and switch III is magenta. The α2/β4 loop is yellow, the α3/β5 loop is orange, and the α4/β6 loop is red. The carboxyl terminus (labeled C) of the α-subunit is magenta. Selected regions of secondary structure in the α-subunit, including the amino-terminal α-helix (αN), are indicated. The β-strands of the β-subunit are orange, and the amino-terminal helix and connecting loops are yellow. The γ-subunit is white. Receptor helices are numbered, and those connected to each other by an intracellular loop are the same color. This figure was drawn using MidasPlus, developed by the Computer Graphics Laboratory at the University of California at San Francisco.
The α-subunits consist of two domains: a GTPase domain that resembles that of EF-Tu and Ras, and a helical domain consisting of α-helices and connecting loops (Fig. 1). The bound nucleotide is buried between these domains. Three regions in the GTPase domain (switches I–III; Fig. 1) assume different conformations in the structures of guanosine 5′-O-(3-thiotriphosphate) (GTPγS)-bound versus GDP-bound α-subunits (Noel et al., 1993; Coleman et al., 1994; Lambright et al., 1994; Mixon et al., 1995). These regions are likely to play a role in receptor-mediated activation, because switches I and II contact βγ (Lambright et al., 1996), with which α must associate to be activated by receptors (Fung, 1983), and mutations that disrupt interactions between switch III and the helical domain impair receptor-mediated activation (Grishina and Berlot, 1998; Marsh et al., 1998; Warner et al., 1998).
Previous studies have identified regions of αsthat play a role in activation by the β2-adrenergic receptor (Hildebrandt et al., 1991; Codina and Birnbaumer, 1994; Iiri et al., 1997; Grishina and Berlot, 1998; Marsh et al., 1998; Warner et al., 1998). Of these regions, many are near the nucleotide, and only the extreme carboxyl terminus is likely to be a receptor contact site. However, the carboxyl terminus is not always necessary or sufficient to confer receptor specificity (Lee et al., 1995; Conklin et al., 1996), indicating that additional receptor-binding sites on the α-subunit have not been identified. Furthermore, it is not clear how receptor-dependent changes in the position of the carboxyl terminus would affect the conformational switch regions or interactions across the domain interface. Identification of the additional receptor binding site or sites is necessary to establish a connection between the receptor, the conformational switch regions, and the nucleotide binding site.
In this report, we evaluate the roles of three adjacent regions of αs, the α2/β4, α3/β5, and α4/β6 loops, in receptor-mediated activation. The location of these regions on the membrane-facing side of the molecule made them viable candidates for being receptor contact sites (Fig. 1). However, they were not tested previously in a comprehensive scanning mutagenesis study of αs that used receptor-stimulated cAMP accumulation as the read-out for receptor-mediated activation (Marsh et al., 1998), because substitutions in these regions with αi2 residues impair the activation of adenylyl cyclase (Itoh and Gilman, 1991; Berlot and Bourne, 1992).
In the crystal structure of αs complexed with the catalytic domains of adenylyl cyclase (Tesmer et al., 1997), the α2/β4 and α3/β5 regions contact adenylyl cyclase, but the α4/β6 loop does not. Therefore, using alanine-scanning mutagenesis, we reevaluate the role of the α4/β6 loop in adenylyl cyclase activation as well as test its role in receptor-mediated activation. None of the alanine substitutions cause specific defects in adenylyl cyclase activation or in receptor-mediated activation, suggesting that the loop plays an indirect role in adenylyl cyclase activation and does not mediate receptor binding.
Replacement of αs residues with αi2 homologs in the α3/β5 loop, but not the α2/β4 loop, decreases receptor-mediated activation and increases affinity for the β2-adrenergic receptor. The effects of the α3/β5 substitutions are not due to a destabilization of the nucleotide-bound state, suggesting that the wild-type residues may be receptor contact sites that are optimized to ensure the reversibility of receptor-G protein association. The correspondence of the α3/β5 region to an exchange factor contact site in both EF-Tu and Ras suggests that the mechanisms by which seven-transmembrane receptors and exchange factors catalyze nucleotide exchange may share common elements.
Experimental Procedures
Materials.
The mammalian expression vector pcDNA I/Amp was obtained from InVitrogen (Carlsbad, CA). The bacterial expression vector pQE60, plasmid maxi kits, and Ni2+ NTA resin were obtained from Qiagen (Santa Clarita, CA). Q Sepharose Fast Flow resin and ECL Western blotting detection reagents were obtained from Amersham Pharmacia Biotech (Piscataway, NJ). Isoproterenol, 1-methyl-3-isobutylxanthine, cAMP, ATP, tosylphenylalanyl chloromethyl ketone-treated trypsin (T-8642), and Lubrol-PX were obtained from Sigma Chemical Co. (St. Louis, MO). Complete, EDTA-free protease inhibitor cocktail tablets were obtained from Boehringer Mannheim (Indianapolis, IN). Nitrocellulose filters for the GTPγS binding assay were obtained from Millipore (Bedford, MA). Norit-SA3 was obtained from Aldrich (Milwaukee, WI). NuPAGE Bis-Tris 4–12% gels were obtained from NOVEX (San Diego, CA). [2-3H]Adenine was obtained from Amersham Pharmacia Biotech (Little Chalfont, UK). [35S]GTPγS, [γ-32P]GTP, and [125I]iodocyanopindolol (ICYP) were obtained from New England Nuclear (Boston, MA).
Construction of αs Mutant Constructs.
For expression in mammalian cells, αs mutant constructs were generated from the rat αs cDNA and contain an epitope, referred to as the EE epitope, that was generated by mutating αs residuesDYVPSD(189–194)1 toEYMPTE (single-letter amino acid code, mutated residues are underlined). For expression inEscherichia coli, αs mutant constructs were generated from the long splice variant of bovine αs containing a carboxyl-terminal hexahistidine tag, which was a generous gift from Alfred Gilman. All mutations were generated by oligonucleotide-directed in vitro mutagenesis using the Bio-Rad Muta-Gene kit except for those in αs(α3/β5), which were produced by subcloning a mutagenic oligodeoxynucleotide cassette. Subcloning and mutagenesis procedures were verified by restriction enzyme analysis and DNA sequencing.
cAMP Accumulation Assay.
To determine the abilities of αs mutant constructs to activate adenylyl cyclase, the constructs were transiently expressed in COS-7 cells using DEAE-dextran under the control of the cytomegalovirus promoter in the expression vector pcDNA 1/Amp, and intracellular cAMP levels in cells labeled with [3H]adenine were determined as described previously (Medina et al., 1996).
To determine the abilities of αs mutant constructs to become activated by endogenous β2-adrenergic receptors, the constructs were introduced by electroporation into a subclone ofcyc− S49 lymphoma cells, which lack endogenous αs (Harris et al., 1985), that stably expresses Simian virus 40 large T antigen. cAMP accumulation in the presence of 1 mM 1-methyl-3-isobutylxanthine (a phosphodiesterase inhibitor) and in the presence or absence of the agonist isoproterenol (0.1 mM) was measured after labeling with [3H]adenine as described previously (Marsh et al., 1998).
Membrane Preparations from COS-7 Cells and Trypsin Assay.
COS-7 cells were transiently transfected with αs mutant constructs using DEAE dextran as described above. Membranes were prepared 48 h after transfection as described previously (Medina et al., 1996). The trypsin resistance assay was performed as described previously (Berlot and Bourne, 1992). Samples were resolved by SDS-polyacrylamide gel electrophoresis (10%), transferred to nitrocellulose, and probed with the anti-EE monoclonal antibody as described previously (Medina et al., 1996). The antigen-antibody complexes were detected using an anti-mouse horseradish peroxidase-linked antibody according to the ECL Western blotting protocol.
Preparation of Stable Cell Lines.
The αs constructs were expressed incyc−kin− S49 lymphoma cells, which lack endogenous αs(cyc−) and in which cAMP-dependent protein kinase is inactivated (kin−), and cell membranes were prepared as described previously (Grishina and Berlot, 1998).
Adenylyl Cyclase Assay.
Adenylyl cyclase activity in membranes of cyc−kin− S49 lymphoma cell lines expressing αs constructs was measured and used to determine EC50 values for stimulation of adenylyl cyclase by GTPγS in the presence and absence of isoproterenol as described previously (Grishina and Berlot, 1998).
Receptor Binding Assay.
Competition between isoproterenol and [125I]ICYP for binding to β2-adrenergic receptors in membranes ofcyc−kin− S49 lymphoma cell lines expressing αs constructs was measured as described by Grishina and Berlot (1998). The experimental data were analyzed for competition at two sites by nonlinear least-squares curve fitting as described by Grishina and Berlot (1998).KL and KH, the low- and high-affinity dissociation constants, were allowed to vary under the two conditions.
Expression and Purification of αs from E. coli.
Both αs and αs(α3/β5) in the plasmid pQE60 were expressed in E. coli strain JM109. Cultures were grown, αs expression was induced, and lysates were produced as described previously (Lee et al., 1994), except that Complete, EDTA-free protease inhibitor cocktail tablets were included in the lysis buffer. The supernatant from a 30-min, 25,000gcentrifugation was applied to a Ni2+ NTA column that had been equilibrated with buffer A (50 mM Tris, pH 8.0, 20 mM 2-mercaptoethanol, 50 μM GDP, 1 mM phenylmethylsulfonyl fluoride, and 1 Complete, EDTA-free protease inhibitor cocktail tablet/50 ml). The column was washed sequentially with buffer A containing 500 mM NaCl and buffer A containing 50 mM NaCl and 10 mM imidazole before elution with buffer A containing 50 mM NaCl, 150 mM imidazole, and 10% glycerol. The protein was concentrated and exchanged into buffer B (50 mM Tris, pH 8.0, 1 mM EDTA, 2 mM dithiothreitol (DTT), and 10% glycerol) containing 50 μM GDP. The protein was then applied to a Q Sepharose Fast Flow column. The column was washed with buffer C (50 mM Tris, pH 8.0, 1 mM EDTA, 5 mM MgCl2, 14.5 mM 2-mercaptoethanol, 25 μM GDP, and 10% glycerol), and αs was eluted with buffer C containing a linearly increasing gradient of NaCl (0–300 mM). Peak fractions were concentrated, exchanged into buffer B containing 10 μM GDP, snap-frozen in liquid nitrogen, and stored at −80°C at a concentration greater than 2 mg/ml.
GTPγS Binding Assays.
To measure association of GTPγS, 100 nM αs or αs(α3/β5) was incubated at 20°C with 1 μM [35S]GTPγS (5 × 104 cpm/pmol) in a buffer containing 25 mM HEPES (pH 8.0), 10 mM MgCl2, 1 mM EDTA, 100 mM NaCl, and 1 mM DTT. At various times, aliquots (50 μl; 5 pmol) were withdrawn and immediately filtered under vacuum on nitrocellulose filters. The filters were rinsed twice with 10 ml of ice-cold Stop Buffer (25 mM Tris, pH 8.0, 100 mM NaCl, and 25 mM MgCl2). Apparent on rates of GTPγS binding (kapp) were calculated by a nonlinear least-squares fit to the equation:
Single Turnover GTPase Assay.
One-hundred nanomolar αs or αs(α3/β5) was incubated at 20°C with 1 μM [32P]GTP (2 × 104 cpm/pmol) in a buffer containing 50 mM HEPES (pH 8.0), 1 mM EDTA, and 1 mM DTT. After 30 min, the first aliquot (50 μl; 5 pmol) was withdrawn, and MgCl2 and GTP were added to final concentrations of 10 mM and 100 μM, respectively. Aliquots were withdrawn at various times and added to 750 μl of ice-cold 5% (w/v) Norit-SA3 in 50 mM NaH2PO4. Samples were microcentrifuged, and [32P]Pi released in the supernatant was determined by liquid scintillation counting. Catalytic rates of GTP hydrolysis (kcat) were calculated by a nonlinear least-squares fit to the equation:
Trypsin Assay Using Purified αs.
αs or αs(α3/β5) (2.7 μM) was incubated at 30°C for 30 min in a buffer containing 20 mM HEPES (pH 8.0), 2 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 0.025% Lubrol-PX, and activators as indicated. Tosylphenylalanyl chloromethyl ketone-treated trypsin was then added to a final concentration of 0.4 mg/ml, and the mixture was incubated for 5 min at 30°C. The digestion was terminated by adding soybean trypsin inhibitor to a final concentration of 1 mg/ml. The samples were resolved by polyacrylamide gel electrophoresis on a NuPAGE Bis-Tris 4–12% gel. Proteins were visualized by staining with Coomassie Blue.
Results
Role of α4/β6 Loop in Adenylyl Cyclase Activation.
Substitutions of αi2 residues for αs residues in the α4/β6 loop were shown previously to decrease activation of adenylyl cyclase by αs (Berlot and Bourne, 1992). However, in the crystal structure of αs complexed with the catalytic domains of adenylyl cyclase (Tesmer et al., 1997), the α4/β6 loop does not contact adenylyl cyclase. In the structure of αs (Sunahara et al., 1997), the α3/β5 and α4/β6 loops are closer together than in the corresponding structures of αt (Noel et al., 1993) and αi1 (Coleman et al., 1994), in part because of an interaction between Trp277 in α3/β5 and His357 in α4/β6. The proximity of these loops suggested that the effects of mutations in the α4/β6 loop might be due to changes in the conformation of the α3/β5 loop, which contacts adenylyl cyclase (Tesmer et al., 1997) and is important for its activation (Itoh and Gilman, 1991; Berlot and Bourne, 1992). To test this hypothesis, we determined the effects of substituting alanine residues in the α4/β6 loop (Fig.2A). The ability of mutant αs constructs to stimulate cAMP accumulation was measured after transient transfection in COS-7 cells. All constructs contained a GTPase-inhibiting mutation, referred to as the RC mutation, that replaces Arg201 with cysteine and causes constitutive activation, facilitating detection of adenylyl cyclase-activating ability above that of αs endogenous to COS-7 cells, as described previously (Berlot and Bourne, 1992).

Adenylyl cyclase activation by αsconstructs with substitutions in the α4/β6 loop. A, panel of αs mutant constructs with substitutions in the α4/β6 region. The top sequence is that of αs. Below that is the sequence of αi2. Residues that are identical with αs residues are represented by dashes. αs(α4/β6)1 and αs(α4/β6)2 are mutant αs constructs with decreased abilities to activate adenylyl cyclase (Berlot and Bourne, 1992). The αi2substitutions in αs(α4/β6)2 were based on an alignment of the αs and αi2 sequences that differs from the current alignment, which is based on the crystal structures of αs (Sunahara et al., 1997) and αi1 (Coleman et al., 1994). The remaining constructs contain alanine substitutions as indicated. B, top, cAMP accumulation in 106 COS-7 cells transfected with 1.5 μg of vector alone or vector containing αsRC or the indicated mutant constructs. cAMP levels in [3H]adenine-labeled cells were determined as described in Experimental Procedures. Each value represents the mean ± S.E. of three independent experiments. Bottom, expression and trypsin sensitivity of these constructs. COS-7 cells (6.25 × 106) were transfected with 3 μg plasmid/106 cells of vector alone or vector containing αsRC or the indicated constructs, and membranes were prepared, treated with trypsin, and immunoblotted as described in Experimental Procedures. The first lane in each set is the control (no trypsin). The second and third lanes show the result of trypsin digestion in the presence or absence, respectively, of GTPγS. Similar results were obtained in two additional experiments.
Figure 2B (top) shows the effects of the alanine substitutions on adenylyl cyclase activation. S349Aαs and T350A/S352Aαs did not exhibit defects in effector activation. These constructs span the region mutated in αs(α4/β6)1 (Fig. 2A), which exhibited reduced activity due to decreased expression levels (Berlot and Bourne, 1992). The remainder of the mutant constructs span the region mutated in αs(α4/β6)2 (Fig. 2A), which exhibited a specific defect in adenylyl cyclase activation (Berlot and Bourne, 1992) (see below). Significant decreases in the ability to activate adenylyl cyclase (P < .05) were caused by alanine substitutions of His357 and of Tyr358 and Tyr360 together (Fig. 2B).
We tested the specificity of the mutations that reduced stimulation of cAMP synthesis by determining the expression levels of the mutants in COS-7 cell membranes and their abilities to undergo an activating conformational change that is measured as the acquisition of resistance to trypsin digestion (Berlot and Bourne, 1992). In the presence of GTPγS, trypsin removes a short segment from the amino terminus of αs but leaves the remainder of the protein intact (Fig. 2B, bottom). In the absence of GTPγS, trypsin cleaves αs into small fragments not seen on the gel. Although all of the constructs assumed the activated conformation, those with decreased activities exhibited decreases in expression level (Fig. 2B, bottom). This qualitative link between activities and expression levels suggests that the alanine substitutions do not cause specific defects in adenylyl cyclase activation.
The effects of alanine substitutions in the α4/β6 loop suggest that residues in this loop are not directly involved in activation of adenylyl cyclase. Instead, the effector-activating defect of αs(α4/β6)2 is most likely due to altered interactions between the α3/β5 and α4/β6 loops. The importance of interactions between α3/β5 and α4/β6 for αs function is underscored by the observation that substitution of cysteine or alanine for Trp277 in the α3/β5 loop (which contacts His357) eliminates adenylyl cyclase activation. Substitution with cysteine does not affect expression level or the ability to assume the activated conformation (Itoh and Gilman, 1991), whereas substitution with alanine results in undetectable levels of expression (data not shown). It is also noteworthy that the α4/β6 loop contains two glycines that were replaced by αi2 residues in αs(α4/β6)2. These glycines may contribute a conformational flexibility to this αs region that is important for activation of adenylyl cyclase.
Role of α4/β6 Loop in Receptor-Mediated Activation.
Because αs mutants with alanine substitutions in the α4/β6 loop did not exhibit defects in ability to activate adenylyl cyclase when expression level was controlled for, we tested their responses to β2-adrenergic receptors by measuring receptor-dependent cAMP accumulation in transiently transfected cyc− S49 lymphoma cells, which lack endogenous αs (Harris et al., 1985). We used receptor-independent cAMP accumulation due to versions of the αs mutants containing the RC mutation (the RC versions) to normalize for expression level, as described previously (Grishina and Berlot, 1998; Marsh et al., 1998). For all of the constructs shown, the plasmid doses required to produce similar receptor-independent activities of the RC versions incyc− cells (Fig.3B) were consistent with their activities and expression levels in COS-7 cells (Fig. 2B). Y358A/Y360Aαs could not be evaluated in this assay due to the unexpectedly low activity of its RC version incyc− cells.

Receptor-mediated activation of αsproteins with mutations in the α4/β6 loop. A, cAMP accumulation incyc− cells transiently transfected with the indicated mutants in the αs context. Cells were electroporated with 30 μg of vector alone and of vector containing αs and S349Aαs, 45 μg of vector containing D354A/R356Aαs and C359A/P361Aαs, and 60 μg of vector containing H357Aαs. At these plasmid doses, similar amounts of receptor-independent cAMP accumulation were produced by versions of the constructs containing the RC mutation (B). cAMP values from unstimulated cells and from cells stimulated with 0.1 mM isoproterenol are dark gray and light gray, respectively. B, receptor-independent cAMP accumulation incyc− cells containing the indicated mutants in the αsRC context. For each mutant, the same amount of plasmid was used as is indicated in A for the corresponding αs mutant. cAMP levels in [3H]adenine-labeled cells were determined as described inExperimental Procedures. All values represent the mean ± S.E. of three independent experiments.
None of the alanine substitution mutants tested exhibited defects in receptor-stimulated cAMP accumulation (Fig. 3A). T350A/S352Aαs was previously tested in this assay and exhibited normal receptor-mediated activation (Marsh et al., 1998). Therefore, we conclude that the α4/β6 loop of αs is not involved in receptor-mediated activation.
Mutations in α3/β5 Region but Not α2/β4 Region of αs Decrease Receptor-Mediated Activation.
To test the roles of the α2/β4 and α3/β5 loop regions in receptor-mediated activation, we examined an αsconstruct referred to as αs(α2/β4), containing three substitutions of αi2 homologs for αs residues (Q236H/N239E/D240G) in the α2 helix and α2/β4 loop, and an αs construct referred to as αs(α3/β5), containing five αi2 homolog substitutions (N271K/K274D/R280K/T284D/I285T) in the α3 helix and α3/β5 loop, after stable expression in cyc− S49 lymphoma cells. (Figure 9 shows the locations of these substitutions in the secondary structure of αs.)

Alignment of GTPase sequences in switch II, the α3/β5 loop, and the α4/β6 loop. A, sequences of αs, αi2, αt, EF-Tu, and Ras in switch II. B, sequences of αs, αi2,αt, EF-Tu, and Ras from the beginning of α3 to the end of β5. C, sequences of αs, αi2, and αt from the α4/β6 loop to the beginning of α5. Residue numbers of αs, αi2,αt, EF-Tu, and Ras are indicated in parentheses. Elements of secondary structure in αs, determined from the structure of αs·GTPγS (Sunahara et al., 1997), are indicated by a (α-helices), b (β-strands), and dashes (turns and loops). In the sequence of αs, mutations of residues with filled circles over them impaired receptor-mediated activation (present report; Iiri et al., 1997; Marsh et al., 1998), mutations of residues with open circles above them did not impair receptor-mediated activation (present report; Masters et al., 1988; Marsh et al., 1998), the effects of mutations in residues with both open and filled circles over them were context-dependent, as described in the text, and the sequence of a peptide that can mimic the effects of αs on the β-adrenergic receptor (Rasenick et al., 1994) is underlined. In the sequence of αt, boxed residues represent residues that contact βγ, based on the structure of a heterotrimeric G protein (Lambright et al., 1996), mutations of residues with filled circles over them impaired receptor-mediated activation (Onrust et al., 1997), and mutations of residues with open circles above them did not impair receptor-mediated activation (Onrust et al., 1997). In the sequence of EF-Tu, boxed residues contact EF-Ts in the structures of EF-Tu·EF-Ts complexes (Kawashima et al., 1996; Wang et al., 1997) and mutations of residues with filled circles over them impaired exchange factor-mediated activation (Jonák et al., 1998; Zhang et al., 1998). In the sequence of Ras, boxed residues contact Sos in the structure of Ras·Sos (Boriack-Sjodin et al., 1998) and mutations of residues with filled circles over them impaired exchange factor-mediated activation (Mistou et al., 1992; Créchet et al., 1996; Leonardsen et al., 1996).
Stimulation of Gs by the β2-adrenergic receptor increases the apparent affinity of αs for GTPγS. This can be measured as an isoproterenol-dependent decrease in the half-maximal effective concentration (EC50) for GTPγS stimulation of adenylyl cyclase (Fig.4A). We used this response to receptor stimulation as the read-out for receptor-mediated activation of αs(α2/β4) and αs(α3/β5).
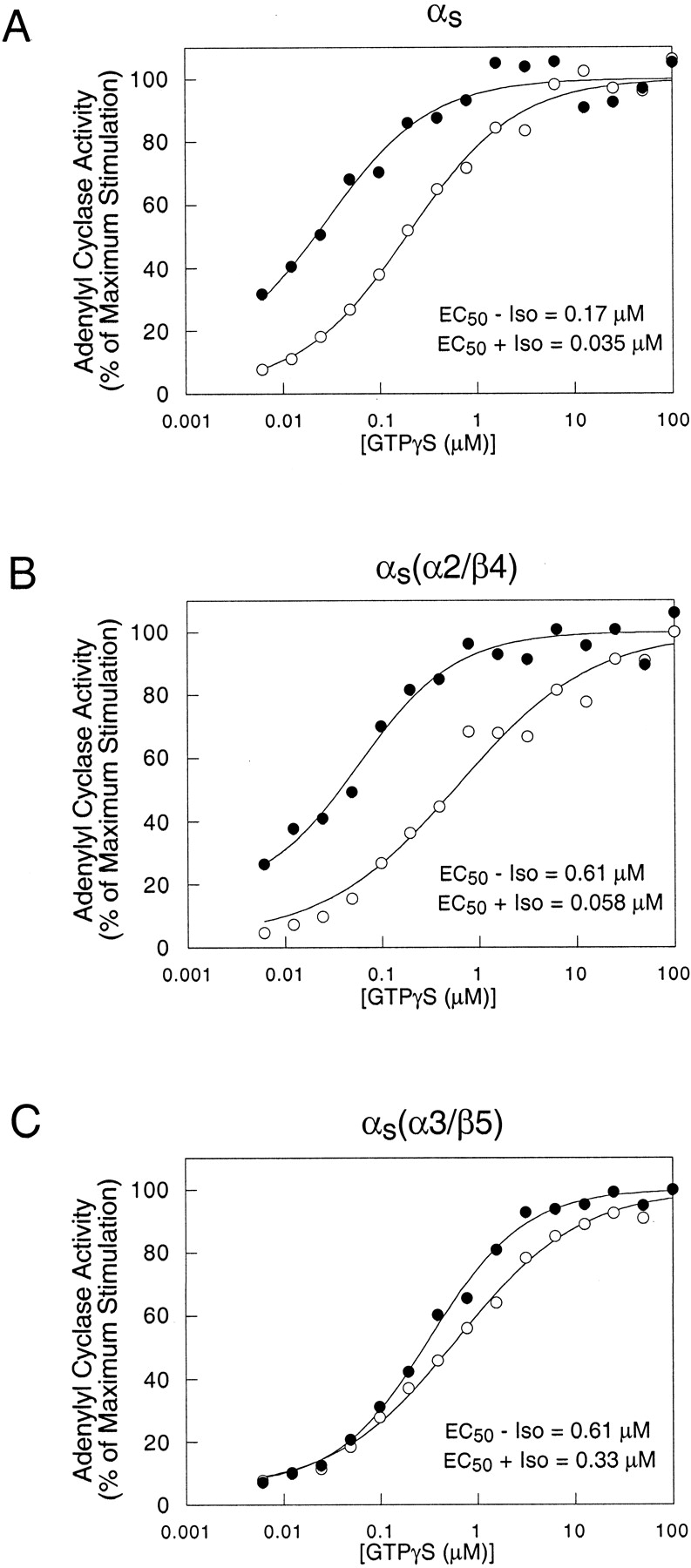
αs(α3/β5) exhibits a defect in receptor-mediated activation. Adenylyl cyclase activities in membranes of cyc− cells stably expressing αs, clone 3 (A), αs(α2/β4), clone 1 (B), or αs(α3/β5), clone 1 (C) were determined in the presence of the indicated concentrations of GTPγS in the presence (●) or absence (○) of 100 μM isoproterenol (Iso). Expression levels of these clones are shown in Fig. 5. Data points represent the mean values from three independent experiments and are expressed as the percentage of the maximum observed adenylyl cyclase activity. EC50 values were calculated as described previously (Grishina and Berlot, 1998).
Although mutations in the α2/β4 and α3/β5 regions decrease adenylyl cyclase activation by αs (Itoh and Gilman, 1991; Berlot and Bourne, 1992), sufficient activity remained to allow determination of the EC50 value for GTPγS activation of αs(α2/β4) and αs(α3/β5). αs(α3/β5) exhibited a substantially smaller isoproterenol-dependent decrease in the EC50 for GTPγS stimulation of adenylyl cyclase (approximately 2-fold) than did αs and αs(α2/β4), which exhibited approximately 5- and 10-fold decreases, respectively (Fig. 4).
Taken together with the results of a previous study, the results with αs(α3/β5) suggest that mutations in the α3/β5 loop rather than the α3 helix are responsible for the defect in receptor-mediated activation. Substitutions of αi2 homologs for the α3 residues Asn271 and Lys274 in combination with αi2 homolog substitutions of Leu266, Gln267, Ala269, and Leu270 (see Fig. 9B) did not impair receptor-mediated activation (Marsh et al., 1998). The remaining residues mutated in αs(α3/β5), Arg280, Thr284, and Ile285, are located in the α3/β5 loop.
Mutations in α3/β5 Region but Not α2/β4 Region of αs Increase Apparent Affinity of αs for β2-Adrenergic Receptor.
To determine whether the decreased receptor-mediated activation of αs(α3/β5) was due to altered receptor binding, we used a competitive binding assay that measures an αs-dependent increase in the affinity of the β2-adrenergic receptor for the agonist isoproterenol (Grishina and Berlot, 1998). The high-affinity isoproterenol-binding state of the receptor, which requires the presence of Gs in the nucleotide-free state, reflects receptor-Gs interaction. Isoproterenol binding is measured in competition with the antagonist ICYP, which binds to the receptor with the same affinity in the presence and absence of Gs.
We measured receptor affinities in membranes from three αs-expressing clones and two clones each expressing αs(α2/β4) and αs(α3/β5) (Fig.5A). The binding results from αs clone 3, αs(α2/β4) clone 1, and αs(α3/β5) clone 1 are shown (Fig. 5, B–D). Similar results were obtained with the other clones, demonstrating that within the range of expression levels examined, binding profiles were independent of expression level (data not shown).

Competition between isoproterenol and [125I]ICYP for binding to the β2-adrenergic receptor. A, immunoblot showing expression levels of αs, αs(α2/β4), and αs(α3/β5) in stablecyc− cell lines. Membranes of αs, clone 3, (B), αs(α2/β4), clone 1 (C), or αs(α3/β5), clone 1 (D), were incubated with [125I]ICYP (75 pM) and the indicated concentrations of isoproterenol in the presence (●) or absence (○) of 300 μM GTP. Values represent the mean values of two independent experiments. The solid lines represent a nonlinear least-squares fit to the data, as described previously (Grishina and Berlot, 1998).KL and KH are the low- and high-affinity dissociation constants, respectively, and % RH is the percentage of receptors in the high-affinity form. In C and D, the binding curves for membranes from αs-expressing cells, from B, are redrawn as dotted lines. Similar results for each construct were obtained in two additional experiments using the other cell lines in A.
In the presence of 300 μM GTP, receptors from αs-expressing cells were predominantly in the low-affinity state (Fig. 5B). In the absence of GTP, αs caused the appearance of high-affinity binding sites for isoproterenol on the receptor (Fig. 5B). In cells expressing αs(α2/β4), the affinity of the receptor for isoproterenol was similar to that in αs-expressing cells (Fig. 5C). However, in αs(α3/β5)-expressing cells, the affinity of the receptor for isoproterenol in both the presence and absence of GTP was greater than that in αs-expressing cells (Fig. 5D). A similar pattern of increased high-affinity binding in cells expressing αs(α3/β5) compared with αs was obtained when the assay was performed using 30 μM GTPγS (data not shown). The simplest explanation for the increased affinity of the receptor for isoproterenol in the presence of αs(α3/β5) compared with αs is that the affinity of αs(α3/β5) for the receptor is greater than that of αs.
αs(α3/β5) Exhibits Normal Guanine Nucleotide Handling Properties.
There is precedent for mutations that both decrease receptor-mediated activation of αs and impair guanine nucleotide binding (Iiri et al., 1997; Warner et al., 1998) and/or hydrolysis (Warner and Weinstein, 1999). In addition, because the affinity of G proteins for receptors is greatest in the nucleotide-free state, changes in nucleotide binding could affect apparent receptor affinity in the presence of nucleotide. Therefore, using purified αs and αs(α3/β5), we investigated whether the mutations in αs(α3/β5) are associated with intrinsic defects in guanine nucleotide handling.
The rates of association of GTPγS to αs and αs(α3/β5) were similar (Fig.6A). Because the rate of GTPγS association is limited by the rate of GDP dissociation, the mutations in αs(α3/β5) do not appear to increase GDP dissociation. The stability of GTPγS binding is also unaffected by the mutations in αs(α3/β5), because dissociation of GTPγS from both αs and αs(α3/β5) was undetectable (Fig. 6B). The intrinsic rates of GTP hydrolysis of αs and αs(α3/β5) were also the same (Fig. 6C). These results indicate that the decreased receptor-mediated activation and increased receptor affinity of αs(α3/β5) are not consequences of a destabilization of the guanine nucleotide-bound state.

Biochemical properties of αs and αs(α3/β5). A, rates of GTPγS binding. αs (●) or αs(α3/β5) (○) (100 nM each) was incubated at 20°C with 1 μM [35S]GTPγS (5 × 104 cpm/pmol). At the times indicated, aliquots (50 μl; 5 pmol) were withdrawn and filtered on nitrocellulose filters as described in Experimental Procedures. Apparent on rates of GTPγS binding (kapp) were calculated as described in Experimental Procedures. Data points represent the mean ± S.E. of six experiments. B, dissociation of GTPγS. αs (●) or αs(α3/β5) (○) (100 nM each) was incubated at 20°C with 1 μM [35S]GTPγS as described in A for 50 min. Dissociation of [35S]GTPγS was initiated by the addition of unlabeled GTPγS to a final concentration of 100 μM. At the times indicated, aliquots (50 μl; 5 pmol) were withdrawn and filtered on nitrocellulose filters as described in Experimental Procedures. Data points represent the mean ± S.E. of four experiments. The values of koff for both αs and αs(α3/β5) were indistinguishable from 0. C, kcat for the hydrolysis of GTP. αs (●) or αs(α3/β5) (○) (100 nM each) was incubated at 20°C with 1 μM [32P]GTP (2 × 104 cpm/pmol) for 30 min in the presence of 1 mM EDTA. After withdrawal of the first aliquot, MgCl2 and GTP were added to final concentrations of 10 mM and 100 μM, respectively. At the times indicated, aliquots (50 μl; 5 pmol) were withdrawn, and [32P]Pi released was determined as described inExperimental Procedures. Catalytic rates of GTP hydrolysis (kcat) were calculated as described in Experimental Procedures. Data points represent the mean ± S.E. of six experiments.
αs(α3/β5) Exhibits Normal Activation by AlF4−.
Some αsmutations (Hildebrandt et al., 1991; Codina and Birnbaumer, 1994; Iiri et al., 1997; Grishina and Berlot, 1998; Warner et al., 1998) that decrease receptor-mediated activation also decrease activation by AlF4−, which activates α-subunits by mimicking the γ-phosphate of GTP in the postulated transition state intermediate of the GTPase reaction (Coleman et al., 1994; Sondek et al., 1994). In contrast, AlF4− induced the trypsin-resistant activated conformation in αs(α3/β5) to the same extent as GTPγS did and to the same extent as in αs (Fig.7). Because activation by AlF4− requires that the nucleotide binding site contain GDP and be in an appropriate conformation, this result further supports the conclusion that the mutations in αs(α3/β5) specifically alter receptor-mediated activation and receptor binding without causing global structural distortions in αs.
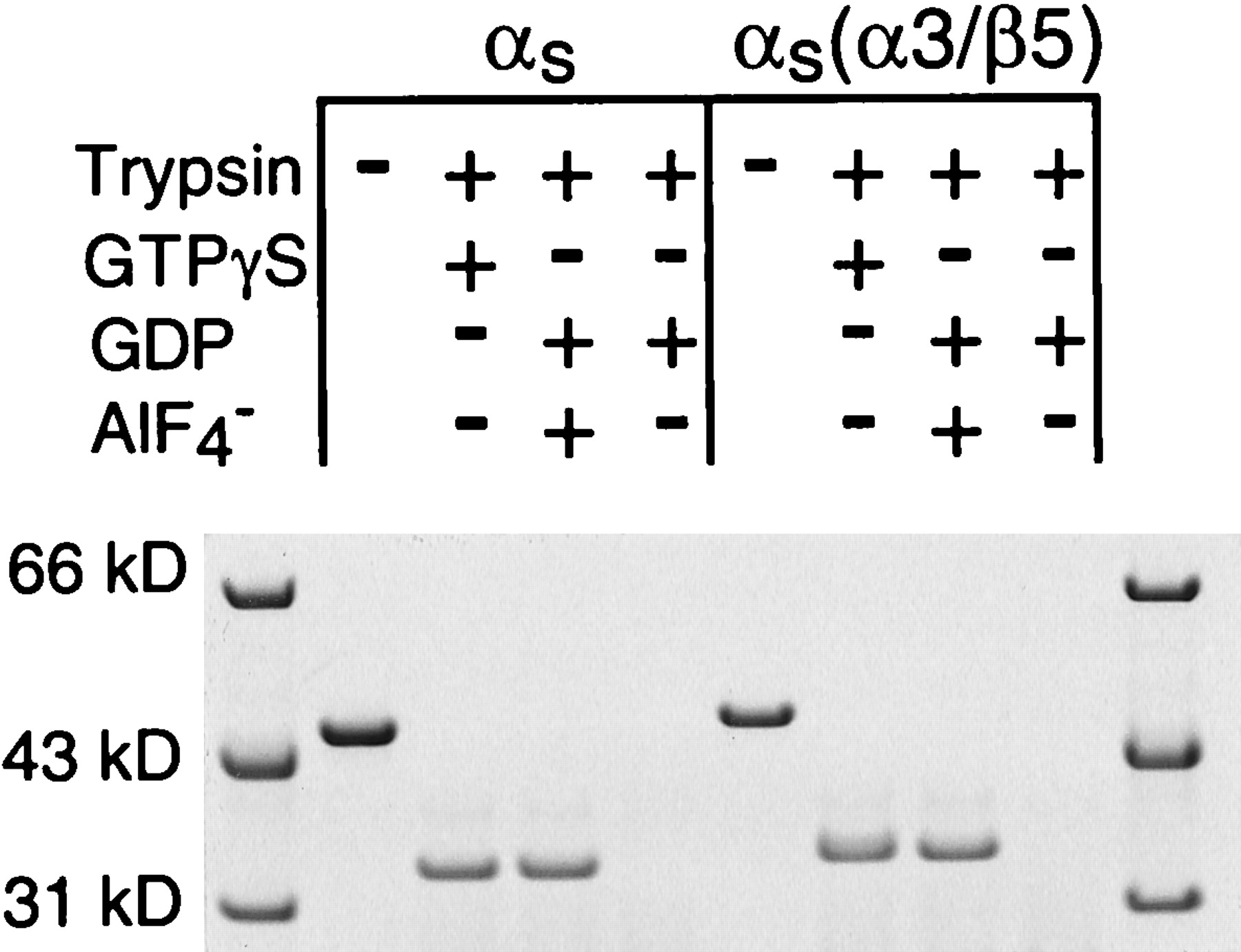
Effects of GTPγS and GDP/AlF4− on tryptic cleavage; 2.7 μM αs or αs(α3/β5) was incubated with 12.5 μM GTPγS, 12.5 μM GDP plus 25 μM AlCl3 and 12.5 mM NaF, or 12.5 μM GDP and then treated with trypsin as described inExperimental Procedures. The αs and trypsin-resistant fragments of αs were resolved by polyacrylamide gel electrophoresis and visualized by staining with Coomassie Blue. Similar results were obtained in two additional experiments.
Mapping of Residues Important for the Gs-β2-Adrenergic Receptor Interaction onto a Receptor-G Protein Model.
We visualized the results of this and previous studies of the αs residues important for interaction with the β2-adrenergic receptor using a model of the receptor-G protein complex with essentially the same constraints as the model proposed by Bourne (1997). The amino terminus of the α-subunit and the carboxyl terminus of the γ-subunit, which contain lipid modifications important for membrane attachment, face the membrane. The third intracellular loop of the receptor is located at the interface between α and βγ, near regions shown by mutagenesis and peptide studies to be important for receptor interaction (Bourne, 1997): the amino and carboxyl termini of α and the carboxyl terminus of β.
In this model, the residues substituted in the α3/β5 loop (280, 284, and 285, red in Fig. 8) are close to both the second and third intracellular loops of the receptor, indicating that they could be receptor contact sites. Substitutions of the residues in the α3 helix (271 and 274, pink in Fig. 8), in combination with additional substitutions, do not impair receptor-mediated activation (Marsh et al., 1998). The α3/β5 loop is in close proximity to both the α2/β4 and α4/β6 loops, suggesting that receptor-initiated signaling between the α3/β5 loop and the nucleotide binding pocket may involve one or both of these adjacent loops. The α3/β5 loop residues exhibit the same degree of solvent exposure in the presence or absence of βγ. However, the residue equivalent to Arg280 is ∼4 Å away from a βt residue (Arg314) with which it forms a water-mediated hydrogen bond. Thus, this region may influence interaction with βγ.

Mapping of residues important for Gs-β2-adrenergic receptor interaction onto a receptor-G protein model. αs residues are mapped onto the model of the receptor-G protein complex shown in Fig. 1A, ribbon diagram. The β-strands of the β-subunit are orange, and the amino-terminal helix and connecting loops are yellow. The γ-subunit is white. The GDP is yellow. αs residues in which substitutions with the homologous αi2 residues were shown previously to leave receptor-mediated activation intact (Masters et al., 1988; Grishina and Berlot, 1998; Marsh et al., 1998) are green. αs residues that have not been tested for their roles in receptor interaction are light blue. The residues mutated in αs(α3/β5) are red spheres (α3/β5 loop) or pink spheres (α3 helix). Additional residues in which mutations alter receptor interaction (Hildebrandt et al., 1991; Codina and Birnbaumer, 1994; Iiri et al., 1997; Grishina and Berlot, 1998; Marsh et al., 1998) are red, except for the carboxyl terminus (labeled C), which is magenta. Residues in the α2/β4 and α4/β6 loops, in which substitutions with αi2 homologs and alanine residues, respectively, do not alter receptor interaction, are green spheres. The numbers on the spheres represent αs residue numbers. Receptor helices are numbered, and those connected to each other by an intracellular loop are the same color. B, space-filling model viewing the heterotrimer surface that faces the receptor. The receptor helices are outlined in black, and the intracellular loops (i.c.1–3) and carboxyl-terminal tail are indicated. Colors are as in A except that β is entirely orange. These figures were drawn using MidasPlus, developed by the Computer Graphics Laboratory at the University of California at San Francisco.
Other than the α3/β5 loop region, the only αs region close to the receptor in which substitutions alter receptor interactions is the carboxyl terminus (Marsh et al., 1998) (C, magenta in Fig. 8). The other αs residues in which substitutions disrupt receptor interactions (red in Fig. 8A), located in α1 (Hildebrandt et al., 1991), the αD/αE loop (Codina and Birnbaumer, 1994; Grishina and Berlot, 1998), α2 (Iiri et al., 1997), the β4/α3 loop including switch III (Grishina and Berlot, 1998; Marsh et al., 1998;Warner et al., 1998), the β5/αG loop (Codina and Birnbaumer, 1994), the αG/α4 loop (Marsh et al., 1998), and the β6/α5 loop (Marsh et al., 1998), are farther from the receptor and in many cases near the nucleotide and/or buried, indicating that they probably are not receptor contact sites.
Shown in green are αs regions that do not specify interaction with the β2-adrenergic receptor. Among these regions is the α4/β6 loop, which does not appear to mediate receptor binding, because alanine substitutions throughout (residues 349, 350, 352, 354,2 356, 357, 359, 361) do not affect receptor-mediated activation (Fig. 3;Marsh et al., 1998). The other regions shown in green were tested by replacing αs residues with αi2 homologs (Masters et al., 1988; Marsh et al., 1998; present study). Homolog substitutions in these regions, which include the amino terminus and the α2/β4 loop (residues 236, 239, 240), do not rule out these regions as being receptor contact sites, because residues that are identical in αs and αi2 were not changed.
Discussion
We have identified a region of the G protein α-subunit, the α3/β5 loop, in which mutations decrease receptor-mediated activation of αs and increase the apparent affinity of Gs for the β2-adrenergic receptor. Based on its location, this region is likely to be a receptor contact site (Fig. 8). This region and the extreme carboxyl terminus are the only regions of αs that map near the receptor in which substitutions have been shown to alter receptor interaction. Of the other regions that map nearby, the α4/β6 loop does not appear to mediate receptor binding, because extensive substitutions with alanines do not affect receptor-mediated activation (Fig. 3). Substitutions of αs residues in the amino terminus (Masters et al., 1988) and the α2/β4 loop (Figs. 4 and 5), which are also near, with the homologous αi2residues indicate that these regions do not specify interaction with the β2-adrenergic receptor, but comprehensive alanine-scanning mutagenesis will be needed to determine whether they are required for receptor binding.
The fact that replacement of αs residues in the α3/β5 loop region with αi2 homologs increases receptor affinity is surprising but suggests that the wild-type residues may be optimized to ensure reversibility of receptor-G protein interaction during the GTPase cycle. This interpretation is consistent with previous observations that substitutions in the sequences of peptides based on the carboxyl terminus of αt (Martin et al., 1996) or a β-subunit region that interacts with phospholipase C (Buck et al., 1999) can cause increased affinities for rhodopsin or phospholipase C, respectively. If the α3/β5 loop region is a receptor contact site, it should be possible to find mutations that decrease receptor affinity. Further studies will be directed at testing the effects of substitutions that change the critical residues in different or opposite ways from the original mutations.
The defects in αs(α3/β5) are different from those of several previously described αs mutants with defects in receptor-mediated activation (shown in red in Fig. 8). Two mutant αs constructs with substitutions at the interface of the GTPase and helical domains, N254D/M255L/I257L/R258Aαs in switch III and N167Rαs in the αD/αE loop also exhibit an apparent increase in receptor affinity and decreased receptor-stimulated binding of GTPγS, but in contrast to αs(α3/β5), these constructs exhibit a decreased response to AlF4−(Grishina and Berlot, 1998). Defects in the response to AlF4− could be due to an altered nucleotide binding site, because the response requires bound GDP. Indeed, R258Aαs exhibits increased rates of both GDP dissociation (Warner et al., 1998) and GTP hydrolysis (Warner and Weinstein, 1999). Other mutations that decrease activation by both receptors and AlF4−include D173K in the αD/αE loop and K293D in the β5/αG loop (Codina and Birnbaumer, 1994), R231H in α2 (Iiri et al., 1997), and S54N in α1 (Hildebrandt et al., 1991). In contrast to the mutations in αs(α3/β5), each of these mutations appears to alter the nucleotide binding site.
Our data, combined with previous results, indicate that switch II does not determine the receptor specificity of αs. The α2/β4 loop is located in the carboxyl-terminal part of switch II. Involvement of the amino-terminal part of switch II in receptor specificity was ruled out previously by an αi2/αs chimera that substituted αi2 residues for αs residues 1 to 235 and that exhibits normal receptor-mediated activation (Masters et al., 1988) (Fig.9A). However, switch II undoubtedly plays an important role in the response to receptor stimulation, because residues in this region bind to βγ, with which α-subunits must be associated to interact with receptors (Fung, 1983). In support of such a role, substitution of histidine for a buried residue in switch II of αs, R231 (Figs. 8 and 9A), reduces receptor-mediated activation, possibly by disrupting the interaction of this residue with α3 and switch III (Iiri et al., 1997). Furthermore, alanine substitutions of αt residues in switch II (Fig. 9A) disrupt activation by rhodopsin (Onrust et al., 1997). Based on their locations, most of these substitutions probably disrupt βγ interaction.
Our data suggest that the α4/β6 loop of αsdoes not regulate binding to or activation by the β2-adrenergic receptor. Others have suggested that this region is a receptor contact site, because a synthetic peptide corresponding to αs residues 354 to 372, which extends from the α4/β6 loop to the beginning of α5 (Fig. 9C), can mimic the effects of αs on the β2-adrenergic receptor (Rasenick et al., 1994). However, within the region spanned by the peptide, the only substitutions that we have found to impair receptor-mediated activation of αs are of buried residues in the β6/α5 loop and the beginning of α5 (Fig. 9C; Marsh et al., 1998), which could not be receptor contact sites. In the case of αt, alanine substitutions of residues in the α4/β6 loop impaired activation by rhodopsin (Onrust et al., 1997; Fig. 9C). Interestingly, the α4/β6 loop is positioned differently in the structures of αs (Sunahara et al., 1997) and αt (Noel et al., 1993; Lambright et al., 1994, 1996). These differences in structure may explain why mutations in this region of αt but not αs impair receptor-mediated activation.
In EF-Tu and Ras, both the α2/β4 and α3/β5 loop regions are exchange factor contact sites (Fig. 9), suggesting that the mechanisms by which seven-transmembrane receptors and exchange factors catalyze nucleotide exchange may share common elements. In the structures of EF-Tu·EF-Ts (Kawashima et al., 1996; Wang et al., 1997), a phenylalanine residue from EF-Ts is inserted in between the helices that correspond to α2 and α3 in the α-subunit. The resulting conformational changes, which involve the phosphate binding loop, disrupt hydrogen bonds to Mg2+ and the β-phosphate of GDP. In the structure of Ras·Sos (Boriack-Sjodin et al., 1998), Sos binds to the phosphate-binding loop, switches I and II, and α3 and the α3/β5 loop of Ras. Binding of Sos to Ras causes conformational changes in switches I and II and introduces residues that block magnesium binding and overlap the site where the α-phosphate of GDP would bind.
By analogy to the actions of EF-Ts and Sos, receptors may alter the relative orientations of α2/switch II (via interaction with βγ) and α3 (via interaction with the α3/β5 loop) to disrupt interactions with the bound GDP and Mg2+. In support of such receptor-stimulated conformational changes, comparison of the structures of the heterotrimer and the activated α-subunit indicates activation-dependent alterations in the positions of α2 and α3. In the heterotrimer (Lambright et al., 1996), binding of βγ to switch II opens a gap between α2 and α3. Upon GTP binding, this gap closes and switch II comes into contact with α3 and switch III (Noel et al., 1993; Coleman et al., 1994). Receptor-dependent changes in the positions of α2 and α3 could then be transmitted across the domain interface via switch III to enable release of the bound nucleotide, which is buried between the GTPase and helical domains. In support of this postulated mechanism, we have shown previously that interactions between switch III and the αD/αE loop at the domain interface are involved in receptor-mediated activation (Grishina and Berlot, 1998). Additional conformational changes induced by binding of the receptor to the carboxyl terminus of the α-subunit could be transmitted to the β6/α5 loop region to loosen contacts with the guanine ring of GDP. Mutations in the β6/α5 loop of αt (Onrust et al., 1997) and αs (Marsh et al., 1998) disrupt receptor-mediated activation, indicating the importance of this region in nucleotide binding.
A more complete understanding of receptor-catalyzed nucleotide exchange by heterotrimeric G proteins awaits the solving of a structure of a seven-transmembrane receptor-heterotrimeric G protein complex. In the meantime, postulated connections between receptor binding sites and the nucleotide binding site can be tested by additional functional studies of α-subunit mutants with substitutions in positions likely to transmit receptor-initiated conformational changes.
Acknowledgments
We thank Thomas Hynes for the computer graphics, helpful discussions, and critical reading of the text.
Footnotes
-
Send reprint requests to: Dr. Catherine H. Berlot, Department of Cellular and Molecular Physiology, Yale University School of Medicine, 333 Cedar Street, New Haven, CT 06520-8026. E-mail:catherine.berlot{at}yale.edu
-
This work was supported by National Institutes of Health Grant GM50369 (C.H.B.) C.H.B. is an Established Investigator of the American Heart Association.
-
↵1 Residue numbering throughout is according to the long splice variant of αs.
-
↵2 The αs residue 354 is not shown because it represents an insertion of sequence relative to that of the αt/αi1 chimera in the model.
- Abbreviations:
- GTPγS
- guanosine 5′-O-(3-thiotriphosphate)
- ICYP
- iodocyanopindolol
- DTT
- dithiothreitol
- Received November 9, 1999.
- Accepted February 10, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}