


Abstract
The protective adaptive response to electrophiles and reactive oxygen species is mediated by the enhanced expression of the phase II detoxifying genes through antioxidant response elements (AREs). The current study was designed to identify the signaling pathways responsible for the expression of rGSTA2 in response to cellular oxidative stress and to establish the molecular mechanistic basis. Deprivation of cystine and methionine caused oxidative stress in H4IIE hepatoma cells as evidenced by a marked decrease in the reduced glutathione (first order rate constant = 0.056 h−1;t 1/2 = 12.6 h) and an increase in pro-oxidant production. Electrophoretic mobility shift assay revealed that the ARE complex, consisting of Nrf-1/2 and Maf proteins, was activated 12 to 48 h after sulfur amino acid deprivation (SAAD). The rGSTA2 mRNA level was elevated by SAAD beginning at 24 h, whereas the rGSTA2 subunit was maximally induced at 48 h. Nuclear ARE activation and rGSTA2 mRNA increase were both completely inhibited by wortmannin or LY294002, the phosphatidylinositol 3-kinase (PI3-kinase) inhibitors. The p38 mitogen-activated protein (MAP) kinase was activated at 0.5 to 3 h after SAAD, followed by sustained diminished activation up to 12 h. Inhibition of p38 MAP kinase by SB203580 prevented the ARE-mediated rGSTA2 induction. The activation of p38 MAP kinase, however, failed to be inhibited by wortmannin or LY294002, showing that PI3-kinase is not involved in the activation of p38 MAP kinase. Data showed that PI3-kinase plays an essential role in the ARE-mediated rGSTA2 induction by oxidative stress after SAAD, which activates the p38 MAP kinase and leads to rGSTA2 induction.
The protective adaptive response to electrophiles and reactive oxygen species (ROS) is mediated by the enhanced expression of the phase II detoxifying genes. The role of antioxidant response elements (AREs) and activator protein-1 (AP-1) in the inducible expression of phase II enzymes (e.g., rGSTA2) by phenolic antioxidants has been studied extensively (Bergelson et al., 1994; Wasserman and Fahl, 1997;Venugopal and Jaiswal, 1998). ARE coordinately regulates the expression of a battery of antioxidant genes. The binding proteins to the ARE consensus sequence involve Nrf proteins and Maf family members (Venugopal and Jaiswal, 1998; Moinova and Mulcahy, 1999). Signals activated by oxidative stress stimulate transduction of Nrf activity and activation of ARE (Venugopal and Jaiswal, 1998; Moinova and Mulcahy, 1999).
Phosphatidylinositol 3-kinase (PI3-kinase) is a lipid kinase that phosphorylates phosphatidylinositols at the 3-position of the inositol ring. This enzyme has been found to be associated with the activation of cellular survival signals in response to several growth factors and has been implicated in mitogenesis and cell transformation (Daulhac et al., 1999). In addition, the phosphorylated forms of phosphatidylinositol act as second messengers for several kinases, including the serine-threonine Akt kinase and ribosomal S6 kinase (Lin et al., 1999). PI3-kinase is also involved in the regulation of the small GTPase Rac by growth factors (e.g., platelet-derived growth factor) and Rac plays an important role in the activation of c-Jun NH2-terminal kinases (JNK; Hawkins et al., 1995;Fritz and Kaina, 1999). In view of the diverse biological effects of PI3-kinase, we were interested in whether PI3-kinase regulated the ARE activation by the oxidative stress after sulfur amino acid deprivation (SAAD) and the subsequent transcriptional induction of rGSTA2 in H4IIE rat hepatoma cells.
A number of cellular stresses and lethal insults (e.g., cytotoxic chemicals) engage the mitogen-activated protein (MAP) kinases and concomitantly induce transactivation of the targeted genes (Amato et al., 1998; Fritz and Kaina, 1999). Three distinct mammalian MAP kinase modules including JNK, extracellular signal-regulated kinase (ERK), and p38 MAP kinase, have been characterized (Cahill et al., 1996; Treisman, 1996). Stress-activated protein kinase cascade involves the activation of JNK, which consequently induces AP-1-mediated transactivation of the genes. Induction of glutathione S-transferase (GST) by chemicals may be mediated through the activation of ERK. The induction of quinone reductase by sulforaphane has been shown to be mediated with the activation of ERK, but not with JNK (Yu et al., 1999). The p38 MAP kinase is a recently identified member of the MAP kinase family. Activity of p38 MAP kinase is involved in apoptosis (Tan et al., 1996). Nonetheless, the role of p38 MAP kinase on the expression of GST in response to oxidative stress has not been explored yet.
GSH as a nonprotein sulfhydryl molecule in cells plays a role as an intracellular protective substance and serves as an effective oxygen radical scavenger. A decrease in cellular reduced GSH content would increase oxidative stress. The present study was designed to investigate the effects of decrease in reduced GSH and resultant oxidative stress on the rGSTA2 expression and the associated signaling pathways. It has been shown that PI3-kinase regulates the activities of JNK and p38 MAP kinase in certain cells (Assefa et al., 1999; Fritz and Kaina, 1999; Hirasawa et al., 2000). For the purposes of this study, we were interested in establishing whether the pathway involving PI3-kinase mediates the p38 MAP kinase activation in association with ARE activation. We showed for the first time that PI3-kinase is essential for the rGSTA2 expression, which requires the activation of p38 MAP kinase, and that PI3-kinase is not involved in the activation of p38 MAP kinase by SAAD.
Experimental Procedures
Materials.
[α-32P]dCTP (3000 mCi/mmol) and [γ-32P]ATP (6000 mCi/mmol) were purchased from New England Nuclear (Arlington Heights, IL). Anti-rGSTA1/2, anti-rGST3/5, anti-rGSTM1, and anti-rGSTM2 antibodies were supplied from Biotrin International (Dublin, Ireland). Biotinylated goat anti-rabbit IgG, minimum essential medium-select amine kit, recombinant protein G-agarose, and 5-bromo-4-chloro-3-indoylphosphate/nitroblue tetrazolium were obtained from Life Technologies (Gaithersburg, MD). Anti-Nrf-1, anti-Nrf-2, and anti-v-Maf antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-p38 MAP kinase antibody (Cat# 9212) and anti-phospho p38 MAP kinase antibody (Cat# 9211) were supplied from New England Biolabs (Beverly, MA). Random prime-labeling kit was purchased from Promega (Madison, WI). PD98059 and LY294002 were obtained from Biomol (Plymouth Meeting, PA). Phosphorylated heat and acid-stable protein-1 (PHAS-1) was purchased from Stratagene (Austin, TX). Wortmannin, 2′,7′-dichlorofluorescein diacetate (DCFH-DA), SB203580 and other reagents in the molecular studies were supplied from Sigma Chemical Co. (St. Louis, MO).
Cell Culture.
H4IIE rat hepatoma cells were obtained from American Type Culture Collection (Rockville, MD) and maintained in Dulbecco's modified Eagle's medium containing 10% fetal calf serum, 50 U/ml penicillin, and 50 μg/ml streptomycin at 37°C in humidified atmosphere with 5% CO2. Sulfur amino acid-deprived minimum essential medium was reconstituted with Earle's balanced salt solution, vitamin mixture, and the amino acids other than cystine and methionine. The H4IIE monolayering cells were cultured for the indicated times in minimum essential medium with or without cystine and methionine.
Reduced GSH Determination.
Cells were washed twice in ice-cold PBS and then scraped into ice-cold 5% metaphosphoric acid. Glutathione was quantified using a commercially available GSH determination kit (Oxis International, Portland, OR).
Assay of Intracellular Peroxides.
Production of intracellular peroxides was monitored spectrofluorometrically using DCFH-DA, a fluorescent dye (Kim and Yurkow, 1996). H4IIE cells were suspended 12 h after incubation in the medium without sulfur amino acids, and then DCFH-DA dissolved in ethanol was added at a final concentration of 10 μM. The dye-loaded cells were further incubated at 37°C for 5, 10, and 30 min after measurement of the initial fluorescence. Oxidation of DCFH by peroxides yielded the fluorescent derivative dichlorofluorescein (DCF). Fluorescence was monitored at the excitation wavelength of 485 nm and the emission wavelength of 530 nm using a fluorescence plate reader (Tecan US Inc., Research Triangle Park, NC). Data were expressed as the relative changes to the initial fluorescence.
Preparation of Nuclear Extracts.
Nuclear extracts were prepared essentially according to Schreiber et al. (1990). Briefly, dishes were washed with ice-cold PBS. The dishes were then scraped and transferred to microtubes. Cells were allowed to swell by adding 100 μl of lysis buffer (10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 1 mM dithiothreitol, and 0.5 mM phenylmethylsulfonylfluoride). Tubes were vortexed to disrupt cell membranes. The samples were incubated for 10 min on ice and centrifuged for 5 min at 4°C. Pellets containing crude nuclei were resuspended in 50 μl of the extraction buffer containing 20 mM HEPES, pH 7.9, 400 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol, and 1 mM phenylmethylsulfonyl fluoride, and then incubated for 30 min on ice. The samples were centrifuged at 15,800g for 10 min to obtain the supernatant containing nuclear extracts. The nuclear extracts were stored at −70°C until use.
Gel Retardation Assay.
A double-stranded DNA probe containing the rGSTA2 gene ARE was used for gel shift analysis after end-labeling of the probe with [γ-32P]ATP and T4 polynucleotide kinase. The sequence of the ARE-containing oligonucleotide was 5′-GATCATGGCATTGCACTAGGTGACAAAGCA-3′. The oligonucleotides for specific protein-1 (SP-1) and AP-1, which were used for competition experiments, were 5′-ATTCGATCGGGGCGGGGCGAGC-3′ and 5′-CGCTTGATGAGTCAGCCGGAA-3′, respectively. The reaction mixtures contained 4 μl of 5× binding buffer containing 20% glycerol, 5 mM MgCl2, 250 mM NaCl, 2.5 mM EDTA, 2.5 mM dithiothreitol, 0.25 mg/ml poly(dI-dC), and 50 mM Tris·Cl, pH 7.5, 5 μg of nuclear extracts, and sterile water in a total volume of 20 μl. The reaction mixtures were preincubated for 10 min. DNA-binding reactions were carried out at room temperature for 30 min after the addition of 1 μl of probe (106 cpm). Specificity of binding was determined by competition experiments, which were carried out by adding a 20-fold excess of an unlabeled ARE, SP-1, or AP-1 oligonucleotide to the reaction mixture before the DNA-binding reaction. Samples were loaded onto 4% polyacrylamide gels at 100 V. The gels were removed, fixed and dried, followed by autoradiography.
In some experiments, 5 μg of nuclear extracts were incubated with 2 μg of highly specific anti-Nrf-1, anti-Nrf-2, or anti-v-Maf antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) on ice for 60 min. For immunodepletion, 10 μl of a 1:1 slurry of recombinant protein G-agarose was added to the reaction mixture and further incubated for 60 min. The immune complexes were removed by centrifugation, and the nuclear extract was assayed for ARE binding activity by electrophoretic mobility shift assay (Handel et al., 1996;Amato et al., 1998). For supershift assay, the antibodies (6 μg each) were added to the reaction mixture after initial 20-min incubation, and additionally incubated for 2 h at 37°C.
To determine the involvement of phosphorylation in the activation of ARE, the nuclei isolated from the cultured cells was dephosphorylated in vitro (Carrier et al., 1992). Briefly, the nuclei were treated with calf intestinal alkaline phosphatase (CIP) (4 units/ml) in the buffer (pH 9.5) containing 100 mM Tris·Cl, 5 mM MgCl2, and 100 mM NaCl for 30 min, and then nuclear extracts were prepared as described above. In this experiment, we could eliminate dephosphorylation of labeled ARE oligonucleotide.
Immunoblot Analysis.
After washing the cells twice with sterile PBS, the cells were scraped and sonicated for disruption. Cytosolic fractions were prepared by differential centrifugation. The subcellular preparations were stored at −70°C until use. SDS-polyacrylamide gel electrophoresis and immunoblot analysis were performed according to previously published procedures (Kim et al., 1997). Cytosolic proteins were separated by 12% gel electrophoresis and electrophoretically transferred to nitrocellulose paper. The nitrocellulose paper was incubated with the form-specific anti-rat GST antibodies, followed by incubation with biotinylated secondary antibody, and developed using 5-bromo-4-chloro-3-indoylphosphate and nitroblue tetrazolium. Specificity of the antibodies for GST subunits has been confirmed by the previous study (Kim et al., 1997). To determine the phosphorylated p38 MAP kinase, total cell lysates were prepared from the scraped cells after addition of 100 μl of boiling lysis buffer containing 0.1% SDS and 10 mM Tris·Cl, pH 7.4. Activation of p38 MAP kinase was immunochemically assessed using the specific antibody, which recognized the active-phosphorylated form, and developed using an enhanced chemiluminescence system (Amersham, Buckinghamshire, UK).
p38 MAP Kinase Activity.
H4IIE cells were lysed in the buffer containing 20 mM Tris·Cl, pH 7.5, 1% Triton X-100, 137 mM sodium chloride, 10% glycerol, 2 mM EDTA, 1 mM sodium orthovanadate, 25 mM β-glycerophosphate, 2 mM sodium pyrophosphate, 1 mM phenylmethylsulfonyl fluoride, and 1 μg/ml leupeptin. Cells were kept on ice for 5 min, vortexed and centrifuged at 10,000g at 4°C for 10 min. Anti-p38 MAP kinase antibody was added to the supernatant (300 μg of protein) and the reaction mixture was incubated with gentle agitation at 4°C for 2 h. The immune complex was allowed to bind protein G-agarose for 2 h and precipitated by centrifugation. The p38 MAP kinase immune complex-protein G-agarose was washed twice with the lysis buffer and once with the kinase buffer containing 25 mM Tris·Cl, pH 7.4, 25 mM β-glycerophosphate, 25 mM magnesium chloride, 1 mM dithiothreitol, and 0.1 mM sodium orthovanadate. The immune complex was again precipitated by centrifugation at 10,000g for 2 min, and resuspended in 25 μl of the kinase buffer. The p38 kinase reaction was initiated by addition of 2 μg PHAS-1 and 5 μCi of [γ-32P]ATP, incubated at 30°C for 30 min, and terminated by addition of 25 μl of 2× SDS-polyacrylamide gel electrophoresis sample dilution buffer. Samples were boiled for 5 min at 95°C and proteins were separated on 12% SDS-polyacrylamide gel electrophoresis. The gels were autoradiographed after fixing and drying.
Preparation of a cDNA Probe for GST.
The specific cDNA probe for the rGSTA2 gene was amplified by reverse transcription-polymerase chain reaction using the selective primers (Kim et al., 1997) and was cloned in the pGEM+T vector (Promega, Madison, WI).
Northern Blot Hybridization.
Total RNA was isolated from H4IIE cells using the improved single-step method of thiocyanate-phenol-chloroform RNA extraction, and Northern blot analysis was carried out according to the procedures described previously (Kim et al., 1997). Briefly, total RNA was resolved by electrophoresis in a 1% agarose gel containing 2.2 M formaldehyde and transferred to nitrocellulose paper. The nitrocellulose paper was baked in a vacuum oven at 80°C for 2 h. The blot was incubated with hybridization buffer containing 50% deionized formamide, 5× Denhardt's solution [0.1% Ficoll, 0.1% polyvinylpyrrolidone, and 0.1% BSA (Pentex Fraction V)], 0.1% SDS, 200 μg/ml of sonicated salmon sperm DNA and 5× SSPE (1× SSPE = 0.15 M NaCl, 10 mM NaH2PO4, and 1 mM Na2EDTA, pH 7.4) at 42°C for 1 h without probe. Hybridization was performed at 42°C for 18 h with a heat-denatured cDNA probe, which was random prime-labeled with [α-32P]dCTP. Filters were washed in 2× standard saline citrate (SSC) and 0.1% SDS for 10 min at room temperature twice and in 0.1× SSC and 0.1% SDS for 10 min at room temperature twice. Filters were washed in the solution containing 0.1× SSC and 0.1% SDS at 60°C for 60 min. After quantification of mRNA levels, the membranes were stripped and rehybridized with a32P-labeled cDNA probe complementary to 18S rRNA to quantify the amount of RNA loaded onto the membranes.
Data Analysis.
The kinetics of cellular GSH level was analyzed using a computer program WinNonlin (version 1.0; Pharsight Inc., Mountain View, CA). Scanning densitometry was performed with a Microcomputer Imaging Device, Model M1 (Imaging Research, St. Catharines, Ontario, Canada). One-way ANOVA procedures were used to assess significant differences among treatment groups. For each significant effect of treatment, the Newman-Keuls test was used for comparisons of multiple group means. The criterion for statistical significance was set at P < .05 or P< .01.
Results
GSH Contents and Pro-Oxidant Production.
The reduced GSH content in H4IIE cells cultured in the complete medium was 3.3 nmol/106 cells. When H4IIE cells were cultured in deficiency of cystine and methionine, the reduced GSH was decreased in a time-dependent manner (Fig. 1A). The first-order rate constant and the half-life time for the decrease in reduced GSH were 0.056 ± 0.007 h−1 and 12.6 ± 1.5 h, respectively (Fig. 1A, inset). To determine whether SAAD induced oxidative stress in cells, peroxide production was assayed using DCFH-DA (Fig. 1B). The intensity of DCF fluorescence was increased 1.2- and 4-fold at 10 and 30 min after incubation of the dye-loaded cells cultured with sulfur amino acids, relative to that of initial fluorescence. The H4IIE cells cultured without sulfur amino acids for 12 h showed significantly greater increases in fluorescence (i.e., 2.5-, 3.0- and 9.7-fold increases at 5, 10, and 30 min, respectively). These results showed that SAAD caused a rapid decrease in cellular reduced GSH and concomitantly induced oxidative stress.

A, the reduced GSH contents in H4IIE cells cultured in deficiency of cystine and methionine. The reduced GSH content was shown as a function of time. Inset shows the logarithmic replot of the GSH contents. Data represent the mean ± S.D. with four separate experiments. One-way ANOVA was used for comparisons of multiple group means followed by Newman-Keuls test (significant compared with the initial GSH content, **P < .01). B, relative DCF fluorescence in H4IIE cells. H4IIE cells were cultured with or without sulfur amino acids for 12 h and loaded with DCFH-DA. Fluorescence was monitored at the excitation wavelength of 485 nm and the emission wavelength of 530 nm using a fluorescence plate reader. Data represent the mean ± S.D. with five separate experiments and are expressed as the relative changes to the initial fluorescence. One-way ANOVA was used for comparisons of multiple group means followed by Newman-Keuls test (significant compared with respective control, **P < .01).
Activation of Nuclear ARE Binding.
The ARE-binding transcription factors transduce the induction signal(s) of rGSTA2 (Liu and Pickett, 1996; Wasserman and Fahl, 1997). The nuclear extracts isolated from H4IIE cells cultured without sulfur amino acids for 12 to 72 h were probed with the radiolabeled rGSTA2 gene ARE to assess whether the nuclear ARE binding proteins were activated in response to the oxidative stress (Fig.2A). The band of slow migrating complex was detected at 12 h after SAAD and extended through 24 h. Minimal activation followed up to 48 h. Competition experiments using excess amounts of unlabeled ARE, AP-1, or SP-1 oligonucleotide confirmed the specificity of ARE binding. Whereas addition of a 20-fold excess of an unlabeled ARE to the nuclear extracts completely abolished the ARE binding, either excess unlabeled AP-1 or SP-1 oligonucleotide failed to inhibit the DNA binding (Fig. 2B).

Gel shift analysis of the ARE transcription complex in the nuclear extracts from H4IIE cells. A, nuclear extracts were prepared from H4IIE cells cultured in the presence (Con) or absence (SAAD) of sulfur amino acids for 12 to 72 h. All lanes contained 5 μg of nuclear extracts and 5 ng of labeled rGSTA2 ARE DNA consensus sequence. B, competition studies were carried out by adding a 20-fold excess of an unlabeled ARE, AP-1, or SP-1 oligonucleotide to the nuclear extracts from the cells stimulated by SAAD for 12 h, and the DNA-binding reactions were performed by gel shift analysis. C, immunodepletion experiments were carried out by incubating the same nuclear extracts from the cells under SAAD for 12 h with the specific polyclonal antibody directed against Nrf-1, Nrf-2, or v-Maf protein. Results were confirmed by repeated experiments. The closed arrow indicates the ARE binding complex. D, supershift analysis was carried out by incubating the same nuclear extracts from the cells under SAAD for 12 h with the antibodies. The closed and open arrows indicate shifted and supershifted ARE binding complexes, respectively. E, the effect of in vitro dephosphorylation of the nuclei isolated from the H4IIE cells stimulated by SAAD for 12 h. The nuclei were incubated in vitro in the presence of CIP before preparation of nuclear extracts. The DNA-binding reactions were carried out as described under Experimental Procedures. Con, control.
The Nrf proteins were shown to be crucial transactivating factors for ARE binding and for activation of the ARE-reponsive genes (Venugopal and Jaiswal, 1998; Moinova and Mulcahy, 1999). Immunodepletion by the specific antibodies has been used to characterize the components of transcriptional factors (Handel et al., 1996; Amato et al., 1998). Immunodepletion of the nuclear extracts produced from sulfur amino acid-deprived H4IIE cells with anti-Nrf-1 or anti-Nrf-2 antibody resulted in significant decreases in the ARE binding (Fig. 2C). Addition of the antibody against anti-v-Maf also inhibited the ARE binding (Fig. 2C). We additionally carried out supershift analysis with anti-Nrf-1, anti-Nrf-2, and v-Maf antibodies. The antibody alone specific for Nrf-1, Nrf-2, or small-Maf reduced formation of the retarded band. Presence of both Nrf-1/2 and small-Maf antibodies induced supershift of the retarded band. Hence, it is likely that the interaction between Nrf-1/2 and Maf induces conformational change that increases their binding affinities to the ARE consensus sequence (Fig.2D). The data indicate that Nrf-1/2 and small-Maf were the components interacting with the rGSTA2 ARE.
We then examined the effect of in vitro dephosphorylation of the nuclei (Fig. 2E). Incubation of the nuclei with CIP completely prevented binding of the ARE-specific complex. CIP was removed after dephosphorylation to exclude the possibility of delabeling of the probe. This was confirmed by the equal intensity of the labeled free probe. This result showed that phosphorylation is required for the ARE binding activity of nuclear proteins.
Selective Induction of rGSTA2 by SAAD.
Expression of GST is affected by oxidative stress (Pinkus et al., 1996). Northern blot analysis was performed to determine whether the rGSTA2 mRNA was increased after the ARE activation. The rGSTA2 mRNA significantly increased 24 h after incubation of the cells in the absence of sulfur amino acids, peaked at 48 h, and then returned to the basal level at 72 h (Fig. 3, A and B).

The rGSTA2 mRNA levels in H4IIE cells. A, Northern blot analysis was performed with the total RNA fraction (30 μg each) prepared from the cells incubated under SAAD for 6 to 72 h. The amount of RNA loaded in each lane was assessed by rehybridization of the stripped membrane with a 32P-labeled probe for 18S rRNA. B, relative changes in the GST mRNA level were assessed by scanning densitometry of the Northern blots. Data represent the mean ± S.D. with four separate experiments. One-way ANOVA was used for comparisons of multiple group means followed by Newman-Keuls test (significant compared with control, **P < .01) (control mRNA level = 1).
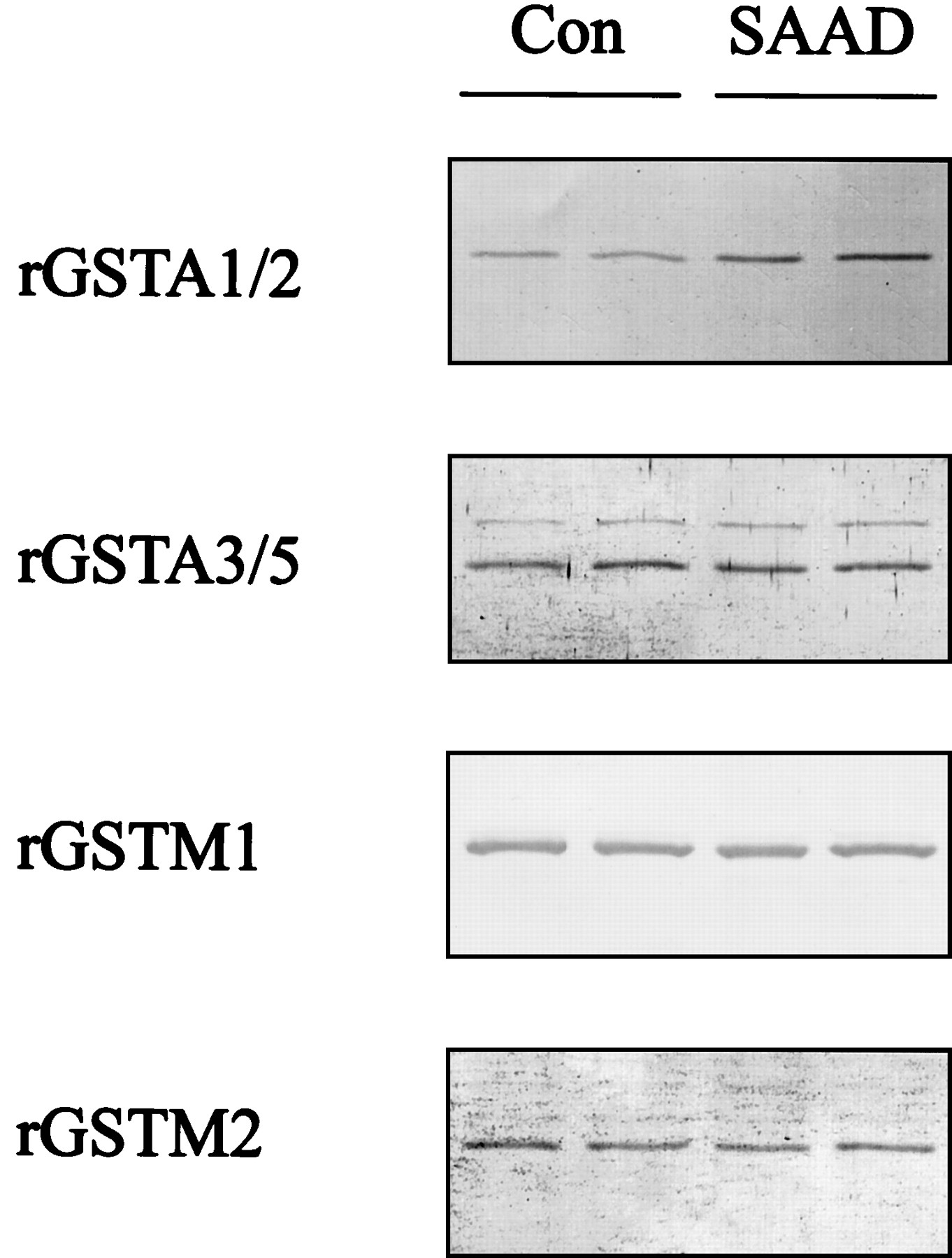
The rGSTA2 subunit was assessed by Western blot analysis to confirm whether ARE activation by SAAD led to induction of the GST subunit. The rGSTA1/2 in the H4IIE cells was induced 48 h after SAAD and extended up to 72 h (Fig. 4, A and B). Whereas rGSTA1/2 was 3.5-fold induced at 48 h, other GST subunits in culture with sulfur amino acids, including rGSTA3/5, rGSTM1, and rGSTM2, failed to be significantly increased (Fig.5). Anti-rGSTA1/2 antibody preferentially recognized the induction of rGSTA2 because the rGSTA2 subunit is inducible.
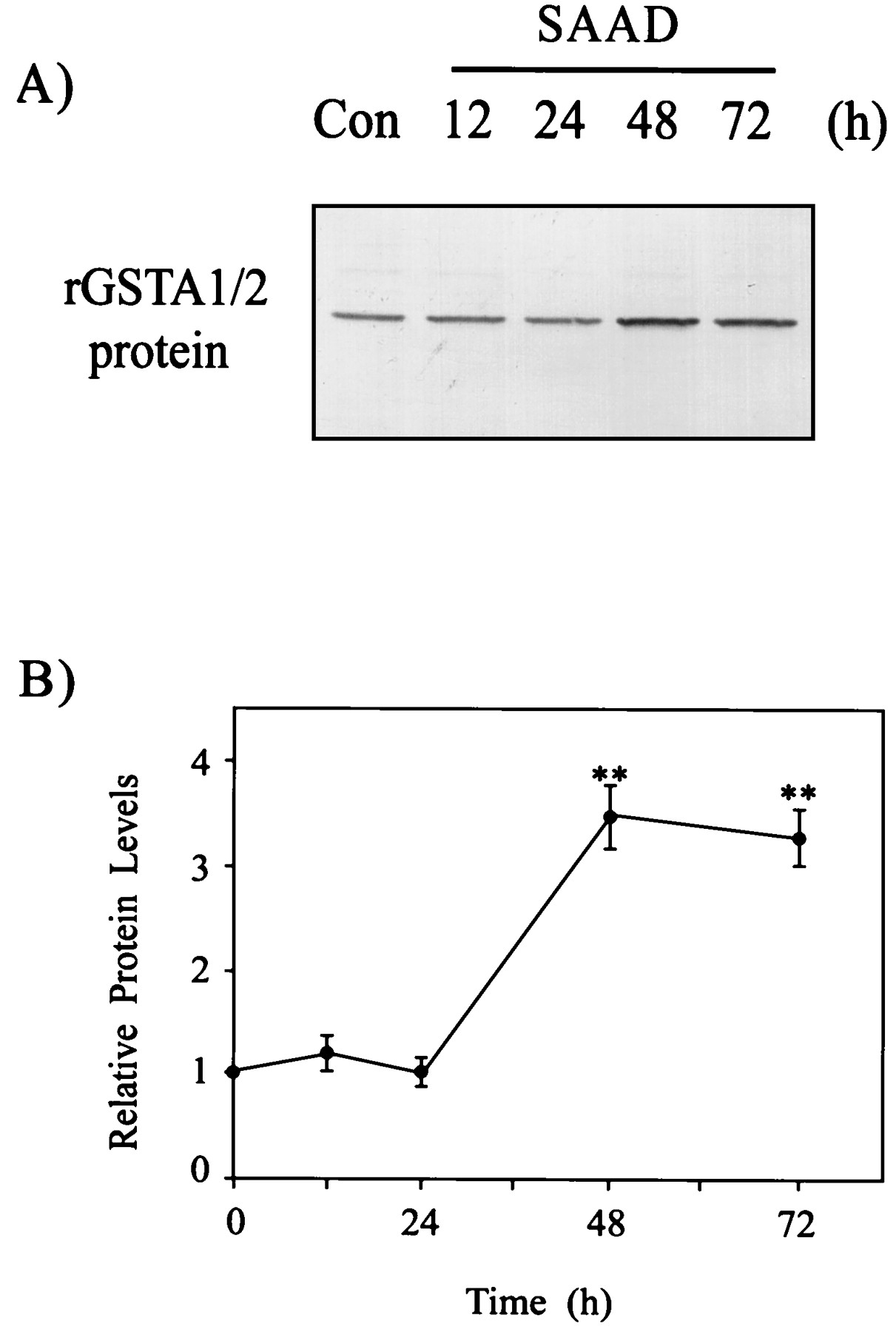
Expression of rGSTA1/2 subunit in H4IIE cells as a function of time. A, a representative immunoblot showing rGSTA1/2 protein in H4IIE cells cultured without sulfur amino acids (SAAD) for 12 to 72 h. Each lane was loaded with 20 μg of proteins. B, relative changes in the rGSTA1/2 subunit were assessed by scanning densitometry. Data represent the mean ± S.D. with four separate experiments. One-way ANOVA was used for comparisons of multiple group means followed by Newman-Keuls test (significant compared with control, **P < .01) (control protein level = 1).

Immunoblot analyses of representative GST subunits in H4IIE cells. The immunoblots show GST protein levels in the cytosol from H4IIE cells cultured in the presence (Con) or absence (SAAD) of sulfur amino acids for 48 h. Each lane was loaded with 20 μg of proteins. Results were confirmed by repeated experiments.
Role of PI3-Kinase in ARE Activation and rGSTA2 mRNA Increase.
To determine whether the PI3-kinase cascade is involved in activation of the ARE-binding transcription factors, H4IIE cells were incubated with 500 nM wortmannin for 12 h in the culture medium without sulfur amino acids (Fig. 6A). Wortmannin completely abolished the activation of ARE, as evidenced by disappearance of the DNA binding with the nuclear extract. The activation of ARE was also inhibited by the presence of 50 μM LY294002 (Fig. 6A). These results clearly showed that the activity of PI3-kinase was essential in the regulatory pathway leading to the activation of ARE.
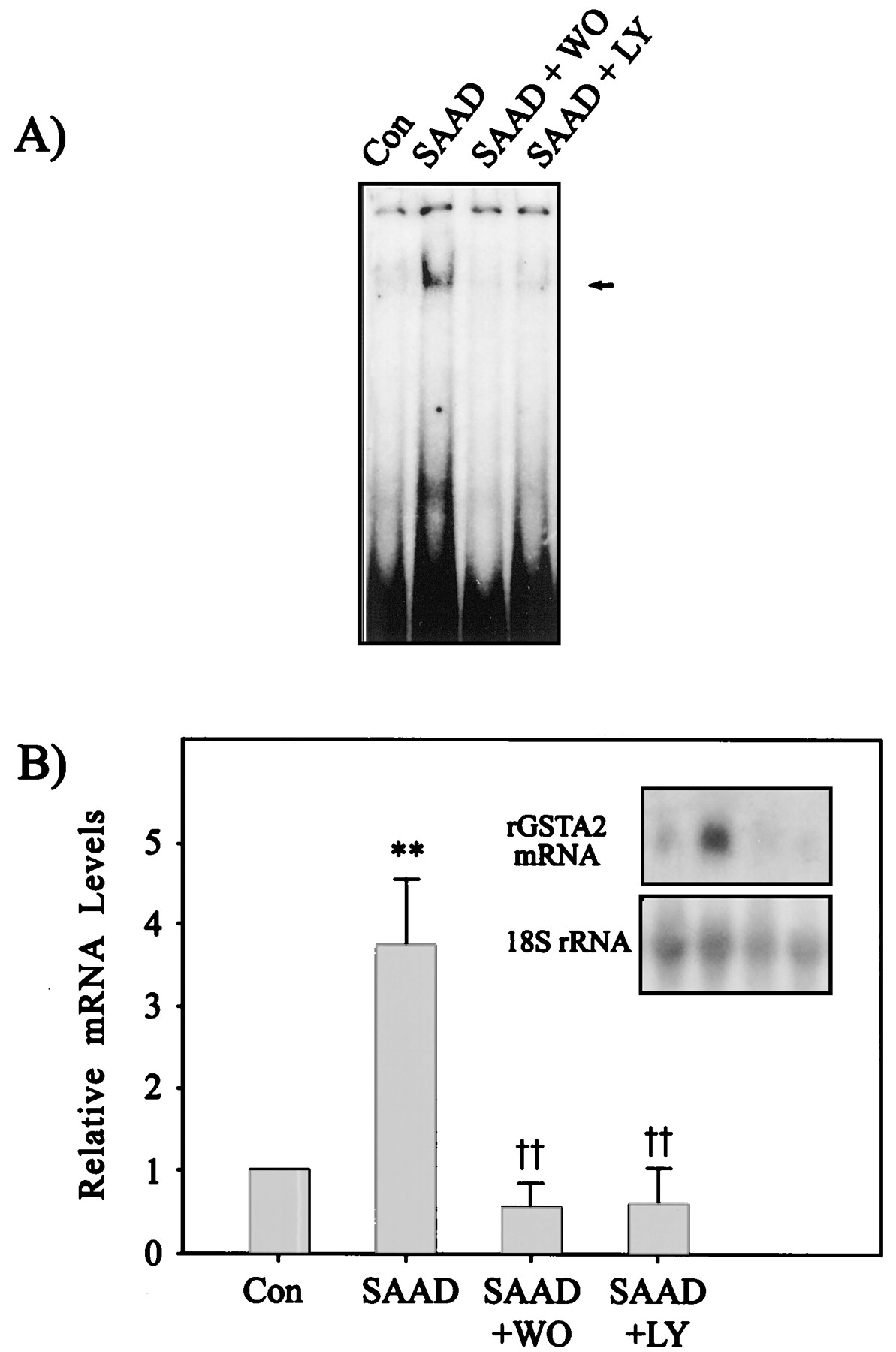
Effects of PI3-kinase inhibitors on the ARE activation and rGSTA2 induction by SAAD. A, effects of wortmannin and LY294002 on the activation of ARE transcription complex in the nuclear extracts from H4IIE cells. Nuclear extracts were prepared from H4IIE cells cultured in the presence (Con) or absence (SAAD) of sulfur amino acids for 12 h. The cells were incubated with 500 nM wortmannin (WO) or 50 μM LY294002 (LY) for 12 h in the culture medium without sulfur amino acids. All lanes contained 5 μg of nuclear extracts and 5 ng of labeled rGSTA2 ARE DNA consensus sequence. B, the rGSTA2 mRNA level in H4IIE cells cultured with the PI3-kinase inhibitors in the absence of the sulfur amino acids. H4IIE cells were incubated with each inhibitor in the absence of sulfur amino acids for 24 h, and the total RNA fractions prepared from the cells were subjected to Northern blot analysis. Data represent the mean ± S.D. with four separate experiments. One-way ANOVA was used for comparisons of multiple group means followed by Newman-Keuls test (significant compared with control, **P < .01; significant compared with SAAD, †† P< .01).
To confirm that inhibition of ARE activation by the PI3-kinase inhibitors prevented rGSTA2 induction, rGSTA2 mRNA level was monitored in the cells incubated with each PI3-kinase inhibitor (Fig. 6B). Either wortmannin or LY294002 completely inhibited the increase in rGSTA2 mRNA at 24 h after SAAD.
p38 MAP Kinase Cascade.
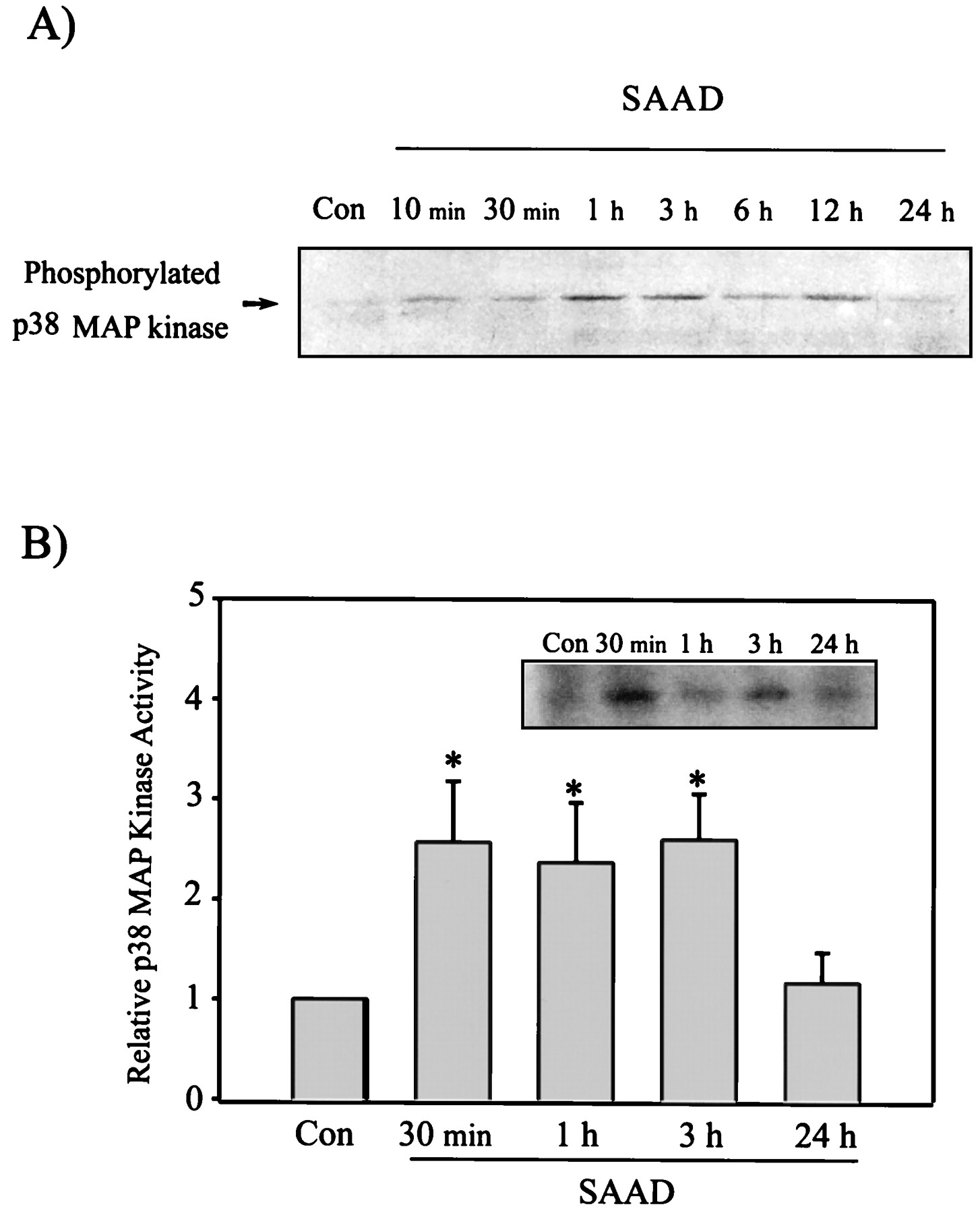
Transcription factors such as c-Fos and c-Jun have been shown to be phosphorylated by the MAP kinase family activated by a variety of the cellular stresses (Luo et al., 1997;Hodge et al., 1998). We measured the activation of the p38 MAP kinase in response to the oxidative stress induced by SAAD. Western blot analysis specifically monitored the phospho-p38 kinase at the residues of Thr180 and Tyr182. The level of phosphorylated p38 MAP kinase was enhanced in cells stimulated by SAAD from 10 min through 12 h, as evidenced by phosphorylation of the kinase (Fig.7A). Activity of the p38 MAP kinase immunoprecipitated using the specific antibody was assayed using PHAS-1 as a substrate (Fig. 7B). The activity of p38 kinase was 2- to 3-fold increased at 0.5 to 3 h after SAAD compared with control (Fig.7B). The activation of p38 kinase was gradually diminished up to 12 h (data not shown), followed by returning toward that of control at 24 h. This was consistent with the level of phosphorylated p38 kinase.
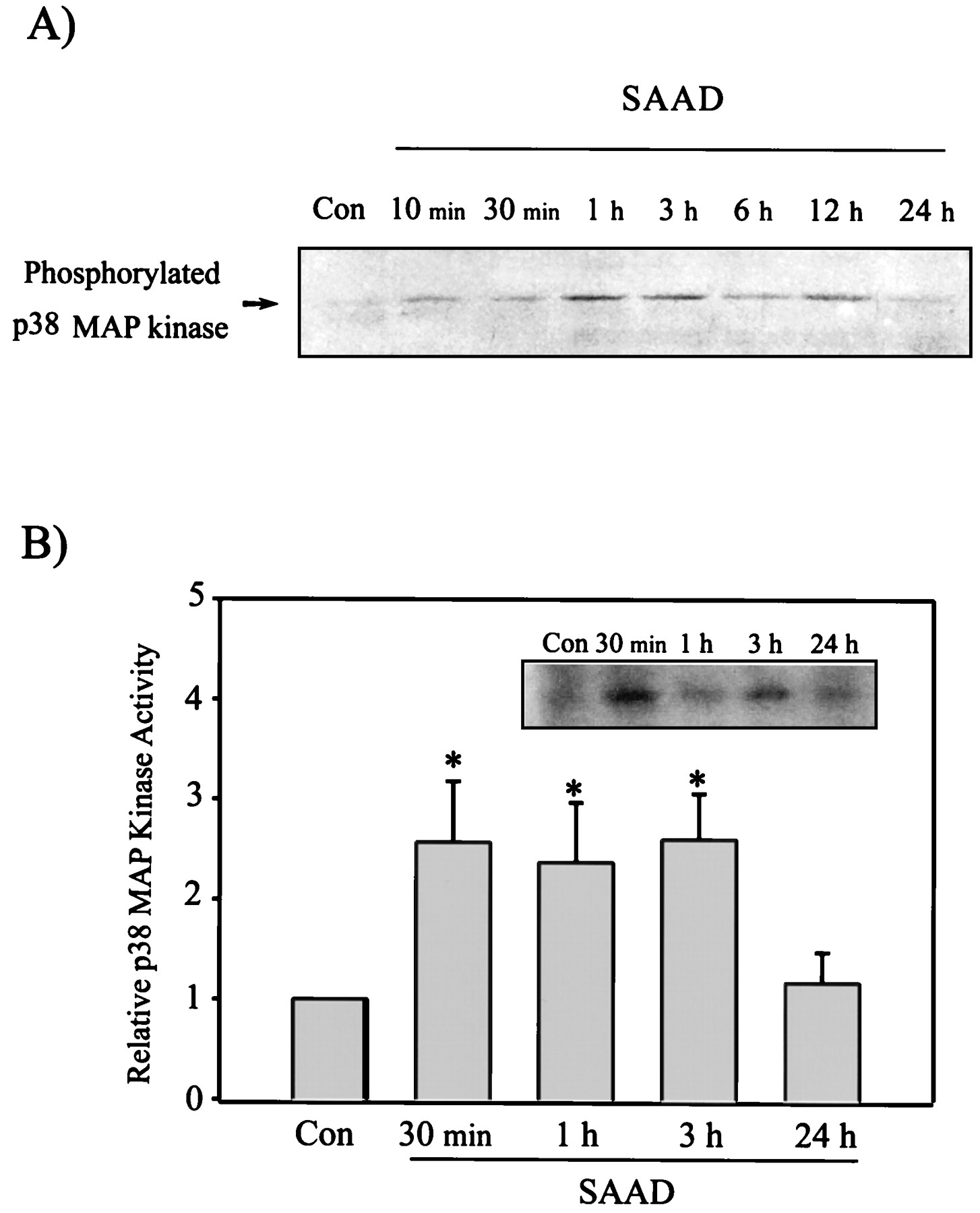
Activation of p38 MAP kinase in H4IIE cells by SAAD. Time-dependent activation of p38 MAP kinase was assessed by immunoblotting of phosphorylated p38 MAP kinase (A), and the p38 MAP kinase activity toward PHAS-1 as a substrate (B) (p38 MAP kinase activity in control cells = 1). Data represent the mean ± S.D. with three separate experiments. One-way ANOVA was used for comparisons of multiple group means followed by Newman-Keuls test (significant compared with control, *P < .05). Con, control.
To test whether blockade of the p38 MAP kinase cascade led to changes in ARE binding activity and rGSTA2 expression stimulated by SAAD, cells were incubated with 10 μM SB203580, a p38 MAP kinase-specific inhibitor. SB203580 suppressed activation of ARE binding 12 h after treatment (Fig. 8A). Inhibition of ARE activation by SB203580 resulted in no increase in the rGSTA2 mRNA level (Fig. 8B).
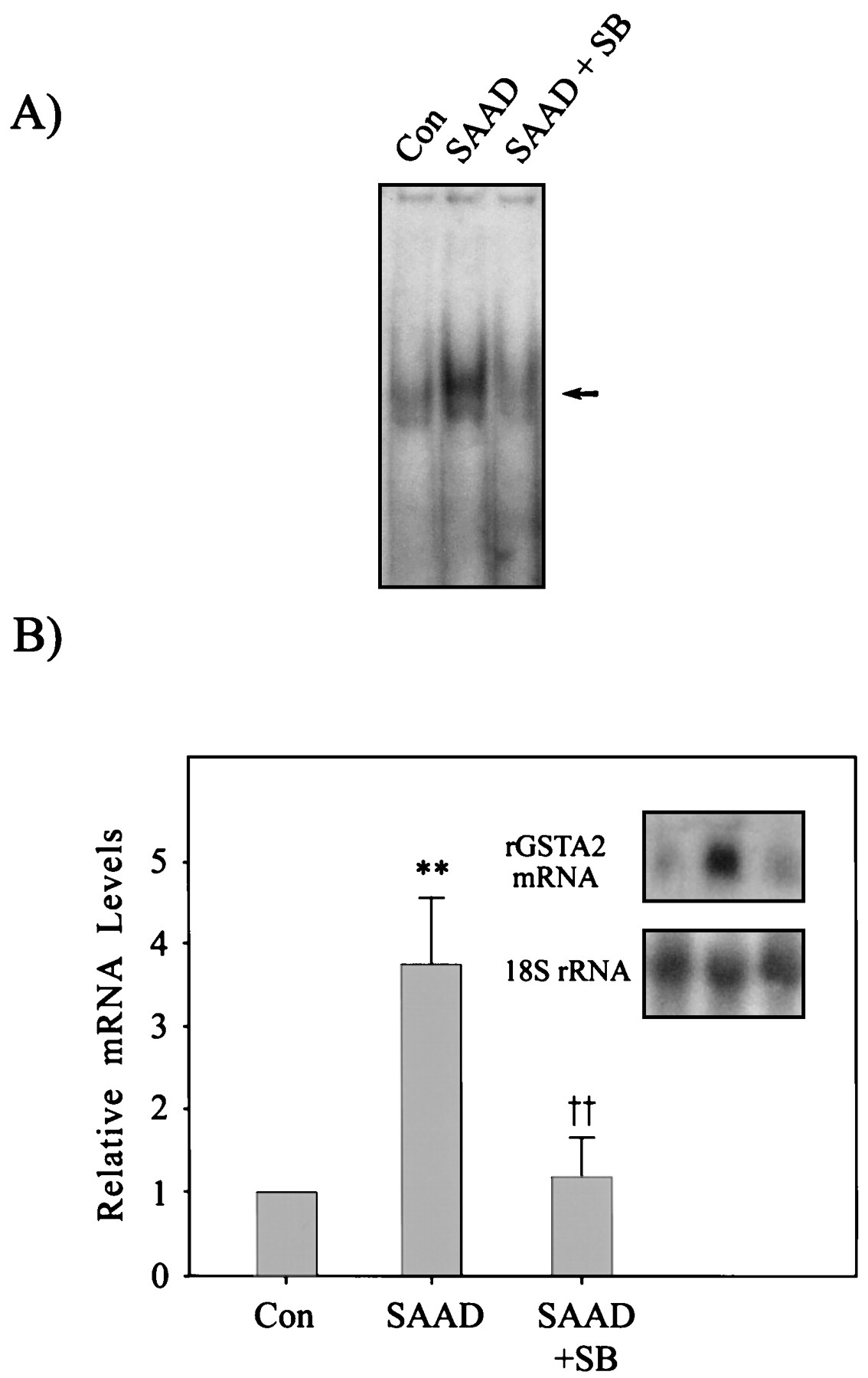
The effects of SB203580 on the ARE binding activity and increase in rGSTA2 mRNA by SAAD in H4IIE cells. A, the effect of SB203580 (SB, 10 μM) on the activation of nuclear ARE transcription complex was assessed in H4IIE cells cultured under SAAD for 12 h. All lanes contained 5 μg of nuclear extracts and 5 ng of labeled rGSTA2 ARE DNA consensus sequence. B, the rGSTA2 mRNA level was determined in H4IIE cells cultured under SAAD in the presence of 10 μM SB203580 for 24 h. Data represent the mean ± S.D. with four separate experiments. One-way ANOVA was used for comparisons of multiple group means followed by Newman-Keuls test (significant compared with control, **P < .01; significant compared with SAAD, †† P < .01).
We then studied whether the PI3-kinase inhibitors affected the activation of p38 MAP kinase. Either wortmannin or LY294002 failed to inhibit the phosphorylation of p38 MAP kinase by SAAD for 1 h (Fig. 9A). Inhibition of PI3-kinase activity also did not affect the activity of p38 kinase in cells induced by SAAD (Fig. 9B). This result supports the conclusion that both PI3-kinase and p38 MAP kinase regulate the ARE-mediated rGSTA2 induction and that PI3-kinase is not involved in the activation of p38 MAP kinase.

The effects of PI3-kinase inhibitors on SAAD-induced p38 MAP kinase activation in H4IIE cells. A, the extents of phosphorylation of p38 MAP kinase were assayed in H4IIE cells cultured in the presence (Con) or absence (SAAD) of sulfur amino acids for 1 h. The cells cultured in deficiency of cystine and methionine were incubated with 500 nM wortmannin (WO) or 50 μM LY294002 (LY). B, activity of the p38 MAP kinase immunoprecipitated using the specific antibody was assayed using PHAS-1 as a substrate. Results were confirmed by repeated experiments.
Discussion
Depletion of hepatic GSH increases the susceptibility of animals to free radical-induced tissue damage because the GSH plays a critical role in the detoxification of oxidative metabolites produced from endogenous and exogenous molecules. Deprivation of sulfur amino acids from the culture medium led to a decrease in the cellular GSH level, which subsequently elevated oxidative stress as evidenced by an increase in the fluorescence of DCF. Because cysteine is a direct precursor of GSH, the lack of sulfur amino acids caused a time-dependent decrease in the reduced GSH, with 50% decrease noted at 12.6 h. The oxidative stress induced by decreased GSH would affect the redox state and the regulation of gene expression. We showed that a decrease in the cellular GSH after SAAD induces rGSTA2 with a concomitant increase in the mRNA as a result of the enhanced ARE-binding activity. GSH-depleting agents, such as buthionine sulfoximine and N-ethylmaleimide, also affect a dynamic equilibrium of the GSH pool by inhibiting the essential proteins involved in GSH synthesis or by direct conjugation. A decrease in the GSH level by buthionine sulfoximine activated the nuclear AP-1, which was consistent with the increase in AP-1 binding by quinones and other phenolic antioxidants (Bergelson et al., 1994; Pinkus et al., 1996). However, the GSH-depleting agents may influence the expression of other genes and may also perturb unknown pathways. Electrophilic chemicals such as phenolic antioxidants induce the phase II detoxifying enzymes and may also exert other biological effects. Although the result of the present experiment was in partial accordance with the AP-1 and ARE activation by electrophilic chemicals, the data differ from those of previous reports in that the decreased GSH and the oxidative stress by SAAD would solely stimulate the associated signaling pathway. Hence, the SAAD experiment would serve as an appropriate model to assess the molecular events and the signaling pathways responsible for the phase II enzyme induction in response to a decrease in the cellular GSH per se.
SAAD-induced oxidative stress activated the nuclear ARE proteins. We found out that the nuclear AP-1 was also activated by SAAD (K. W. Kang and S. G. Kim, unpublished observations). However, the ARE activation seemed to be distinct from the activation of AP-1, as supported by the selective competition experiments. Immunodepletion and supershift analyses provided evidence that the ARE binding proteins involve Nrf-1/2 and Maf proteins. Both Nrf-1 and Nrf-2 play as general regulators of phase II enzyme expression, whereas the small Maf proteins are required for the high affinity ARE sequence-specific binding activity (Venugopal and Jaiswal, 1998). Activation of nuclear ARE-binding proteins by SAAD led to induction of rGSTA2. The ARE activation preceded the increase in the mRNA and persisted up to 48 h. The inducible expression of the γ-glutamylcysteine synthetase light chain and rGSTA2 by phenolic antioxidants has also been shown to be dependent on the ARE element (Purford and Hayes, 1996;Moinova and Mulcahy, 1998). The present data support the notion that oxidative stress induced by SAAD subsequently activated the ARE binding proteins, consisting of Nrf and Maf proteins, and transcriptionally stimulated rGSTA2 gene expression.
A number of transcription factors are phosphorylated by distinct members of kinase family triggered in response to a variety of stimuli (Gupta et al., 1995; Tan et al., 1996). Importance of phosphorylation of the nuclear proteins was evidenced by the lack of ARE activation after in vitro dephosphorylation of the nuclei. Either wortmannin or LY294002 suppressed the ARE activation and rGSTA2 mRNA expression. The inhibition of both ARE activation and rGSTA2 mRNA increase by wortmannin or LY294002 represents the essential role of PI3-kinase in the regulation of phase II detoxifying enzyme expression.
Activation of JNK or p38 kinase is an early response of cells upon exposure to a variety of stresses including heat, UV irradiation, and DNA-damaging agents (Zanke et al., 1996; Wesselborg et al., 1997; Fritz and Kaina, 1999; Treinies et al., 1999). JNK-induced phosphorylation of c-Jun activates AP-1 and increases the expression of AP-1-targeted genes (Wesselborg et al., 1997; Fritz and Kaina, 1999). The inhibitory effects of JNK activation on the expression of the target genes have not been analyzed yet because of the lack of suitable pharmacological JNK inhibitors (Fritz and Kaina, 1999). The p38 MAP kinase is activated by osmotic shock (Wiese et al., 1998). Activation of p38 MAP kinase precedes the induction of apoptosis. Although a variety of stressful stimuli concomitantly activate p38 MAP kinase and JNK, the p38 MAP kinase represents the distinct stress-activated pathway. An increase in p38 MAP kinase activity was paralleled by an antioxidant-induced activation of AP-1 (Wesselborg et al., 1997) and seemed to be activated by prooxidants such as hydrogen peroxide or nitric oxide (Bhat and Zhang, 1999; Jun et al., 1999). Nonetheless, the MAP kinase family responsible for activation of the transcriptional factors involved in the induction of phase II detoxifying enzymes by oxidative stress per se has not been clearly identified. Whether SAAD led to activation of p38 MAP kinase was assessed in the present study to address the physiological significance of this kinase. We showed for the first time that the cellular oxidative stress induced by SAAD stimulates the activation of p38 kinase at early times, which subsequently leads to the activation of nuclear ARE binding to its target gene, rGSTA2. This was further supported by the observation that inhibition of p38 kinase by SB203580 led to prevention of ARE activation and hence of rGSTA2 mRNA increase. Thus, signaling cascades involving the p38 MAP kinase for the induction of phase II antioxidant enzymes may serve as a defense mechanism of the cells.
PI3-kinase regulates the activation of MAP kinase cascade by a range of receptors (Assefa et al., 1999; Sasaoka et al., 1999; Smalley et al., 1999). PI3-kinase is also a critical factor in the regulation of p38 MAP kinase activation for interleukin-4 production in mast cells (Hirasawa et al., 2000). We raised a further question: whether the pathway mediated by PI3-kinase might involve stimulation of p38 MAP kinase in association with the ARE activation. Initially, we expected that p38 kinase regulates the gene expression in the downstream of PI3-kinase. PI3-kinase inhibitors, however, failed to block SAAD-induced activation of p38 MAP kinase. Thus, the p38 MAP kinase may represent the distinct pathway responsible for the activation of ARE transcriptional factors. Inhibition of either PI3-kinase or p38 kinase simultaneously blocked the activation of ARE and subsequent induction of rGSTA2. This result raised the possibility that PI3-kinase and p38 MAP kinase are both involved in the activation of ARE. Because p38 kinase activity was not affected by the inhibition of PI3-kinase, the pathways activated by the kinases may target different component(s) consisting of ARE complexes or other factor(s) interacting with the ARE binding proteins. This raised the possibility that multiprotein complexes simultaneously bind to the enhancer, involving ARE as the transcription factors, and that nucleoprotein complexes that assemble from transcription factors may cooperatively bind to the ARE binding sites in an enhancer. Taken together, the present study revealed that the oxidative stress by SAAD induces rGSTA2 through the activation of ARE involving the pathway mediated with PI3-kinase, and p38 MAP kinase is activated separately by GSH depletion, which is required for the induction of rGSTA2.
Recently, it has been reported that inhibition of p38 MAP kinase activity augmented the increase in ARE-reporter activity byt-butylhydroquinone (Yu et al., 2000). The discrepancy in the role of p38 MAP kinase on the ARE-mediated phase II enzyme expression may result from the difference in cell type or in the gene of interest. It has been shown that a battery of genes have multiple ARE elements. However, the presence of ARE(s) does not always lead to activation and the subsequent transcriptional activation of ARE-containing genes. Preliminary studies in this laboratory have shown that the MAP kinase responsible for the expression of ferritin light chain gene, which has ARE, differed from that of rGSTA2 in response to SAAD or t-butylhydroquinone.
Whether the cellular oxidative stress per se stimulates ERK is not clear. The induction of quinone reductase by sulforaphane has been shown to be mediated with the activation of ERK (Yu et al., 1999). We also found out that SAAD induces activation of ERK (M. H. Son, K. W. Kang, C. H. Lee, and S. G. Kim, unpublished observations). However, PD98059 failed to inhibit the SAAD-induced increase in rGSTA2 expression (K. N. Kang and S. G. Kim, unpublished observations). The expression of rGSTA2 was rather elevated by the ERK inhibitor. Thus, the activation of ERK is unlikely to be responsible for the induction of rGSTA2 by SAAD. Preliminary studies also showed that JNK was also activated by SAAD. Curcumin suppressed the rGSTA2, which showed the possibility that JNK might also be involved in the induction of rGSTA2 by SAAD. The role of JNK in rGSTA2 induction by SAAD is being studied using dominant negative mutant and overexpression vectors of JNK.
Previous studies showed that protein-calorie malnutrition elicits oxidative stress with concomitant activation of the antioxidant gene expression (Cho et al., 2000a,b). The nuclear ARE complex was activated in the livers of rats with protein-calorie malnutrition (Cho et al., 2000b). Oxidative stress during protein-calorie malnutrition was supported by the reversal of ARE activation in response to cysteine supplementation. The present data provide evidence for the essential role of PI3-kinase and of p38 MAP kinase in the activation of ARE and the subsequent rGSTA2 gene expression by SAAD. Hence, these results would also help us understand the basic mechanism associated with the pathophysiology of protein-calorie malnutrition.
Footnotes
- Received January 24, 2000.
- Accepted August 1, 2000.
-
Send reprint requests to: Sang Geon Kim, Ph.D., College of Pharmacy, Seoul National University, Sillim-dong, Kwanak-gu, Seoul 151-742, South Korea. E-mail: sgk{at}snu.ac.kr
-
This work was supported by Grant No. 2000-2-21700-002-5 from the Basic Research Program of the Korea Science and Engineering Foundation (KOSEF), Republic of Korea.
Abbreviations
- ROS
- reactive oxygen species
- ARE
- antioxidant response element
- AP-1
- activator protein-1
- PI3
- phosphatidylinositol 3
- JNK
- c-Jun N-terminal kinase
- SAAD
- sulfur amino acid deprivation
- MAP
- mitogen-activated protein
- ERK
- extracellular signal-regulated kinase
- GST
- glutathione S-transferase
- PHAS-1
- phosphorylated heat and acid-stable protein-1
- DCFH-DA
- 2′,7′-dichlorofluorescein diacetate
- DCF
- dichlorofluorescein
- CIP
- calf intestinal alkaline phosphatase
- SP-1
- specific protein-1
- SSC
- standard saline citrate
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}