


Abstract
Allosteric modulation is a mechanism for modifying pharmacological receptor activity that has largely been ignored in terms of therapeutic drug design, although benzodiazepine receptor ligands are an example of the serendipitous discovery of this class of compound. The current mathematical models of allosteric interactions at (particularly G-protein-coupled) receptors concentrate on the effects of the allosteric ligand on orthosteric ligand binding and ignore potential effects of these compounds on the ability of orthosteric ligands to cause receptor activation. In this report a mathematical model of allosteric interactions at pharmacological receptors has been investigated that explicitly includes effects of the allosteric ligand on receptor activation. This model uses the two-state model of receptor activation as its basis and is qualitatively consistent with currently reported behavior of allosteric modulators. The predictions of this model suggest a series of criteria that should be tested before the effects of an allosteric modulator can be quantified in a nonsystem-dependent manner. It has also been used to provide a potential mechanistic explanation for the functional effects of the A1 adenosine receptor allosteric enhancer PD 81,723 and a recently reported allosteric modulator of type 1 metabotropic glutamate receptors.
The majority of the agonists and antagonists used to study pharmacological receptors (and for therapeutic intervention) are competitive, i.e., they bind to the same site as the endogenous ligand on the receptor, the orthosteric site. Allosteric modulators are molecules that exert their effects via a site on the receptor protein that is distinct from the orthosteric site. With the notable exception of muscarinic receptors (e.g., Lazareno et al., 1998), allosteric modulators have largely been ignored as targets for drug discovery programs. The allosteric effects of benzodiazepine receptor ligands at γ-aminobutyric acidA receptors were discovered after their in vivo effects were known (Ehlert et al., 1983). There are arguments, however, that suggest that allosteric modulators may, under certain circumstances, provide better drugs than their orthosteric counterparts. In particular, the effects of allosteric modulators are saturable and thus there is less likelihood of adverse effects from overdose. This property can be capitalized upon to provide increased duration of action, by increasing dosage, while still avoiding unwanted effects. Also, allosteric activators of receptors may have advantages over direct agonist molecules. The effects of this type of compound require the presence of the endogenous agonist and are thus likely to mimic more closely the normal physiological effects of that agonist. Allosteric sites are also likely to be less well conserved than the ligand binding site in a particular receptor family thus potentially allowing the design of ligands with greater subtype selectivity (Tuček and Proška, 1995).
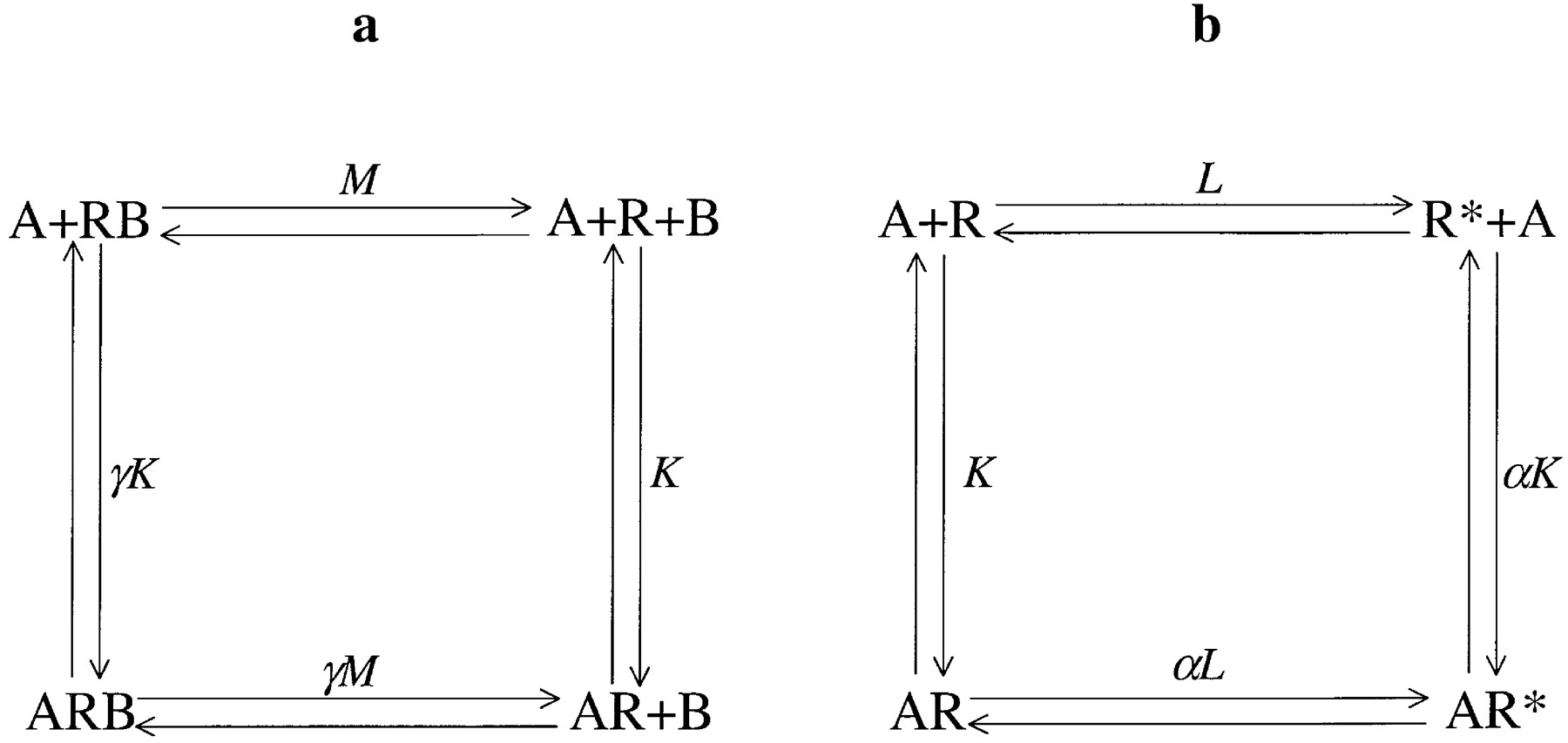
The muscarinic acetylcholine receptor family has been widely studied as a model system for the interaction of allosteric modulators of G-protein-coupled receptors (Stockton et al., 1983; Ellis et al., 1991;Lee and El-Fakahany, 1991; Lazareno and Birdsall, 1995; Proška and Tuček, 1995). In this case, a ternary complex model (Fig.1a) has been proposed to describe the effects of these allosteric modulators on the binding of ligands to the orthosteric site (Stockton et al., 1983; Tuček and Proška, 1995). A number of adaptations of this basic model have also been proposed to explain more complex behaviors (Waelbroeck, 1994; Lazareno and Birdsall, 1995; Proška and Tuček, 1995; Hoare and Strange, 1996). However, there are, to my knowledge, no reports of models of allosteric modulation in which modern theoretical models of receptor activation are explicitly considered, although Ehlert's work is important in terms of the more classical models (Ehlert, 1988). The two-state model of receptor activation (originally presented in Karlin, 1967; Thron, 1972; and Colquhoun, 1973; and recapitulated by Leff, 1995) (Fig. 1b) is one of the simpler models of receptor activation that qualitatively describes our current understanding of receptor behavior. In particular, unlike earlier models (Stephenson, 1956;Furchgott, 1966; Black and Leff, 1983), it allows receptors to have a low level of activity in the absence of agonist. Another advantage of this model, for the present discussion, is that it models only the behavior of the receptor and makes no assumptions about subsequent signal transduction steps, thus making it potentially applicable to any type of receptor. This is in contrast with models such as the ternary complex (De Lean et al., 1980) and cubic ternary complex models (Weiss et al., 1996a–c), which are specifically formulated to describe the behavior of G-protein-coupled receptors.

a, the ternary complex model of allosteric modulation. The two ligands, A and B, bind to different sites on the same receptor, R. This results in two binary complexes AR and RB and the ternary complex ARB. The affinity of ligand for the complementary binary complex may be different from its affinity for the free receptor. The equilibrium constants are explained in the text and defined in Table 1. b, the two-state model of receptor activation. The receptor exists in two forms, an inactive state, R, and an active state, R*. The ligand, A, can bind to either of these forms, to generate AR or AR*, and may discriminate between them. Again, the equilibrium constants are defined in Table 1.
In this report, therefore, the principles of the two-state model of receptor activation have been combined with those of the ternary complex model of allosteric modulation to generate a general qualitative model of the interaction between allosteric and orthosteric ligands at pharmacological receptors both in terms of binding and of functional activation of the receptor. Models of this form have previously been published to aid discussion of the effects of allosteric modulators in specific receptor contexts (Bruns and Fergus, 1990; Ehlert et al., 1983), however, the mathematical analyses were not presented and the possibility of cooperativity at the level of receptor activation was not considered.
Formulation of the Model
In the ternary complex model (Fig. 1a), the receptor (R) can bind reversibly to an (arbitrarily assigned) orthosteric ligand (A) with affinity constant K, or to an allosteric ligand (B) with affinity constant M, resulting in two binary complexes AR and RB (the order denoting the binding site). The unbound ligand may subsequently bind to the complementary binary complex (e.g., A to RB), its affinity being modified by the allosteric constant γ. This parameter is therefore analogous to the cooperativity factor designated α by Ehlert (1988) and Tuček and Proška (1995). In this formulation of the model, a value of γ greater than unity indicates positive cooperativity and a value of γ less than unity indicates negative cooperativity between the two ligands. When γ = 1 there is no interaction between the two compounds.
In the two-state model (Fig. 1b), the inactive receptor (R) enters the active state by undergoing a conformational change to form the active receptor (R*). Functional responses are assumed to be proportional to the amount of R*-containing species within the system (Leff, 1995), hence, strictly, it is the behavior of the pharmacological stimulus rather than the response that is determined in this model. In the absence of ligand, the proportion of receptors in the R* state is governed by the equilibrium constant L. The ligand (A) binds to the inactive state of the receptor with an affinity constant,K, and to the active state of the receptor with a modified affinity, αK. Binding of the ligand shifts the isomerization equilibrium constant to αL. In this model the allosteric constant α can be regarded as the intrinsic efficacy of the ligand. If A has a value of α greater than unity it favors the formation of R* and is an agonist. A value of α equal to unity indicates a neutral antagonist (i.e., no receptor state selectivity) and a value of α less than unity indicates a molecule that stabilizes the R state, i.e., an inverse agonist.
As shown in Fig. 1, both of these models can be represented by cyclic equilibria and appear to contain the common step of a ligand binding to the endogenous agonist binding site (A binding to R). However, the equilibrium constants governing these events differ in character between the two models. In the ternary complex model, Krepresents a macroscopic (and therefore experimentally measurable) equilibrium constant for the binding of the orthosteric ligand to the receptor. In contrast, in the two-state model, K represents the microscopic binding affinity of the ligand for the inactive state of the receptor, a quantity that cannot be determined experimentally [the measured affinity constant of A in the two-state model is given by K(1 + αL)/(1 + L)]. The approach inherent in the ternary complex model can, however, be applied to the two-state model on the basis of an effect of the allosteric ligand on the affinity of the orthosteric ligand for the inactive receptor.
Inserting an allosteric ligand into the two-state model and completing the equilibrium to allow for all of the potential interactions leads to the cubic model shown in Fig. 2 (a ternary complex model and the two-state model form, respectively, the front and left-hand faces of this new model). In the combined model, the allosteric two-state model, the allosteric modulator could be an agonist or an inverse agonist in its own right. This extra property of the allosteric modulator is denoted by the intrinsic efficacy term β and forms the upper face of the cube. The allosteric interaction of the two ligands at the level of binding (to the inactive receptor) is again described by γ. However, it is also possible for the binding of one of the ligands to modify the ability of the other ligand to activate the receptor (i.e., change its intrinsic efficacy). This potential activation cooperativity is modeled by the parameter δ. The parameters of the model and their definitions are summarized in Table1. It should be noted that, although it is based on the two-state model, this model allows the two ligands to bind to the binary complexes with affinities that are different from their affinities for R and R*. Strictly, therefore, it should be described as a multistate rather than a two-state model. This also brings the resulting model to a level of analysis that is intermediate between that of the two-state and ternary complex models. Its equilibrium constants are neither macroscopic because they are not measurable nor microscopic because conformational changes in the receptor are implicit in nonunit values of the allosteric constants γ and δ (e.g., when γ ≠ 1 the conformation of the receptor in AR must be different from that of R otherwise B would not distinguish between them). The cubic ternary complex model (Weiss et al., 1996a) is also formulated at this level of analysis.

The allosteric two state model. The free receptor exists in inactive and active forms, R and R*, as in the two-state model. The two ligands, A and B, bind to different sites on the receptor and may discriminate between the two states and between the free receptor and the binary complexes. In the model, A is assumed to bind to the orthosteric site and B to the allosteric site; however, this assignment is entirely arbitrary due to the symmetry of the model. The equilibrium constants are defined in Table 1.
Summary of the equilibrium constants of the allosteric two-state model
Behavior of the Model
The derivation of expressions for the binding of the orthosteric ligand to the receptor in the presence of an allosteric ligand and for the functional activation of the receptor are shown under , as are expressions for the midpoints of the saturation binding, competition binding, and activation curves and for the maxima and minima of these curves. These expressions were used to investigate the effects of the various parameters of the model on binding and activation curves. The effects of varying parameters β, γ, or δ in turn were investigated in simulations of binding and functional experiments. These parameters (and α) may take any nonnegative value; a value of unity implies the parameter has no effect, i.e., it removes any selectivity between the states that parameter governs. The magnitudes of values above and below unity should be considered in log terms, thus the converse of α = 100 is α = 0.01. In the simulations, the two allosteric constants that were not being varied were set to unity to remove their effects from the system.
Simulations of the effects of varying the two affinity constants (K and M) are not shown because they are entirely as would be expected from a change in the affinity of the radioligand or the allosteric modulator. Similarly, varying α or Lhave effects analogous to those of the original two-state model (Leff, 1995) and are not discussed. Obviously, because α is the intrinsic efficacy of the orthosteric ligand, it was necessary to vary this parameter when simulations required an orthosteric agonist or antagonist ligand. The value of L in the simulations shown below was chosen to produce very little activity in the absence of agonist (unless otherwise stated), because this most closely mimics the behavior of most experimental systems.
Simulations of Competition Binding Curves.
In these simulations the concentration of the orthosteric ligand, which adopts the role of radioligand, was chosen to provide a control fractional occupancy of ∼0.5, i.e., at the apparentK D. This concentration is most frequently used in radioligand binding experiments and allows effects of the allosteric ligand in either direction to be seen clearly.
Effect of β (Intrinsic Efficacy of B).
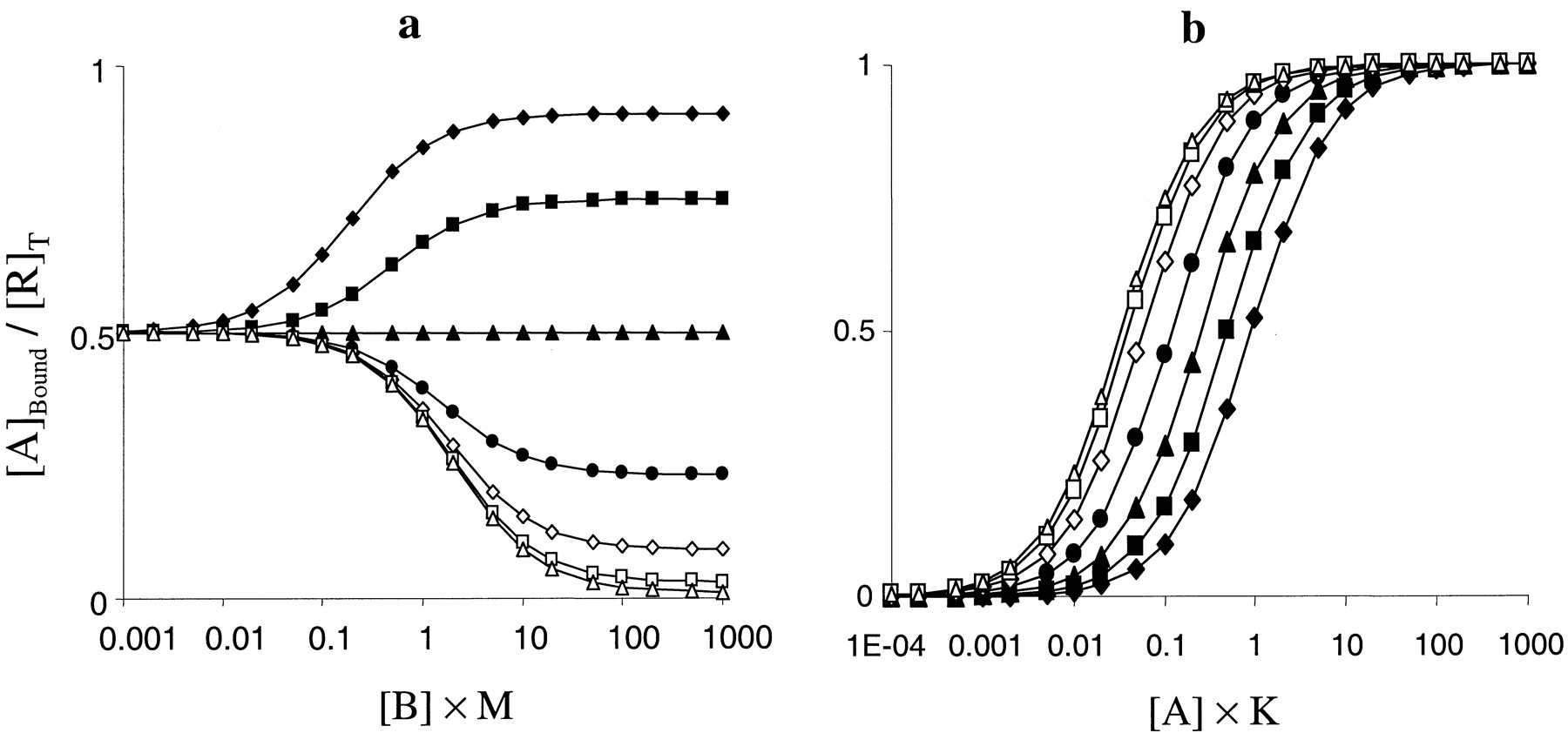
The effects of this parameter on binding are dependent on the pharmacological activity of the radioligand that is used (Fig. 3). With an agonist radioligand (α > 1), binding is increased with increasing concentrations of an allosteric agonist (β > 1) and is decreased if B is an inverse agonist (β < 1) (Fig. 3a). When B is neutral (β = 1), it has no effect on the binding of A. These effects can be explained in terms of the effect of B on the conformation of the receptor. An allosteric agonist will increase the level of R* in the system and make it energetically more favorable for an orthosteric agonist to convert more receptors into this state. R* is the state of the receptor with a high affinity for agonists and an increase in the concentration of this form of the receptor will therefore increase the amount of binding of an orthosteric agonist due to an increase in its apparent affinity (Fig. 3b).

Effects of parameter β (the intrinsic efficacy of B) on binding curves. a, effect of varying β on competition binding curves against an agonist radioligand (α = 1000). Other parameters were [A] = 0.1/K, K =M = 1, L = 0.01, α = 1000, γ = δ = 1, and β = 100 (♦), 30 (▪), 10 (▴), 3 (●), 1 (∗), 0.3 (⋄), 0.1 (■), 0.03 (▵), or 0.01 (○). b, effect of various concentrations of an allosteric agonist (β = 100) on saturation binding curves (presented on a log scale) to an agonist radioligand (α = 1000). Note the increase in apparent affinity of the radioligand. Other parameters wereK = M = 1, L = 0.01, γ = δ = 1, and [B] = 0 (♦), 0.01 (▪), 0.03 (▴]), 0.1 (●), 0.3 (⋄), 1 (■), and 3/M (▵). c, effect of β on competition binding curves against an orthosteric inverse agonist radioligand (α = 0.001). Other parameters are as described in a.
In an analogous way an allosteric inverse agonist (β < 1) increases the concentration of R in the system and makes it energetically less favorable for an orthosteric agonist to convert the receptor into the R* state. The R state has a low affinity for agonists and so an allosteric inverse agonist decreases the binding of an orthosteric agonist by reducing the apparent affinity of the receptors for it. When the allosteric ligand is neutral it has no effect on the distribution of the receptor between R and R* and thus has no effect on the binding of an orthosteric ligand (Fig. 3a, asterisks).
If the orthosteric ligand is a neutral antagonist (α = 1), then it is not sensitive to the conformational state of the receptor (it has the same affinity for R and R*) and its binding is unaffected by compounds whose only effect is due to their intrinsic efficacy at the allosteric site (i.e., their ability to perturb the R ↔ R* equilibrium). The effects of β on inverse agonist radioligands (α < 1) are basically the opposite of those on agonist radioligands, that is, an allosteric agonist (β > 1) will decrease the binding of an orthosteric inverse agonist by increasing [R*], the low-affinity state for inverse agonists. A caveat to this statement is that a quiescent system is already overwhelmingly in the R state. Thus, although in theory an allosteric inverse agonist should increase the binding of an orthosteric inverse agonist, the effect may be so small that it cannot be seen (as demonstrated in Fig. 3c).
Effect of γ (Binding Cooperativity).
The effects of this parameter are independent of the pharmacological activity of the radioligand used (Fig. 4a). Thus, positive cooperativity (γ > 1) results in an increase in the level of binding of the radioligand and negative cooperativity (γ < 1) results in a decrease. These effects are again due to changes in the apparent affinity of the receptor for the orthosteric ligand caused by the allosteric ligand (Fig. 4b). This is of course exactly the behavior of the analogous parameter in the ternary complex model of allosterism (Tuček and Proška, 1995). It should be noted that negative allosteric modulators do not inhibit binding to the nonspecific level unless the negative cooperativity is very strong, that is if γ ≪ 1 (the maximal reduction of binding will of course depend on the concentration of the radioligand).

Effects of parameter γ (the binding cooperativity between A and B) on binding curves. a, effect of varying γ on competition binding curves against a neutral antagonist radioligand (α = 1) (for illustration, the effects of this parameter are independent of α). Other parameters were [A] = 1/K,K = M = 1,L = 0.01, β = δ = 1, and γ = 10 (♦), 3 (▪), 1 (▴), 0.3 (●), 0.1 (⋄), 0.03 (■), or 0.01 (▵). b, effect of various concentrations of a positive allosteric modulator (γ = 30) on saturation binding curves (presented on a log scale) to an antagonist radioligand (α = 1). Note the increase in affinity of the radioligand. Parameters wereK = M = 1,L = 0.01, β = δ = 1, and [B] = 0 (♦), 0.03 (▪), 0.1 (▴), 0.3 (●), 1 (⋄), 3 (■), and 10/M (▵).
Effect of δ (Activation Cooperativity).
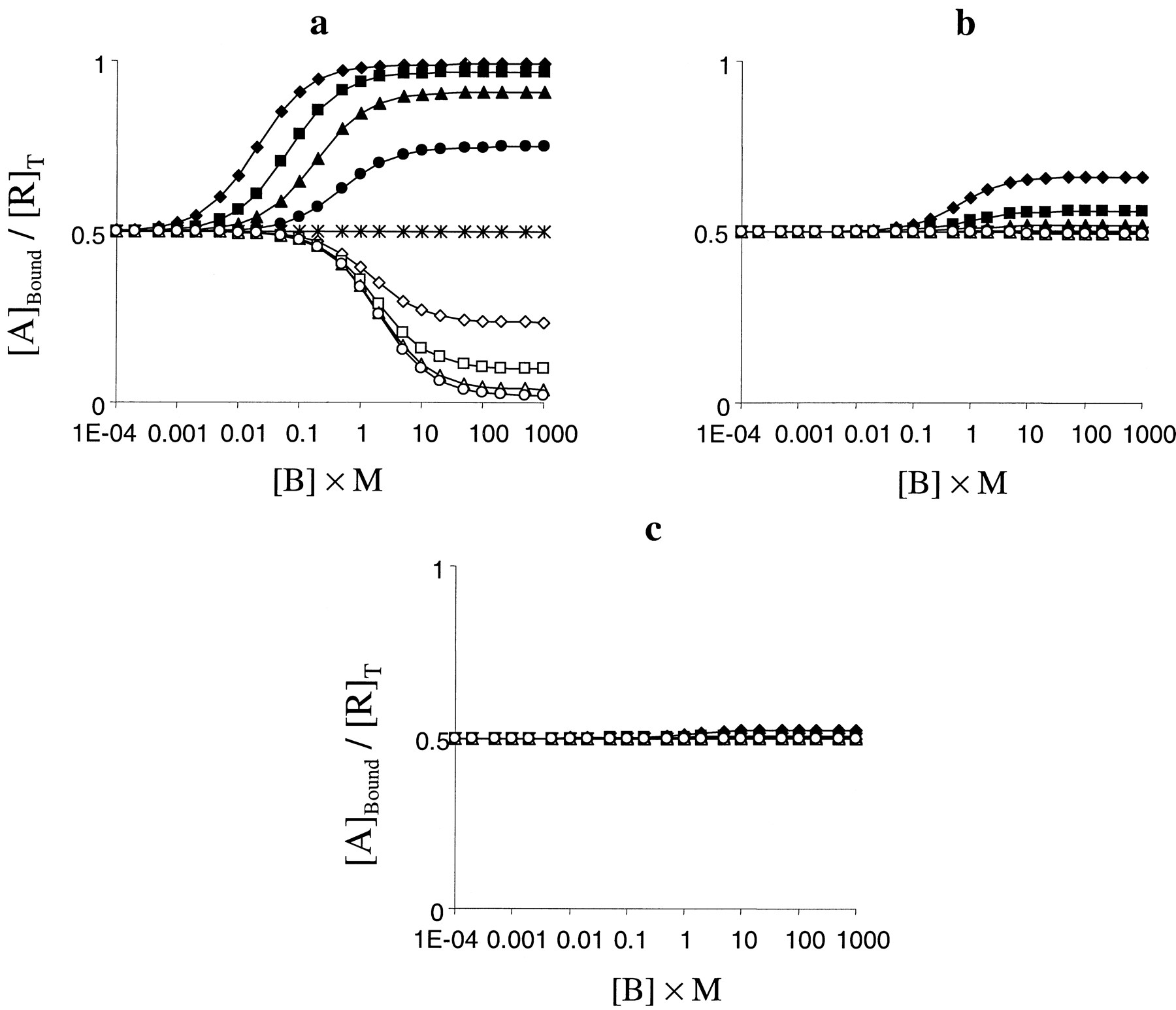
The qualitative effects of this parameter are again independent of the pharmacological activity of the radioligand; however, the magnitude of the effect is affected. Thus, positive activation cooperativity (δ > 1) results in an increase in the level of binding of a given ligand, whereas negative activation cooperativity (δ < 1) can decrease it (Fig. 5). The magnitude of the effect of this parameter varies with α, however. Thus, highly efficacious agonist ligands show very marked changes in their level of binding (Fig. 5a), whereas the effects are less pronounced as α decreases and only extremely large values of δ change the binding characteristics of antagonists and inverse agonists (Fig. 5, b and c). As with β, inhibitory effects seem more sensitive to α than potentiative effects.
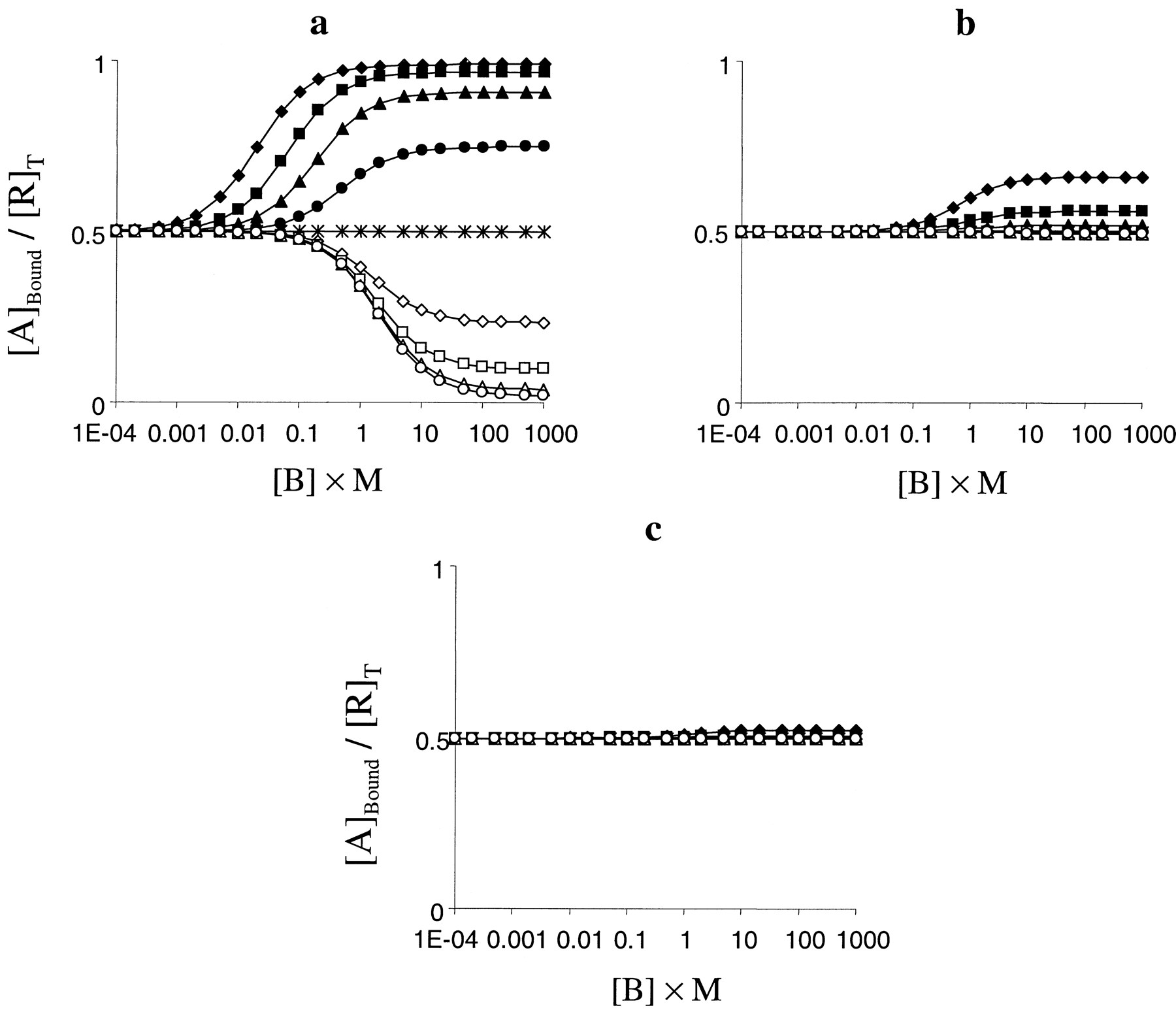
Effect of parameter δ (the activation cooperativity between A and B) on binding curves. a, effect of varying δ on competition binding curves against an agonist radioligand (α = 10 000; [A] = 0.01/K). Other parameter wereK = M = 1,L = 0.01, β = γ = 1, and δ = 100 (♦), 30 (▪), 10 (▴), 3 (●), 1 (∗), 0.3 (⋄), 0.1 (■), 0.03 (▵), or 0.01 (○). b, effect of varying δ on competition binding curves against a neutral antagonist radioligand (α = 1; [A] = 1/K). Other parameters were as described in a. c, effect of varying δ on competition binding curves against an inverse agonist radioligand (α = 0.1; [A] = 1/K). Other parameters were as described in a.
The effect of δ in the model (in terms of A) is to change the relative affinity of A for the RB and R*B complexes. Thus, high values of δ increase the affinity of A for R*B relative to RB, whereas values of δ less than unity decrease the affinity of A for R*B relative to RB. For any ligand, therefore, a value of δ > 1 increases its affinity for one of the forms of the receptor in the system and hence its overall affinity (and therefore level of binding at a given concentration) and a value less than unity has the opposite effect. The greater effect on agonists is due to the already greater affinity of an agonist for R*B compared with RB. The more marked potentiating effect compared with inhibition is due to the quiescence of the system (L < 1) and thus greater prevalence of R.
Simulations of Activation Curves.
In these simulations, the effects of various concentrations of the allosteric modulator were determined on concentration-response curves to an orthosteric agonist. This mimics the experimental protocol that would be used when intending to perform a Schild analysis. The effects of the different parameters can be distinguished by their effects on the asymptotes and midpoints of the concentration-response curves (CRCs).
Effect of β.
The parameter β is an intrinsic efficacy term in the allosteric two-state model and it behaves in the same way as α in terms of the activation state of the receptor. Thus, when β > 1, the allosteric ligand is an agonist and causes activation of the receptor in its own right. When β < 1, the allosteric ligand is an inverse agonist, it therefore inhibits any constitutive activity in the system. These effects can be seen as changes in the lower asymptotes of the CRCs to A in Fig. 6.

Effect of parameter β on activation curves to A. a, effects of various concentrations of an allosteric agonist (β = 100) on CRCs to A (α = 1000). Other parameters wereK = M = 1,L = 0.01, γ = δ = 1, and [B] =0 (♦), 0.01 (▪), 0.03 (▴), 0.1 (●), 0.3 (∗), 1 (⋄), 3 (■), 10 (▵), or 30/M (○). b, effects of various concentrations of an allosteric inverse agonist (β = 0.01) on CRCs to A (α = 1000). Other parameters wereK = M = 1,L = 0.01, γ = δ = 1, and [B] = 0 (♦), 0.1 (▪), 0.3 (▴), 1 (●), 3 (∗), 10 (⋄), 30 (■), 100 (▵), or 300/M (○).
The interesting effects of β occur in the presence of an orthosteric agonist. When β > 1 (B is an agonist), the effects of A are both potentiated and augmented, that is, the midpoint is left-shifted and the upper asymptote of the curve is increased (Fig. 6a). These effects are due to the allosteric ligand increasing the energetic favorability of R* formation and therefore synergizing with the activating effects of A, which (as an agonist itself) also favors R* formation (however, see Discussion for an important caveat to this prediction). When β < 1 (inverse agonist), B right-shifts the CRC to A and depresses the maximal response (Fig. 6b). In this case, the presence of B decreases the energetic favorability of R* formation and thus counteracts the activating effects of A. These two effects can be seen mathematically under ; the midpoint of A ([A]50) (eq. 4) is hyperbolically dependent on both [B] and β, as is the upper asymptote of the CRC (eq. 11).
Effect of γ.
The only effect of γ is on the position of the CRCs; it has no effects on the upper or lower asymptotes of the curves (Fig. 7). Thus, values of γ > 1 result in left-shifting of the curves (potentiation), whereas values of γ < 1 cause right-shifting. The lack of effect of this parameter on the asymptotes is a logical consequence of its function in the model. It represents the binding cooperativity of the two ligands. However, it doesn't affect the ability of the ligands to discriminate between R and R*, thus it does not affect the maximal extent of functional activation. Again, this is borne out mathematically under where γ is present in the denominator of the midpoint function for A (eq. 4). Thus, high values of this parameter decrease [A]50 (increase potency), whereas values less than unity increase it. However, the limiting value of the upper asymptote of the CRC to A is αβδL/(1 + αβδL), which does not contain γ and is therefore unaffected by it.

Effect of parameter γ on activation curves to A. a, effects of various concentrations of a positive allosteric modulator (γ = 100) on CRCs to A (α = 1000). Other parameters wereK = M = 1,L = 0.01, β = δ = 1, and [B] = 0 (♦), 0.01 (▪), 0.03 (▴), 0.1 (●), 0.3 (∗), 1 (⋄), 3 (■), 10 (▵), or 30/M (○). b, effect of various concentrations of a negative allosteric modulator (γ = 0.01) on CRCs to A (α = 1000). Other parameters wereK = M = 1,L = 0.01, β = δ = 1, and [B] = 0 (♦), 0.1 (▪), 0.3 (▴), 1 (●), 3 (∗), 10 (⋄), 30 (■), 100 (▵), or 300/M (○).
Effect of δ.
This parameter allows the allosteric ligand to modify the intrinsic efficacy of the orthosteric ligand without having any intrinsic efficacy of its own. In other words the modulator is not an agonist (or inverse agonist) in its own right and therefore has no effect on the lower asymptote of the CRC to A. When δ > 1 the effective intrinsic efficacy of the orthosteric ligand is increased and the effects of A are potentiated and augmented (Fig.8a). When δ < 1, the effective intrinsic efficacy of the orthosteric ligand is decreased and the curves are right-shifted and the maximal effect is depressed (Fig. 8b). In common with binding, these effects are related to the effect of δ on the ability of a ligand to convert RB to R*B. Thus, when δ > 1 an agonist will convert RB to R*B more efficiently, R*B is an active form of the receptor thus a greater level of activation will result. The converse is true when δ < 1.

Effect of parameter δ on activation curves to A. a, effects of various concentrations of an allosteric activator (δ = 100) on CRCs to A (α = 1000). Other parameters wereK = M = 1,L = 0.01, β = γ = 1, and [B] = 0 (♦), 0.003 (▪), 0.01 (▴), 0.03 (●), 0.1 (∗), 0.3 (⋄), 1 (■), 3 (▵), or 10/M (○). b, effect of various concentrations of an allosteric inhibitor (δ = 0.01) on CRCs to A (α = 1000). Other parameters were K =M = 1, L = 0.01, β = γ = 1, and [B] = 0 (♦), 0.1 (▪), 0.3 (▴), 1 (●), 3 (∗), 10 (⋄), 30 (■), 100 (▵), or 300/M (○).
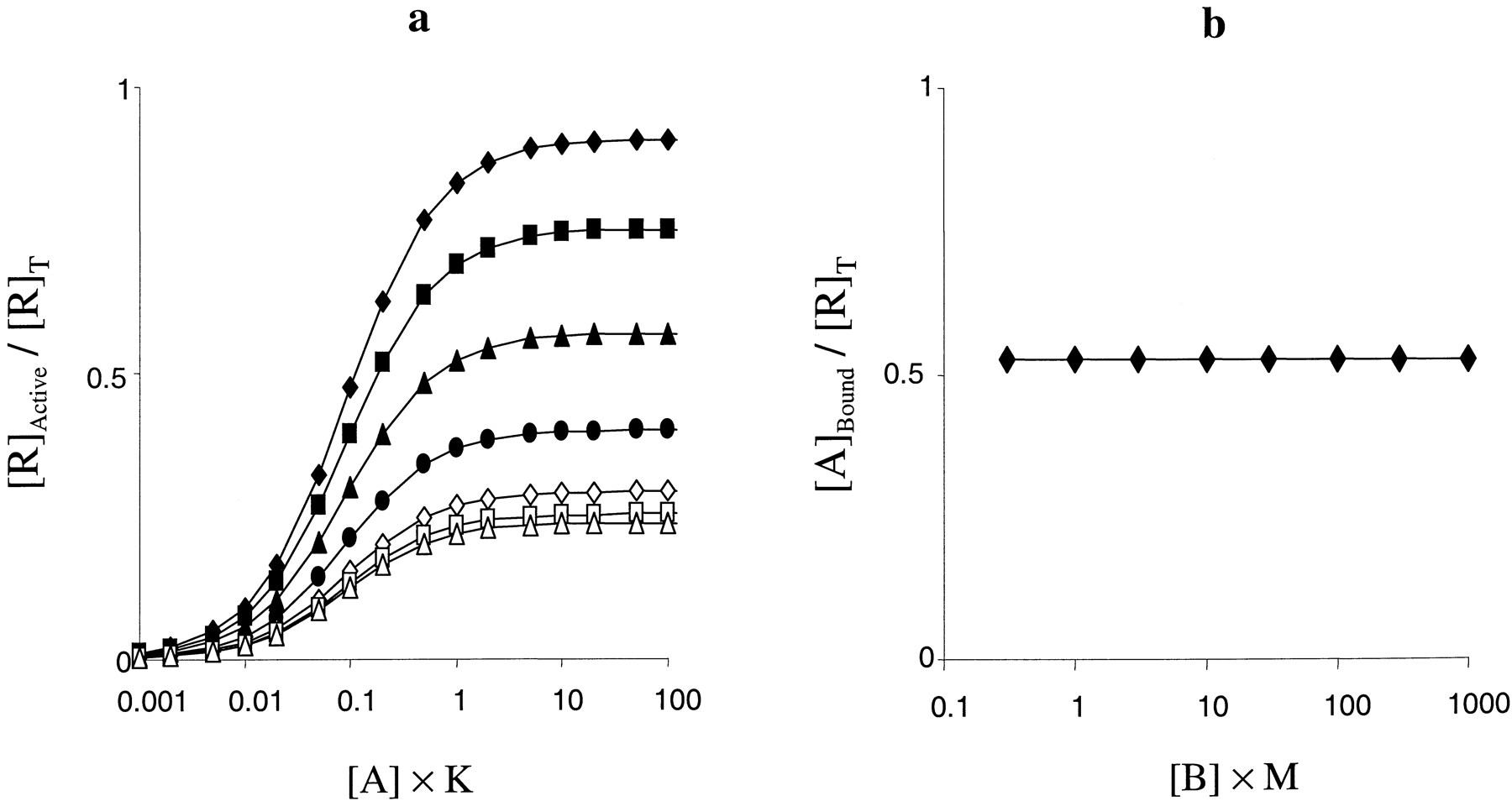
The property described above is not only true of agonists, it is true of all ligand classes, which gives rise to an interesting effect of δ. If neither the orthosteric nor the allosteric ligand have intrinsic efficacy (α = β = 1) but are positively cooperative for activation (δ > 1) then agonist activity results if both ligands are present. This is illustrated in Fig.9. A similar effect will also occur for inverse agonists (α,β < 1) but δ must be correspondingly greater to see a measurable effect. Initially this seems a rather strange finding: two apparently inactive ligands that, when coapplied, produce a functional response. However, this effect corresponds to the phenomenon of coagonism, the requirement for two compounds to be present to see a functional response. This has been well described forN-methyl-d-aspartate receptors (Corsi et al., 1996), in this case, both glutamate and glycine must be present to induce gating of this ligand-gated ion channel.

Effect of various concentrations of an allosteric activator (δ = 100) on activation curves to a neutral antagonist (α = 1). Other parameters were K =M = 1, L = 0.01, β = γ = 1, and [B] = 0 (♦), 0.003 (▪), 0.01 (▴), 0.03 (●), 0.1 (×), 0.3 (⋄), 1 (■), 3 (▵), or 10/M (○).
The behavior of this parameter emphasizes the multistate nature of the model. It allows for ligands that do not discriminate between R and R* to discriminate between AR and AR* or BR and BR* implying that the conformations of the receptors in the complexes differ from those of the free receptors.
Discussion
The model described in this report is a simple and logical extension of the two-state model when considering allosteric ligands. As in the two state model (and indeed other more recent models), this model highlights the inter-relatedness of the pharmacological concepts of affinity and efficacy (Leff, 1995; Weiss et al., 1996a–c). It predicts that the measurable affinity of a pharmacological agent depends not only on its binding affinity for the receptor but also on its ability to induce conformational changes within the receptor and the resistance of the receptor to those conformational changes. The model is, of course, also a modification of the ternary complex model of allosteric interactions. In this case, the model provides a framework in which each of the receptor species in the ternary complex model can cause downstream functional effects with different efficacies.
The behavior of the model is intuitively reasonable and consistent with published experimental observations (Bruns and Fergus, 1990; Lazareno and Birdsall, 1995; Kollias-Barker et al., 1997; Litschig et al., 1999). It predicts that an allosteric ligand that only affects orthosteric ligand binding (γ ≠ 1, β = δ = 1) will only effect the midpoint of an orthosteric agonist with no effect on the maximal response. Only allosteric agents that can affect receptor activation, either through their own intrinsic efficacy (β≠ 1) or through cooperative modification of the orthosteric ligand's intrinsic efficacy (δ ≠ 1), can affect the maximal response induced by an orthosteric ligand. This potential cooperativity at the level of receptor activation (parameter δ) also predicts the phenomenon of coagonism (Corsi et al., 1996). As described under Applicability of the Model the distinct behavior of each parameter in the model can be used to suggest potential mechanisms for the action of allosteric compounds.
An important practical prediction of this model is that it is only possible to generate a system-independent measure of the cooperativity between allosteric and orthosteric ligands when certain conditions are met (under ). Specifically, when using functional assays to characterize an allosteric ligand, the allosteric ligand must not have any agonist (or inverse agonist) activity in its own right or its effects become system-dependent. Agonism is of course easy to test for in a functional assay. Inverse agonism is more difficult to determine because not all experimental systems are constitutively active. Fortunately, the inaccuracies introduced by inverse agonist activity are likely to be very small. In eq. 12, under , when β < 1, the terms in L(L is also less than 1) approximate to unity and the expression reduces to βγδ, which is still an independent characteristic of the ligand pair.
When using a ligand binding assay to characterize allosteric interactions, the requirements for system-independent quantification are even more stringent. The allosteric ligand must not only be devoid of agonist activity (β = 1) but must also have no effects on the efficacy of the orthosteric ligand (δ = 1). Functional assays would, therefore, still be required even when characterizing an allosteric agent in binding assays (to allow appropriate interpretation of the data generated) because ligand binding assays cannot give meaningful information about agonist efficacy. If characterization were to be performed in ligand binding assays, the endogenous agonist would obviously be the preferred radioligand if available (and of course suitable).
Effects of an allosteric ligand on an orthosteric agonist's efficacy would most likely be manifest as a change in the maximal response that it elicits in a functional assay. However, care must be exercised when testing effects on maximal responses because the limiting factor to response size in many experimental systems is not the activation of the receptor but the activation of the signaling cascade (a manifestation of receptor reserve). This would make it impossible to detect increases in the maximal response to a full agonist and may truncate increases in the maximal response to partial agonists. It would also result in unreliable data on depressions of the maximal response. It may be possible to diagnose these problems by comparing the effects of a modulator on the midpoint of the agonist CRC and on its maximal effect. Discrepancies between the midpoints of the effects of the allosteric agent on these two parameters would indicate that the data could not be interpreted reliably. The most suitable assay system may therefore be one with very low receptor expression where even highly efficacious agonists are unable to fully activate the signal transduction cascade (see the concluding paragraph under ). When an allosteric ligand does not meet the above-mentioned criteria (i.e., when it is also an agonist), the least complicated parameter that could be used to quantify its effect seems to be the ratio of the asymptotes of the maximal effect ratios and the dose ratios (under ) taken from a functional assay.
Comparison with the Cubic Ternary Complex Model.
The model described in this report has very close parallels with the cubic ternary complex model proposed by Weiss et al. (1996a–c). Indeed, if B were a G-protein the scheme in Fig. 2 would be identical with that used to describe the cubic ternary complex model. There is, however, one important difference between these two models in terms of the behavior of the allosteric constants and the activation curves. This difference is due to the way the two models quantify functional activity. In the allosteric two-state model, as in the two-state model, functional activity is quantified as the stimulus, i.e., the proportion of the receptor complexes that contain R*. In the cubic ternary complex model, the functional response is assumed to be proportional to the concentration of activated receptors that are bound to G-protein (Weiss et al., 1996a). This would be equivalent to [R*B] + [AR*B] in Fig.2. This difference radically changes the effect of parameter γ. In the present model, the only effect of γ is on the affinity of the orthosteric ligand; however, in the cubic ternary complex model, γ modifies the ability of the receptor to interact with the G-protein and is therefore able to modify the maximal response induced by the ligand. Thus, in the cubic ternary complex model, γ is an efficacy defining parameter along with α, β, and δ, whereas in the allosteric two-state model it is not. Another difference is that, in the cubic ternary complex model, it is possible to define a simple condition for agonism (δ > (1 + αL + γM[B]/αγ(1 +L + M[B])) whereas it is not meaningful to derive an equivalent expression in the allosteric two-state model.
Applicability of the Model.
The acid test of a mathematical model is that it can be applied, at least qualitatively, to real, experimental data. A number of reports have been published on binding interactions between allosteric and orthosteric ligands (Ellis et al., 1991; Lee and El-Fakahany, 1991; Lazareno and Birdsall, 1995;Proška and Tuček, 1995; Hoare and Strange, 1996; Leppik et al., 1998). Because it is based on the most basic equilibrium model of these binding interactions, the allosteric two-state model is also compatible with these data, assuming that the allosteric and orthosteric ligands are site specific, of course. In terms of function, the prediction of coagonism by the allosteric two-state model has already been mentioned.
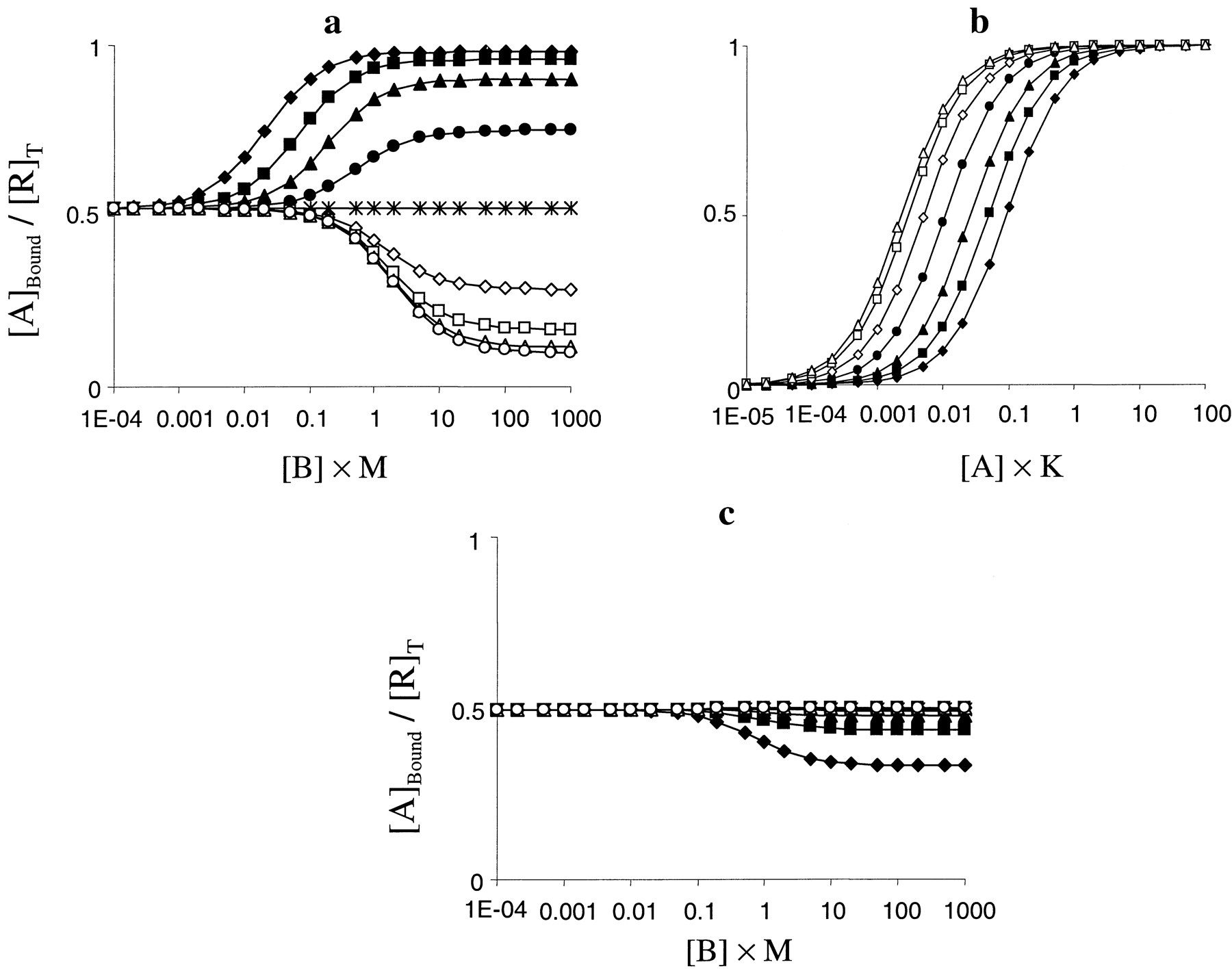
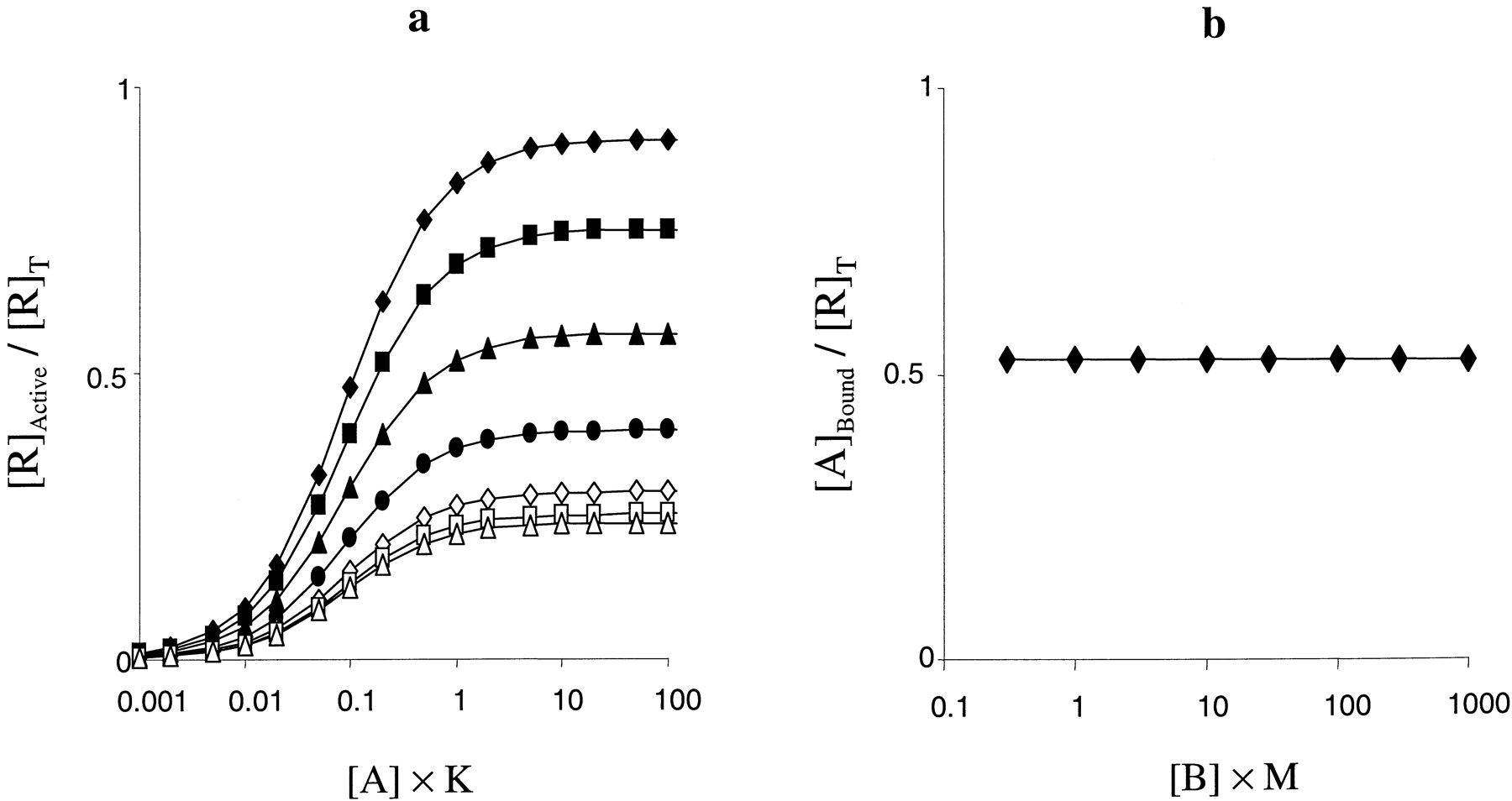
Interestingly, the characterization of the binding and functional effects of an allosteric modulator at type 1 metabotropic glutamate receptors, CPCCOEt (7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester), has recently been published (Litschig et al., 1999). This compound caused concentration-dependent decreases in the maximal response to glutamate at human mGluR1b (the limiting maximal effect was ∼0.25 of the control) without causing a significant shift in the midpoint of the CRCs. There was no evidence that this compound had any agonist or inverse agonist activity. Interestingly, this compound also inhibited inositol phosphate formation in response to glutamate at rat mGluR1a but showed no effects on [3H]glutamate binding to this receptor at similar concentrations. Assuming that the functional effects of this compound are similar at the two receptors, this behavior can be mimicked using the allosteric two-state model (Fig. 10). The inhibitory effect on the maximal response is indicative of negative activation cooperativity (δ < 1). However, a pure δ effect would be accompanied by a right shift in the CRCs (Fig. 8b). This indicates that the compound also shows positive binding cooperativity (γ > 1) with glutamate, which offsets the right-shift caused by δ. Values of γ and δ that mimicked these functional data were 8.5 and 0.03, respectively (Fig. 10a). Importantly, when the same parameters were entered into a competition binding simulation, increasing concentrations of the allosteric modulator had no effect on binding (Fig. 10b), again mimicking the experimental data. Thus, it would appear that these data are, at least qualitatively, consistent with the allosteric two-state model and that CPCCOEt is positively cooperative with the binding of glutamate but negatively cooperative with its functional activation of the receptor.
a, simulations of the effect of CPCCOEt on glutamate-induced inositol phosphate production. Parameters were defined such that the allosteric ligand decreased the maximal response to the agonist to 25% of the control level without altering the midpoint of the CRCs. The parameters were K =M = 1, L = 0.001, α = 10 000, β = 1, γ = 8.5, and δ = 0.03. The concentrations of B were 0 (♦), 0.3 (▪), 1 (▴), 3 (●), 10 (⋄), 30 (■), 100 (▵), or 300/M (○). b, simulated allosteric ligand competition binding curve using the parameters defined in a. The concentration of A was 0.1/K, however, there was no displacement of A over this range of concentrations of B at any concentration of A simulated (1 × 10−5− 1000/K).
Another system that has been extensively characterized is the interaction of the compound (2-amino-4,5-dimethyl-3-thienyl)-[3-(trifluoromethyl)phenyl]methanone (PD 81,723) with agonists at the A1 adenosine receptor (Bruns and Fergus, 1990; Janusz et al., 1991; Kollias-Barker et al., 1994, 1997). This compound has been characterized against the receptor from several species, however, this discussion will concentrate on data from the (recombinant) human receptor (Kollias-Barker et al., 1997). In this study, PD 81,723 appeared to be an agonist at A1 receptors when added alone. It also caused an increase in the binding of the agonists [3H]N 6-cyclohexyladenosine and of R-N 6-(2-phenylisopropyl)adenosine and potentiated the effects of R-N 6-(2-phenylisopropyl)adenosine in both adenylate cyclase and [35S]GTPγS binding assays. This is inconsistent with the behavior of an orthosteric agonist but is consistent with the behavior of an allosteric agonist (although the compound did appear to have some affinity for the orthosteric site at high concentrations). However, the effects of this compound on binding appeared to be due to an increase in the maximal level rather than the affinity of binding. This is inconsistent with the allosteric two-state model's predictions and probably highlights the lack of G-protein effects in the model (an “allosteric cubic ternary complex model” is currently being investigated). The allosteric two-state model may be better applied to agonist binding to 7-transmembrane receptors in the presence of guanine nucleotides to remove the complicating effects of G-protein interactions. Interestingly, PD 81,723 did not appear to have an effect on the binding of the inverse agonist [3H]8-cyclopentyl-1,3-dipropylxanthine in a saturation binding assay. However, this may simply be because an insufficiently high concentration of PD 81,723 (10 μM) was used to determine this effect. In the [35S]GTPγS binding assays, [3H]8-cyclopentyl-1,3-dipropylxanthine at approximately 50 times its K D for the orthosteric site (100 nM) completely abolished the response to PD 81,723. This is consistent with the interaction between an orthosteric inverse agonist and an allosteric agonist in the allosteric two-state model. Thus, the simplest explanation of the effects of PD 81,723 is that it is an allosteric agonist at A1 adenosine receptors.
As noted in the Introduction, a similar scheme has also been proposed for the allosteric modulatory effects of benzodiazepine receptor ligands on γ-aminobutyric acidA receptors (Ehlert et al., 1983). It is important to stress, however, that the predictions of this model are qualitative and can be used only to provide plausible mechanisms for the behavior of allosteric ligands. There may, however, be situations where this model cannot distinguish between possible mechanisms. For example, although allosteric agonism is the simplest explanation for the effects of PD 81,723, it is not possible to rule out cooperative effects of this compound. Contributions from γ and δ would alter the magnitude of the effects due to the compound's agonism rather than change the pattern of behavior.
Concluding Remarks.
This report describes a mathematical model of the interaction between orthosteric and allosteric ligands at pharmacological receptors that explicitly includes an effect of the allosteric modulator on the activation of the receptor. To my knowledge this is the first detailed report of such a system, particularly in its application to G-protein-coupled receptors (with the caveat of the lack of G-protein already mentioned). The behavior of this model is qualitatively consistent with the behavior of allosteric modulators in radioligand binding assays. Importantly, this model can also simulate and therefore suggest a potential mechanism for the effects of an allosteric modulator of metabotropic glutamate receptors that inhibits functional activity but does not affect the binding of the agonist. It also suggests a number of caveats that should be considered when analyzing the interaction between allosteric and orthosteric ligands at pharmacological receptors.
Acknowledgments
I thank Drs. Heather Giles, Terry Kenakin, and Mike Sheehan for helpful comments during the preparation of this manuscript.
Binding.
From the definitions of the equilibrium constants given in Table 1 and the law of conservation of mass (applied to the receptor; eq. 1), it is possible to derive
From the model, the fractional occupancy of the receptor by A can be derived from
The limiting affinity of A (at a saturating concentration of B) is then given by
The level of binding of A in the absence of B is given by
which, after rearrangement and factorization, simplifies to
Functional Activation.
Using the same approach as above, it is possible to derive an expression for the fraction of the total receptor pool in the active state. Thus, This is the equation of a rectangular hyperbola. The midpoint of this curve is again given by eq. 4; that is, in this model, the binding and activation curves have the same midpoints, a property that it shares with the original two-state model. However, the limiting values of eq. 10 are not zero and unity as they are for the binding curves. The value of eq. 10 in the absence of any ligands (the basal activity) is L/(1 + L). The upper asymptote of eq. 10 can be stated for a variety of conditions, depending on the details of the experiment that is being simulated. Thus, in the presence of A or B alone it is αL/(1 + αL) or βL/(1 + βL), respectively. When both ligands are present it can be stated as
This is the equation of a rectangular hyperbola. The midpoint of this curve is again given by eq. 4; that is, in this model, the binding and activation curves have the same midpoints, a property that it shares with the original two-state model. However, the limiting values of eq. 10 are not zero and unity as they are for the binding curves. The value of eq. 10 in the absence of any ligands (the basal activity) is L/(1 + L). The upper asymptote of eq. 10 can be stated for a variety of conditions, depending on the details of the experiment that is being simulated. Thus, in the presence of A or B alone it is αL/(1 + αL) or βL/(1 + βL), respectively. When both ligands are present it can be stated as
It is interesting to consider the midpoints of the effects of B on the upper asymptote and midpoint of the activation curves of A. Taking the effect of B on the upper asymptote of the activation curve of A, the midpoint of this effect again occurs at the value that is the average of the upper asymptotes for the control curve (αL/(1 + αL)) and that in the presence of saturating B (αβδL/(1 + αβδL)). Thus, substituting this into the left-hand side of eq. 11,
If B affects the upper asymptote of A's activation curve, then the ratio of the maximal effect in the presence and absence of a maximally active concentration of B is
Affinity Ratio Analysis.
Allosteric effects can also be quantified by analysis of the changes in the macroscopic affinity of the orthosteric ligand in the presence of a variety of concentrations of the allosteric ligand (Lazareno and Birdsall, 1995). Thus, from eq.4
This type of analysis could also be performed on functional data. In this case, the potency ratios (PR) result in the same expressions for midpoint and asymptote as the affinity ratios (potency and affinity are the same in this model). However, for functional data, the effect of the allosteric agent on the maximal effect of the orthosteric ligand also provides useful data. The ratio of the maximal effect of the agonist in the presence and absence of allosteric ligand (MR) can be derived from eq. 11
This analysis of course depends on the maximal response of the system being limited by the receptor and not by the signal transduction system; that is, it relies on a system with a low (or nonexistent) receptor reserve, a rather unusual situation. Under these conditions the potencies of agonists should be identical with their affinities. In terms of G-protein-coupled receptors, the system that is most likely to conform to this restriction is the GTPγS binding assay. However, recombinant cell lines with very low levels of receptor expression (the opposite of that which is usually required) may also furnish suitable systems. For ligand-gated ion channels, ion flux measurements may provide a suitable system because maximal flux should occur only with maximal channel opening. In this case, however, the model may require modification to account for steep activation curves.
Footnotes
- Received February 22, 2000.
- Accepted August 28, 2000.
-
Send reprint requests to: David A. Hall, Receptor Pharmacology Unit, In Vitro Pharmacology Department, Medicines Research Centre, Glaxo Wellcome Research and Development, Gunnells Wood Road, Stevenage, Hertfordshire, SG1 2NY UK. E-mail:dah40302{at}glaxowellcome.co.uk
Abbreviations
- CRC
- concentration-response curve
- CPCCOEt
- 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester
- GTPγS
- guanosine-5′-O-(3-thio)triphosphate
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}