


Abstract
Cocaine causes cardiac arrhythmias, sudden death, and occasionally long QT syndrome in humans. We investigated the effect of cocaine on the human K+ channels HERG and KvLQT1+minK that encode native rapidly (IKr) and slowly (IKs) activating delayed rectifier K+ channels in the heart. HERG and KvLQT1+minK channels were heterologously expressed in human embryonic kidney 293 cells, and whole-cell currents were recorded. Cocaine had no effect on KvLQT1+minK current in concentrations up to 200 μM. In contrast, cocaine reversibly blocked HERG current with half-maximal block of peak tail current of 7.2 μM. By using a protocol to quickly activate HERG channels, we found that cocaine block developed rapidly after channel activation. At 0 mV, the time constants for the development of block were 38.2 ± 2.1, 15.2 ± 0.8, and 6.9 ± 1.1 ms in 10, 50 and 200 μM cocaine, respectively. Cocaine-blocked channels also recovered rapidly from block after repolarization. At −100 mV, recovery from block followed a biphasic time course with fast and slow time constants of 3.5 ± 0.7 and 100.3 ± 15.4 ms, respectively. UsingN-methyl-cocaine, a permanently charged, membrane-impermeable cocaine analog, block of HERG channels rapidly developed when the drug was applied intracellularly through the patch pipette, suggesting that the cocaine binding site on the HERG protein is located on a cytoplasmic accessible domain. These results indicate that cocaine suppresses HERG, but not KvLQT1+minK, channels by preferentially blocking activated channels, that it unblocks upon repolarization, and does so with unique ultrarapid kinetics. Because the cocaine concentration range we studied is achieved in humans, HERG block may provide an additional mechanism for cocaine-induced arrhythmias and sudden death.
Cocaine is a widely used drug that is capable of causing cardiac arrhythmias and sudden death in otherwise healthy persons (Kloner et al., 1992;Bauman et al., 1994). It has been thought that cocaine has two principal pharmacological properties that affect the heart and vascular systems (Kloner et al., 1992; Bauman et al., 1994). First, cocaine blocks the reuptake and increases the release of catecholamines from central and peripheral stores, causing catecholamine accumulation at postsynaptic receptors and intense sympathomimetic stimulation. Second, cocaine exerts local anesthetic effects by blocking Na+ channels to slow cardiac action potential conduction.
In addition to the above mechanisms, in isolated myocytes, cocaine has been shown to block delayed rectifier K+ current, prolong ventricular action potential duration, and trigger early afterdepolarizations (Kimura et al., 1992;Clarkson et al., 1996), and in humans to induce long QT syndrome and torsades de pointes (Schrem et al., 1990; Perera et al., 1997; Khan et al., 1999). In cardiac ventricular cells, the principal K+ channels activated during action potential repolarization are the rapidly (IKr) and slowly (IKs) activating delayed rectifier K+ currents, encoded by the human ether-a go-go-related gene (HERG) (Sanguinetti et al., 1995;Trudeau et al., 1995) and KvLQT1+minK genes (Barhanin et al., 1996; Sanguinetti et al., 1996), respectively. These K+ channels are common targets for drug block or mutations that cause acquired and congenital long QT syndrome. The aim of this work was to investigate the effect of cocaine on human cloned HERG and KvLQT1+minK channels heterologously expressed in a mammalian cell line. We found that cocaine had no effect on KvLQT1+minK channels, whereas it potently blocked HERG channels. Cocaine preferentially blocked activated (open or inactivated) HERG channels, and drug block and unblock occurred with unusual ultrarapid kinetic properties.
Materials and Methods
DNA Constructs and Transfection of HEK293 Cells.
HERG channels were stably expressed in a HEK293 cell line as described previously (Zhou et al., 1998; Zhang et al., 1999). KvLQT1and minK cDNAs were provided by Dr. Mark Keating (University of Utah, Salt Lake City, UT) and Dr. Richard Swanson (Merck Research Laboratories, West Point, PA). For KvLQT1+minKexpression, native HEK293 cells were seeded at 5 × 105 cells/60-mm diameter dish. The cells were transiently transfected using lipofectamine with 2.5 μg pCDNA3-KvLQT1 and 2.5 μg pCDNA3-minK. After 48 h, 30 to 50% of cells expressed a slowly activating outward current characteristic of KvLQT1+minK channels. Transfection with the individual pCDNA3-minK or pCDNA3-KvLQT1 vector did not produce this current confirming previous reports (Barhanin et al., 1996; Sanguinetti et al., 1996).
Patch-Clamp Recording Method.
Membrane currents were recorded in the whole-cell, patch-clamp configuration as described previously (Chouabe et al., 1998; Zhou et al., 1998; Zhang et al., 1999). Patch electrodes typically had resistances of 1 to 3 MΩ. Capacitance compensation was used in all experiments. Data were sampled at 20 kHz and filtered (8-pole Bessel) at 5 kHz. For HERG current recording, the extracellular solution contained 137 mM NaCl, 4 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and 10 mM HEPES/NaOH, pH 7.4. The internal pipette solution contained 130 mM KCl, 1 mM MgCl2, 5 mM EGTA, 5 mM MgATP, 10 mM HEPES/KOH, pH 7.2. For KvLQT1+minK current recording, the extracellular solution contained 150 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, and 10 mM HEPES/NaOH, pH 7.4. The internal pipette solution contained 150 mM KCl, 0.5 mM MgCl2, 5 mM EGTA, 10 mM HEPES/KOH, pH 7.2. All experiments were performed at 23 ± 1oC.
Drugs and Chemicals.
Cocaine was obtained from Sigma Chemicals (St. Louis, MO). N-Methyl-cocaine iodide was generated from cocaine. Methyl iodide (200 mg) was added to free base cocaine (50 mg) in anhydrous diethyl ether (25 ml). The mixture was stirred at room temperature for 12 h. The precipitate was filtered and dissolved in the 10 ml of distilled water. The resulting aqueous solution was extracted with anhydrous diethyl ether (3 × 10 ml) and the aqueous phase lyophilized to afford 22 mg ofN-methyl-cocaine iodide as a white powder. TheN-methyl-cocaine was determined to be completely free of cocaine as assessed by silica gel thin-layer chromatography in methylene chloride/methanol/ammonium hydroxide (10:2:0.1). Proton NMR in deuterated water confirmed the chemical shift for the added methyl group compared with cocaine. Mibefradil, which blocks several ion channels including KvLQT1+minK and HERG, was a gift from Hoffmann-La Roche Ltd (Basel, Switzerland). E-4031, which selectively blocks IKr and HERG channels, was a gift from Eisai Ltd (Ibaraki, Japan). Fenpropimorph was obtained from Crescent Chemical Co. (Islandic, NY) and iodoazidococaine was synthesized as described previously (Kahoun and Ruoho, 1992a). Each drug was dissolved in distilled water and the final drug concentrations were made by diluting stock solution with extracellular solution. Solution exchanges in the chamber were complete within 1 to 2 min (see Fig. 2B).

Cocaine reversibly blocks HERG current in a concentration-dependent manner. A, voltage protocol is shown above current traces. HERG current was recorded for control conditions and in the presence of 2, 10, 40, and 200 μM cocaine, and steady-state current traces are superimposed. B, HERG tail current peak amplitude is plotted versus time during control and increasing concentrations of cocaine. Block was reversed with drug washout. C, the concentration-response relation for block of HERG tail current. At cocaine concentrations of 2, 10, 40, and 200 μM, the relative tail current peak amplitude was 0.78 ± 0.01, 0.42 ± 0.02, 0.15 ± 0.02, and 0.03 ± 0.01, respectively. D, HERG current recorded during an action potential clamp. The ventricular action potential command voltage was applied every 5 s and is shown above HERG current recorded in control and with steady-state block by 10 μM cocaine.
Curve Fitting and Statistical Methods.
Data are given as a mean ± S.E.M. Concentration-dependent effects were fit to the Hill equation (Idrug/Icontrol = 1 / [1 + (D / IC50)nH], where D is the drug concentration, IC50 is the drug concentration for 50% block, and n H is the Hill coefficient. Statistical significance was analyzed using a Student's t test or analysis of variance, where appropriate. A p value < 0.05 was considered statistically significant.
Results
Cocaine Blocks HERG but Not KvLQT1+minK Potassium Channels.
The effect of cocaine on HERG and KvLQT1+minK currents is shown in Fig.1. Figure 1A shows control HERG current elicited from a holding potential of −80 mV by 4-s depolarizing steps to between −70 and 70 mV in 10 mV increments applied every 15 s. Tail current was recorded with a step to −50 mV. HERG current activated at voltages positive to −50 mV, maximum current was reached at 0 mV, and at more positive voltages inward rectification was present (Smith et al., 1996). Tail current amplitude was maximal after voltage steps to >10 mV. Figure 1, B and C, shows example records of the effect of 10 and 200 μM cocaine in different cells. Figure 1D shows averaged I-V relations for HERG current measured at the end of the depolarizing step (Istep) and peak tail current amplitude (Itail), for control conditions and with 10 and 200 μM cocaine (n = 6 cells at each concentration). Cocaine (200 μM) completely blocked both Istep and Itail at all voltages tested. Figure 1, A and B, also shows that 10 μM cocaine, in addition to reducing current amplitude, slowed the tail current decay. To analyze this, the current decay was fit as the sum of two exponential values. For control conditions and following exposure to 10 μM cocaine, the rapid time constant of decay (τ1) at −50 mV was 335.3 ± 16.9 ms versus 973.8 ± 75.6 ms (p < 0.05), and the slow time constant of decay (τ2) was 2000.8 ± 84.7 ms versus 7953.3 ± 1362.6 ms (p < 0.05), respectively (n = 9 cells). This finding can be explained by time-dependent cocaine unbinding from HERG channels with the drug unbound channels then opening before deactivating (for discussion, see Zhang et al., 1999). Figure 1E shows control KvLQT1+minK current elicited from a holding potential of −80 mV by 4-s depolarizing steps to between −60 and 120 mV in 20 mV increments applied every 15 s. Deactivating tail current was recorded upon repolarization to −50 mV. KvLQT1+minK current activated as a time-dependent outward current at voltages positive to −20 mV with its amplitude increased at more positive voltages. Similarly, tail current amplitude was increased after depolarizing steps up to 120 mV. These properties are characteristic of KvLQT1+minK current (Barhanin et al., 1996; Sanguinetti et al., 1996; Chouabe et al., 1998). As shown in Fig.1F, cocaine in concentrations studied up to 200 μM had no effect on KvLQT1+minK current. Figure 1G shows the averaged I-V relations for KvLQT1+minK current measured at the end of the depolarizing step (Istep) and for the peak tail current amplitude (Itail) for control conditions and with 200 μM cocaine (n = 5 cells), indicating the lack of cocaine block of KvLQT1+minK channels. To confirm that the KvLQT1+minK current had the expected pharmacological sensitivity, mibefradil, which blocks KvLQT1+minK current with an IC50 value of 11.8 μM (Chouabe et al., 1998), was tested. Mibefradil (100 μM) suppressed KvLQT1+minK current by >95% (n = 5 cells, data not shown). E-4031 (5 μM), which we have shown previously to block HERG current with an IC50 value of 7.7 nM (Zhou et al., 1998) had no effect on KvLQT1+minK current (n = 3 cells, data not shown).

Effect of cocaine on HERG and KvLQT1+minK K+ channel current. A to C, HERG current elicited by the voltage protocol for control conditions (A) and with 10 (B) or 200 μM (C) cocaine. D, averaged I-V relations for HERG Istep and Itail for control, 10 and 200 μM cocaine. E and F, KvLQT1+minK current elicited by the voltage protocol for control conditions (E) and with 200 μM cocaine (F). G, averaged I-V relations for KvLQT1+minK Istep and Itail for control and 200 μM cocaine.
Cocaine reversibly blocked HERG current in a concentration-dependent manner. Figure 2A shows the voltage protocol with HERG current activated by a depolarizing step from −80 to 20 mV and tail current recorded at −50 mV, with the voltage protocol repeated at 15-s intervals. The current records show superimposed traces with steady-state block in different cocaine concentrations. Figure 2B shows HERG tail current peak amplitude plotted versus time for one cell. Exposure to cocaine caused concentration-dependent block of HERG current, which was reversed after drug washout. In Fig. 2C, HERG tail current peak amplitude at steady-state block in each cocaine concentration was normalized to the control value and plotted as relative current amplitude (n = 8, 11, 7, and 3 cells at cocaine concentrations of 2, 10, 40, and 200 μM, respectively). The calculated IC50 value for block of tail current was 7.2 μM with a Hill coefficient of 1.0, consistent with cocaine binding to a single receptor site.
To examine further the effect of cocaine on HERG current, an action potential clamp method was used as shown in Fig. 2D (Zhou et al., 1998). HERG channels rapidly inactivate at positive voltages after action potential depolarization, causing the control HERG current amplitude to be small initially. As the action potential repolarizes, HERG channels rapidly recover from inactivation to reopen and slowly deactivate, thus increasing their occupancy in the open state and HERG current amplitude. The current then deactivates with the final phase of repolarization. With steady-state block by cocaine (10 μM), using the action potential clamp protocol, the peak current during the action potential plateau was reduced by 67.7 ± 3.1% (n= 7 cells, p < 0.05).
Intracellularly Applied N-Methyl-cocaine Blocks HERG Channels.
N-Methyl-cocaine, a quaternary, permanently charged, membrane-impermeable cocaine analog, was used to test whether cocaine acts on HERG channels from the inside or outside of the cell membrane. N-Methyl-cocaine was included in the pipette solution to diffuse into the cell and, after obtaining whole-cell clamp, HERG current was recorded at 15-s intervals using the voltage clamp protocol shown in Fig. 2A. Figure3A shows superimposed current traces recorded with the first step (1st step) after obtaining whole-cell clamp conditions, and 2 and 10 min later. N-Methyl-cocaine rapidly blocked HERG current and results showing the time course of the decrease in tail current peak amplitude are summarized in Fig. 3B (n = 4 cells at each time bar). Figure 3B also shows the effect of adding N-methyl-cocaine (50 μM) to the bath. HERG current was recorded at 15-s intervals using the voltage clamp protocol shown in Fig. 2A. After obtaining control current records drug wash-in was completed within 1 to 2 min as shown in Fig. 2B. Exposure to N-methyl-cocaine in the bath caused only a minimal reduction in HERG tail current amplitude (n = 4 or 5 cells at each time bar, p > 0.05 at each time compared with control). The data also show that the tail current amplitude obtained with the 1st step during the intracellular application ofN-methyl-cocaine is smaller than the control value obtained before the extracellular application of N-methyl-cocaine (p < 0.05), suggesting that significant block of HERG channels had already developed before the first depolarizing step was applied. Figure 3C shows results obtained with a higher concentration of N-methyl-cocaine in the pipette.N-Methyl-cocaine (500 μM) nearly completely blocked HERG current within 120 s. These findings suggest that cocaine rapidly accesses a binding site on the HERG channel from the cytoplasmic side of the cell membrane.

N-Methyl-cocaine rapidly blocks HERG current when applied intracellularly through the pipette. The voltage protocol was the same as used in Fig. 2A with depolarizing steps applied at 15-s intervals. A, data shown were recorded with the first depolarizing step (1st step) after obtaining intracellular access, 2 and 10 min later with 50 μM N-methyl-cocaine in the pipette. B, averaged peak tail current amplitude from cells exposed to 50 μM N-methyl-cocaine in the pipette or bath. See text for discussion. C, a higher N-methyl-cocaine concentration (500 μM) in the pipette produced nearly complete block of HERG current within 120 s.
Use-Dependent Block of HERG Channels by Cocaine.
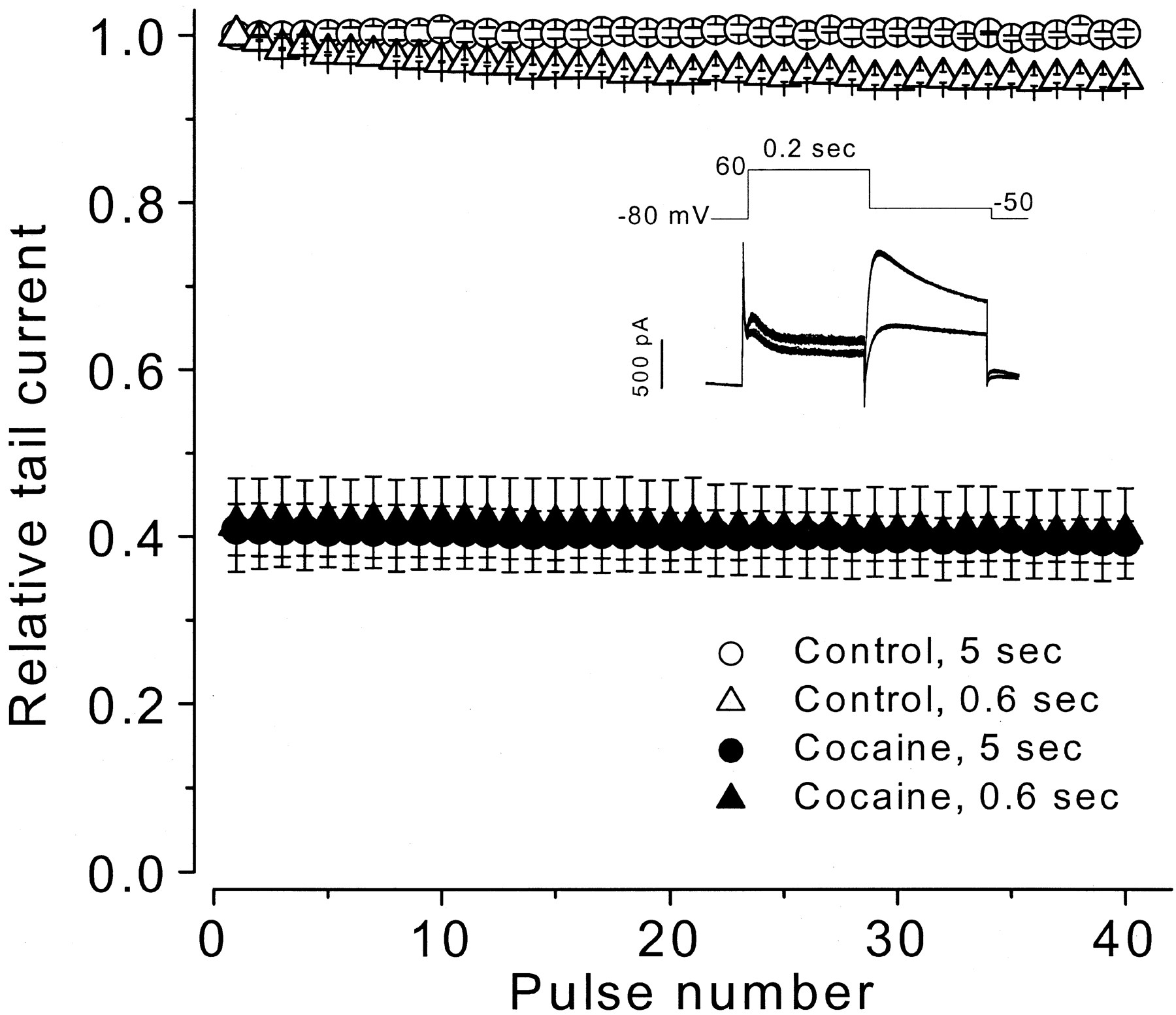
Drugs that block HERG channels do so by preferentially interacting with activated channels in the open or inactivated states to produce use-dependent block. To evaluate the state-dependence of cocaine block of HERG current, a concentration (10 μM) near the IC50value was applied to evaluate use-dependent properties. From a holding potential of −80 mV, HERG current was activated by a 0.2-s depolarizing step to 60 mV and tail current was recorded with a repolarizing step to −50 mV for 0.2 s. Trains of 40 pulses were applied at an interval of 5 or 0.6 s. After obtaining control data, 10 μM cocaine was washed into the bath for 10 min while the cell was held at −80 mV to maintain HERG channels in a closed state, and the pulse train was repeated. Each cell was studied at only one pulse rate. In Fig. 4, averaged tail current peak amplitude data are plotted versus pulse train number. For control conditions tail current amplitude was constant during pulse trains applied at a 5-s (○) interval and declined by 5% during pulse trains applied at a 0.6-s (▵) interval. With 10 μM cocaine present in the bath, the data show that cocaine-induced block of HERG tail current was fully developed with the first pulse of the train. Tail current amplitude was reduced by an average of 59% at both the 5 (●,n = 5 cells) and 0.6 (▴, n = 4 cells) second interval pulse trains and additional block did not accumulate.

Cocaine effects during trains of voltage-clamp pulses. Voltage clamp pulse trains (protocol shown in inset) were applied at intervals of 5 and 0.6 s. Tail current peak amplitude for each pulse was normalized to its control value for each cell, averaged, and plotted versus pulse number. Inset shows example superimposed control current (larger) and cocaine exposure current (smaller) traces at the 0.6-s pulse interval.
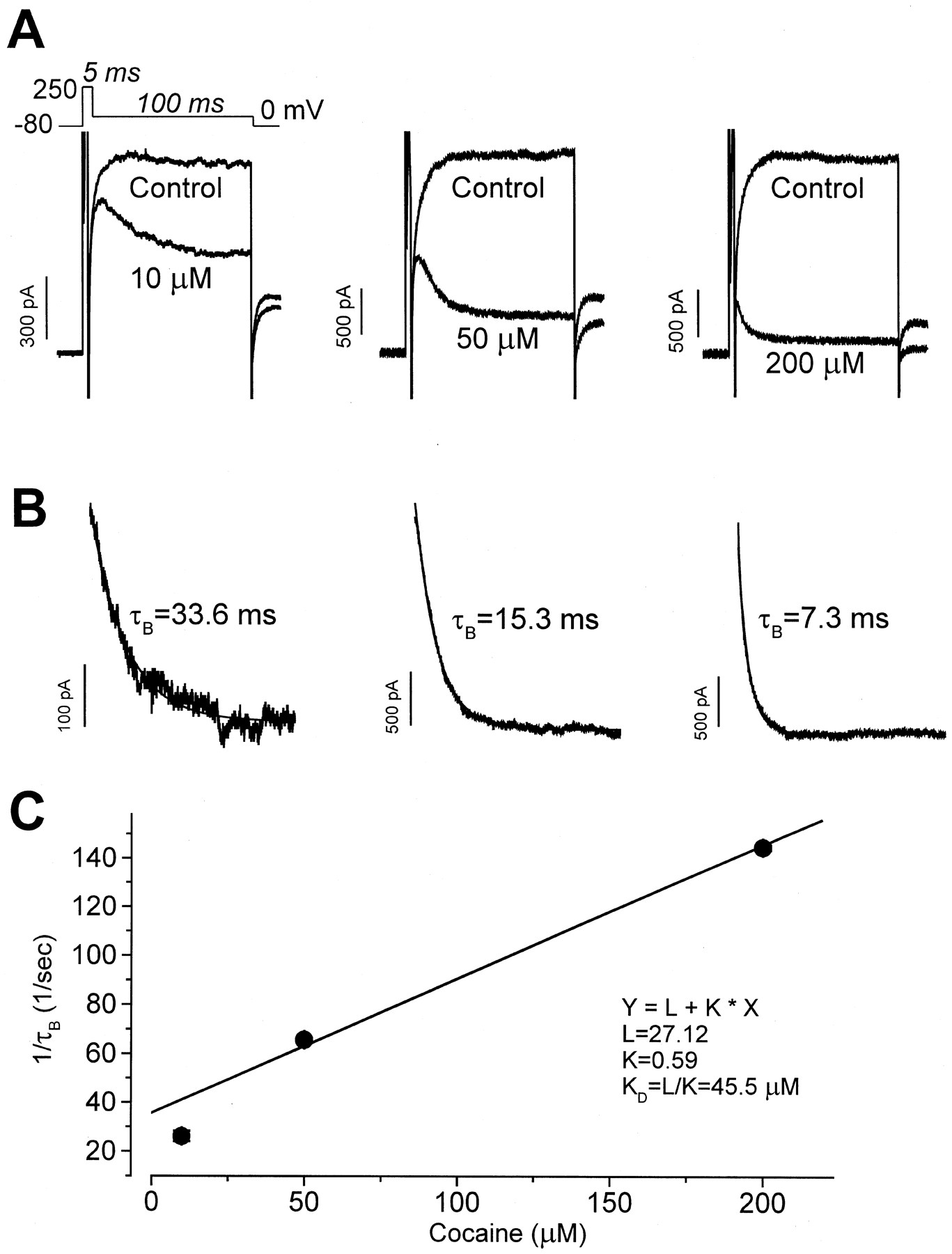
The above findings suggest that cocaine could block with similar affinity the rested (closed) and activated states of HERG channels, or that cocaine block and unblock kinetics could be state-dependent and ultrarapid. To study further cocaine block of HERG channels, we used the voltage protocol shown in Fig. 5A. Because activation of HERG channels is strongly voltage-dependent, from a holding potential of −80 mV HERG channels were rapidly activated by a 5-ms step to 250 mV followed by a 100-ms step to 0 mV, before returning to the holding potential. With this protocol, the control HERG current amplitude at 0 mV was nearly constant, and tail current was recorded with the subsequent repolarizing step to −80 mV. After obtaining control recordings, cells were held continuously at −80 mV for 10 min to maintain channels in a closed state during cocaine (10, 50, or 200 μM) wash-in. With 10 μM cocaine present, following the step to 0 mV, a large amplitude HERG current was present that then declined to a steady level as additional block developed. This pattern is consistent with preferential cocaine binding to activated channels. With the higher cocaine concentrations, the initial HERG current recorded at 0 mV was reduced in amplitude, and the development of additional block occurred more rapidly. Subtraction of current recorded at 0 mV during control conditions from the current recorded in the presence of cocaine gives a cocaine-sensitive current. The cocaine-sensitive component is shown in Fig. 5B as a declining current as drug block developed. This current was then fit as a single exponential decay (solid lines in Fig. 5B), with the time constants (τB) being concentration-dependent. These data suggest that cocaine preferentially blocks activated HERG channels and that the block occurs with ultrarapid kinetic properties. Our findings, however, do not distinguish between open or inactivated state block, and the possibility of weak block to the closed state cannot be excluded.
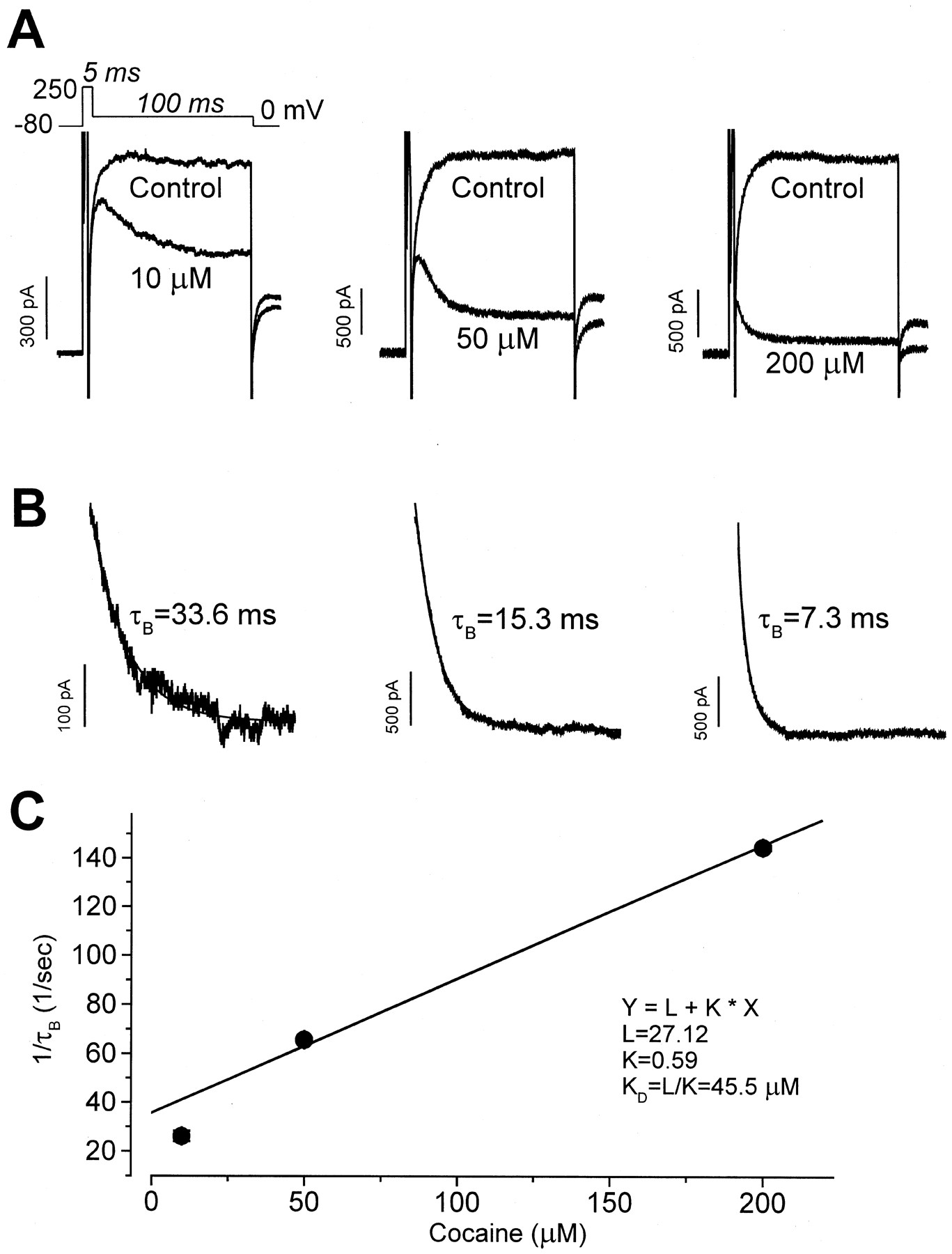
Block by cocaine requires channel activation. A, using the voltage protocol shown at the top, HERG current was rapidly activated from a holding potential of −80 mV by a 5-ms depolarizing step to 250 mV followed by a step to 0 mV for 100 ms. Tail current was observed upon repolarization to −80 mV. The control current amplitude became constant during the step to 0 mV. After wash-in of 10 μM cocaine, depolarization to 0 mV initially caused minimal block, which then accumulated. Superimposed HERG current traces for control conditions and in the presence of 10, 50, and 200 μM cocaine are shown. B, the cocaine-sensitive current during the step to 0 mV with each drug concentration was fit as a single exponential decay to obtain the time constant (τB) for the development of block. C, the inverse of the averaged τB is plotted versus the cocaine concentration. See text for discussion.
The time course of the cocaine-induced HERG current decay can be used to estimate drug-channel interaction kinetic properties (Snyders et al., 1992). As shown in Fig. 5C, τB was obtained at cocaine concentrations of 10 (n = 5 cells), 50 (n = 4 cells), and 200 μM (n = 5 cells), averaged, and are plotted as 1 /τBversus the cocaine concentration, D. From the equation 1 / τB = k D +l, the least-squares fit to the data gives an apparent association rate constant, k, of 0.59 × 106 M-1s-1 and an apparent dissociation rate constant,l, of 27.12 s−1. The apparentK D (l / k) was 45.5 μM, which is close to the IC50 value of 7.2 μM.
Cocaine Unblocks during Repolarization.
The finding that cocaine preferentially blocks activated HERG channels (Fig. 5), yet use-dependent block was not observed during pulse train protocols (Fig.4), suggests that cocaine may rapidly unbind from HERG channels. To examine drug unblocking properties, the voltage clamp protocol shown in Fig. 6A was used. Cells were held at 60 mV, conditions where HERG channels are predominantly inactivated. A repolarizing step to −100 mV for a variable duration (2 to 100 ms) was then applied. This step causes HERG channels to recover from inactivation to reopen rapidly and then to deactivate slowly (for discussion of HERG gating properties, see Trudeau et al., 1995; Smith et al., 1996; Spector et al., 1996; Zhou et al., 1998). A test step to 60 mV was then applied to elicit a large amplitude outward current that rapidly decayed as HERG channels inactivated. For control conditions (Fig. 6A, solid current traces), the peak current elicited with the test step initially increased in amplitude because of increased occupancy of the open state by channels rapidly recovering from inactivation at −100 mV. As the recovery interval at −100 mV lengthened beyond 10 ms, the peak current amplitude elicited with the test step decreased because of time-dependent channel deactivation. In the presence of cocaine (10 μM; Fig. 6A, dashed current traces), HERG current elicited with the test step to 60 mV was reduced in amplitude compared with control current. In addition, the amount of drug block decreased as the duration of the repolarizing step to −100 mV increased. By the end of the 100-ms recovery interval at −100 mV, the control and cocaine exposure currents elicited by the test step were nearly identical, suggesting rapid time-dependent unblocking of the channels in the presence of cocaine. To obtain the rate of recovery of HERG current from cocaine block during the step to −100 mV, we measured the cocaine-sensitive component of HERG current as the ratio of peak current in the presence of cocaine versus the control current at the same recovery interval (2–100 ms) at −100 mV (n = 7 cells). The averaged data are plotted as relative current in Fig. 6B and show that HERG current amplitude rapidly recovered from cocaine block at −100 mV. These data were fit as the sum of two exponential values, giving time constants (τ1 and τ2) of 3.5 ± 0.7 and 100.3 ± 15.4 ms with amplitudes (A1 and A2) of 0.25 ± 0.02 and 0.46 ± 0.02, respectively. We also measured the rate of HERG current inactivation at 2 ms of recovery at −100 mV with a monoexponential fit to the current decay after the test step to 60 mV (see Zhou et al., 1998). The control time constant was 2.5 ± 0.1 ms (n = 4 cells) and with 10 μM cocaine present the time constant was 2.1 ± 0.2 ms (n = 7 cells), which is not different from the control value (p > 0.05).

Unblock of HERG channels with repolarization during exposure to cocaine. A, the voltage protocol is shown above superimposed current traces recorded in the absence (solid lines) and presence of (dashed lines) of 10 μM cocaine. B, the time-dependent recovery from block of HERG current in 10 μM cocaine. Data were fit as a double exponential function with τ1 of 3.5 ± 0.7 ms (A1=0.25 ± 0.02) and τ2 of 100.3 ± 15.4 ms (A2=0.46 ± 0.02).
Discussion
These data provide new information about cocaine block of human cardiac K+ channels. Previously, Clarkson et al. (1996) showed that cocaine blocked delayed rectifier K+ current and that the cocaine-sensitive component was also suppressed by E-4031, consistent with cocaine block of IKr. Our finding that cocaine blocks HERG channels, but not KvLQT1+minK channels, provides direct evidence that cocaine suppresses IKr, not IKs. This provides a molecular mechanism for previously reported action potential and QT interval prolonging effects of cocaine.
The drug binding domain on the HERG protein has been studied for relatively few drugs. Our experiments with theN-methyl-cocaine quaternary compound, a membrane impermeant derivative, showed that when added to the patch pipette and after obtaining whole-cell clamp, it resulted in the rapid block of HERG current as it diffused into the cell. In the bath,N-methyl-cocaine exerted minimal effects. These findings suggest that N-methyl-cocaine accesses a binding site on the HERG channel protein from the cytoplasmic side of the cell surface membrane. If cocaine binds to same site, these findings suggest that it permeates the cell membrane in the neutral form and then binds to a cytoplasmic accessible site. Drug binding to a site accessible from the cell interior has been proposed for HERG channel block by class III methanesulfonanilide antiarrhythmic drugs (Ficker et al., 1998;Lees-Miller et al., 2000; Mitcheson et al., 2000), verapamil (Zhang et al., 1999), and terfenadine and cisapride (Mitcheson et al., 2000) and is thought to involve drug binding to S6 aromatic residues lining the channel pore (Mitcheson et al., 2000). Whether the cocaine-binding domain is shared with that for other drugs or is distinct is not known.
Although some evidence suggests that sigma-1 receptors can regulate certain K+ channels and cocaine can serve as a sigma-1 receptor ligand (Sharkey et al., 1988; Wilke et al., 1999; Lupardus et al., 2000; Aydar et al., 2000), the inhibition of HERG channels as reported in this work is very likely to be caused by direct cocaine binding to the channel protein without involvement of the sigma-1 receptor. Droperidol and haloperidol, both ligands with nanomolar affinity for the sigma-1 receptor (Tam and Cook, 1984) have 30–1,000 times less affinity for HERG channels expressed in HEK 293 cells (Drolet et al., 1999) or in Xenopus laevisoocytes (Suessbrich et al., 1997). In addition, the high affinity sigma-1 receptor ligands fenpropimorph (Moebius et al., 1997) and iodoazidococaine (Kahoun and Ruoho., 1992a,b) inhibit these HERG channels in micromolar concentrations (data not shown) whereas their affinities for the sigma-1 receptor are in the picomolar range. Furthermore, sigma-1 receptors cannot be detected in HEK 293 cell membranes by the photoaffinity probe [125I]iodoazidococaine under conditions that readily detect the sigma-1 receptor in rat neurohypophysial terminals and DMS-114 tumor cell membranes (Wilke et al., 1999; Lupardus et al., 2000). The cocaine binding site on the HERG protein may be similar to that for some monoamine transporters (Ritz et al., 1987) or L-type Ca2+ channels (Renard et al., 1994) because the IC50 values for cocaine on HERG, the monoamine transporters, and the L-type Ca2+ channels are all in the same range (1–50 μM).
Our results suggest that block of HERG channels by cocaine required channel activation with drug binding to the open and/or inactivated states of the channel. Activated state block, as proposed in the modulated receptor model (see Hille, 1977; Hondeghem and Katzung, 1977), has been a consistent observation with drugs that interact with HERG channels. For most drugs the development of HERG channel block is slow and pulse train protocols require many steps to achieve a steady state of use-dependent block (for example, see Snyders and Chaudhary, 1996; Zhang et al., 1999; Tie et al., 2000). Recovery of HERG channels from block by drugs such as highly charged methanesulfonanilides (e.g., E-4031, dofetilide, and MK-499) is extremely slow, which has been attributed to trapping of the charged drug moiety within the inner vestibule of the channel by voltage-dependent closure of the activation gate (see Carmeliet, 1992; Mitcheson et al., 2000). Recovery of HERG channels from block by drugs such as verapamil, which exist in both charged and neutral forms at physiological pH, occurs slowly during membrane hyperpolarization even in the presence of drug. Under these conditions, drug trapping of the charged moiety may still occur but with drug progressively diffusing away from the drug-binding site in its neutral form (for discussion, see Zhang et al., 1999). A striking difference between the present findings and previous reports with HERG channel blocking drugs is that with cocaine the development of block and recovery from it had uniquely rapid kinetics. Block developed within milliseconds of depolarization. Recovery of HERG current from cocaine block at −100 mV followed a multiexponential time course and was nearly complete within 100 ms. The mechanisms accounting for these ultrarapid kinetic properties are not certain, however, because cocaine is a relatively small molecule present in both neutral and charged forms at physiological pH (pK a ∼8.6;Crumb and Clarkson, 1990), it is likely to easily access to the channel binding site with depolarization to give the rapid apparent association constant. The rapid recovery phase may represent unbinding of the neutral form of cocaine, whereas the slower recovery phase may represent unblock by the charged moiety of cocaine trapped in the channel pore before it diffuses away from the drug binding site as its charge is neutralized. Alternatively, the rapid recovery phase corresponds with the rapid recovery of HERG channels from inactivated to open states, thus drug may be able to rapidly escape the channel pore before deactivation results in drug trapping, producing the slower recovery phase.
Clinical Implications.
The mechanisms underlying cocaine-induced arrhythmias and sudden death remains speculative (Kloner et al., 1992; Bauman et al., 1994). Proarrhythmia has been attributed to the sympathomimetic effects of cocaine. In toxicity, cocaine elicits intense vasoconstriction along with increased heart rate and myocardial contractility, which have been postulated to cause myocardial ischemia and consequent arrhythmias. In patients who die suddenly of cocaine toxicity, however, autopsy studies have generally not shown evidence of acute myocardial infarction (Bauman et al., 1994). Cocaine also has potent local anesthetic properties. It blocks cardiac Na+ channels with estimated IC50 values of 328, 19, and 8 μM for channels in rested, activated, and inactivated states, respectively (Crumb and Clarkson, 1990). By blocking Na+ channels in cardiac cells, cocaine has been postulated to slow cardiac impulse conduction and enhance reentrant arrhythmias (Kloner et al., 1992;Bauman et al., 1994).
Cocaine in humans also can prolong the normal QT interval (Perera et al., 1997), and induce torsades de pointes in patients with underlying long QT syndrome (Schrem et al., 1990; Khan et al., 1999). In isolated ventricular myocytes, cocaine has been shown to block delayed rectifier K+ current, prolong action potential duration and trigger arrhythmogenic early afterdepolarizations (Kimura et al., 1992;Clarkson et al., 1996). Our data show that cocaine blocks HERG channels, which are thought to produce the pore forming subunit of the IKr channel. Because of the ultrarapid kinetic properties of cocaine block of HERG channels, drug binding (particularly at physiological temperature) should occur during each action potential upstroke and drug unbinding should occur between action potentials.
In humans, cocaine use has been reported to produce peak plasma concentrations of 1 to 3 μM (Paly et al., 1982). In cocaine-associated sudden death, average post mortem plasma cocaine concentrations of 20 μM have been reported, with the highest values reaching 80 μM (Mittleman and Wetli, 1984). Although cocaine binds to serum proteins, binding is concentration-dependent, with the free-fraction increasing from 16 to 68% over a cocaine range of 0.003 to 300 μM (Parker et al., 1995). Our results show that cocaine blocks HERG channels with an IC50 value of 7.2 μM. This value is within a drug concentration range that can be achieved in humans, at least during cocaine toxicity. Because suppression of IKr has well-described arrhythmogenic potential (e.g., causes long QT syndrome), our findings support the hypothesis that HERG channel block causing the generation of triggered cardiac arrhythmias may participate in the arrhythmogenic actions of cocaine.
In conclusion, we found that cocaine blocks HERG but not KvLQT1+minK K+ channels. Block occurs from the inside of the cell surface membrane. Cocaine preferentially blocks activated HERG channels and it unblocks upon repolarization. The ultrarapid kinetics of block and unblock are unique. HERG channel block provides an additional mechanism for cocaine-induced arrhythmias and sudden death.
Acknowledgments
We thank Drs. Gail A. Robertson and Jonathan C. Makielski for helpful discussion.
Footnotes
- Received October 16, 2000.
- Accepted January 16, 2001.
-
Send reprint requests to: Craig T. January, M.D., Ph.D., Section of Cardiology, Room H6/354, University of Wisconsin Hospital, 600 Highland Ave., Madison, WI 53792. E-mail: ctj{at}medicine.wisc.edu
-
This work was supported, in part, by National Institutes of Health Grants HL60723 and GM33138. S.Z. and S.R. are supported by postdoctoral fellowship awards from the American Heart Association, Northland Affiliate. Z.Z. is the recipient of a Scientist Development Grant from the American Heart Association.
-
Part of this work has been reported in abstract form: Zhang S, Zhou Z, Chen Y, Gong Q, Ruoho AE, January CT. Cocaine blocks HERG potassium channels (Abstract). Circulation 100(Suppl):I-424, 1999 and Zhang S, Rajamani S, Robertson GA, January CT. Mechanism of cocaine block of HERG potassium channels (Abstract). Biophys J 78(Suppl):221A, 2000.
Abbreviations
- HERG
- human ether-a go-go-related gene
- HEK
- human embryonic kidney
- I-V
- current-voltage
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}