


Abstract
The effect of endocytosis inhibitors on 5-hydroxytryptamine2A (5-HT2A) receptor desensitization and resensitization was examined in transiently transfected human embryonic kidney (HEK) 293 cells and in C6 glioma cells that endogenously express 5-HT2A receptors. In HEK-293 cells, 5-HT2A receptor desensitization was unaffected by cotransfection with a dominant-negative mutant of dynamin (DynK44A), a truncation mutant of arrestin-2 [Arr2(319–418)], or by two well-characterized chemical inhibitors of endocytosis: concanavalin A (conA) and phenylarsine oxide (PAO). In contrast, β2-adrenergic receptor desensitization was significantly potentiated by each of these treatments in HEK-293 cells. In C6 glioma cells, however, DynK44A, Arr2(319–418), conA, and PAO each resulted in the potentiation of 5-HT2A and β-adrenergic receptor desensitization. The cell-type-specific effect of Arr2(319–418) on 5-HT2Areceptor desensitization was not related to the level of GRK2 or GRK5 expression. Interestingly, although β2-adrenergic receptor resensitization was potently blocked by cotransfection with DynK44A, 5-HT2A receptor resensitization was enhanced, suggesting the existence of a novel cell-surface mechanism for 5-HT2Areceptor resensitization in HEK-293 cells. In addition, Arr2(319–418) had no effect on 5-HT2A receptor resensitization in HEK-293 cells, although it attenuated the resensitization of the β2-adrenergic receptor. However, in C6 glioma cells, both DynK44A and Arr2(319–418) significantly reduced 5-HT2A receptor resensitization. Taken together, these results provide the first convincing evidence of cell-type-specific roles for endocytosis inhibitors in regulating GPCR activity. Additionally, these results imply that novel GRK and arrestin-independent mechanisms of 5-HT2A receptor desensitization and resensitization exist in HEK-293 cells.
5-Hydroxytryptamine2A(5-HT2A) receptors are important for mediating a large number of physiologic processes in the periphery and in the central nervous system including platelet aggregation, smooth muscle contraction, and the modulation of mood and perception (Roth et al., 1998b). Many drugs of diverse therapeutic classes mediate their actions, at least in part, by interactions with 5-HT2A receptors. Hallucinogens, such as lysergic acid diethylamide and N,N′-dimethyltryptamine, function as agonists at 5-HT2A receptors (Glennon et al., 1984), whereas atypical antipsychotic drugs are potent 5-HT2A receptor antagonists (Meltzer et al., 1989). It has been known for some time that acute and chronic exposure to 5-HT2A receptor-active drugs (either agonists or antagonists) causes a decrease in 5-HT2Areceptor number and activity in vivo (for review, see Roth et al., 1990, 1998a). In culture, studies have demonstrated that short-term agonist exposure results in the desensitization of 5-HT2A receptor-mediated phosphoinositide (PI) hydrolysis in NIH 3T3 cells (Roth et al., 1995) and P11 cells (Ivins and Molinoff, 1991).
Many prior studies have suggested a general mechanism of G protein-coupled receptor (GPCR) desensitization involving the phosphorylation of intracellular domains of the receptor by second-messenger kinases and specific G protein-coupled receptor kinases (GRKs) leading to G protein uncoupling (for review, seeFreedman and Lefkowitz, 1996). G protein uncoupling is further promoted by the binding of arrestins to the third intracellular loops and carboxyl-terminal tails of agonist-activated GPCRs (for review, seeFerguson, 2001). Additionally, the interaction of GPCRs with arrestins is thought to promote the targeting of desensitized receptors to clathrin-coated pits for their subsequent internalization by the interaction of the carboxy-terminal portions of arrestin with both the clathrin heavy chain and the β2-adaptin subunit of AP-2 (Krupnick et al., 1997; Laporte et al., 1999, 2000). After endocytosis and sorting to the endosomal compartment, GPCRs may be rapidly dephosphorylated and recycled back to the plasma membrane (a process termed resensitization) or targeted to lysosomes for degradation.
It has been clear, however, that this is by no means universal for all GPCRs. Thus 5-HT2A receptors, for example, are subject to alternative modes of regulation compared with other GPCRs. For example, both agonists and antagonists induce down-regulation and internalization of 5-HT2A receptors in vitro and in vivo (Berry et al., 1996; Willins et al., 1999). Additionally, we have recently demonstrated that 5-HT2A receptor internalization follows the clathrin-mediated endocytic pathway and is dynamin-dependent (Berry et al., 1996; Bhatnagar et al., 2001). However, we also found that 5-HT2A receptor endocytosis is independent of the action of arrestins and, interestingly, results in a redistribution of arrestins into intracellular vesicles and plasma membrane compartments distinct from those containing internalized 5-HT2A receptors (Bhatnagar et al., 2001).
In the present studies, we examined the effects of various inhibitors of endocytosis on the desensitization and resensitization of 5-HT2A receptors in two cell lines: HEK-293 cells and C6 glioma. Endocytic blockade was accomplished using dominant negative mutants of dynamin (DynK44A) and arrestin-2 [Arr2(319–418)] and well-characterized chemical inhibitors of clathrin-mediated endocytosis. As we report, in HEK-293 cells, blocking 5-HT2A receptor endocytosis by any means tested had no effect on agonist-induced desensitization. However, in C6 glioma cells, each endocytosis inhibitor induced a potentiation of 5-HT2A receptor desensitization and an attenuation of resensitization. Conversely, in HEK-293 cells, cotransfection of the 5-HT2A receptor with DynK44A resulted in an unexpected increase in receptor resensitization. Additionally, the cell-type-specific effect of Arr2(319–418) on 5-HT2A receptor desensitization is unrelated to the levels of GRK2 and GRK5 because overexpression of GRK2 and GRK5 have no effect on 5-HT2A receptor desensitization in HEK-293 cells. Taken together, these results demonstrate a novel arrestin-independent mode of 5-HT2A receptor desensitization and resensitization that is cell-type specific and unrelated to the levels of GRK2 and GRK5.
Experimental Procedures
Materials and Constructs.
HEK-293 and C6 glioma cells were purchased from the American Type Culture Collection (Manassas, VA). [3H]Inositol (21.0 Ci/mmol), [3H]cAMP (37.2 Ci/mmol), and [3H]ketanserin (63.3 Ci/mmol) were obtained from PerkinElmer Life Sciences (Boston, MA). Quipazine, 5-hydroxytryptamine (5-HT), (−)-isoproterenol, 3-isobutyl-1-methylxanthine (IBMX), adenosine 3′:5′-cyclic monophosphate (cAMP), phenylarsine oxide (PAO), concanavalin A (ConA), and cyclohexamide were purchased from Sigma Chemical (St. Louis, MO). Phenoxybenzamine and spiperone were purchased from Sigma/RBI (Natick, MA). An amino-terminal FLAG epitope-tagged rat 5-HT2A receptor was constructed as described previously (Bhatnagar et al., 2001). The carboxy-terminal arrestin-2 dominant negative mutant [Arr2(319–418)] cDNA and anti-arrestin2-ct antibody were described previously (Bhatnagar et al., 2001). The cDNA of the dominant negative mutant of dynamin I (DynK44A) was the generous gift of Dr. M. G. Caron (Duke University Medical Center, Durham, NC). Anti-GRK2 and anti-dynamin I antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal anti-GRK5 antibody was purchased from Upstate Biotechnology (Lake Placid, NY). Polyclonal anti-green fluorescent protein antibody was purchased from CLONTECH (Palo Alto, CA). The amino terminus-specific polyclonal 5-HT2A receptor antibody (Ab51) was described previously (Berry et al., 1996). Goat anti-rabbit-HRP, horse anti-goat-HRP, horse anti-mouse-HRP, and goat anti-rabbit-Texas Red were purchased from Vector Laboratories (Burlingame, CA), whereas goat anti-rabbit-BODIPY-FL secondary antibody was from Molecular Probes (Eugene, OR).
Cell Culture and Transfection.
HEK-293 cells were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal calf serum, 1 mM sodium pyruvate, 100 U/ml penicillin, and 100 μg/ml streptomycin. HEK-293 cells were transfected in 10-cm dishes at 60 to 80% confluence with 2 μg of receptor DNA and 4 μg of cotransfected DNA using Fugene6 (Roche Molecular Biochemicals, Indianapolis, IN) exactly as described by the manufacturer. For transfection of receptor alone, total amount of DNA transfected was kept constant with the addition of empty vector (pcDNA3). At 24 h after transfection, cells were split into poly-l-lysine-coated 24-well plates and grown for an additional 24 h in DMEM supplemented with 5% dialyzed fetal calf serum. C6 glioma cells were grown in F-12K nutrient mixture-Kaighn's modification supplemented with 15% horse serum, 2.5% fetal calf serum, 1 mM sodium pyruvate, 100 U/ml penicillin, and 100 μg/ml streptomycin as recommended by the supplier. Cell cultures were grown at 37°C in 5% CO2. C6 glioma cells were transfected in 10-cm dishes at 50% to 80% confluency with 2 μg of DNA using Effectene transfection reagent (QIAGEN, Valencia, CA) exactly as described by the manufacturer. At 24 h after transfection, cells were split into poly-l-lysine-coated 24-well plates and grown for an additional 48 h in F-12K supplemented with 5% dialyzed fetal calf serum, adding fresh medium after 24 h.
Determination of PI Hydrolysis.
Twenty-four hours after cells were split into 24-well plates, the cells were washed with inositol-free DMEM and incubated for an additional 18 h with inositol-free DMEM containing 1 μCi/ml [3H]inositol and 5% dialyzed fetal calf serum. To assess desensitization, cells were preincubated for the indicated times in the absence or presence of 300 μM quipazine and then washed twice on ice with inositol-free DMEM. To assess resensitization, cells were first treated for 2 h with 300 μM quipazine, washed twice with inositol-free DMEM, placed in inositol-free DMEM containing 1 μCi/ml [3H]inositol and 5% dialyzed fetal calf serum for the indicated times, and then washed twice on ice with inositol-free DMEM. After all pretreatments, medium was replaced with a modified Krebs-bicarbonate buffer containing 118 mM NaCl, 4.7 mM KCl, 1.2 mM CaCl2, 1.2 mM MgCl2, 25 mM NaHCO3, and 11 mM glucose. Before use, buffer was equilibrated to 37°C and 5% CO2. Cells were then incubated in the modified Krebs-bicarbonate buffer with 10 mM LiCl for 1 h at 37°C in the presence or absence of 10 μM 5-HT. The reaction was terminated by aspiration and the addition of 1 ml of methanol/water/HCl (25:25:0.1). Cells were harvested into glass tubes and membranes were extracted by the addition of 0.5 ml of chloroform and vortexed vigorously. After phase separation, the 0.6 ml of the upper aqueous phase was removed and added to columns containing 1 ml of anion exchange resin (formate form) and washed with 12 ml of water followed by 10 ml of 5 mM sodium borate/50 mM sodium formate (Roth et al., 1986). Total phosphoinositides (PIs) were eluted with 10 ml of 0.1 M formic acid/0.2 M ammonium formate into vials containing 3a70B liquid scintillation cocktail (Research Products International, Elk Grove Village, IL), and radioactivity was measure by liquid scintillation counting.
Determination of cAMP Production.
To assess desensitization, cells were preincubated for the indicated times in the absence or presence of 10 μM (−)-isoproterenol and 100 μM ascorbic acid and then washed twice on ice with ice-cold Ham's F12 nutrient mixture (F12). To assess resensitization, cells were first treated for 30 min with 10 μM (−)-isoproterenol and 100 μM ascorbic acid, washed twice with F12, and incubated at 37°C in F12 equilibrated to 37°C and 5% CO2 for the indicated times and then washed twice on ice with ice-cold F12. After all pretreatments, medium was replaced with fresh F12 containing 100 μM IBMX and 100 μM ascorbic acid and incubated for 30 min at 37°C in the presence or absence of 10 μM (−)-isoproterenol. The reaction was terminated by aspiration and the addition of 0.5 ml of ice-cold 3% trichloroacetic acid. Plates were chilled for 1 h at 4°C and spun at 1000g for 15 min. cAMP was quantified using a competitive binding assay adapted with minor modifications (Nordstedt and Fredholm, 1990). Briefly, the trichloroacetic acid extracts (10–40 μl) were added to reaction tubes containing cAMP assay buffer (100 mM Tris-HCl, pH 7.4, 100 mM NaCl, and 5 mM EDTA). [3H]cAMP (1 nM final concentration) was added to each tube, followed by cAMP-binding proteins (approximately 100 μg of crude extract from bovine adrenal cortex in 500 μl of cAMP buffer). The reaction tubes were incubated on ice for 2 h then harvested with a Brandel cell harvester onto Whatman GF/C filters. Filters were allowed to dry, and radioactivity bound was quantified by liquid scintillation counting. The concentration of cAMP in each sample was estimated from a standard curve ranging from 0.1 to 100 pmol of cAMP/assay.
Radioligand Binding Assay.
Saturation binding assays were performed with [3H]ketanserin in total volumes of 0.25 ml at 25°C for 1 h with 5 to 20 μg of membrane protein, as described previously (Choudhary et al., 1992), in 50 mM Tris-Cl buffer, pH 7.4. Nonspecific binding was defined as radioactivity bound in the presence of 10 μM spiperone and represented less than 10% of total binding. Membranes were harvested with a Brandel cell harvester followed by three ice-cold washes onto polyethyleneimine-pretreated (0.3%) Whatman GF/C filters. Radioactivity bound to filters was quantified by liquid scintillation counting. Membrane protein concentrations were determined using an assay kit from Bio-Rad (Hercules, CA) with bovine serum albumin as the standard.
Immunocytochemistry and Confocal Microscopy.
After various treatments, cells were fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 30 min, lightly permeabilized on ice (0.2% Triton X-100 in PBS) for 20 min, and incubated with blocking buffer (5% milk in PBS) for 1 h. Cells were then incubated overnight with the following antibodies with either a 5-HT2A receptor amino terminus-specific antibody (Ab51; 1:3000 dilution; Berry et al., 1996) or polyclonal anti-green fluorescent protein antibody (1:5000 dilution) each diluted in blocking buffer. Cells were then washed twice with PBS and incubated with a 1:200 dilution of either Texas Red-labeled goat anti-rabbit antibody or BODIPY-FL-labeled goat anti-rabbit antibody for 1 h in blocking buffer. Cells were then washed with PBS and mounted for fluorescent confocal microscopic evaluation as previously detailed (Berry et al., 1996). For confocal microscopy, all images were taken at an overall optical magnification of 1000×; and in selected images, electronic magnification greater than 1000× was obtained.
Western Blotting.
Twenty-four hours after transfection, HEK-293 cells or C6 glioma cells were split into poly-l-lysine-coated 6-well plates in DMEM (HEK-293 cells) or F-12K (C6 glioma cells) supplemented with 5% dialyzed fetal calf serum, adding fresh medium after 24 h and grown for an additional 18 h. Cells were placed on ice, washed three times with ice-cold PBS, and lysed with 1 ml of SDS sample buffer (50 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 0.1% bromphenol blue, and 100 mM dithiothreitol). Samples were drawn twice through a 22-gauge needle and incubated at 95°C for 10 min. Total protein concentrations were determined using the Bio-Rad assay kit with bovine serum albumin as the standard. Samples were resolved by 12% SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose. Blots were blocked in blocking buffer (tris-buffered saline with 0.01% Tween-20 and 5% nonfat dried milk) overnight at 4°C. The blots were then incubated for 1 h at room temperature with anti-arrestin2-ct (1:10,000), anti-dynamin I (1:1,000), anti-GRK2 (1:1,000), or anti-GRK5 (1:1,000) in blocking buffer. Blots were washed three times for 10 min each at room temperature with tris-buffered saline containing 0.05% Tween-20 and incubated with secondary antibody (goat anti-rabbit-HRP, horse anti-goat-HRP, or horse anti-mouse-HRP) at 1:2000 dilutions for 1 h at room temperature in blocking buffer. Blots were washed and visualized with the use of Lumi-light Western Blotting Substrate (Roche Molecular Biochemicals).
Data Analysis.
PI hydrolysis assays were performed in triplicate and repeated at least three times. cAMP experiments were performed in triplicate or quadruplicate and were repeated at least three times. Dose response curves for cAMP were analyzed by nonlinear regression using Prism 3.0 software (GraphPad, San Diego, CA). Statistical significance of the data was determined by two-tailed paired t test, was defined as p < 0.05, and was analyzed by GraphPad Prism. Binding data were analyzed using the weighted, nonlinear least-squares curve-fitting program LIGAND (Munson and Rodbard, 1980).
Results
Effect of Endocytic Blockade on 5-HT2A Receptor Desensitization in HEK-293 Cells.
In initial studies, we characterized the expression levels of FLAG-epitope tagged 5-HT2A receptors transiently transfected into HEK-293 cells. Cotransfection of HEK-293 cells with 2 μg of 5-HT2A receptor plasmid DNA and 4 μg of empty vector consistently yielded 200 to 600 fmol of 5-HT2A receptor/mg of total protein (data not shown). Additionally, preincubation of transfected HEK-293 cells with 0.1 μM phenoxybenzamine for 30 min before determination of the dose response of 5-HT on PI hydrolysis resulted in a significant reduction of maximal PI hydrolysis without a shift in the dose-response curve (data not shown). Taken together, these results demonstrate that 5-HT2A receptor expression levels in HEK-293 cells did not create a situation of receptor reserve (Sanders-Bush and Breeding, 1990). The transfection efficiency of HEK-293 cells in these experiments was 55% as assessed by confocal microscopy of HEK-293 cells transfected with pEGFP-N1 (data not shown). We next cotransfected 5-HT2A receptor cDNA and a dynamin I dominant-negative mutant (DynK44A) cDNA to investigate the role of clathrin-mediated endocytosis on 5-HT2A receptor desensitization in HEK-293 cells. In prior studies (Bhatnagar et al., 2001), we demonstrated that DynK44A inhibited 5-HT2A receptor internalization in HEK-293 cells, whereas the C-terminal tail of arrestin-2 [Arr2(319–418)] had no effect on 5-HT2A receptor internalization. As shown in Fig. 1A, DynK44A had no effect on the extent of the desensitization of agonist-stimulated PI hydrolysis in HEK-293 transiently transfected with 5-HT2A receptors versus cells transfected with receptor alone. Likewise, overexpression of Arr2(319–418), previously shown to inhibit the endocytosis of the β2-adrenergic receptor (Orsini and Benovic, 1998), also had no effect on the extent of 5-HT2A receptor desensitization (Fig. 1A). Western blot analysis of transfected cells from the same experiments demonstrated that the DynK44A and Arr2(319–418) constructs were overexpressed in HEK-293 cells (Fig. 2).
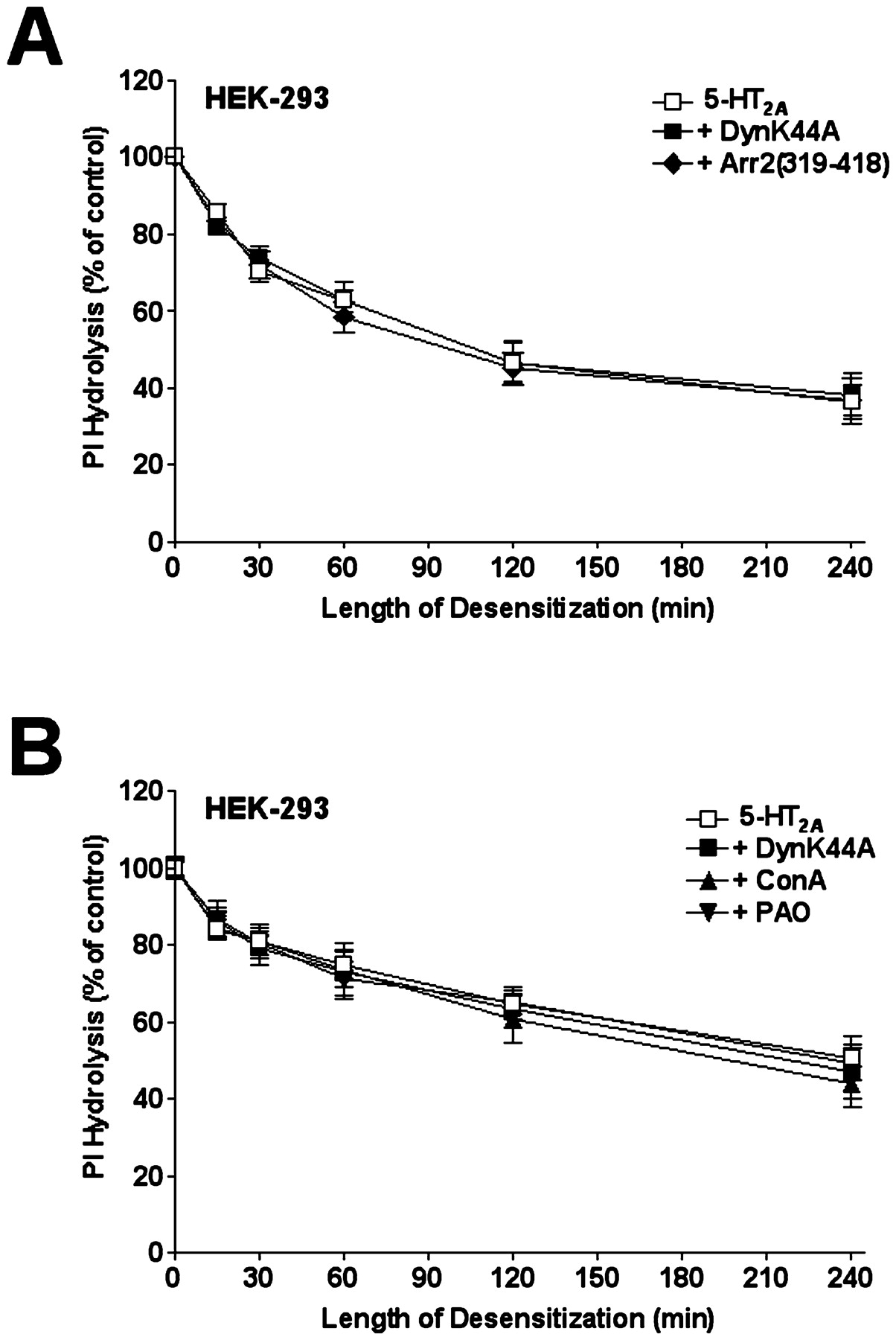
Effect of DynK44A, Arr2(319–418), and chemical inhibitors of endocytosis on the time course of quipazine-induced desensitization of 5-HT2A receptor-mediated PI hydrolysis in HEK-293 cells. Transiently transfected HEK-293 cells were preincubated with vehicle or 300 μM quipazine for various times. Cells were then washed extensively and stimulated with 10 μM 5-HT in the presence of 10 mM LiCl, and the levels of PI hydrolysis were measured as described under Experimental Procedures. A, time course of 5-HT2A receptor desensitization when cotransfected with empty vector, DynK44A or Arr2(319–418). B, time course of 5-HT2A receptor desensitization in the presence or absence of ConA (250 μg/ml for 2 h) or PAO (10 μM for 30 min) when cotransfected with empty vector or DynK44A. The data are expressed as a percentage of the response observed in the control (vehicle-treated) group of cells. The data represent the mean ± S.E.M. values from at least three independent experiments performed in triplicate.

Overexpression of DynK44A and Arr2(319–418) in HEK-293 cells. Detection of overexpressed DynK44A or Arr2(319–418) in total cell lysates of HEK-293 cells transiently transfected with 2 μg of 5-HT2A receptor plasmid DNA and 4 μg of empty pcDNA3 vector (−) or 4 μg of DynK44A or Arr2(319–418) vector (+) as described under Experimental Procedures. For the Arr2(319–418) blot, the doublets near the 47.5-kDa marker correspond to endogenous wild-type arrestin, whereas the band around 10 kDa corresponds to transfected Arr2(319–418).
To further examine the role of clathrin-mediated endocytosis on 5-HT2A receptor desensitization in HEK-293 cells, the cells were treated with either concanavalin A (250 μg/ml) or phenylarsine oxide (10 μM) before adding the desensitizing stimulus. As shown in Fig. 1B, blocking endocytosis by chemical means also had no significant effect on the extent of 5-HT2Areceptor desensitization in HEK-293 cells versus cells transfected with receptor alone. As shown in Fig. 3, concanavalin A and phenylarsine oxide inhibited 5-HT2A receptor endocytosis in HEK-293 cells as assessed by fluorescence confocal microscopy. Taken together, these results demonstrate that 5-HT2A receptor desensitization is independent of clathrin-mediated endocytosis in HEK-293 cells.

Effect of concanavalin A and phenylarsine oxide on 5-HT2A receptor internalization in HEK-293 cells. Transiently transfected HEK-293 cells subjected to various treatments then prepared for immunofluorescent confocal microscopy as described under Experimental Procedures. A, control cells demonstrating characteristic surface expression of transfected 5-HT2A receptors in HEK-293 cells (arrow). B, cells treated with 100 μM quipazine for 5 min; arrowhead shows location of intracellular receptors, whereas arrow depicts plasma-membrane immunoflourescence. C, cells incubated with concanavalin A (250 μg/ml) for 2 h then treated with 100 μM quipazine for 5 min. D, cells incubated with 10 μM phenylarsine oxide for 30 min then treated with 100 μM quipazine for 5 min. Images shown representative of two independent experiments.
Effect of Endocytic Blockade on β2-Adrenergic Receptor Desensitization in HEK-293 Cells.
Because β2-adrenergic receptor desensitization has been shown previously to be affected by dominant negative mutants of dynamin and arrestin-2 in HEK-293 cells (Zhang et al., 1997), we used β2-adrenergic receptors as a positive control. Thus, cotransfection of HEK-293 cells with β2-adrenergic receptor cDNA and DynK44A or Arr2(319–418) cDNAs resulted in a potentiation of the isoproterenol-induced desensitization of receptor-stimulated cAMP accumulation (Fig. 4A). Additionally, the DynK44A mutant resulted in a greater potentiation of desensitization than did Arr2(319–418) (Fig. 4A). Furthermore, blocking β2-adrenergic receptor internalization with concanavalin A (250 μg/ml) or phenylarsine oxide (10 μM) potentiated β2-adrenergic receptor desensitization to an extent similar to DynK44A (Fig. 4B).

Effect of DynK44A, Arr2(319–418), and chemical inhibitors of endocytosis on the time course of isoproterenol-induced desensitization of β2AR-mediated cAMP accumulation in HEK-293 cells. Transiently transfected HEK-293 cells were preincubated with vehicle or 10 μM isoproterenol for various times. Cells were then washed extensively and stimulated with 10 μM isoproterenol in the presence of 100 μM IBMX, and the levels of cAMP accumulation were assessed as described under Experimental Procedures. A, time course of β2AR desensitization when cotransfected with empty vector, DynK44A, or Arr2(319–418). All points except Arr2(319–418) at 5 min are significantly different from receptor alone withp < 0.05. B, time course of β2AR desensitization in the presence or absence of ConA (250 μg/ml for 2 h) or PAO (10 μM for 30 min) when cotransfected with empty vector or DynK44A. All points are significantly different from receptor alone withp < 0.05. The data are expressed as a percentage of the response observed in the control (vehicle-treated) group of cells. The data represent the mean ± S.E.M. values from at least three independent experiments performed in quadruplicate.
Resensitization of 5-HT2A Receptors in HEK-293 Cells.
To determine whether 5-HT2A receptor resensitization is endocytosis-dependent or arrestin-dependent, we cotransfected the 5-HT2A receptor with either DynK44A or Arr2(319–418), treated the cells for 2 h with a desensitizing stimulus (300 μM quipazine), washed the cells extensively, and then assayed the cells at various times for the recovery of agonist-induced PI hydrolysis. Cotransfection with DynK44A resulted in a significant increase in the amount of 5-HT2A receptor resensitization compared with receptor alone at 30 min, 1 h, 2 h, and 4 h after removal of the desensitizing treatment (Fig.5). However, cotransfection with Arr2(319–418) had no significant effect on the level of 5-HT2A receptor resensitization at any of the times assayed (Fig. 5). Taken together, these results suggest a role for a cell-surface mechanism of 5-HT2A receptor resensitization in HEK-293 cells that is arrestin-independent.

Effect of DynK44A and Arr2(319–418) on the time course of the resensitization of 5-HT2A receptor-mediated PI hydrolysis after quipazine-induced desensitization in HEK-293 cells. Transiently transfected HEK-293 were preincubated with vehicle or 300 μM quipazine for 2 h (Des), then washed extensively with inositol-free DMEM, and incubated for various times in inositol-free DMEM containing 1 μCi/ml [3H]inositol (Res). Cells were then washed and stimulated with 10 μM 5-HT in the presence of 10 mM LiCl, and the levels of PI hydrolysis were measured as described underExperimental Procedures. The data are expressed as a percentage of the control (vehicle-treated) response (100%) recovered after 2-h desensitization (0%). The data represent the mean ± S.E.M. values from at least three independent experiments performed in triplicate. *, Significantly different from receptor alone atp < 0.05.
Resensitization of β2-Adrenergic Receptors in HEK-293 Cells.
β2-adrenergic receptor resensitization has previously been shown to be blocked by dominant negative mutants of dynamin and arrestin-2 in HEK-293 cells (Zhang et al., 1997). Similarly, here we cotransfected the β2-adrenergic receptor with either DynK44A or Arr2(319–418), treated the cells with a desensitizing stimulus (10 μM isoproterenol), washed extensively the cells, then assayed at various times for the recovery of agonist-induced cAMP accumulation. Cotransfection with DynK44A resulted in a large attenuation of the extent of β2-adrenergic receptor resensitization at 15, 30, and 60 min after removal of the desensitizing treatment (Fig.6). Cotransfection with Arr2(319–418) resulted in the attenuation of β2-adrenergic receptor resensitization only at 30 min and 60 min after removal of the desensitizing stimulus (Fig. 6). Thus, overexpression of DynK44A and Arr2(319–418) effectively blocks β2-adrenergic receptor resensitization in HEK-293 as expected from previous studies (Zhang et al., 1997; Orsini and Benovic, 1998).

Effect of DynK44A and Arr2(319–418) on the time course of the resensitization of β2AR-mediated cAMP accumulation after isoproterenol-induced desensitization in HEK-293 cells. Transiently transfected HEK-293 were preincubated with vehicle or 10 μM isoproterenol for 30 min (Des) and then washed extensively and incubated for various times in fresh medium (Res). Cells were then washed and stimulated with 10 μM isoproterenol in the presence of 100 μM IBMX, and the levels of cAMP accumulation were measured as described under Experimental Procedures. The data are expressed as a percentage of the control (vehicle-treated) response (100%) recovered after 2-h desensitization (0%). The data represent the mean ± S.E.M. values from at least three independent experiments performed in quadruplicate. *, Significantly different from receptor alone at p < 0.05.
Effect of Endocytosis Inhibitors on 5-HT2A Receptor Desensitization in C6 Glioma Cells.
To determine whether the effects of endocytosis inhibitors on 5-HT2Areceptor desensitization were cell-type specific, we repeated the time course of 5-HT2A receptor desensitization in C6 glioma cells that endogenously express low levels of the 5-HT2A receptor (∼100 fmol/mg protein) as determined by [3H]ketanserin saturation binding (Elliott et al., 1995). As shown in Fig.7A, overexpression of DynK44A potentiated desensitization of the agonist-stimulated PI hydrolysis in C6 glioma cells. Additionally, Arr2(318–418) also potentiated 5-HT2A receptor desensitization (Fig. 7A). Western blot analysis of transfected cells from the same experiments demonstrated that the DynK44A and Arr2(319–418) constructs were overexpressed in C6 glioma cells (Fig.8). The transfection efficiency of C6 glioma cells in these experiments was 46% as assessed by confocal microscopy of C6 glioma cells transfected with pEGFP-N1 (data not shown). To further examine the effect of known endocytosis inhibitors on 5-HT2A receptor desensitization in C6 glioma cells, the cells were treated with either concanavalin A (250 μg/ml) or phenylarsine oxide (10 μM) before adding the desensitizing stimulus. As shown in Fig. 7B, treatment with well-characterized chemical inhibitors of endocytosis resulted in the potentiation of 5-HT2A receptor desensitization. For PAO-treated cells, 5-HT2A receptor desensitization was measured only to 30 min due to the toxic effects of PAO on C6 glioma cells. These results demonstrate a differential role of endocytosis inhibitors and dominant-negative arrestin constructs in 5-HT2A desensitization in C6 glioma cells compared with HEK-293 cells.

Effect of DynK44A, Arr2(319–418), and chemical inhibitors of endocytosis on the time course of quipazine-induced desensitization of endogenous 5-HT2A receptor-mediated PI hydrolysis in C6 glioma cells. Transiently transfected HC6 glioma cells were preincubated with vehicle or 300 μM quipazine for various times. Cells were then washed extensively and stimulated with 10 μM 5-HT in the presence of 10 mM LiCl, and the levels of PI hydrolysis were measured as described under Experimental Procedures. A, time course of 5-HT2A receptor desensitization when transfected with empty vector, DynK44A, or Arr2(319–418). All points except Arr2(319–418) at 10 min are significantly different from empty vector with p < 0.05. B, time course of 5-HT2A receptor desensitization in the presence or absence of ConA (250 μg/ml for 2 h) or PAO (10 μM for 30 min) when transfected with empty vector or DynK44A. All points are significantly different from empty vector with p < 0.05. Note: PAO treatments were only extended to the 30 min due to toxic effects on C6 glioma cells. The data are expressed as a percentage of the response observed in the control (vehicle-treated) group of cells. The data represent the mean ± S.E.M. values from three independent experiments performed in triplicate.

Overexpression of DynK44A and Arr2(319–418) in C6 glioma cells. Detection of overexpressed DynK44A or Arr2(319–418) in total cell lysates of C6 glioma cells transiently transfected with 2 μg of empty pcDNA3 vector (−) or 2 μg of DynK44A or Arr2(319–418) vectors (+) as described under Experimental Procedures. For the Arr2(319–418) blot, the doublets near the 47.5-kDa marker correspond to endogenous wild-type arrestin, whereas the band around 10 kDa corresponds to transfected Arr2(319–418).
Effect of Endocytosis Inhibitors on the Desensitization of β-Adrenergic Receptors in C6 Glioma Cells.
C6 glioma cells endogenously express both β1-adrenergic receptors and β2-adrenergic receptors at a total density of approximately 800 fmol/mg protein as determined by 125I-cyanopindolol saturation binding (Zhong and Minneman, 1995), with β1-adrenergic receptors and β2-adrenergic receptors expressed at a 4:1 ratio (Homburger et al., 1981). For these experiments, C6 glioma cells were transfected with either DynK44A or Arr2(319–418) and the isoproterenol-induced desensitization of β-adrenergic receptor stimulated cAMP accumulation was measured. Transfection with DynK44A resulted in an increase in the extent of desensitization, as did Arr2(319–418) (Fig. 9A). As in HEK-293 cells (Fig. 4A), the DynK44A mutant resulted in a greater potentiation of desensitization than did Arr2(319–418) (Fig. 9A). Furthermore, treatment with ConA (250 μg/ml) or PAO (10 μM) also potentiated β-adrenergic receptor desensitization to an extent similar to DynK44A (Fig. 9B). These results are important in that they suggest cell-type-specific modes of regulating 5-HT2Areceptors but not β-adrenergic receptors.

Effect of DynK44A, Arr2(319–418), and chemical inhibitors of endocytosis on the time course of isoproterenol-induced desensitization of endogenous βAR-mediated cAMP accumulation in C6 glioma cells. Transiently transfected C6 glioma cells were preincubated with vehicle or 10 μM isoproterenol for various times. Cells were then washed extensively and stimulated with 10 μM isoproterenol in the presence of 100 μM IBMX, and the levels of cAMP accumulation were assessed as described under Experimental Procedures. A, time course of βAR desensitization when transfected with empty vector, DynK44A, or Arr2(319–418). All points are significantly different from empty vector with p < 0.05. B, time course of βAR desensitization in the presence or absence of ConA (250 μg/ml for 2 h) or PAO (10 μM for 30 min) when transfected with empty vector or DynK44A. All points are significantly different from empty vector with p < 0.05. The data are expressed as a percentage of the response observed in the control (vehicle-treated) group of cells. The data represent the mean ± S.E.M. values from three independent experiments performed in quadruplicate.
Resensitization of 5-HT2A Receptors in C6 Glioma Cells.
To determine whether the cell-type-specific difference in the sensitivity of 5-HT2A receptor desensitization to DynK44A and Arr2(319–418) is caused by blockade of receptor resensitization, we examined 5-HT2Areceptor resensitization in C6 glioma cells. C6 glioma cells transfected with either DynK44A or Arr2(319–418) were treated for 2 h with a desensitizing stimulus (300 μM quipazine), were washed extensively, and were then assayed at various times for the recovery of agonist-induced PI hydrolysis. Transfection with DynK44A and Arr2(319–418) each resulted in a significant decrease in the amount of 5-HT2A receptor resensitization compared with receptor alone at 30 min, 1 h, 2 h, and 4 h after removal of the desensitizing treatment (Fig.10). These results, combined with those presented in Fig. 7, suggest that treatment of C6 glioma cells with well-characterized inhibitors of endocytosis reduces the ability of desensitized 5-HT2A receptors to resensitize in this cell line.
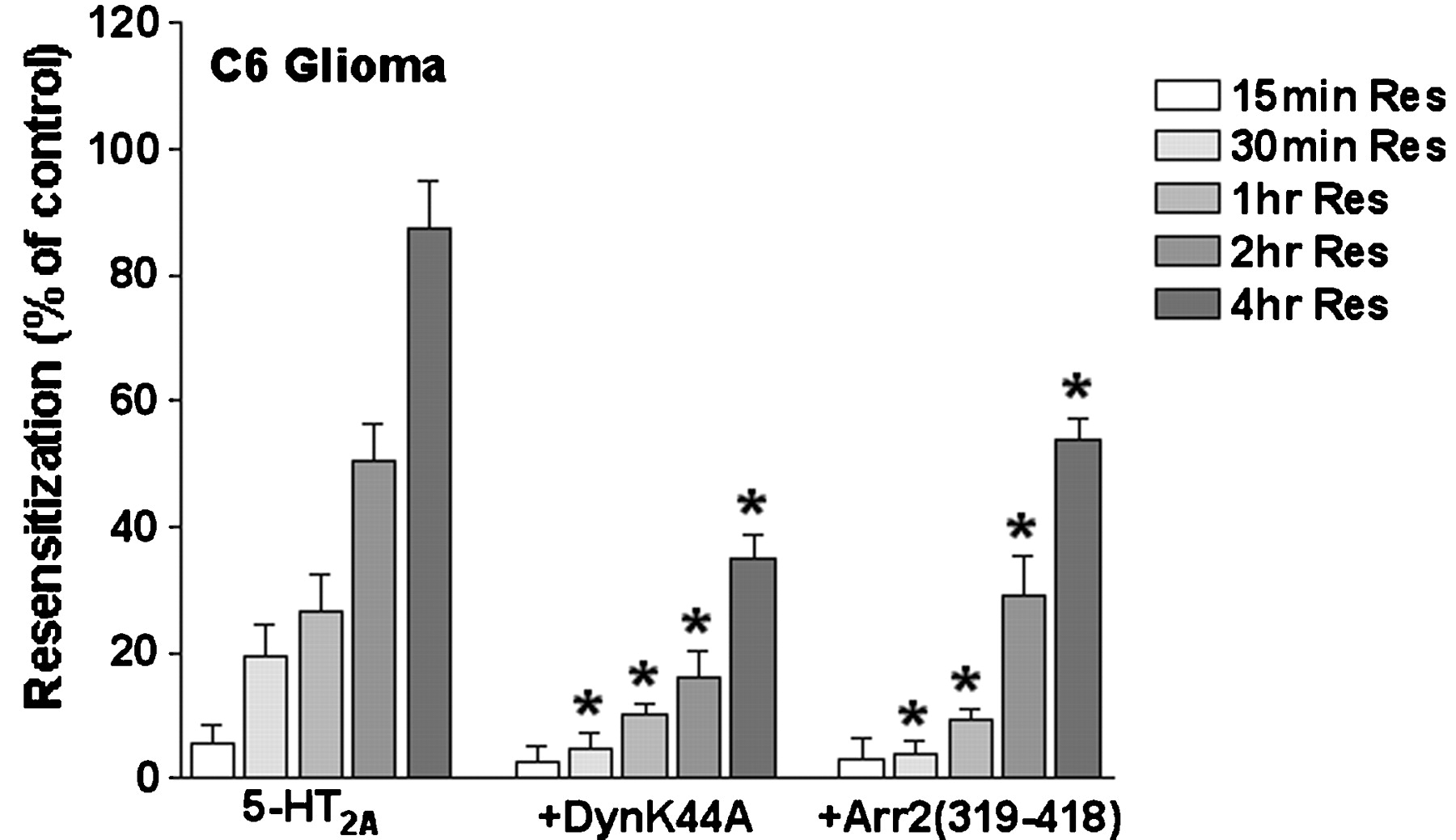
Effect of DynK44A and Arr2(319–418) overexpression on 5-HT2A receptor resensitization in C6 glioma cells. Transiently transfected C6 glioma cells were preincubated with vehicle or 300 μM quipazine for 2 h (Des), then washed extensively with inositol-free DMEM, and incubated for various times in inositol-free DMEM containing 1 μCi/ml [3H]inositol (Res). Cells were then washed and stimulated with 10 μM 5-HT in the presence of 10 mM LiCl, and the levels of PI hydrolysis were measured as described underExperimental Procedures. The data are expressed as a percentage of the control (vehicle-treated) response (100%) recovered after 2-h desensitization (0%). The data represent the mean ± S.E.M. values from two independent experiments performed in triplicate. *, Significantly different from receptor alone at p< 0.05.
Expression Levels of Arrestins, GRK2, and GRK5 in HEK-293 Cells and C6 Glioma Cells.
To examine the basis of the cell-type-specific effects of Arr2(319–418) on 5-HT2A receptor desensitization, we measured the expression levels of endogenous arrestins, GRK2, and GRK5 in C6 glioma cells and HEK-293 cells. Cell lysates of untransfected HEK-293 cells and C6 glioma cells were prepared and 20 μg of total protein from each lysate was analyzed by Western blotting. Arrestin and GRK2 expression in HEK-293 cells is greater than in C6 glioma cells (Fig.11). GRK5 expression, however, is similar in HEK-293 cells and C6 glioma cells, although C6 glioma cells may express slightly more GRK5 (Fig. 11). These results demonstrate that the cell-type-specific effects of Arr2(319–418) on 5-HT2A receptor desensitization are unrelated to the levels of arrestins and GRK2, but may be related to GRK5 expression.

Expression levels of Arrestins, GRK2, and GRK5 in HEK-293 cells and C6 glioma cells. Endogenous arrestin GRK2 and GRK5 levels were measured from total cell lysates of untransfected HEK-293 cells and C6 glioma cells. Total protein levels in each lysate were determined, and 20 μg of each was resolved and blotted as described under Experimental Procedures.
Overexpression of GRK2 and GRK5 in HEK-293 Cells Reveals Differential Effects on 5-HT2A and β2-Adrenergic Receptor Desensitization.
We then further examined the role of GRK2 and GRK5 in 5-HT2A receptor desensitization in HEK-293 cells. Cotransfection of 5-HT2A receptors with wild-type GRK2, wild-type GRK5, or a kinase-deficient GRK2 mutant (GRK2-K220R) had no effect on the extent or time course of 5-HT2A receptor desensitization in HEK-293 cells (Fig. 12A). In contrast, β2-adrenergic receptor desensitization is significantly enhanced by overexpression of wild-type GRK2 and GRK5 in HEK-293 cells (Fig. 12B). Additionally, the kinase-deficient GRK2 mutant results in a significant attenuation of β2-adrenergic agonist-induced desensitization at 10 min, 15 min, and 30 min, but not at 5 min (Fig. 12B). Taken together, these results further suggest that the cell-type-specific effects of arrestins on 5-HT2A receptor desensitization are unrelated to the levels of GRK2 or GRK5.

Differential effects of GRK2, GRK2-K220R, and GRK5-overexpression on agonist-induced desensitization of 5-HT2A and β2-adrenergic receptors in HEK-293 cells. A, transiently transfected HEK-293 cells were preincubated with vehicle or 300 μM quipazine for various times. Cells were then washed extensively and stimulated with 10 μM 5-HT in the presence of 10 mM LiCl, and the levels of PI hydrolysis were measured as described underExperimental Procedures. B, transiently transfected HEK-293 cells were preincubated with vehicle or 10 μM isoproterenol for various times. Cells were then washed extensively and stimulated with 10 μM isoproterenol in the presence of 100 μM IBMX, and the levels of cAMP accumulation were assessed as described underExperimental Procedures. All points except GRK2-K220R at 5 min are significantly different from receptor alone withp < 0.05. The data are expressed as a percentage of the response observed in the control (vehicle-treated) group of cells. The data represent the mean ± S.E.M. values from two independent experiments performed in triplicate (A) or quadruplicate (B).
Discussion
The major findings of this study are that 1) well-characterized inhibitors of endocytosis have cell-type-specific effects on 5-HT2A receptor desensitization and resensitization, 2) the cell-type-specific roles of arrestins in 5-HT2A receptor regulation are unrelated to the levels of GRK2 and GRK5 expression, and 3) blocking endocytosis by DynK44A in HEK-293 significantly increases 5-HT2Areceptor resensitization in HEK-293 cells. These results demonstrate that novel modes of regulation exist for 5-HT2Areceptors and that the arrestin-independent endocytosis seen in HEK-293 cells (Bhatnagar et al., 2001) is unrelated to the levels of GRK2 or GRK5 expression. Additionally, our findings suggest a role for a novel cell-surface mechanism of 5-HT2A receptor resensitization in HEK-293 cells. Taken together, these results provide the first convincing evidence of cell-type-specific roles for endocytosis inhibitors in regulating GPCR activity.
The responsiveness of a GPCR to agonist exposure represents the proportion of receptors that are in an active state on the cell surface and thus is governed by the mechanisms underlying receptor signaling, internalization, desensitization, and resensitization. Desensitization is the attenuation of receptor responsiveness to agonist after continued or repeated exposure and represents an important feedback mechanism for preventing receptor overstimulation. Classically, based on analogies with model systems like the β2-adrenergic receptor, GPCR desensitization is a result of the uncoupling of receptors from their heterotrimeric G proteins via phosphorylation of the intracellular domains of the receptor by second messenger kinases and specific GRKs (for review, see Freedman and Lefkowitz, 1996). For many but not all receptors, phosphorylation allows the receptors to become better substrates for arrestin binding, an event that further uncouples the G protein from the receptor and targets the desensitized receptor to clathrin-coated pits for subsequent internalization (for review, seeFerguson, 2001). Although GPCR internalization was initially thought to be the basis of receptor desensitization, to date this has been demonstrated only for the somatostatin receptor (Beaumont et al., 1998). For most GPCRs, internalization mediates the recycling of desensitized receptors back to the plasma membrane in a reactivated, or resensitized, state (Yu et al., 1993; Pippig et al., 1995).
For many years, it has been clear that a large number of GPCRs, including opiate receptors (Roth et al., 1981; Bennett et al., 1985) and β-adrenergic receptors (Chuang et al., 1986; von Zastrow and Kobilka, 1992), are found in coated vesicles and various intracellular vesicles associated with endocytic pathways. More recently, 5-HT2A receptors have been shown to be internalized via the endosomal pathway after agonist exposure in vitro (Berry et al., 1996; Bhatnagar et al., 2001). Although 5-HT2A receptor endocytosis is dynamin-dependent, it is arrestin-independent because dominant negative mutants of arrestin-2 and arrestin-3 have no effect on 5-HT2A receptor endocytosis in HEK-293 cells (Bhatnagar et al., 2001). Interestingly, in HEK-293 cells, arrestins are sorted to intracellular and plasma membrane compartments that are distinct from those containing internalized 5-HT2A receptors. These results revealed novel sorting pathways for 5-HT2A receptors and arrestins and implied internalization-independent functions for arrestin-2 and arrestin-3 (see Bhatnagar et al., 2001).
In the present studies, we demonstrated that in HEK-293 cells, the agonist-induced desensitization of 5-HT2Areceptor-mediated PI hydrolysis was unaffected by various well-characterized inhibitors of clathrin-mediated endocytosis, including the overexpression of a GTPase-deficient dynamin mutant (DynK44A) and treatment with ConA or PAO. Importantly, DynK44A also blocks caveolae-mediated endocytosis (Oh et al., 1998). ConA binds terminal sugar residues resulting in the agglutination of cell surface receptors and has been used to block GPCR internalization (Luttrell et al., 1997), whereas PAO reacts with sulfhydral groups forming stable ring structures and is widely used as a general inhibitor of receptor-mediated endocytosis (Beaumont et al., 1998). As an essential positive control, we showed in parallel experiments performed simultaneously that blocking clathrin-mediated endocytosis resulted in the potentiation of β2-adrenergic receptor desensitization, consistent with prior studies (Zhang et al., 1997). Additionally, overexpression of a dominant negative arrestin did not affect 5-HT2A receptor desensitization but potentiated β2-adreneric receptor desensitization. This was as predicted from previous studies in which the overexpression of Arr2(319–418) had no effect on 5-HT2A receptor internalization in HEK-293 cells (Bhatnagar et al., 2001) but inhibited β2-adrenergic receptor internalization (Orsini and Benovic, 1998).
Based on classical models of GPCR regulation, blocking receptor internalization should inhibit the rapid recycling and resensitization of GPCRs, but not directly affect receptor desensitization. Thus, the potentiation observed by blocking the internalization of β2-adrenergic receptors is probably caused by the inability of desensitized receptors to resensitize (Zhang et al., 1997), resulting in an increase in the proportion of desensitized receptors on the cell surface. Accordingly, we compared 5-HT2A and β2-adrenergic receptor resensitization in the presence of DynK44A and Arr2(319–418) in HEK-293 cells. As expected, β2-adrenergic receptor resensitization was potently inhibited by overexpression of DynK44A and Arr2(319–418). Also, as predicted from our prior studies (Bhatnagar et al., 2001), Arr2(319–418) had no effect on 5-HT2A receptor resensitization in HEK-293 cells. Unexpectedly, the coexpression of 5-HT2Areceptors and DynK44A resulted in a significant increase in the extent of 5-HT2A receptor resensitization, suggesting the existence of a novel cell surface mechanism for 5-HT2A receptor resensitization. These results are reminiscent of recent studies demonstrating that, in some cell lines, β2-adrenergic receptors can be down-regulated in the absence of endocytosis (Jockers et al., 1999), providing additional evidence for the existence of novel cell surface mechanisms of GPCR regulation.
It has been proposed previously that the cellular background in which 5-HT2A receptors are expressed determines the regulatory properties of the receptor (Pauwels et al., 1990; Grotewiel and Sanders-Bush, 1994). Accordingly, we examined the effect of endocytic inhibitors on 5-HT2A and β-adrenergic receptor desensitization in C6 glioma cells that endogenously express low levels of β1-adrenergic, β2-adrenergic, and 5-HT2A receptors (Zhong and Minneman, 1995;Elliott et al., 1995). In contrast to HEK-293 cells, 5-HT2A receptor desensitization in C6 glioma cells was potentiated by overexpression of DynK44A and Arr2(319–418) as well as treatment with ConA and PAO. Correspondingly, 5-HT2A receptor resensitization was significantly blocked by DynK44A and Arr2(319–418). These results suggest that in C6 glioma cells, unlike in HEK-293 cells, 5-HT2A receptor endocytosis might be important for receptor recycling and resensitization. However, although DynK44A, Arr2(319–418), ConA, and PAO are well-established inhibitors of endocytosis in many cell lines, their ability to block 5-HT2A receptor internalization in C6 glioma cells remains to be determined, because 5-HT2Areceptors are expressed at levels too low to be studied biochemically or by confocal microscopy. Nonetheless, the data obtained with four well-characterized inhibitors of endocytosis provide strong support for the hypothesis that endocytosis has been effectively blocked in C6 glioma cells.
To examine the mechanism responsible for the differential effects of Arr2(319–418) on 5-HT2A receptor desensitization in HEK-293 cells versus C6 glioma cells, we measured the expression levels of GRK2 and GRK5 in both cell lines and in brain lysates. HEK-293 cells express more GRK2 than do C6 glioma cells suggesting that the level of GRK2 expression is unrelated to the differential effects of Arr2(319–418) seen with the two cell lines. GRK5 expression, however, was slightly higher in C6 glioma cells versus HEK-293 cells. Thus, we demonstrated that overexpression of GRK2, GRK5, and a kinase-deficient GRK2 mutant in HEK-293 cells had no effect on 5-HT2A receptor desensitization, suggesting that the cell-type-specific effects of Arr2(319–418) are unrelated to GRK expression levels. As a parallel control, we demonstrated that GRK2 and GRK5 overexpression significantly enhanced β2-adrenergic receptor desensitization and GRK2-K220R blocked desensitization in HEK-293 cells. Taken together, these results imply novel roles for arrestins and GRKs in 5-HT2A trafficking and regulation.
Although these results offer the first convincing example of cell-type-specific effects of endocytosis inhibitors on GPCR desensitization and resensitization, cell-type-specific differences in GPCR internalization have been noted previously. For example, the chemokine receptor CXCR1 internalizes well in a neutrophil-like cell line but does not internalize in HEK-293 cells (Barlic et al., 1999), a difference probably caused by the higher expression levels of GRK2 and arrestin-3 in the neutrophil-like cell line (Barlic et al., 1999). Indeed, internalization of CXCR1 in HEK-293 cells requires the overexpression of both GRK2 and arrestins (Barlic et al., 1999). Another study demonstrated a correlation between the levels of expression of GRKs and arrestins in different cell lines with the extent of β2-adrenergic receptor internalization in those cells (Ménard et al., 1997). Together, these studies demonstrate that the cellular complement of regulatory proteins, such as arrestins and GRKs, may dictate that patterns of GPCR regulation in the cell lines studied. Interestingly, our current studies indicate that GRK2 and GRK5 expression levels are unrelated to 5-HT2Areceptor regulation by arrestins.
The present studies demonstrate clearly that 5-HT2A receptors are differentially regulated in HEK-293 cells compared with β2-adrenergic receptors in that 5-HT2A receptor internalization is not necessary for receptor resensitization. Importantly, blocking 5-HT2A receptor internalization with DynK44A in HEK-293 cells results in an increase in the extent of resensitization, suggesting a cell surface mechanism for resensitization. Additionally, our results demonstrate novel cell-type-specific modes of regulation for 5-HT2A receptors and suggest differing roles for arrestins and possibly GRKs in 5-HT2Areceptor regulation depending on the expression system.
Footnotes
- Received March 22, 2001.
- Accepted July 25, 2001.
-
This work was supported in part by National Institutes of Health Grants RO1-MH61887, RO1-MH57635, and KO2-MH01366 and by a NARSAD independent investigator award (to B.L.R.).
Abbreviations
- 5-HT
- 5-hydroxytryptamine (serotonin)
- PI
- phosphoinositide
- GPCR
- G protein-coupled receptor
- GRK
- G protein-coupled receptor kinase
- HEK
- human embryonic kidney
- DynK44A
- Dynamin K44A
- Arr2(319–418)
- arrestin-2 (319–418)
- IBMX
- 3-isobutyl-1-methylxanthine
- PAO
- phenylarsine oxide
- ConA
- concanavalin A
- HRP
- horseradish peroxidase
- BODIPY-FL
- 4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-pentanoic acid, succinimidyl ester
- DMEM
- Dulbecco's modified essential medium
- F12
- Ham's F12 nutrient mixture
- PBS
- phosphate-buffered saline
- β2AR
- β2-adrenergic receptor
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}