


Abstract
It is unknown why the potencies and efficacies of long-chained guanidine-type histamine H2-receptor (H2R) agonists are lower at the H2R of human neutrophils than at the H2R of the guinea pig atrium. To elucidate these differences, we analyzed fusion proteins of the human H2R (hH2R) and guinea pig H2R (gpH2R), respectively, and the short splice variant of Gsα(GsαS) expressed in Sf9 cells. The potencies and efficacies of small H2R agonists in the GTPase assay and the potencies of antagonists at inhibiting histamine-stimulated GTP hydrolysis by hH2R-GsαS and gpH2R-GsαS were similar. In contrast, the potencies and efficacies of guanidines were lower at hH2R-GsαS than at gpH2R-GsαS. Guanidines bound to hH2R-GsαS with lower affinity than to gpH2R-GsαS, and high-affinity binding of guanidines at gpH2R-GsαS was more resistant to disruption by GTPγS than binding at hH2R-GsαS. Molecular modeling suggested that the nonconserved Asp-271 in transmembrane domain 7 of gpH2R (Ala-271 in hH2R) confers high potency to guanidines. This hypothesis was confirmed by Ala-271→Asp-271 mutation in hH2R-GsαS. Intriguingly, the efficacies of guanidines at the Ala-271→Asp-271 mutant and at hH2R/gpH2R chimeras were lower than at gpH2R. Our model suggests that a Tyr-17/Asp-271 H-bond, present only in gpH2R-GsαS but not the other constructs studied, stabilizes the active guanidine-H2R state. Collectively, our data show 1) distinct interaction of H2R species isoforms with guanidines, 2) that a single amino acid in transmembrane domain 7 critically determines guanidine potency, and 3) that an interaction between transmembrane domains 1 and 7 is important for guanidine efficacy.
HIS (1) is a biogenic amine (Fig.1) that functions as a neurotransmitter and autacoid (Hill et al., 1997). HIS exerts its effects through at least four receptor subtypes, designated H1, H2, H3, and H4, respectively (Hill et al., 1997; Hough, 2001). HIS receptors belong to the superfamily of GPCRs that possess seven transmembrane domains, three extracellular and three intracellular loops. The H2R couples to Gs-proteins to activate adenylyl cyclase. Numerous H2R agonists and antagonists have been developed; the guinea pig atrium has been the standard model for ligand design for decades (Ganellin, 1982; Hill et al., 1997). Figure 1 shows the structures of prototypical H2R agonists and antagonists. Among agonists, DIM (2), AMT (3), and BET (4) are similar to HIS (1). BET is a nonselective H2R partial agonist (Ganellin, 1982;Burde et al., 1989) and is therefore an interesting experimental tool. Compared with compounds 1 to 4, the guanidines5 to 13 are long-chained and more bulky. IMP (5), ARP (8), and several ARP analogs are much more potent in the guinea pig atrium than HIS (Durant et al., 1978;Buschauer, 1989). H2R antagonists are divided into five chemical classes: imidazoles such as CIM (14), furans such as RAN (15), thiazoles such as FAM (16), and TIO (17), piperidinomethylphenoxy derivatives such as ZOL (18), and (benzamidoalkyl)cyanoguanidines such as APT (19) (Hill et al., 1997). H2R antagonists are of great importance for the treatment of gastroduodenal ulcer disease (Hill et al., 1997). H2R agonists may be useful as positive inotropic drugs for the treatment of heart failure (Felix et al., 1995), as differentiation-inducing agents in acute myelogenous leukemia (Seifert et al., 1992), and as anti-inflammatory drugs (Burde et al., 1990).

Structures of H2R agonists and antagonists. 1 to 13, agonists; 14 to19, antagonists. 6 to 13 represent arpromidine-derived guanidines.
Guanidine-type compounds are less potent and/or efficient agonists at the H2R of human neutrophils than at the H2R of the guinea pig atrium (Burde et al., 1989,1990; Buschauer, 1989). Additionally, several GPCR species isoforms, including the H3R, differ from each other in their pharmacological properties as assessed by the analysis of recombinant GPCRs (Kopin et al., 2000; Ligneau et al., 2000; Lovenberg et al., 2000). There are relatively few amino acid differences between hH2R and gpH2R (Gantz et al., 1991; Traiffort et al., 1995) (Fig.2), particularly in the established ligand-binding domains TM3 and TM5, but even a single amino acid exchange between GPCR species isoforms can strongly affect their pharmacological properties (Kopin et al., 2000; Ligneau et al., 2000). Based on these findings, the hypothesis arose that the H2R exhibits species-specific pharmacological properties as well.

Comparison of the amino acid sequences of hH2R and gpH2R. The amino acid sequences of the cloned hH2R (Gantz et al., 1991) and gpH2R (Traiffort et al., 1995) are given in the one-letter code. Dots in the gpH2R sequence indicate identity with hH2R. TM domains are shown in bold. Amino acids shown in green in TM3 and TM5 represent the interaction sites of HIS with the H2R (Gantz et al., 1992; Nederkoorn et al., 1996). Amino acids shown in black in the gpH2R sequence represent conservative exchanges. Amino acids shown in red in the gpH2R sequence represent nonconservative exchanges. The arrow indicates the cleavage site ofKpnI, present in the cDNA of both gpH2R and hH2R. The KpnI site allowed us to construct reciprocal hH2R/gpH2R chimeras (see Fig. 10).N-term, extracellular N-terminal domain of H2Rs; C-term, intracellular C-terminal domain of H2Rs.; i1, i2, and i3; 1st, 2nd, and 3rd intracellular loop, respectively; e1, e2, and e3, 1st, 2nd, and 3rd extracellular loop, respectively; TM1–7, transmembrane domains 1–7.
To test our hypothesis, we constructed fusion proteins of the hH2R and gpH2R, respectively, and GsαS and expressed the fusion proteins in Sf9 cell membranes. GPCR-Gα fusion proteins ensure a defined 1:1 stoichiometry of the signaling partners and efficient coupling (Seifert et al., 1999; Milligan, 2000). The measurement of GTP hydrolysis in GPCR-Gα fusion proteins is presumably the most precise method currently available for the analysis of ligand potencies and efficacies, because the GTPase assay is a steady-state method, is extremely sensitive in GPCR-Gsα fusion proteins, assesses GPCR/G-protein coupling directly at the G-protein level, and is independent of the expression level of the components (Seifert et al., 1999; Milligan, 2000). Finally, the analysis of H2R species isoforms in the same host cell membrane annihilates the impact of pharmacokinetic differences between different test systems. Here, we report that hH2R and gpH2R exhibit distinct pharmacological properties, particularly with respect to interaction with guanidines.
Experimental Procedures
Materials.
The cDNA for the hH2R was kindly provided by Dr. I. Gantz (University of Michigan Medical School and Ann Arbor VA Medical Center, Ann Arbor, MI) (Gantz et al., 1991). The cDNA for the gpH2R was kindly provided by Drs. E. Traiffort and J.-C. Schwartz (Department of Neurobiology and Pharmacology, Center Paul Broca, Institut National de la Santé et de la Recherche Médicale, Paris, France) (Traiffort et al., 1995). The generation of the baculovirus encoding β2AR-GsαL had been described previously (Seifert et al., 1998a). APT was synthesized as described previously (Hirschfeld et al., 1992). IMP was prepared as described previously (Durant et al., 1978). Guanidines 6 to11 were synthesized as described previously (Buschauer, 1989). Guanidines 12 and 13 (Schalkhausser, 1998) were prepared by analogy to the procedures described for guanidines6 to 11 (Buschauer, 1989). The structures of the synthesized compounds were confirmed by analysis (C, H, N),1H NMR, and mass spectroscopy spectra. Purity of compounds was >98% as determined by high-performance liquid chromatography or capillary electrophoresis (Schuster et al., 1997). The anti-FLAG Ig (M1 monoclonal antibody) was from Sigma (St. Louis, MO). The anti-Gsα Ig (C-terminal) was from Calbiochem (La Jolla, CA). [γ-32P]GTP (6000 Ci/mmol), [35S]GTPγS (1100 Ci/mmol), [3H]DHA (85–90 Ci/mmol), and [3H]TIO (90 Ci/mmol) were from PerkinElmer Life Sciences (Boston, MA). All unlabeled nucleotides were from Roche (Indianapolis, IN). HIS, BET, CIM, RAN, and FAM were from Sigma. AMT, TIO, and ZOL were from Tocris Cookson (Ballwin, MO). DIM was from RBI (Natick, MA). All restriction enzymes and T4 DNA ligase were from New England Biolabs (Beverly, MA). Cloned Pfu DNA polymerase was from Stratagene (La Jolla, CA).
Construction of FLAG Epitope- and Hexahistidine-Tagged cDNA for hH2R-GsαS.
A DNA sequence encoding the cleavable signal peptide from influenza hemagglutinin (S) followed by the FLAG epitope (F), which is recognized by the M1 antibody, was placed 5′ of the start codon of the hH2R to enhance GPCR expression and allow immunological detection. We also added a hexahistidine tag to the C terminus of hH2R to allow future purification and to provide additional protection against proteolysis (Seifert et al., 1998a). The GPCR modifications were generated by sequential overlap-extension PCRs. In PCR 1A, the DNA sequence of the N-terminal portion of the hH2R was amplified using CMVneo-hH2R as template. The sense primer annealed with the first 18 bp of the 5′-end of the hH2R and included the last 18 bp of the SF in its 5′-extension. The antisense primer encoded the sequence GAGCTGTTGATATCCGGTGCGGAAGTCTCTG to generate a silent mutation yielding a new EcoRV site. In PCR 1B, the DNA sequence of the C-terminal portion of the hH2R was amplified using CMVneo-hH2R as template. The sense primer encoded the sequence TTCCGCACCGGATATCAACAGCTCTTCTGCTGC to generate the newEcoRV site. The antisense primer encoded the five C-terminal amino acids of the hH2R, a hexahistidine tag, the stop codon and an XbaI site. In PCR 2, the products of PCRs 1A and 1B annealed in the region encoding the newly createdEcoRV site. In PCR 2, the sense primer of PCR 1A and the antisense primer of PCR 1B were used. In this way, a fragment encoding the signal sequence, the FLAG epitope, hH2R cDNA with a new EcoRV site and a hexahistidine tag followed by anXbaI site was obtained. This fragment was digested withNcoI and XbaI and cloned into pGEM-3Z-SF-human formyl peptide receptor-6His digested with NcoI andXbaI. In PCR 3A, the C-terminal portion of the H2R was amplified using pGEM-3Z-SF-hH2R as template, a sense primer annealing 5′ of the newly created EcoRV site and an antisense primer annealing with the hexahistidine tag. In PCR 3B, the sequence of GsαS was amplified, using pGEM-3Z-SF-β2AR-GsαS as template, a sense primer annealing with the hexahistidine tag and an antisense primer annealing with the 5 C-terminal amino acids of Gsα, the stop codon, and an XbaI site. In PCR 4, the products of PCRs 3A and 3B annealed in the hexahistidine region, and the sense primer of PCR 3A and the antisense primer of PCR 3B were used. In this way, a fragment encoding the C-terminal portion of the hH2R, a hexahistidine tag, GsαS, a stop codon and an XbaI site was created. This fragment was digested with EcoRV andXbaI and cloned into pGEM-3Z-SFhH2R digested with EcoRV and XbaI. In this way, the full-length cDNA for hH2R-GsαS was created. pGEM-3Z-SF-hH2R-GsαS was digested with NcoI and XbaI to recover the fusion protein cDNA and cloned into the baculovirus transfer vector pVL 1392-SF-β2AR-Giα2digested with NcoI and XbaI. PCR-generated DNA sequences were confirmed by extensive restriction enzyme analysis and enzymatic sequencing.
Construction of FLAG Epitope- and Hexahistidine-Tagged cDNA for gpH2R-GsαS.
The strategy for creation of gpH2R-GsαS cDNA was analogous to the strategy for creation of hH2R-GsαS cDNA. In PCR 1A, the DNA sequence of the N-terminal portion of gpH2R was amplified using pGEM4Z-gpH2R as template. The sense primer annealed with the first 20 bp of the 5′-end of the gpH2R and included the last 8 bp of the SF in its 5′-extension. The antisense primer encoded the sequence CTCATGGGAGTTGTGGCTAGCGAGCCTGCAGCAGAAGAGC to create a silent mutation yielding a new NheI site. In PCR 1B, the sequence of the C-terminal portion of gpH2R was amplified using pGEM4Z-gpH2R as template. The sense primer encoded the sequence GCTCTTCTGCTGCAGGCTCGCTAGCCACAACTCCCATGAG to create the newNheI site. The antisense primer encoded the five C-terminal amino acids of the gpH2R, a hexahistidine tag, the stop codon, and an XbaI site. In PCR 2, the products of PCRs 1A and 1B annealed in the region encoding the newly createdNheI site. In PCR 2, the sense primer of PCR 1A and the antisense primer of PCR 1B were used. In this way, a fragment encoding the signal sequence, the FLAG epitope, gpH2R cDNA with a new NheI site, and a hexahistidine tag followed by anXbaI site was obtained. This fragment was digested withNcoI and XbaI and cloned into pGEM-3Z-SF-human formyl peptide receptor-6His digested with NcoI andXbaI. In PCR 3A, the C-terminal portion of the gpH2R was amplified using pGEM-3Z-SF-gpH2R as template, a sense primer annealing 5′ of the newly created NheI site, and an antisense primer annealing with the hexahistidine tag. In PCR 3B, the sequence of GsαS was amplified, using pGEM-3Z-SF-β2AR-GsαS as template, a sense primer annealing with the hexahistidine tag, and an antisense primer annealing with the 5 C-terminal amino acids of Gsα, the stop codon, and an XbaI site. In PCR 4, the products of PCRs 3A and 3B annealed in the hexahistidine region, and the sense primer of PCR 3A and the antisense primer of PCR 3B were used. In this way, a fragment encoding the C-terminal portion of the gpH2R, a hexahistidine tag, GsαS, a stop codon, and an XbaI site was created. This fragment was digested with NheI andXbaI and cloned into pGEM-3Z-SFgpH2R digested with NheI and XbaI. In this way, the full-length cDNA for gpH2R-GsαS was created. pGEM-3Z-SF-hH2R-GsαS was digested with NcoI and XbaI to recover the fusion protein cDNA and cloned into the baculovirus transfer vector pVL 1392-SF-β2AR-Giα2digested with NcoI and XbaI. PCR-generated DNA sequences were confirmed by extensive restriction enzyme analysis and enzymatic sequencing.
Construction of the cDNA for hH2R-A271D-GsαS.
The Ala-271 → Asp-271 exchange in hH2R was generated by sequential overlap-extension PCRs. In PCR 1A, the DNA sequence of the N-terminal portion of hH2R was amplified using pGEM-3Z-SF-hH2R-GsαS as a template. The sense primer annealed with the first 18 bp of the 5′ end of hH2R and included the last 18 bp of the SF in its 5′ extension. The antisense primer encoded the sequence CAGAACGATATCTTCTAACACCTCATTGATGGCATC to generate the Ala-271 → Asp-271 exchange and a new EcoRV site at the position of the mutated amino acid. In PCR 1B, the DNA sequence of the C-terminal portion of the hH2R and the entire sequence of Gsαs was amplified using pGEM-3Z-SF-hH2R-GsαS as a template. The sense primer encoded the sequence GTTAGAAGATATCGTTCTGTGGCTGGGCTATGCCAAC to generate the Ala-271 → Asp-271 exchange and a new EcoRV site at the position of the mutated amino acid. The antisense primer encoded the five C-terminal amino acids of Gsα, the stop codon and an XbaI site. In PCR 2, the products of PCR 1A and 1B annealed in the region encoding the newly created Ala-271 → Asp-271 exchange and the EcoRV site. In the PCR 2, the sense primer of PCR 1A and the antisense primer of PCR 1B were used. In this way, a fragment encoding the entire hH2R-A271D-GsαS fusion protein was created. This fragment was digested with EcoRI and NcoI and cloned into pGEM-3Z-SF-hH2R-GsαSdigested with EcoRI and NcoI. pGEM-3Z-SF-hH2R-A271D-GsαSwas digested with SacI and EcoN I and cloned into the baculovirus transfer vector pVL 1392-SF-hH2R-GsαSdigested with SacI and EcoN I. PCR-generated DNA sequences were confirmed by extensive restriction enzyme analysis and enzymatic sequencing.
Construction of the cDNAs for NgpChH2R-GsαS and NhCgpH2R-GsαS.
For construction of hH2R/gpH2R chimeras, we took advantage of the KpnI site present at the same position of the cDNAs of both receptors. KpnI cleaves hH2R- and gpH2R cDNA in the center of the second intracellular loop (Fig. 2). pGEM-3Z-SF-hH2R-GsαS and pGEM-3Z-SF-gpH2R-GsαSwere digested with KpnI and XbaI so that the C-terminal halves of H2Rs and the fused GsαS were cut out. The fragments obtained were reciprocally cloned back into pGEM-3Z-SF-hH2R-GsαS and pGEM-3Z-SF-gpH2R-GsαS. As a result of this exchange, we created pGEM-3Z-SF-NgpChH2R-GsαSand pGEM-3Z-SF-NhCgpH2R-GsαS. These plasmids were digested with NcoI and XbaI and cloned into the baculovirus transfer vector pVL 1392-SF-gpH2R-GsαSdigested with NcoI and XbaI. The chimeric H2R-GsαS DNA sequences were confirmed by extensive restriction enzyme analysis.
Generation of Recombinant Baculoviruses, Cell Culture and Membrane Preparation.
Recombinant baculoviruses encoding the H2R-Gsα fusion proteins were generated in Sf9 cells using the BaculoGOLD transfection kit (BD PharMingen, San Diego, CA) according to the manufacturer's instructions. After initial transfection, high-titer virus stocks were generated by two sequential virus amplifications. Sf9 cells were cultured in 250-ml disposable Erlenmeyer flasks at 28°C under rotation at 125 rpm in SF 900 II medium (Invitrogen, Carlsbad, CA) supplemented with 5% (v/v) fetal calf serum (BioWhittaker, Walkersville, MD) and 0.1 mg/ml gentamicin (BioWhittaker). Cells were maintained at a density of 0.5 to 6.0 × 106cells/ml. For infection, cells were sedimented by centrifugation and suspended in fresh medium. Cells were seeded at 3.0 × 106 cells/ml and infected with a 1:100 dilution of high-titer baculovirus stocks encoding H2R-GsαS fusion proteins. Cells were cultured for 48 h before membrane preparation. Sf9 membranes were prepared as described previously (Seifert et al., 1998a), using 1 mM EDTA, 0.2 mM phenylmethylsulfonyl fluoride, 10 μg/ml benzamidine, and 10 μg/ml leupeptin as protease inhibitors. Membranes were suspended in binding buffer (12.5 mM MgCl2, 1 mM EDTA and 75 mM Tris/HCl, pH 7.4) and stored at −80°C until use.
Receptor Ligand Binding Assays.
Membranes were thawed and sedimented by a 15-min centrifugation at 4°C and 15,000gto remove residual endogenous guanine nucleotides as much as possible. Membranes were resuspended in binding buffer (12.5 mM MgCl2, 1 mM EDTA and 75 mM Tris/HCl, pH 7.4). In [3H]TIO binding assays, each tube (total volume, 250 μl) contained 200 to 250 μg of protein. Nonspecific binding was determined in the presence of [3H]TIO at various concentrations plus 100 μM unlabeled TIO. Incubations were conducted for 90 min at 25°C and shaking at 250 rpm. In saturation binding experiments, tubes contained 1 to 20 nM [3H]TIO plus unlabeled TIO to obtain final ligand concentrations of up to 300 nM. Competition binding experiments were carried out in the presence of 10 nM [3H]TIO and unlabeled ligands at various concentrations without or with GTPγS (10 μM). Bound [3H]TIO was separated from free [3H]TIO by filtration through GF/C filters, followed by three washes with 2 ml of binding buffer (4°C). Filter-bound radioactivity was determined by liquid scintillation counting. The experimental conditions chosen ensured that not more than 5% of the total amount of [3H]TIO added to binding tubes was bound to filters. The expression level of β2AR-GsαL was determined with 10 nM [3H]DHA as radioligand as described previously (Seifert et al., 1998a).
[35S]GTPγS Binding Assay.
Membranes were thawed, sedimented, and suspended as for receptor ligand binding assays. Reaction mixtures (total volume, 500 μl) contained Sf9 membranes expressing H2R-Gsα fusion proteins (15 μg of protein/tube) in binding buffer supplemented with 0.05% (w/v) bovine serum albumin, 1 μM GDP, and 1 nM [35S]GTPγS plus 9 nM unlabeled GTPγS. Reaction mixtures additionally contained distilled water (basal) and HIS at a saturating concentration (100 μM). Incubations were conducted for 90 min at 25°C and shaking at 250 rpm. Bound [35S]GTPγS was separated from free [35S]GTPγS by filtration through GF/C filters, followed by three washes with 2 ml of binding buffer (4°C). Filter-bound radioactivity was determined by liquid scintillation counting. The experimental conditions chosen ensured that no more than 10% of the total amount of [35S]GTPγS added was bound to filters.
Steady-State GTPase Activity Assay.
Membranes were thawed, sedimented, and resuspended in 10 mM Tris/HCl, pH 7.4. Assay tubes contained Sf9 membranes expressing H2R-Gsα fusion proteins (10 μg of protein/tube), 1.0 mM MgCl2, 0.1 mM EDTA, 0.1 mM ATP, 100 nM GTP, 1 mM adenylyl imidodiphosphate, 5 mM creatine phosphate, 40 μg of creatine kinase, and 0.2% (w/v) bovine serum albumin in 50 mM Tris/HCl, pH 7.4, and H2R ligands at various concentrations. Reaction mixtures (80 μl) were incubated for 3 min at 25°C before the addition of 20 μl of [γ-32P]GTP (0.2–0.5 μCi/tube). All stock and work dilutions of [γ-32P]GTP were prepared in 20 mM Tris/HCl, pH 7.4. Reactions were conducted for 20 min at 25°C. Preliminary studies under basal conditions and with HIS, IMP, and ARP showed that under these conditions, GTP hydrolysis was linear. Reactions were terminated by the addition of 900 μl of slurry consisting of 5% (w/v) activated charcoal and 50 mM NaH2PO4, pH 2.0. Charcoal absorbs nucleotides but not Pi. Charcoal-quenched reaction mixtures were centrifuged for 15 min at room temperature at 15,000g. Seven hundred microliters of the supernatant fluid of reaction mixtures were removed, and32Pi was determined by liquid scintillation counting. Enzyme activities were corrected for spontaneous degradation of [γ-32P]GTP. Spontaneous [γ-32P]GTP degradation was determined in tubes containing all of the above described components plus a very high concentration of unlabeled GTP (1 mM) that, by competition with [γ-32P]GTP, prevents [γ-32P]GTP hydrolysis by enzymatic activities present in Sf9 membranes. Spontaneous [γ-32P]GTP degradation was <1% of the total amount of radioactivity added using 20 mM Tris/HCl, pH 7.4, as solvent for [γ-32P]GTP. The experimental conditions chosen ensured that not more than 10% of the total amount of [γ-32P]GTP added was converted to32Pi.
SDS-PAGE and Immunoblot Analysis.
Membrane proteins were separated on SDS polyacrylamide gels containing 8% (w/v) acrylamide. Proteins were then transferred onto Immobilon-P transfer membranes (Millipore, Bedford, MA). Membranes were reacted with M1 antibody or anti-Gsα Ig (1:1000 each). Immunoreactive bands were visualized by sheep anti-mouse IgG (M1 antibody) and donkey anti-rabbit IgG (anti-Gsα Ig), respectively, coupled to peroxidase, using o-dianisidine and H2O2 as substrates.
Molecular Modeling.
Models of the seven TM helices were taken from the PDB bovine rhodopsin file 1f88 (Palczewski et al., 2000). The starting structure of gpH2R TM domains was constructed from a multiple sequence-alignment of bovine rhodopsin with hβ2AR (Palczewski et al., 2000), gpH1R, hH2R, and gpH2R (Gantz et al., 1991; Traiffort et al., 1995; Hill et al., 1997). The resulting TM helices in hH2R and gpH2R are highlighted in bold (Fig. 2). First, the model was roughly minimized by the steepest descent method to remove bad contacts due to the mutated residues. In the first 100 steps, the backbone was fixed. Energy calculations were based on the Kollman all-atom force field (Kollman charges, distant dependent dielectricity constant of 4). Then IMP (5) and ARP (8) were manually docked into the model in a conformation suggested to be active from 3D QSAR results (Dove and Buschauer, 1998, 1999). The selection of amino acids interacting with the imidazolylpropylguanidine moiety based on studies with hH2R (Gantz et al., 1992), hβ2AR (Wieland et al., 1996; Isogaya et al., 1999), and hH1R (Wieland et al., 1999) mutants. The docking with respect to TM 6 and 7 was only roughly suggested by seeking a pocket near the “hot” region around Asp-271 that may accommodate the imidazole and the pyridine moiety of IMP and ARP, respectively. Kollman all-atom types were assigned to IMP and ARP by analogy, including definition of the new atom type F (fluorine). Missing parameters (e.g., for bonds CA—NB, CC—CC, F—CA, and a number of bond angles) were derived from similar types or from the Tripos force field. As hydrogen bonding parameters for F-H3, the respective values for O and N were assigned. Both ligands were provided with Gasteiger-Hueckel charges. The complexes were fully minimized (distant dependent dielectricity constant of 1) without constraints by the Powell method down to an RMS gradient of less than 0.05. All calculations were performed with SYBYL 6.7 (Tripos, St. Louis, MO) on an SGI Octane workstation (SGI, Mountain View, CA)
Miscellaneous.
Protein concentrations were determined using the DC protein assay kit (Bio-Rad, Hercules, CA). All analyses of experimental data were performed with the Prism III program (GraphPad Software, San Diego, CA).
Results and Discussion
Immunological Detection of hH2R-GsαS and gpH2R-GsαS in Sf9 Cell Membranes.
Monomeric nonfused H2R expressed in Sf9 cells migrates as ∼33-kDa band in SDS-PAGE (Fukushima et al., 1997), and the apparent molecular mass of GsαS is ∼45 kDa. Thus, the molecular mass of H2R-GsαS fusion proteins was expected to be ∼78 kDa. In fact, the anti-FLAG Ig detected a ∼78-kDa band in immunoblots (Fig. 3). The intensities of immunologically detected bands in membranes expressing hH2R-GsαS and gpH2R-GsαS were similar to the band intensities in membranes expressing β2AR-GsαL fusion protein at 7.0 pmol/mg as determined by [3H]DHA saturation binding. The ∼44-kDa band in membranes expressing β2AR-GsαL represents a degradation product of the fusion protein that was generated as the result of incidental freeze/thaw cycles. The membranes expressing H2R-GsαS did not undergo such cycles, and accordingly, we did not observe degradation products. In membranes expressing hH2R-GsαS and, to a much lesser extent, in membranes expressing gpH2R-GsαS, we also observed ∼160-kDa bands. The H2R is known to form homodimers (Fukushima et al., 1997), and thus the ∼160-kDa bands most probably represent H2R-GsαS fusion protein homodimers.

Analysis of the expression of H2R-GsαS fusion proteins in Sf9 cell membranes. Various Sf9 cell membrane preparations (SP followed by number) expressing β2AR-GsαL (7.0 pmol/mg as assessed by [3H]DHA saturation binding) and H2R-Gsα fusion proteins were separated by SDS-PAGE using a gel that contained 8% (w/v) acrylamide. Fusion proteins were probed with the anti-FLAG Ig (M1 antibody). Each membrane preparation was analyzed in three different amounts (25, 50, and 100 μg of protein, respectively, from left to right). Numbers on the left of the immunoblot indicate molecular masses of marker proteins. Shown is the horseradish peroxidase-reacted Immobilon P membrane of a representative gel. Similar results were obtained with four other membrane preparations of hH2R-GsαS and gpH2R-GsαS each.
[3H]TIO- and [35S]GTPγS Saturation Binding to hH2R-GsαS and gpH2R-GsαS Expressed in Sf9 Cell Membranes.
Native gpH2R binds [3H]TIO with a K dvalue of ∼17 nM (Gajtkowski et al., 1983). However, the use of [3H]TIO in native organs is severely limited by the fact that nonspecific binding with saturating [3H]TIO concentrations amounts to ∼85 to 90% of total [3H]TIO binding. In Sf9 membranes, only ∼55 to 65% nonspecific [3H]TIO binding occurred with saturating radioligand concentrations. Therefore, a more precise determination of the kinetics of specific [3H]TIO binding was possible (Fig.4). H2R-GsαS fusion proteins expressed in Sf9 membranes bound [3H]TIO according to monophasic saturation curves: hH2R-GsαS with aK d value of 32.0 ± 4.6 nM and aB max value of 0.43 ± 0.02 pmol/mg (SP166); gpH2R-GsαS with a K d value of 34.4 ± 8.4 nM and aB max value of 0.72 ± 0.02 pmol/mg (SP391). Although the K d values for [3H]TIO fit well to theK B values for TIO in the GTPase competition studies with HIS (Table 1), we would have expected B max values of ∼5 to 7 pmol/mg for H2R-GsαS fusion proteins.

[3H]TIO saturation binding in Sf9 membranes expressing hH2R-GsαS and gpH2R-GsαS. Membranes expressing hH2R-GsαS (A, SP166) or gpH2R-GsαS (B, SP391) were incubated in the presence of [3H]TIO at the concentrations indicated on the abscissa as described under Experimental Procedures. Nonspecific binding is the [3H]TIO binding not competed for by 100 μM unlabeled TIO. Specific binding is the difference between total [3H]TIO binding and nonspecific [3H]TIO binding for a given [3H]TIO concentration. Data were analyzed by nonlinear regression and are the means ± S.D. of three experiments performed in duplicate. Similar results were obtained with four other membrane preparations of hH2R-GsαS and gpH2R-GsαS each.
Potencies and inverse agonist efficacies of antagonists at hH2R-GsαS and gpH2R-GsαS expressed in Sf9 cell membranes
There are two possible explanations for the lowB max values of [3H]TIO binding to H2R-GsαS fusion proteins. [3H]TIO could either bind to only a fraction of the functionally active fusion protein molecules or the majority of the expressed molecules could be inactive. To discriminate between these alternatives, we took advantage of the fact that in GPCR-Gα fusion proteins, 1 mol of protein binds ∼1 mol of [35S]GTPγS when fully activated (Wenzel-Seifert and Seifert, 2000; Liu et al., 2001). TheB max value of HIS-stimulated [35S]GTPγS binding was 5.2 ± 0.6 pmol/mg for hH2R-GsαS(SP166) and 8.7 ± 1.1 pmol/mg for gpH2R-GsαS (SP391). These values fit well to the immunological data (Fig. 3) and suggest that almost all of the expressed H2R-Gsα molecules are functionally active. Accordingly, the majority of ligand-free H2R-Gsα molecules exist in a conformation that does not bind [3H]TIO. Future studies will have to determine whether H2R-Gsα differentially binds [3H]TIO and another H2R radioligand, [125I]iodoaminopotentidine (Hill et al., 1997).
Similar Antagonist Potencies at hH2R-GsαSand gpH2R-GsαS Expressed in Sf9 Cell Membranes and Evidence that RAN and APT Differentially Stabilize an Inactive Conformation in H2R Species Isoforms.
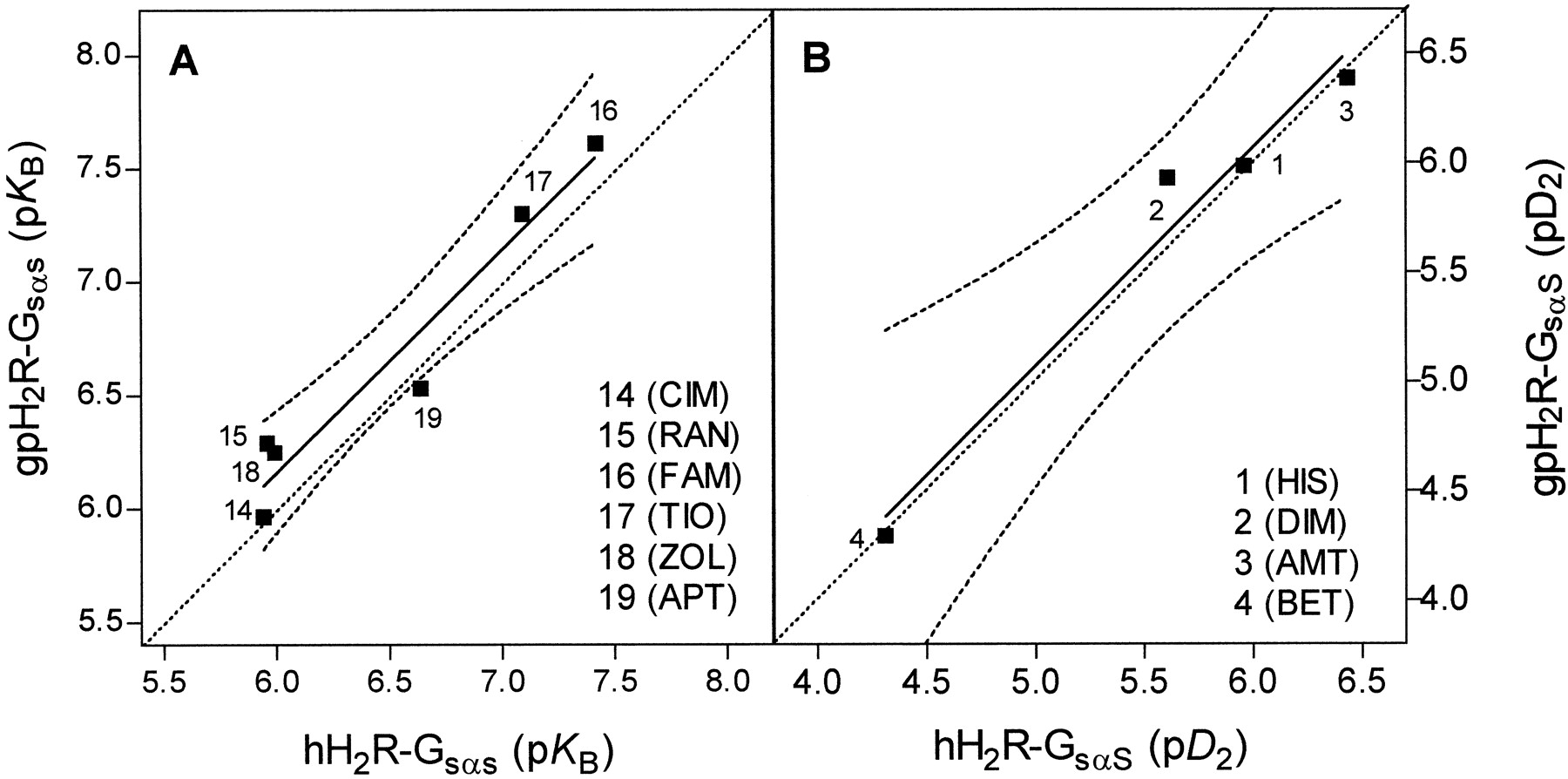
In GPCR-Gα fusion proteins, the GTPase assay is a highly sensitive method to determine ligand potencies and efficacies (Seifert et al., 1999; Milligan, 2000). GTP hydrolysis in Sf9 membranes expressing H2R-GsαSfusion proteins was stimulated with HIS at a submaximally effective concentration, and the HIS-stimulated GTP hydrolysis was inhibited by H2R antagonists of various chemical classes. TheK B values for CIM (14), RAN (15), ZOL (16), TIO (17), FAM (18), and APT (19) did not vary by more than a factor of 2 between hH2R-GsαS and gpH2R-GsαS (Table 1). We correlated pK B values of antagonists at hH2R-GsαS versus gpH2R-GsαS. If the antagonist-affinities of hH2R-GsαS and gpH2R-GsαS were identical, we would expect a linear correlation with a slope of 1.00 that follows the dotted line in Fig. 5A. Indeed, we obtained a highly significant correlation with a slope of 0.98 close to the theoretical curve (Fig. 5A), indicating that the antagonist-binding properties of hH2R-GsαS and gpH2R-GsαS are very similar. This is an important finding, because for other GPCR species isoforms, including the H3R, differences in antagonist-binding properties were observed (Kopin et al., 2000;Ligneau et al., 2000; Lovenberg et al., 2000).

Relations between pK Bvalues for antagonists and between pD 2values for HIS-like agonists at hH2R-GsαS and gpH2R-GsαS. pK Bvalues were derived from K B values shown in Table 1. pD 2 values were derived from EC50 values shown in Table 2. Solid lines represent the actual correlations obtained. Dashed lines represent the 95% confidence intervals of the correlations. The straight dotted lines represent the theoretical correlations that would have been obtained if pK B values and pD 2 values, respectively, had been identical in the two systems compared with each other. The theoretical curves have a slope of 1.00. A, correlation of pK Bvalues for hH2R-GsαS versus gpH2R-GsαS. Slope, 0.98 ± 0.13; r2, 0.94; p = 0.0015 (significant). B, correlation of pD 2 values for agonists1 to 4 at hH2R-GsαS versus gpH2R-GsαS. Slope, 0.99 ± 0.13; r2, 0.97; p = 0.0172 (significant).
Previous studies had shown that the hH2R is constitutively active (i.e., H2R antagonists decrease the activity of the agonist-free hH2R) (Alewijnse et al., 1998). In fact, RAN (15) had a consistent inverse agonist effect at hH2R-GsαS (Table 1). APT (19), which had not been studied in this respect previously, was a much more efficient inverse agonist than RAN (15) at hH2R-GsαS. At gpH2R-GsαS, RAN showed the greatest inverse agonist effect among the antagonists studied, and the effect of RAN at gpH2R-GsαS was also significantly greater than at hH2R-GsαS. Conversely, APT was considerably more efficient as an inverse agonist at hH2R-GsαS than at gpH2R-GsαS. These data show that RAN and APT differentially stabilize an inactive conformation in hH2R and gpH2R. In support of the hypothesis that different H2R antagonists stabilize unique conformations in H2Rs from different species is the finding that at the rat H2R, burimamide is a neutral antagonist, whereas at hH2R, it is a partial agonist (Alewijnse et al., 1998).
The absolute inverse agonist activities of APT at hH2R-GsαS and of RAN at gpH2R-GsαS, respectively, were similar, indicating that both GPCRs exhibit a similar degree of constitutive activity. In comparison, the rat H2R is less constitutively active than hH2R (Alewijnse et al., 1998). These data show that H2R species isoforms differ from each other in their constitutive activity. Differences in constitutive activity among GPCR species isoforms are not restricted to the H2R (Kopin et al., 2000). We also noted that, except for RAN (15) and APT (19), the inverse agonist effect of each individual antagonist was quite variable (Table1). This variability does not reflect insensitivity of the GTPase assay. Rather, we assume that in our system, CIM (14), ZOL (16), TIO (17), and FAM (18) casually act as neutral antagonists or inverse agonists at H2Rs, resulting in considerable data variability. Stochastic actions of weak inverse agonists were also reported for the β2AR (Chidiac et al., 1996). Another factor that could have contributed to the relatively small inverse agonist effects of RAN and FAM at H2R in this study relative to a previous study (Alewijnse et al., 1998) could be that we studied coupling of H2Rs to GsαS and not GsαL (seeExperimental Procedures). Specifically, it is known that GsαL confers the properties of constitutive activity to the β2AR; i.e., GsαL increases inverse agonists effects, whereas GsαS does not have these effects (Seifert et al., 1998b). Thus, it is possible that in the Chinese hamster ovary cells studied by Alewijnse et al. (1998), the H2R was preferentially coupled to GsαL, thereby increasing inverse agonist effects.
Agonist Efficacies, Potencies and Affinities at hH2R-GsαS and gpH2R-GsαS Expressed in Sf9 Cell Membranes: Evidence that Guanidines Stabilize an Active Conformation in gpH2R More Efficiently and Potently Than in hH2R.
We determined the efficacies and potencies of the small H2R agonists 1 to4 and of the larger H2R agonists5 to 13 in the GTPase assay (Table2). The efficacies of HIS (1), DIM (2), AMT (3), and BET (4) were similar at hH2R-GsαS and gpH2R-GsαS, respectively. In contrast, the efficacies of the guanidines 5 to13 at hH2R-GsαS were all significantly lower than at gpH2R-GsαS. The differences in efficacy were most prominent for BU-E-43 (7), BU-E-48 (10), and D281 (13). Specifically, elongation of the alkyl chain between the guanidino group and the phenyl ring (6 → 7) and introduction of a bulky Br atom (6 → 10) or of multiple Cl atoms into the phenyl ring (12 → 13) had pronounced negative effects on agonist efficacy at hH2R-GsαS but not at gpH2R-GsαS. Accordingly, the slope of the correlation of the efficacies of agonists at hH2R-GsαS and gpH2R-GsαS was very shallow, and the theoretical curve assuming pharmacological identity of H2R species isoforms was not approached within the data interval (Fig.6A). Particularly informative is the comparison of the efficacies of BET (4) and guanidines 8 to 11. At hH2R-GsαS, these compounds possess efficacies of 0.73 to 0.87, but only guanidines have increased efficacies at gpH2R-GsαS (Table 2). Taken together, these results indicate that the hH2R-GsαS and gpH2R-GsαS conformations stabilized by one of the small agonists 1 to 4similarly promote GDP/GTP exchange. In contrast, the guanidines5 to 13 stabilize a hH2R-GsαS conformation considerably less efficient for GDP/GTP exchange than the corresponding gpH2R-GsαS conformation.
Agonist efficacies and potencies at hH2R-GsαS and gpH2R-GsαS expressed in Sf9 cell membranes

Relations between efficacies and potencies of guanidines at hH2R-GsαS, hH2R-A271D-GsαS, NgpChH2R-GsαS and NhCgpH2R-GsαS, respectively, versus gpH2R-GsαS. Agonist efficacies were taken from Tables 2 and 4, and pD 2 values were derived from the EC50 values shown in Tables 2 and 4. Solid lines represent the actual correlations obtained. Dashed lines represent the 95% confidence intervals of the correlations. The straight dotted lines represent the theoretical correlations that would have been obtained if efficacies and pD 2values, respectively, had been identical in the two systems compared with each other. The theoretical curves have a slope of 1.00. A, correlation of efficacies of agonists 5 to 13 at hH2R-GsαS versus gpH2R-GsαS. Slope, 0.41 ± 0.10; r2, 0.72; p = 0.0038 (significant). B, correlation of pD 2values for agonists 5 to 13 at hH2R-GsαS versus gpH2R-GsαS. Slope, 0.89 ± 0.32; r2, 0.53; p = 0.0270 (significant). C, correlation of efficacies of agonists5 to 8, 10, 11, and13 at hH2R-A271D-GsαS versus gpH2R-GsαS. Slope, 0.50 ± 0.12; r2, 0.77; p =0.0096 (significant). D, correlation of pD 2 values for agonists 5 to8, 10, 11, and 13 at hH2R-A271D-GsαS versus gpH2R-GsαS. Slope, 0.81 ± 0.16; r2, 0.83; p = 0.0041 (significant). E, correlation of efficacies of agonists5 to 8, 10, 11, and13 at NgpChH2R-GsαS versus gpH2R-GsαS. Slope, 0.34 ± 0.04; r2, 0.94; p = 0.0003 (significant). F, correlation of pD 2 values for agonists 5 to8, 10, 11, and 13 at NgpChH2R-GsαS versus gpH2R-GsαS. Slope, 0.67 ± 1.12; r2, 0.07; p = 0.57 (not significant). G, correlation of efficacies of agonists5 to 8, 10, 11, and13 at NhCgpH2R-GsαS versus gpH2R-GsαS. Slope, 0.36 ± 0.06; r2, 0.88; p = 0.0015 (significant). H, correlation of pD 2values for agonists 5 to 8, 10,11, and 13 at NhCgpH2R-GsαS versus gpH2R-GsαS. Slope, 0.73 ± 0.28; r2, 0.58; p = 0.0471 (significant).
The potencies of small agonists (1–4) differed by not more than a factor of 2 between hH2R-GsαS and gpH2R-GsαS (Table 2). The correlation of the pD 2 values of compounds1 to 4 at both H2R-GsαS was highly significant with a slope close to the theoretical curve assuming pharmacological identity of H2R species isoforms (Fig. 5B). Except for BU-E-43 (7), the potencies of guanidines were all significantly lower at hH2R-GsαS than at gpH2R-GsαS. Differences in potency between hH2R-GsαS and gpH2R-GsαS were particularly large for BU-E-75 (11) (6.6-fold), BU-E-42 (6) (6.0-fold), and IMP (5) (5.0-fold). The slope of the correlation between the pD 2 values of guanidines at hH2R-GsαS and gpH2R-GsαS was 0.89, and the curve nearly paralleled the theoretical curve assuming pharmacological identity of H2R species isoforms (Fig. 6B), indicating a constant contribution of the guanidinoalkylaryl moiety to the ligand/GPCR interaction difference between hH2R and gpH2R. Notably, agonist potency decreased almost 3-fold at gpH2R-GsαS by elongation of the alkyl chain between the guanidino group and the phenyl ring (6 → 7) (Fig. 1), but slightly increased at hH2R-GsαS. These data indicate that the guanidine binding pocket in gpH2R is smaller or less flexible than in hH2R. Taken together, guanidines stabilize an active conformation in gpH2R not only more efficiently but also more potently than in hH2R, and the structure-activity relationships for guanidines at hH2R and gpH2R are slightly different.
To further corroborate the concept of species-specific H2R conformations stabilized by guanidines, we competed [3H]TIO binding to H2R-GsαS fusion proteins with unlabeled guanidines. In the absence of guanine nucleotides, agonist, GPCR, and G-protein form a ternary complex that is characterized by high agonist affinity (Seifert et al., 1998a). Typically, ternary complex formation is not complete; i.e., a certain fraction of GPCRs display low agonist-affinity (Seifert et al., 1998a). Consequently, agonist-competition curves are biphasic. GTPγS reduces agonist affinity, presumably reflecting ternary complex dissociation. However, in some cases, high-affinity agonist binding is GTPγS-insensitive, indicative of tight GPCR/G-protein coupling (Seifert et al., 1998a).
Fig. 7 shows the [3H]TIO competition curves with IMP (5), ARP (8), and BU-E-48 (10) in membranes expressing hH2R-GsαS and gpH2R-GsαS in the absence and presence of GTPγS, and Table 3provides a summary of the nonlinear regression analysis. TheK l-values of guanidines 5,8, and 10 at hH2R-GsαS were all higher than at gpH2R-GsαS. AllK l values were much more similar to the corresponding EC50 in the GTPase assay than theK h values (Tables 2 and 3). These data suggest that H2Rs in a conformation with low affinity for guanidines can efficiently mediate GDP/GTP exchange. An explanation for the moderate divergence betweenK l and EC50 values could be that individual guanidines may interact differently with [3H]TIO-bound and ligand-free H2R. Dissociations between agonist-affinities in binding assays and agonist potencies in functional assays have been observed for several GPCRs (Wenzel-Seifert et al., 1999; Seifert et al., 2001), and the reader is referred to these articles and references cited therein for a detailed discussion on this topic.

Competition of [3H]TIO binding by guanidines in Sf9 membranes expressing hH2R-GsαS and gpH2R-GsαS. [3H]TIO binding in Sf9 membranes was performed as described under Experimental Procedures. Reaction mixtures contained membranes expressing fusion proteins, 10 nM [3H]TIO and agonists at the concentrations indicated on the abscissa. Reaction mixtures additionally contained distilled water (control) or GTPγS (10 μM). A and B, IMP (5); C and D, ARP (8); E and F, BU-E-48 (10). Data were analyzed for best fit to monophasic or biphasic competition curves (F test). Data shown are the means ± S.D. of three to five experiments performed in duplicate.
Binding properties of guanidines at hH2R-GsαS and gpH2R-GsαS expressed in Sf9 cell membranes
At hH2R-GsαS and gpH2R-GsαS the IMP-competition curves in the absence of GTPγS were monophasic (Fig.7, A and B). GTPγS shifted the IMP-competition curves at both fusion proteins to the right, indicating that despite our inability to resolve high- and low-affinity binding components, IMP still formed a ternary complex with H2R-GsαS. Intriguingly, with gpH2R-GsαS, the IMP-competition curve in the presence of GTPγS was biphasic, suggesting partial stability of the ternary complex of IMP-liganded gpH2R with GTPγS-liganded GsαS. The ternary complex in the hH2R-GsαS system seems to be less stable as indicated by the monophasic IMP-competition curve in the presence of GTPγS. ARP was highly efficient at stabilizing the ternary complex in both H2R-GsαS fusion proteins (Fig. 7, C and D). In hH2R-GsαS, GTPγS abolished ternary complex formation with ARP, as reflected by the monophasic and strongly rightward-shifted agonist competition curve (Fig. 7C). In marked contrast, at gpH2R-GsαS, GTPγS had only a small effect on the ARP-competition curve (Fig. 7D), indicating high stability of the ternary complex in the presence of GTPγS-liganded GsαS. Similar to ARP, the binding of BU-E-48 (10) at hH2R-GsαS was highly GTPγS-sensitive, and at gpH2R-GsαS it was largely GTPγS-insensitive (Fig. 7, E and F). Collectively, these data indicate that the conformations of gpH2R stabilized by guanidines interact more tightly with GsαS than the corresponding conformations of hH2R. Because of this different GPCR/G-protein interaction, guanidines promote steady-state GDP/GTP exchange through gpH2R more efficiently than through hH2R (Fig. 6 and Table 2).
Molecular Analysis of Guanidine/H2R Interactions.
The crystal structure of bovine rhodopsin (Palczewski et al., 2000) has improved the reliability of GPCR models with bound ligands. To elucidate the structural basis for the differences in interactions of guanidines with H2R species isoforms, we built three-dimensional models of the seven TM helices of gpH2R starting from the PDB rhodopsin file 1f88and using the alignment with the β2AR (Palczewski et al., 2000). Additionally, our previous 3D QSAR data obtained by comparative molecular field analysis, correlating pD 2 values of guanidines at gpH2R-atrium with electrostatic and steric field variables, were considered (Dove and Buschauer, 1998, 1999), in particular for defining the conformation and superposition of structures.
Figs. 8 and9 show the putative binding of IMP (5) and ARP (8), respectively, to gpH2R. Presumably, the imidazolylpropylguanidine moiety binds to H2R like HIS (1). Studies with H2R mutants proved an ionic interaction of the protonated amino group with Asp-98 (TM3) (see also Fig. 2) (Gantz et al., 1992). The second and third site of the widely accepted three-point model for biogenic amine/GPCR interaction could principally be formed by the couples Asp-186/Thr-190 (Gantz et al., 1992) or Tyr-182/Asp-186 in TM5 (Nederkoorn et al., 1996). Based on a pure α-helical TM5, the proposed two hydrogen bonds of the imidazole ring with H2R are only possible with Tyr-182 and Asp-186. This assumption is also in agreement with a pH-dependent model of H2R activation that suggests tautomerization of the imidazole into the Nπ-H form caused by neutralization of HIS upon binding and accompanied by proton transfers from Tyr-182 to Nπ and from Nτ to Asp-186, respectively (Giraldo, 1999). Interactions of nontautomeric agonists with H2R are compatible with this model, too. Asn-293 of the β2AR (Wieland et al., 1996) and Phe-436 of the H1R (Wieland et al., 1999) have been suggested to interact with the β-OH group of epinephrine and with the imidazolylethyl side chain of HIS, respectively. The corresponding residue in TM6 of the H2R, Phe-254, is near imidazolylpropyl side chain only if agonists do not deeply penetrate into the GPCR core. The selected orientation of the guanidines in Figs.8 and 9, therefore, is in agreement with our present knowledge on biogenic amine/GPCR interactions.

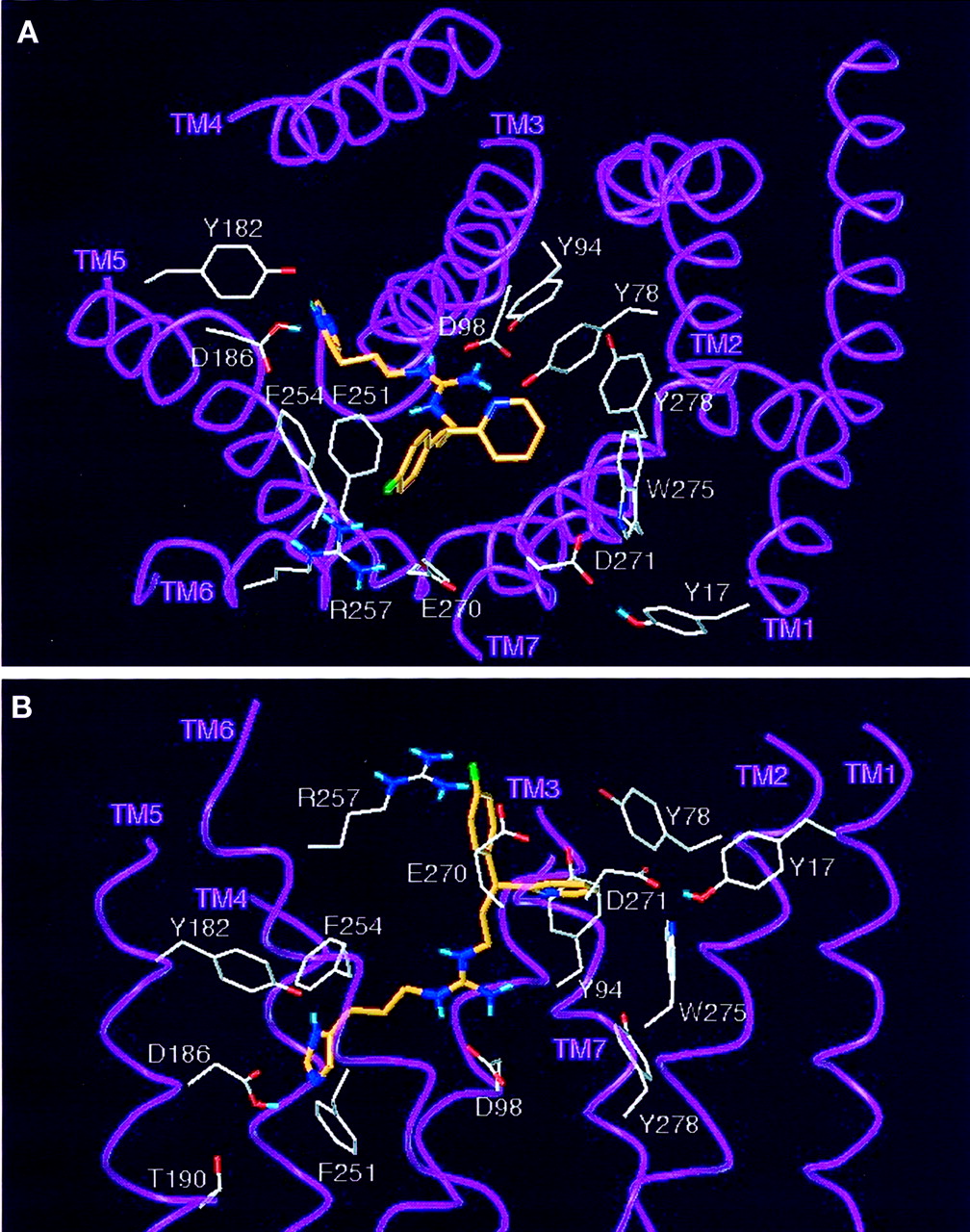
Model of the interaction of IMP with the gpH2R. The carbon atoms of IMP (5) are shown in green. Side chains and α-carbon atoms of the putative agonist-binding site and at the top of TM1 and TM7 are drawn. A, view into the GPCR core; B, lateral view. The imidazolylpropylguanidine moiety interacts with Asp-98 in TM3 (Gantz et al., 1992) and the Tyr-182/Asp-186 pair in TM5 (Nederkoorn et al., 1996). Asn-293 of the β2AR (Wieland et al., 1996) and Phe-436 of the H1R (Wieland et al., 1999) have been suggested to interact with the β-OH group of epinephrine and with the imidazolylethyl side chain of HIS, respectively. The corresponding residue of the H2R, Phe-254, is close to the imidazolylpropyl side chain only if agonists do not deeply penetrate into the GPCR core. Major differences in amino acid sequence between hH2R and gpH2R occur on the top of TM1 and TM7. The gpH2R model proposes H-bonds of Asp-271 (hH2R: Ala-271) in TM7 to the Nτ hydrogen of the 5-methyl-1H-imidazol-4-yl group of IMP and to the OH group of Tyr-17 (hH2R: Cys-17) in TM1. Because of the different amino acid substitutions in hH2R, these H-bonds are not possible in this GPCR.

Model of the interaction of ARP with the gpH2R. The carbon atoms of ARP (8) are shown in orange. Side chains and α-carbon atoms of the putative agonist-binding site and at the top of TM1 and TM7 are drawn. A, view into the GPCR core; B, lateral view. For description of the binding of the imidazolylpropyl moiety, see Fig. 8. Asp-271 (hH2R: Ala-271) in TM7 again forms an H-bond to the OH group of Tyr-17 (hH2R: Cys-17) in TM1. For most favorable interaction with the H2R, the pyridyl- and the phenyl group must be arranged as shown. Attempts to roughly exchange the position of both rings by rotation of the propyl side chain bonds either resulted in high-energy conformations or in collision with the helices. Significantly different QSAR with respect to substituents at the phenyl ring of derivatives of ARP (8) and of unbranched phenyl as well as benzylthioalkyl compounds, respectively, suggest that the aryl or heteroaryl ring of structures such as IMP must be superimposed with the pyridyl ring of the pyridylphenylpropyl analogs. In particular, bulk is unfavorable for potency around the “pyridyl site” and allowed in metaand para position of the “phenyl site”. This agrees with a model where the latter site points outside the GPCR core. The suggested binding mode of the “pyridyl site” of ARP derivatives further considers that 2- and 3-pyridylphenylpropyl structures are on average 0.3 to 0.4 pD 2 units more potent than the corresponding diphenyl analogs. The model in Fig. 9 assumes the pyridyl group of ARP to be surrounded by a pocket of the aromatic residues Tyr-78, Tyr-94, Trp-275, and Tyr-278, where ligand bulk is restricted. Corresponding to Tyr-78, His-93 of the β2AR is probably involved in the binding of thep-methoxy group of formoterol (Isogaya et al., 1999). One of these hydrogen-donating aromatic residues might form an H-bond with the pyridyl nitrogen of ARP, but the model rather suggests an ion-dipole interaction of Asp-271 with a positively charged region of the pyridyl group. Studies with β2AR mutants (Isogaya et al., 1999) point to an interaction of Tyr-308, corresponding to Glu-270 in H2Rs, with the arylalkyl side chain of formoterol. In the gpH2R model, a salt bridge between Glu-270 and Arg-257 (TM6) positions both side chains for additional interaction with the phenyl moiety of ARP. In agreement with our 3D QSAR studies, indicating favorable effects of negatively chargedmeta and para substituents, ion-dipole interactions or, as anticipated in the case of ARP, a F—HN H-bond with Arg-257 could be possible. The field effect of Arg-257 might additionally amplify interactions of Glu-270 with positively charged phenyl regions.
Disregarding sequence differences deeply within the GPCR core, amino acid exchanges between hH2R and gpH2R occur only on the top of TM1 and TM7. Direct IMP- and ARP-TM1 interaction is impossible. However, the minimized gpH2R models consistently result in an interhelical TM1–TM7 H-bond between Tyr-17 (hH2R: Cys-17) and Asp-271. In TM7, there are three amino acid differences between hH2R and gpH2R (Leu-269 → Phe-269, Ala-271 → Asp-271, and Ile-272 → Val-272) (Fig. 2). With our rhodopsin-based alignment of TM7, only Asp-271 is capable of directly participating in ligand binding. Intriguingly, the Ala-271→Asp-271 switch is the only nonconserved amino acid exchange between hH2R and gpH2R in TM7. The model in Fig. 8 suggests that the preference of IMP for gpH2R relative to hH2R is caused by an H-bond of the NτH function to Asp-271. For the pyridyl moiety of ARP, an ion-dipole interaction with Asp-271 is possible (Fig. 9) in agreement with our 3D QSAR results (Dove and Buschauer, 1998, 1999), indicating that a positive charge in the pyridyl region increases potency at gpH2R. Ala-271 in hH2R cannot take part in these H-bond and ion-dipole interactions.
To test the predictions of the 3D QSAR and computer modeling studies, we constructed hH2R-A271D-GsαS in which Ala-271 is exchanged against Asp-271 (Fig.10A). Additionally, we took advantage of a KpnI site present in the cDNA of both hH2R and gpH2R (Fig. 2).KpnI cleaves the cDNAs of hH2R and gpH2R at the same position localized in the middle of the second intracellular loop (Fig. 2). Thus, we could readily construct chimeras in which the N- and C-terminal portions of hH2R and gpH2R were exchanged against each other (Fig. 10A). NhCgpH2R-Gsα bears Cys-17 and Asp-271, and NgpChH2R-Gsα bears Tyr-17 and Ala-271. The chimeras could be used 1) to study the role of Asp-271/Ala-271 in the interaction with guanidines and 2) to study the importance of the suggested H-bond between Tyr-17 and Asp-271 for the effects of guanidines at gpH2R. In hH2R-Gsα, both chimeras and hH2R-A271D-Gsα this interaction cannot take place anymore because either of the two amino acids is replaced (Fig. 1).
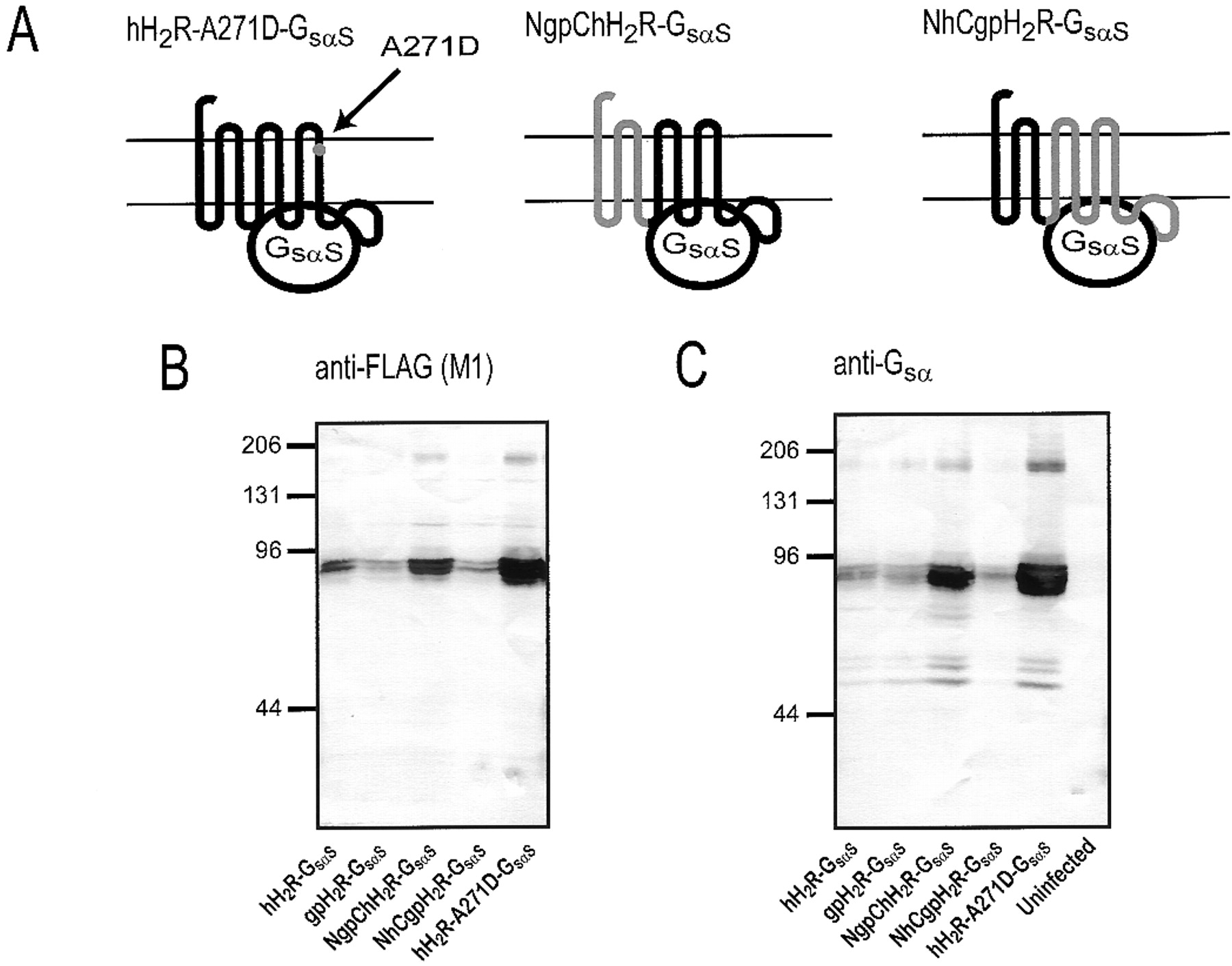
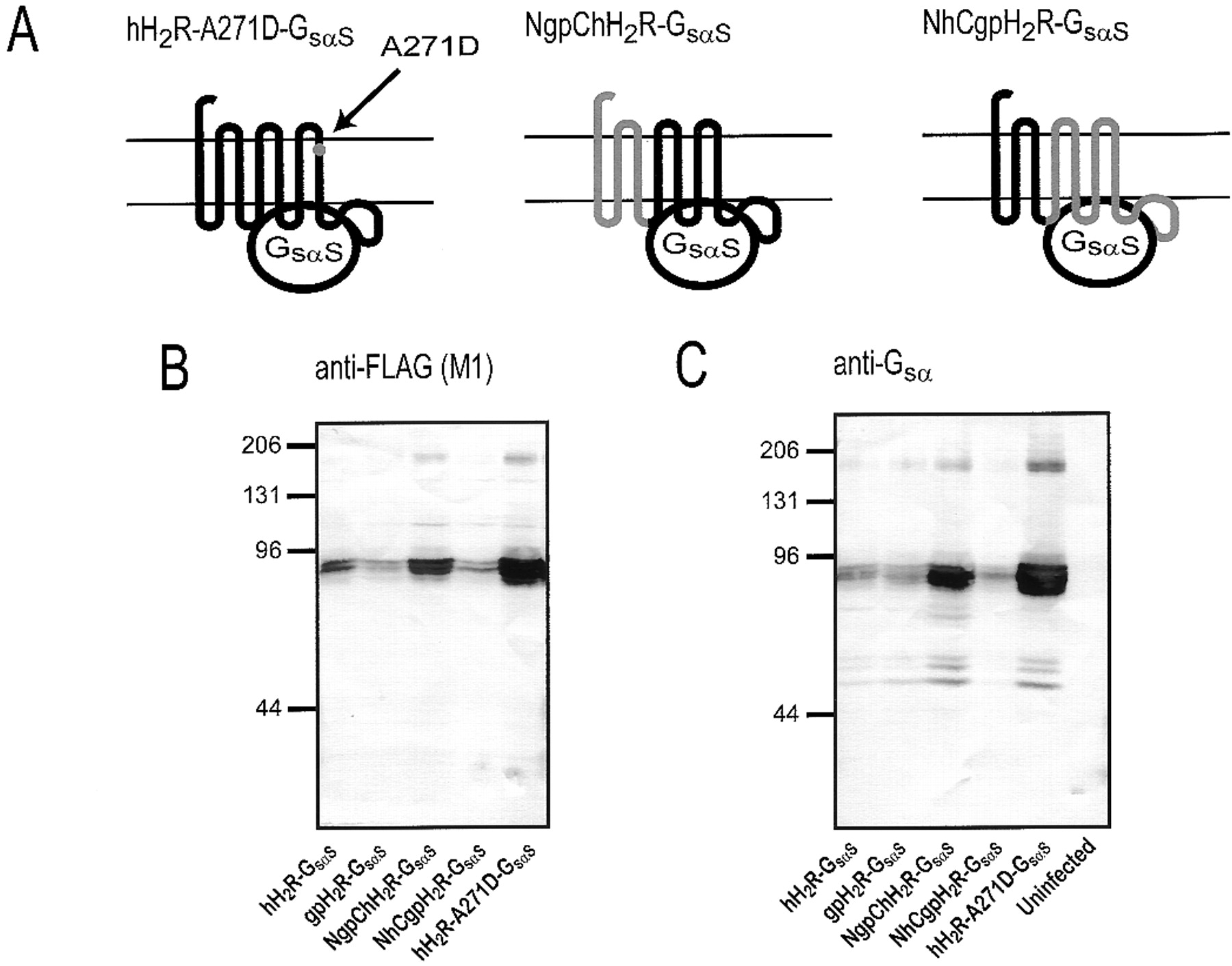
Analysis of the expression of hH2R-A271D-GsαS, NgpChH2R-Gsα and NhCgpH2R-Gsα fusion proteins in Sf9 cell membranes. A, schematic structures of hH2R-A271D-GsαS, NgpChH2R-Gsα and NhCgpH2R-Gsα fusion proteins. For precise location of the KpnI site used for construction of chimeras, see Fig. 2. Sf9 cell membranes expressing various H2R-Gsα fusion proteins or membranes from uninfected Sf9 cells were separated by SDS-PAGE using a gel that contained 8% (w/v) acrylamide. Fusion proteins were probed with the anti-FLAG Ig (M1 antibody) (B) or the anti-Gsα Ig (C). For each membrane preparation, 100 μg of protein was applied to each lane. Numbers on the left of the immunoblots indicate molecular masses of marker proteins. Shown are the horseradish peroxidase-reacted Immobilon P membranes of representative gels. Similar results were obtained with two other membrane preparations of hH2R-A271D-GsαS, NgpChH2R-Gsα, and NhCgpH2R-Gsα fusion proteins.
Immunoblots with the anti-FLAG Ig showed that hH2R-A271D-GsαS, NgpChH2R-GsαS, and NhCgpH2R-GsαS, like hH2R-GsαS and gpH2R-GsαS, had a molecular mass of ∼78 kDa (Fig. 10B). The expression levels of the membrane preparations shown in Fig. 10 were quite different, but this does not reflect intrinsic differences in the expression levels of constructs. Specifically, Fig. 3 contains immunoblots in which hH2R-GsαS and gpH2R-GsαS were expressed at higher levels than in those displayed in Fig. 10, and similar data were obtained for NhCgpH2R-GsαS (data not shown). Membranes with different expression levels were intentionally chosen for the immunoblot in Fig. 10 because certain features, namely the double bands of H2R-Gsα fusion proteins corresponding to differently glycosylated species (Liu et al., 2001), become much more obvious with constructs at low expression levels. The immunoblot with the anti-FLAG Ig also shows the dimers of NgpChH2R-GsαS and hH2R-A271D-GsαS migrating at ∼160 kDa. The immunoblot with anti-Gsα Ig confirmed that H2R-Gsαfusion proteins migrate as a monomer of ∼78 kDa and a dimer of ∼160 kDa (Fig. 10C). Three additional bands at ∼50 to 60 kDa not identified with the anti-FLAG Ig are seen in the immunoblot with anti-Gsα. They do not correspond to the endogenous Gsα-like G-protein of insect cells because the anti-Gsα Ig did not recognize an antigen in membranes from uninfected Sf9 cells (Fig. 10C). The bands also do not represent degradation products since the immunoblot with the anti-FLAG Ig did not provide an indication (compare also with the partial degradation of β2AR-GsαL shown in Fig.3). Rather, we assume that the ∼50- to 60-kDa proteins are atypically migrating H2R-Gsα fusion proteins that, because of specific N-terminal glycosylation patterns, are recognized only by the C-terminally reacting anti-Gsα Ig but not with the N-terminally reacting anti-FLAG Ig. Glycosylation-dependent reactions of fusion proteins with anti-FLAG Ig and anti-Gsα Ig and atypical migrations of GPCRs in SDS-PAGE have been observed earlier (Grünewald et al., 1996; Liu et al., 2001). Collectively, the immunoblots with the anti-FLAG Ig and anti-GsαIg show that hH2R-A271D-GsαS, NgpChH2R-GsαS, and NhCgpH2R-GsαS are expressed in Sf9 cell membranes and that the electrophoretic mobility of those fusion proteins is similar to the mobility of fusion proteins containing wild-type H2Rs.
Table 4 summarizes the potencies and efficacies of HIS (1) and guanidines 5 to8, 10, 11, and 13 at the GTPase of hH2R-A271D-GsαS, NgpChH2R-GsαS, and NhCgpH2R-GsαS. Figure 6, C-H, shows correlations of the efficacies and potencies, respectively of guanidines at hH2R-A271D-GsαS, NgpChH2R-GsαS and NhCgpH2R-GsαS, respectively, versus gpH2R-GsαS and allow direct comparison with the properties of hH2R-GsαS versus gpH2R-GsαS (Fig. 6, A and B). Most strikingly, the correlation between the pD 2 values of guanidines at hH2R-A271D-GsαS and gpH2R-GsαS almost exactly followed the theoretical curve assuming pharmacological identity of the two fusion proteins (Fig. 6D). Thus, the Ala-271 → Asp-271 mutation increased the potency of hH2R for guanidines to the level of gpH2R (compare Fig. 6, B and D, and Tables 2 and 4). These findings confirm the results of the 3D QSAR (Dove and Buschauer, 1998, 1999) and modeling studies (Figs. 8 and 9) suggesting that the potency of guanidines is increased by ion-dipole or H-bond interactions with Asp-271. Such interactions cannot occur with Ala-271 in hH2R, which explains why at hH2R guanidines exhibit substantially lower potencies than at gpH2R (compare Fig. 6, B and D).
Agonist efficacies and potencies at hH2R-A271D-GsαS, NgpChH2R-GsαS, and NhCgpH2R-GsαSexpressed in Sf9 cell membranes
The chimera NgpChH2R-GsαScontains Ala-271 (Figs. 1 and 10A). As expected, the potencies of guanidines at NgpChH2R-GsαS did not approach the theoretical curve assuming pharmacological identity of NgpChH2R-GsαS and gpH2R-GsαS (Fig. 6F). Like hH2R-A271D-GsαS, NhCgpH2R-GsαS contains Asp-271. Thus, the potencies of guanidines at NhCgpH2R-GsαS were expected to be higher than at hH2R-GsαS and similar to the potencies at hH2R-A271D-GsαS and gpH2R-GsαS. Overall, the data fulfilled the predictions (Tables 2 and 4). As a result of the increase in guanidine potency at NhCgpH2R-GsαS the correlation between the pD 2 values at NhCgpH2R-GsαS versus gpH2R-GsαS approached the theoretical function assuming pharmacological identity (compare Fig. 6, B and H), but not as close as in the case of hH2R-A271D-GsαS versus gpH2R-GsαS (compare Figs.6, D and H). Such slightly reduced potencies were not unexpected, because in NhCgpH2R-GsαS, there is not only the Ala-271 → Asp-271 exchange, but also 32 additional amino acid exchanges in various regions of the C-terminal half of the GPCR (Fig. 2). These exchanges can have multiple effects on the arrangements of TM domains and thereby partially annihilate the effect of the Ala-271 → Asp-271 exchange on guanidine potency.
Regarding the properties of some specific agonists, it becomes obvious that elongation of the alkyl chain between the guanidino group and the phenyl ring (6 → 7) (Fig. 1) decreased agonist potency at hH2R-A271D-GsαS and NhCgpH2R-GsαS by ∼4-fold (Table 4). This decrease in potency is similar to that observed for compounds 6→7 at gpH2R-GsαS (Table 2). Conversely, at NgpChH2R-GsαS and hH2R-GsαS the longer alkyl chain slightly increased agonist potency (Tables 2 and 4). These data indicate that the amino acid at position 271 of H2R affects the size and flexibility of the guanidine binding pocket. With Ala-271, the binding pocket is wider and more flexible and accommodates the longer (7) as well as the shorter guanidine (6). In contrast, with Asp-271, the fit of the longer guanidine (7) must be probably enforced by conformational strain.
Among all guanidines studied, the amino acid substitution at position 271 had the greatest and most consistent impact on the potency of IMP (5). Specifically, with Asp-271 the potencies of IMP were consistently ∼5- to 7-fold higher than with Ala-271, regardless of whether fusion proteins with wild-type H2Rs, mutated H2R, or chimeric H2Rs were considered (Tables 2 and 4). For other guanidines, the impact of the amino acid substitution at position 271 was less consistent. These data indicate that the binding of IMP to H2R is considerably more dependent on interaction with Asp-271 than the binding of other guanidines to H2R. Major structural features of IMP (5) compared with the other guanidines studied (6–13) are the possibility of a H-bond to Asp-271 (see Fig. 8) and the absence of a phenyl branch (Fig. 1). In the case of the guanidines 6–13, the weaker ion-dipole interaction with Asp-271 may be compensated by electrostatic interactions of the substituted phenyl group with Arg-257 and Glu-270 and by better fit of the pyridyl moiety into a pocket of aromatic residues (see Fig. 9).
Another striking result of the studies with hH2R-A271-D-GsαS, NgpChH2R-GsαS, and NhCgpH2R-GsαS was that the correlations of the efficacies of guanidines at the aforementioned constructs versus gpH2R-GsαS remained almost unchanged (Fig. 6, A, C, E, and G), indicating that neither Asp-271 nor all 33 amino acids specific for the C-terminal half of gpH2R restored high efficacy of guanidines observed for gpH2R-GsαS. These data demonstrate that guanidine potency and efficacy are independent H2R properties. Intriguingly, the models in Figs. 8 and 9 point to the existence of a H-bond between Tyr-17 and Asp-271, a couple of residues present only in gpH2R-GsαS. Thus, our modeling and experimental data suggest that the Tyr-Asp H-bond stabilizes the agonistic conformation of the gpH2R bound to guanidines.
It was also surprising that HIS is 2- to 3.5-fold more potent at hH2R-A271D-GsαS and NhCgpH2R-GsαS (both with Cys-17 and Asp-271) than at gpH2R-GsαS (Tables 2 and4). TM1 and TM7 do not belong to the binding site of HIS, although it cannot be ruled out that an unfixed Asp side chain in a more flexible TM7 can slightly improve the association kinetics of small amines by “dynamic escorting”.
Conclusions
In previous studies we observed that several guanidine-type H2R agonists are less potent and/or less efficient at the H2R of human neutrophils than at the H2R of the guinea pig atrium (Burde et al., 1989, 1990; Buschauer, 1989). Taking advantage of the cloned hH2R and gpH2R (Gantz et al., 1991; Traiffort et al., 1995) and the GPCR-Gα fusion protein technique (Seifert et al., 1999; Milligan, 2000) we were able to analyze the coupling of hH2R and gpH2R to GsαS under identical experimental conditions. Using this approach, we have dissected pharmacological differences between hH2R and gpH2R with respect to the inverse agonist efficacies of RAN (16) and APT (19) and the agonist potencies and efficacies of guanidines 5–13. Thus, our present data clearly show that hH2R and gpH2R possess, indeed, different pharmacological properties.
Guanidines stabilize an active conformation in gpH2R more efficiently and potently than in hH2R. Site-directed mutagenesis studies and analysis of chimeric hH2R/gpH2R receptors confirmed computer modeling proposing that Asp-271 accounts for the high potency of guanidines. The gpH2R model also suggests that high guanidine efficacy depends on an H-bond between Asp-271 and Tyr-17 that is not possible in any other H2R-Gsα construct studied. Thus, our data show that hH2R and gpH2R selectively interact with a single class of synthetic agonists, that high guanidine potency critically depends on interaction with a single single amino acid, and that agonist potency and efficacy are regulated independently of each other. The inverse order of potency of compounds 6 and 7 at hH2R and gpH2R, respectively, indicates that it is possible to develop guanidines with potency at hH2R. Such compounds may be useful for treating cardiac failure, acute myelogenous leukemia, and inflammatory diseases.
Our present study adds the H2R to the growing list of GPCR species isoforms that interact similarly with their endogenous ligand, but quite differently with synthetic ligands (Kopin et al., 2000; Ligneau et al., 2000; Lovenberg et al., 2000). Many GPCR species isoforms, including the H3R, differ from each other primarily in antagonist pharmacology (Kopin et al., 2000; Ligneau et al., 2000; Lovenberg et al., 2000). From a historical perspective, it is very fortunate that the pharmacological differences between hH2R and gpH2R mainly concern agonists and not antagonists (Fig. 5). Otherwise, it would have been much more difficult if not impossible to develop potent H2R antagonists for treatment of gastroduodenal ulcer disease in man relying on animal models at a time when recombinant hH2R had not yet been available. However, for future development of potent and selective H2R agonists it will be crucial to analyze hH2R and not gpH2R.
Acknowledgments
We thank Dr. I. Gantz (Ann Arbor, MI) for providing the cDNA of hH2R, Drs. E. Traiffort and J.-C. Schwartz (Paris, France) for providing the cDNA of gpH2R and Dr. F. Schalkhausser (Department of Pharmacy, University of Regensburg, Germany) for the synthesis of guanidine 13. We also thank Dr. U. Gether (Department of Medical Physiology, The Panum Institute, Copenhagen, Denmark) for stimulating discussions. Finally, we thank the reviewers for their very helpful critique and suggestions.
Footnotes
- Received March 16, 2001.
- Accepted July 3, 2001.
-
↵1 Present address: Quintiles Inc., Kansas City, Missouri.
-
This work was supported by a New Faculty Award of The University of Kansas (R.S.), the National Institutes of Health COBRE award 1-P20-RR15563, matching support from the State of Kansas and the University of Kansas (R.S.), grants of the Fonds der Chemischen Industrie (A.B.), and the Deutscher Akademischer Austauschdienst within the international network “Medicinal Chemistry” (A.B.). M.T.K. and T.B. contributed equally to this work.
Abbreviations
- HIS
- histamine
- AMT
- amthamine
- DIM
- dimaprit
- BET
- betahistine
- GPCR
- G-protein-coupled receptor
- HxR
- histamine Hx-receptor, where x is 1, 2, 3, or 4
- IMP
- impromidine
- ARP
- arpromidine
- CIM
- cimetidine
- RAN
- ranitidine
- FAM
- famotidine
- TIO
- tiotidine
- ZOL
- zolantidine
- APT
- aminopotentidine
- h
- human
- gp
- guinea pig
- Gs-proteins
- family of G-proteins that mediates adenylyl cyclase activation
- GsαS
- short splice variant of the Gs-protein Gsα
- GsαL
- long splice variant of the Gs-protein Gsα
- β2AR
- β2-adrenoceptor
- β2AR-GsαL
- fusion protein containing the β2AR and the long splice variant of Gsα
- DHA
- dihydroalprenolol
- PCR
- polymerase chain reaction
- bp
- base pair(s)
- 3D QSAR
- three-dimensional quantitative structure-activity relationship
- gpH2R-GsαS
- fusion protein of the guinea pig histamine H2-receptor and the short splice variant of Gsα
- hH2R-A271D-GsαS
- fusion protein of the human histamine H2-receptor bearing an Ala→Asp mutation at position 271 and the short splice variant of Gsα
- hH2R-GsαS
- fusion protein of the human histamine H2-receptor and the short splice variant of Gsα
- NhCgpH2R-GsαS
- fusion protein consisting of the N-terminal half of the human histamine H2-receptor, the C-terminal half of the guinea pig histamine H2-receptor, and the short splice variant of Gsα
- NgpChH2R-GsαS
- fusion protein consisting of the N-terminal half of the guinea pig histamine H2-receptor, the C-terminal half of the human histamine H2-receptor, and the short splice variant of Gsα
- TM
- transmembrane domain of a G-protein-coupled receptor
- PAGE
- polyacrylamide gel electrophoresis
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}