


Abstract
S23906-1 is a diester derivative of 1,2-dihydrobenzo[b]acronycine with an unknown mechanism of action. This cytotoxic compound was 20-fold more potent than acronycine in inhibiting the proliferation of six tumor cell lines. Using a clonogenic assay of cell survival, the HT29 human colon carcinoma cell line was 100-fold more sensitive to S23906-1 than acronycine. Cell cycle analysis, by flow cytometry, showed thatS23906-1 induced a partially reversible arrest of HT29 cells in G2+M at 1 μM and below and an irreversible arrest in S phase at 2.5 μM and above. These cell cycle effects were followed by cell death through apoptosis, quantified by annexin-V labeling. Inhibition of DNA synthesis was observed by complete prevention of bromodeoxyuridine (BrdU) incorporation after only 4 h of incubation with 5 μM S23906-1. Interestingly, under the same experimental conditions, a significant increase of cyclin E protein level was observed without any modification of cyclins D1, D2, D3, or A. This overexpressed cyclin E protein was not complexed with Cdk2, as shown by western blotting for Cdk2 in immunoprecipitates of cyclin E. Similar inhibition of BrdU incorporation and elevation of cyclin E protein were observed after treatment with cytosine arabinoside, which reversibly inhibited progression into S phase, but not after DNA damage induced by cisplatin. S23906-1 thus has a novel mechanism of action. A cell line resistant to S23906-1 showed that overexpression of cyclin E was implicated in the novel cytotoxic activity of this compound.
Although novel anticancer therapeutic strategies are emerging that are based on targeting genes implicated in the tumor pathology, there is a continued need for cytotoxic agents with novel mechanisms of action. Acronycine, an acridone alkaloid isolated from Acronychia baueri (Hugues et al., 1948), was found to be active in experimental models of murine tumors, including several leukemias, sarcomas, carcinomas, a myeloma, a melanoma (Svoboda et al., 1966), and, more recently, a human breast cancer xenograft (Dorr et al., 1989). On the basis of this broad spectrum of antitumor activity, acronycine was subjected to limited clinical trials, which were inconclusive (Scarffe et al., 1983), possibly because of the very low solubility in aqueous solution and the moderate potency of this drug, both in vitro and in vivo.
To improve the potency and antitumor activity of this interesting alkaloid, we synthesized a new series of diester derivatives of 1,2-dihydroacronycine (Elomri et al., 1996) that were recently optimized by the addition of an aromatic ring fused to the acronycine skeleton (Costes et al., 2000). One of these derivatives, S23906-1, has now been selected on the basis of its potency in vitro and antitumor activity in vivo against the murine C38 colon adenocarcinoma (Costes et al., 2000). It has been further evaluated against human orthotopic models of ovarian, lung, and colon carcinomas, where it has a broad and unusual spectrum of activity (Guilbaud et al., 2001;Pierré et al., 2000).
The precise mechanism of action of acronycine itself is still unknown, although early experiments have suggested DNA binding properties for this alkaloid (Dorr et al., 1989). Its effects on the cell cycle, which reflects its molecular mechanism of action at the cellular level, are not clear in the literature, because accumulation of treated cells in the G1, G2+M, or S+G2+M phases of the cell cycle were reported (Reddy et al., 1977; Shieh et al., 1992). Our cytotoxic derivatives, exemplified by S23906-1, induced a marked accumulation of L1210 murine leukemic cells in the S phase of the cell cycle. Moreover, a good relation was found in the series between cytotoxicity and the ability of these compounds to induce an irreversible accumulation of cells in S phase (Costes et al., 2000). This suggested that cell death might be the consequence of an arrest in S phase.
The aim of the present study was to investigate the mechanism of action, at the molecular and cellular level, of S23906-1. The sensitivity of human tumor cells to S23906-1 and acronycine was assessed by a standard proliferation assay and by the clonogenic assay after a brief exposure of the cells. On the basis of the marked antitumor activity of S23906-1 observed in orthotopic models of colon cancer (Guilbaud et al., 2001), the HT29 cell line was chosen to analyze, by flow cytometry and biochemical analysis, the cell cycle effects and apoptosis induced by S23906-1.
Materials and Methods
Compounds.
Acronycine and S23906-1 were synthesized as described previously (Costes et al., 2000) (Fig.1). Both compounds were solubilized at 10−2 M in dimethyl sulfoxide (DMSO), aliquoted and stored at −20°C. Cytosine arabinoside (Ara-C, 10−1 M in water) was provided by Pharmacia & Upjohn SA (St-Quentin en Yvelines, France), gemcitabine (dFdC, 10−1 M in water) by Lilly (Saint-Cloud, France), cisplatin (CDDP, 10−2 M in water) by Bellon-Aventis (Montrouge, France), methotrexate (MTX, 10−2 M in water) by Wyeth-Lederle (Puteaux, France), and 5-fluorouracil (FUra, 10−1 M in water) by Roche (Neuilly sur Seine, France). All compounds were aliquoted and stored at −20°C. The solutions were thawed only once, just before the experiments. Carmustin (BCNU, 10−1 M in ethanol) was provided by Bristol Myers Squibb (La Défense, France) and was stored at 4°C.

Chemical structures of acronycine and S23906-1.
Cell Lines.
The human cell lines A549 (lung carcinoma), NIH-OVCAR3 (ovary adenocarcinoma), HT29 (colon adenocarcinoma), and MCF7 (breast adenocarcinoma) and the murine cell line L1210 (lymphocytic leukemia), were obtained from the American Type Culture Collection (Manassas, VA).
The human KB-3-1 epidermoid carcinoma (Cornwell et al., 1986) was kindly provided by Dr. M. Gottesman (Bethesda, MD). The KB/S23-500 cells were made resistant to S23906-1 by stepwise exposure to the drug up to 500 nM. This cell line, for which the resistance to S23906-1 was stable (at least 2 months), was not positive for P-glycoprotein overexpression. It was not cross-resistant to intercalating drugs or topoisomerase I or II inhibitors, (data not shown). Cells were maintained in RPMI 1640 medium supplemented with 10% decomplemented fetal calf serum (FCS), 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10 mM HEPES, pH 7.4. Cells were grown at 37°C in 5% CO2/95% air. All media and supplements were from Invitrogen (Cergy-Pontoise, France) except FCS, which was purchased from Sigma Chemical, Co. (St. Louis, MO).
Standard Proliferation Assay.
This assay has been described previously (Pierré et al., 1991; Léonce et al., 1996). Briefly, adherent cells were seeded in 96-well microplates and incubated for 2 days. Compounds were then added and plates were incubated for four doubling times (continuous exposure). For short duration of exposure, HT29 cells were incubated with the drugs for 1 h, then washed and incubated in drug-free medium for an overall duration of four doubling times (96 h). The nonadherent L1210 cells were directly incubated for 48 h with the compounds. At the end of this period, 15 μl of 5 mg/ml 3-(4,5 dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma) were added to each well and the plates were incubated for 4 h at 37°C. The medium was aspirated and the formazan solubilized by 100 μl of DMSO. The IC50 [concentration reducing by 50% the absorbance at 540 nm] was calculated by a linear regression performed on the linear zone of the dose-reponse curve. All the measurements were performed in triplicate.
Clonogenic Assay.
HT29 cells in exponential phase of growth were exposed for 1 h to various concentrations of compounds, then washed, detached with trypsin and seeded at a density of 2.5 × 103 cells/well (six-well plates) in complete culture medium containing 0.4% agarose type VII (Sigma). The plates were incubated for 14 days and the cells were then stained with MTT. The number of colonies and their surfaces were determined with a computer assisted image analyzer (Biocom, France). All the measurements were performed in triplicate. Results are expressed as percentages of colonies with respect to untreated cells, which have a plating efficiency of 22%. The IC50 and IC90, the concentrations that reduced by 50 and 90% the number of colonies, were determined by a linear regression as described above.
Cell Cycle Analysis.
HT29 cells in exponential phase of growth were exposed to S23906-1 for the indicated times, then washed, harvested, fixed by 70% ethanol and incubated for 30 min in PBS containing 100 μg/ml RNase and 50 μg/ml propidium iodide (PI; Sigma). For each sample, 104 cells were analyzed on an Epics XL/MCL flow cytometer (Beckman Coulter, France). Results are expressed as the percentage of cells in each phase of the cell cycle.
Detection of Apoptosis by Annexin-V Labeling.
Cells were exposed to S23906-1 for 24 h, washed and incubated in drug-free medium for further 24 h. Labeling of cells was performed as described previously (Sabatini et al., 2000). Briefly, the culture medium was collected and centrifuged and the cell pellet was pooled with adherent cells harvested by trypsin/EDTA. Cells were washed twice with culture medium, pelleted by centrifugation, and resuspended at a density of 106 cells/ml in culture medium containing 20% FCS. After 1 h at 37°C, cells were washed twice with cold PBS and resuspended in 200 μl of binding buffer (100 μM HEPES, 14 mM NaCl, and 25 μM CaCl2) containing 10 μl of annexin-V-FITC (BD Pharmingen, San Diego, CA) and 10 μg/ml PI. After 15 min at 20°C in the dark, 800 μl of cold binding buffer was added and samples were kept at 4°C before flow cytometric analysis. For each sample, 104 cells were analyzed by flow cytometry. FITC and PI fluorescences were collected through 520- and 630-nm bandpass filters, respectively. Results are displayed as biparametric histograms of annexin-V-FITC and PI fluorescences allowing discrimination between viable cells, apoptotic cells with an intact membrane and cells undergoing secondary necrosis. For each sample, the remaining cells were fixed by 70% ethanol, washed twice with PBS, and incubated for 30 min in PBS containing 100 μg/ml RNase and 50 μg/ml PI. Samples were analyzed again to measure the biparametric detection of annexin-V versus DNA content.
BrdU Incorporation.
HT29 cells were incubated at 37°C with the drugs for the indicated times, washed, and 10 μM BrdU (Sigma) was added for 1 h. Cells were then harvested and fixed by 70% ethanol at 4°C for at least 4 h. Samples were washed twice with PBS and incubated for 30 min in 2N HCl at room temperature, then washed twice with PBS containing 0.5% Tween 20, and incubated for 45 min with 20 μl of anti-BrdU-FITC (BD Pharmingen). After being washed twice with PBS, cells were incubated for 30 min with 100 μg/ml Rnase, 10 μg/ml PI, and analyzed by flow cytometry. Results are displayed as bivariate distribution of BrdU content versus DNA content.
Flow Cytometric Detection of Cyclins.
HT29 cells were drug-treated for the times indicated in Figs. 9 and 10, washed, harvested, and fixed by 70% ethanol at −20°C for at least 1 h (for d-type cyclins, cells were fixed by 1% formaldehyde for 15 min on ice before fixation with ethanol). Samples were washed twice with PBS, and incubated for 5 min in PBS containing 0.5% triton X-100 at 4°C. After 2 washes with PBS, cells were incubated for 2 h at room temperature with 5 μl of anti-cyclin E (clone HE12, Pharmingen), or 20 μl of anti-cyclin D2 (clone G132–43, Pharmingen), or 20 μl of FITC-conjugated anti-cyclin A, D1, or D3 (clone BF-683, G124–326, G107–565, respectively, Pharmingen). For cyclin E and cyclin D2, cells were washed twice and incubated for 1 h with 20 μl of FITC-conjugated goat anti-mouse IgG (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). After being washed twice with PBS, cells were incubated for 30 min with 100 μg/ml RNase, 10 μg/ml PI, and analyzed by flow cytometry. Results are displayed as bivariate linear distribution of cyclin level versus DNA content.

Increase of cyclin E expression induced by S23906-1. HT29 cells were treated for 4 or 24 h with 5 μM S23906-1. A, cells were fixed and labeled with PI and with a FITC-conjugated anti-cyclin E antibody before flow cytometric analysis. In control cells, expression of cyclin E is maximal in late G1 and early S phases and decreases during progression through S. B, immunoprecipitation with anti-cyclin-E and probed either with anti-cyclin E or anti-Cdk2 antibodies; Western blots were performed as described under Materials and Methods.

Western Blot Analysis.
Whole-cell extracts were prepared with extraction buffer (25 mM Tris-HCl, pH 7.5, 25 mM NaCl, 5 mM EDTA, 0.1% Nonidet P-40, 2 mM phenylmethylsulfonyl fluoride, and 2 μg/ml aprotinin). After a brief sonication at 4°C, the protein concentration was determined by the Coomassie Plus protein assay kit (Pierce, Rockford, IL). Similar amounts of proteins were subjected to immunoprecipitation with affinity-purified antibody to cyclin E (Novocastra Laboratories Ltd, Newcastle, UK) coupled to Sepharose. Samples were mixed with 2× Laemmli sample buffer (Bio-Rad Laboratories, Richmond, CA), denatured at 100°C for 3 min, and resolved on 10% SDS-polyacrylamide gels. After gel transfer, anti-cyclin E (clone HE12; BD Pharmingen) or anti-Cdk2 (Santa Cruz Biotechnology) antibodies were added for 1 h at room temperature and membranes were blotted for 1 h at room temperature with an appropriate horseradish peroxidase-linked secondary antibody (Amersham Pharmacia Biotech, Orsay, France). Proteins were visualized with a chemiluminescence assay system (Amersham Pharmacia Biotech).
Results
Inhibition of Cellular Proliferation.
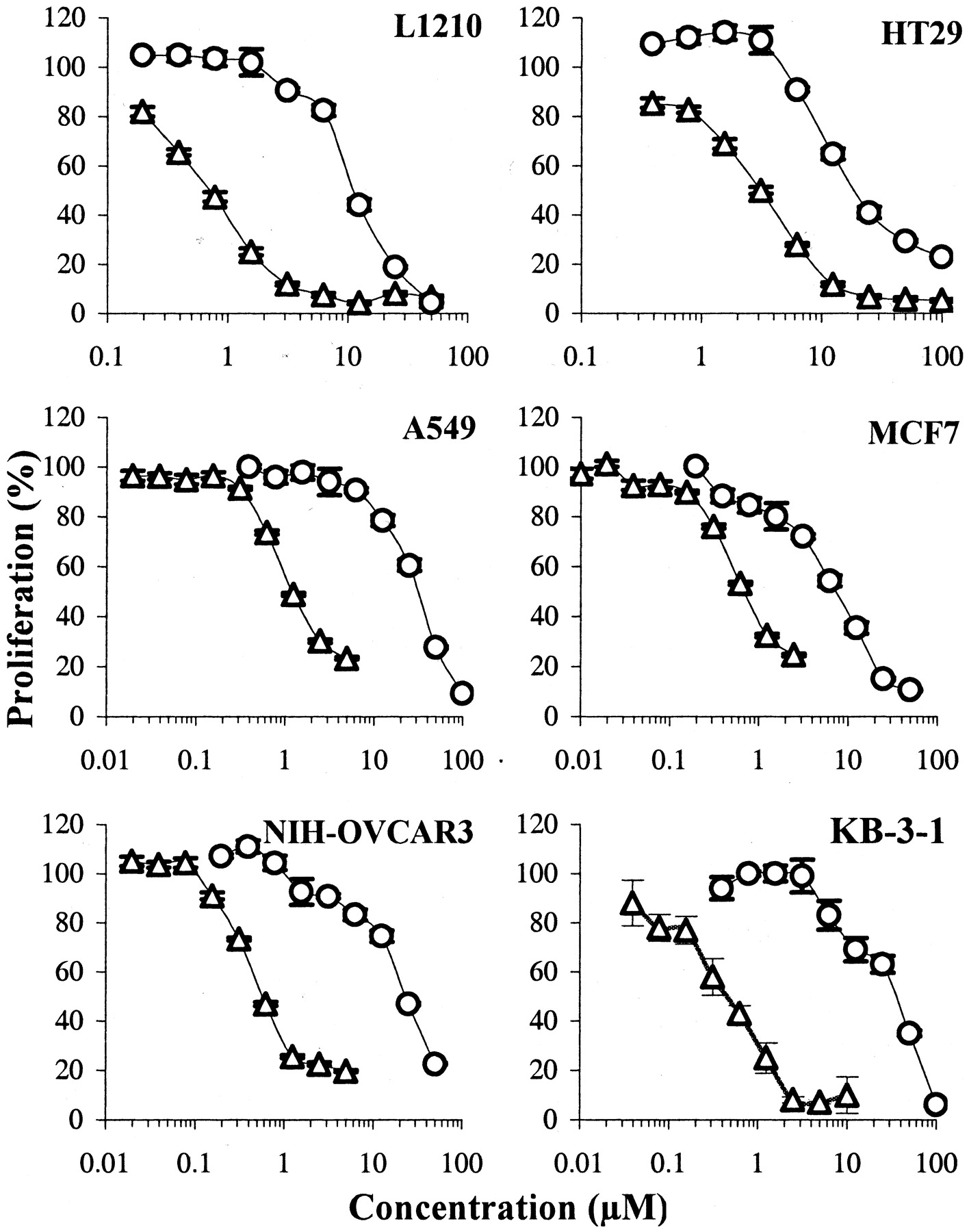
The inhibition of cell proliferation by S23906-1 and acronycine was first measured with the MTT assay. A murine leukemia (L1210) and five human cell lines (A549, NIH-OVCAR3, HT29, MCF7, and KB-3-1) representative of different pathologies and routinely used for the evaluation of antitumor drugs were used. In the first set of experiments, tumor cells were continuously exposed to the compounds. As shown on Fig.2, S23906-1 was significantly more potent than acronycine on the six cell lines tested, with IC50 values ranging from 0.5 to 2.2 μM (mean, 1.23 μM) versus 6.3 to 60.8 μM (mean, 30.1 μM), respectively. The range of S23906-1 IC50 values seems to be relatively narrow, suggesting that cells derived from different histological types could be equally sensitive to S23906-1. Compared with KB-3-1, the KB/S23-500 cells were approximately 13-fold resistant to S23906-1 although they were equally sensitive to acronycine (not shown).

Inhibition of proliferation by S23906-1 (▵) and acronycine (○). Cells were incubated for four doubling times with various concentrations of drugs, and cell viability was determined by the MTT assay. Results are expressed as percentages proliferation with respect to untreated cells (mean ± S.E.M., n= 6).
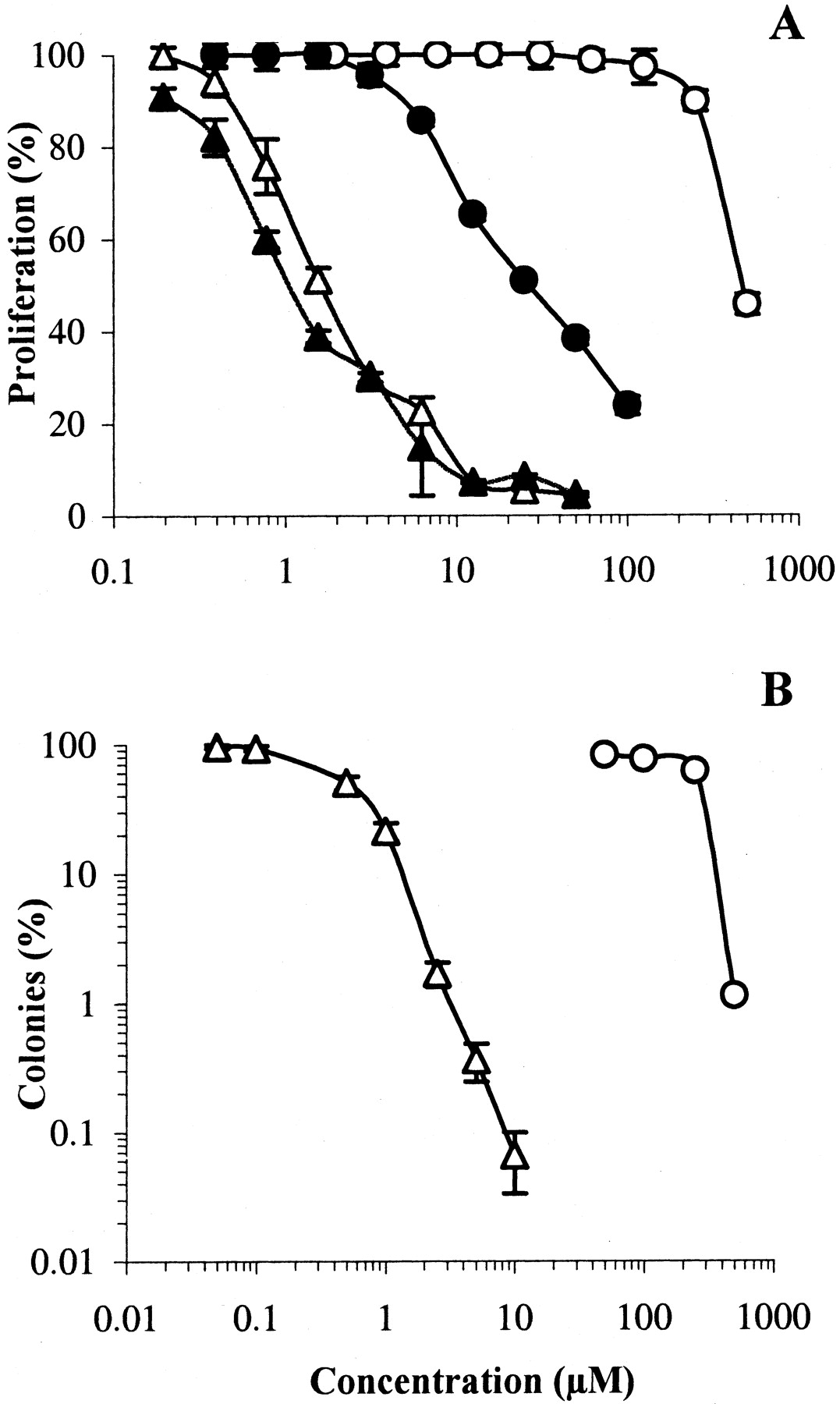
The HT29 cell line was then chosen for a more in-depth evaluation of the cytotoxic properties of S23906-1. The effect of duration of drug exposure was first investigated. Exponentially growing HT29 cells were incubated with S23906-1 or acronycine for 96 h or for 1 h, then washed and incubated in drug-free medium for an overall duration of 96 h. The inhibition of proliferation by S23906-1 was quite similar whatever the duration of exposure (Fig.3A); the IC50values were 1.7 μM (1 h) and 1.1 μM (96 h), in contrast to acronycine, which was 15-fold less potent after 1 h. To confirm the potent and irreversible cytotoxicity of S23906-1, adherent HT29 cells were treated for 1 h with acronycine or S23906-1, then washed and incubated for 14 days in drug-free semisolid medium. Figure3B shows that S23906-1 was about 100-fold more potent (in term of ratio of IC50 values) and markedly more active than acronycine, because more than 99.9% of the cells were killed by 10 μM S23906-1. This result demonstrates that the lesions induced byS23906-1 were completely irreversible, even after 14 days of tridimensional growth in drug-free medium. Interestingly, S23906-1 was approximately 5-fold more potent, after 1 h exposure, in inhibiting the formation of colonies (IC50 = 0.48 μM) than the proliferation of HT29 cells when grown as a monolayer culture (IC50 = 1.7 μM). This unusual observation was confirmed with four different human tumor cell lines (not shown). Because a delayed cell death might have occurred in treated cells (i.e., the cells died after a few mitoses), the surface of colonies was measured. This delayed death seems unlikely, because no aborted colonies were observed and the curves expressing either the surface area or the number of colonies as a function of drug concentration were superimposable (not shown). Anchorage-independent growth is thus more sensitive to the damage induced by S23906-1 than anchorage-dependent growth.
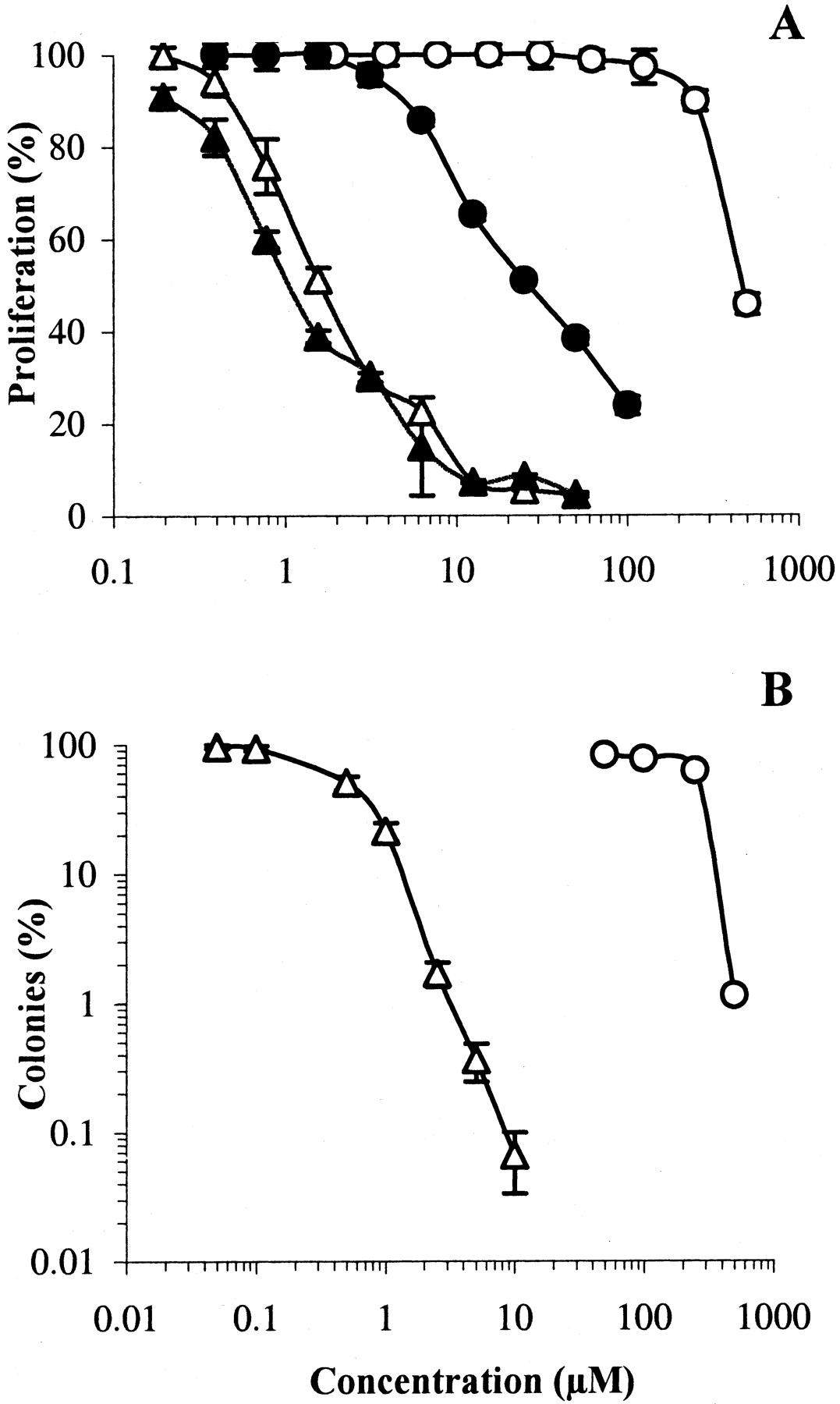
Cytotoxicity of S23906-1 and acronycine. A, proliferating HT29 cells were treated for 1 h and then washed (open symbols) or maintained for 96 h (closed symbols) with various concentrations of S23906-1 (▴, ▵) or acronycine (●, ○); cell viability was determined by the MTT assay. Results are expressed as percentages proliferation with respect to untreated cells (mean ± S.E.M., n = 6). B, proliferating HT29 cells were treated for 1 h with different concentrations of S23906-1 (▵) or acronycine (○), washed and plated for 14 days in agar for colony formation. Results are expressed as percentages of colonies with respect to untreated cells (mean ± S.E.M., n= 3).
Effect on the Cell Cycle.
The analysis of the perturbations of the cell cycle induced by a novel compound not related structurally to existing antitumor drugs gives valuable information on its mechanism of action. We therefore studied the cell cycle of HT29 cells treated for 24 h with S23906-1.
As shown on Fig. 4, the distribution of HT29 cells in the cell cycle was modified differently as the concentration of S23906-1 increased: from 0.1 to 1 μM S23906-1 induced a dose-dependent increase of cells in the G2+M phases whereas 2.5 and 5 μM resulted in the accumulation of more than 70% of cells in the S phase. At 10 μM, cells were arrested at the G1-S boundary. Under the same experimental conditions, no significant perturbation of the cell cycle could be detected with 50 μM acronycine (not shown). Because of the cytotoxicity of DMSO at concentrations above 0.5% and the very low solubility of acronycine, it was impossible to investigate the effects of higher concentrations on the cell cycle.

The kinetics of the effects of S23906-1 on the cell cycle was then studied at 1 and 5 μM, concentrations for which cells accumulated in G2+M and S phases, respectively. The percentages of HT29 cells in each phase of the cell cycle, as a function of time of contact, are shown in Fig. 5. From 4 h, cells treated with 1 μM S23906-1 progressed from G1 to S (Fig. 5A), with the maximum of S-phase cells being reached at 14 h (60%). From 14 to 24 h, the percentage of cells in S decreased (60 to 26%) and cells moved to G2+M (68%) where they arrested for up to 32 h. Thus S23906-1, at 1 μM, delayed but did not inhibit the progression of cells from S to G2+M.

Kinetics of cell cycle perturbations induced byS23906-1. Cells were incubated for the indicated times with 1 μM (A) or 5 μM (B) S23906-1 then fixed and labeled with PI before flow cytometric analysis. After 24 h incubation with 1 μM (C) or 5 μM (D) S23906-1, cells were washed and incubated in drug-free medium for 4, 6, 8, 10, and 24 h, before flow cytometric analysis.
At 5 μM and from 14 h, S23906-1 increased the number of cells in S phase. This was associated with a simultaneous decrease of cells in G1 phase (Fig. 5B). The highest number of cells in S phase (73%) was reached after 24 h and was maintained for up to 32 h. At 5 μM, the progress from S to G2+M and from G2+M to G1 was completely blocked.
To determine whether the accumulation of cells in G2+M or S phases was reversible, HT29 cells previously incubated for 24 h with 1 or 5 μM S23906-1 were washed and incubated in drug-free medium for 4 to 24 h. At 5 μMS23906-1, the percentages of cells in G1, S, or G2+M measured from 2 to 24 h after drug removal remained almost constant, showing that the S phase arrest was not reversible (Fig. 5D). However, for cells treated with 1 μMS23906-1, a decrease of cells in the G2+M phases (20%) was associated with an increase of the number of cells in G1, measured 24 h after drug removal (Fig.5C). The blockade in G2+M induced by 1 μMS23906-1 was thus partially reversible.
Induction of Apoptosis by S23906-1.
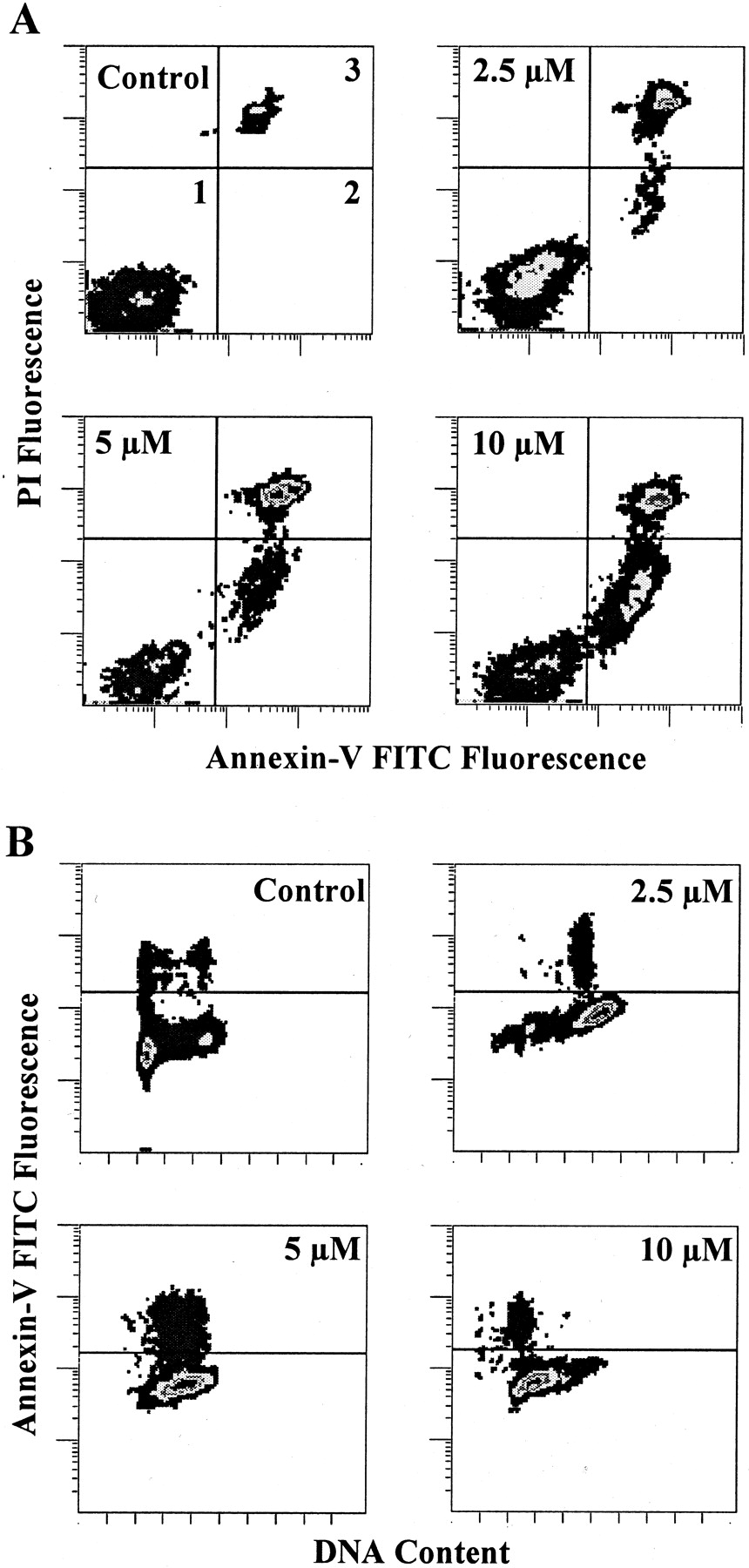
After 24 h of exposure to S23906-1, cells with condensed chromatin were detected by microscopic observation (not shown), suggesting that S23906-1 induced apoptosis. To investigate whether the G2+M or S blockade resulted in apoptosis, HT29 cells were incubated with S23906-1 for 24 h, washed and incubated in drug-free medium for a further 24 h. Cells were then labeled with annexin-V-FITC and PI and analyzed by flow cytometry as described under Materials and Methods. Biparametric histograms are shown in Fig.6A. S23906-1 induced a concentration-dependent annexin-V labeling of cells: 12, 22, and 30% of cells were annexin-V positive and PI negative at 2.5, 5, and 10 μM, respectively, versus 2% for untreated cells. Because the annexin-V labeling was maintained when cells were subsequently fixed and incubated with RNase and PI, it was possible to study the distribution of the annexin-V positive cells in the different phases of the cell cycle. Biparametric histograms of DNA content versus annexin-V (Fig. 6B) show that apoptotic cells were found in the phases in which treated cells accumulated: late S G2+M at 2.5 μM, S at 5 μM, and G1-S boundary at 10 μM. The induction of apoptosis by S23906-1 seemed to be a consequence of the blockade in the cell cycle rather than an early event after the induction of damage.

Induction of apoptosis by S23906-1. HT29 cells were exposed to S23906-1 for 24 h, washed and incubated in drug-free medium for 24 h. A, biparametric analysis of annexin-V-FITC and PI labeled cells. Quadrant 1, viable cells; quadrant 2, apoptotic cells with an intact membrane; quadrant 3, apoptotic cells undergoing secondary necrosis. B, biparametric analysis of annexin-V-FITC labeled cells subsequently fixed by ethanol before incubation with RNase and PI labeling.
Inhibition of BrdU Incorporation.
The accumulation of cells in S phase by cytotoxic concentrations of S23906-1 suggested that DNA synthesis was inhibited, directly or indirectly, by the drug. The effect of S23906-1 on DNA synthesis was thus investigated by measuring BrdU incorporation by flow cytometry with an anti BrdU-FITC monoclonal antibody. The first experiments were performed under standard conditions: HT29 cells were incubated with 5 μM S23906-1 for 24 h, washed, and exposed to BrdU for 1 h. Biparametric histograms of BrdU-FITC fluorescence versus PI fluorescence are shown in Fig.7. Cells accumulated in middle S phase, and BrdU incorporation was completely inhibited. To determine whether inhibition of DNA synthesis is an early event after a brief exposure of cells to S23906-1, cells were incubated for 1 or 4 h with S23906-1 before the BrdU pulse. The inhibition of BrdU incorporation was observed after 1 h of exposure, and was complete after 4 h of incubation (Fig. 7). Interestingly, the inhibition of BrdU incorporation was observed before the modification of the cell cycle (DNA content) became apparent, demonstrating that accumulation of cells in S phase is the result of the inhibition of DNA synthesis.
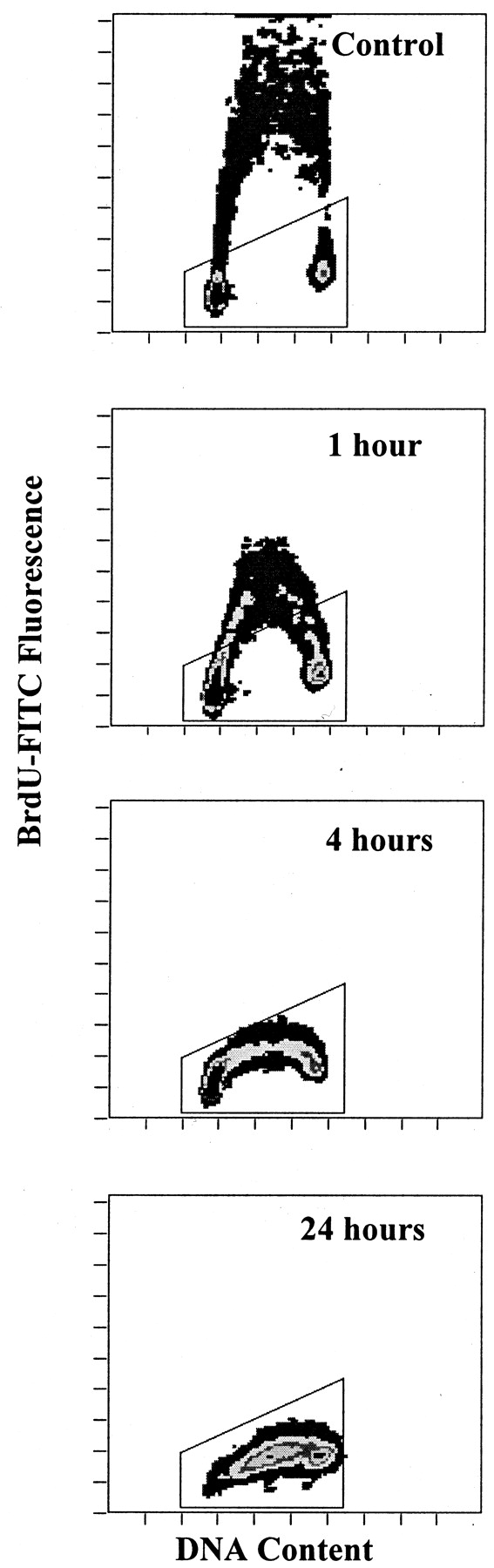
Inhibition of BrdU incorporation by S23906-1. HT29 cells were treated for 1, 4, or 24 h with 5 μM S23906-1, washed and incubated for 1 h with BrdU. BrdU labeling and flow cytometric analysis were performed as described under Materials and Methods. Results are displayed as linear biparametric histograms of BrdU incorporation versus DNA content. The indicated gates correspond to BrdU negative cells. The gates were established by the analysis of a control sample incubated without BrdU.
Other cytotoxic agents known to interfere with DNA synthesis were tested to compare their effects with those of S23906-1. HT29 cells were incubated for 4 h with 10 μM CDDP, 250 nM MTX, 500 nM Ara-C, 25 nM dFdC, 50 μM FUra, or 100 μM BCNU; these concentrations induced an accumulation of HT29 cells in S phase after 24 h. Among these drugs, only Ara-C produced effects on the cell cycle and DNA synthesis that were similar to those of S23906-1 (Fig.8). Cells with a DNA content corresponding to the S phase were not synthesizing DNA, and cells that were in late G1 during the labeling period were not able to move out of G1 phase. The effect of gemcitabine was also near that of S23906-1, except that a fraction of G1 cells were able to move into early S phase during BrdU incorporation.
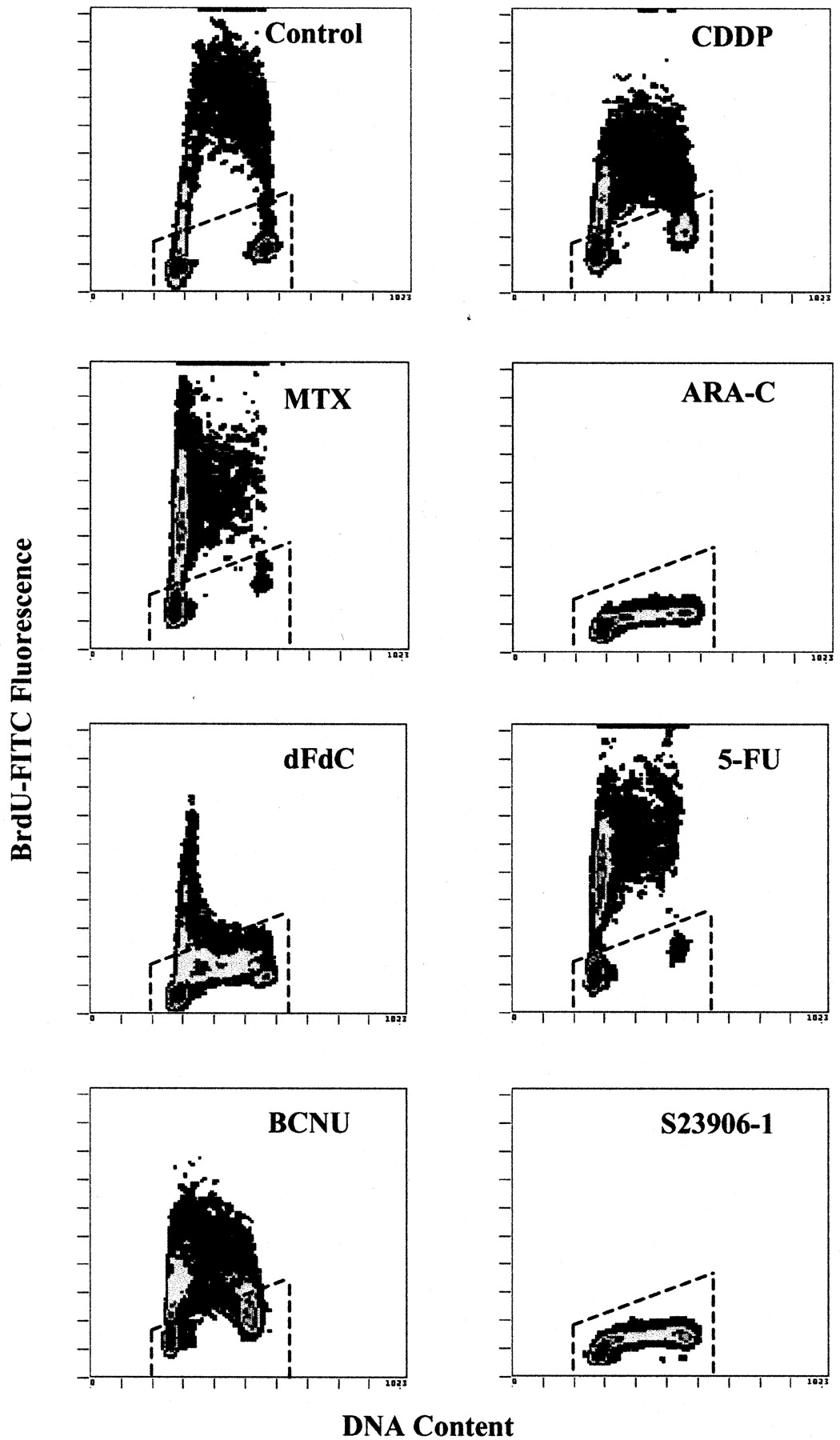
Effect of DNA synthesis inhibitors on BrdU incorporation. HT29 cells were treated for 4 h with 10 μM CDDP, 250 nM MTX, 500 nM Ara-C, 25 nM dFdC, 50 μM FUra, 100 μM BCNU, or 5 μM S23906-1, washed and incubated for 1 h with BrdU. BrdU labeling and flow cytometric analysis were performed as described underMaterials and Methods.
Induction of Cyclin E Expression.
Because of the unusual effects of S23906-1 on progression into and through S-phase, we investigated the effect of S23906-1 on the level of cyclins implicated in the G1-S transition. HT29 cells were exposed to the drugs for the times indicated in Figs. 9 and 10, washed, and labeled with specific antibodies. Whereas the level of cyclins D1, D2, D3, or A proteins were not modified (not shown), cyclin E expression was increased after exposure to S23906-1. As shown in Fig.9A, 5 μM S23906-1 induced a 3-fold increase of cyclin E level after 24 h compared with untreated HT29 cells. This increase of cyclin E expression was also observed after only 4 h of contact (i.e., before modification of the cell cycle became apparent; compare Figs. 9A and 5B). With respect to inhibition of BrdU incorporation, an increase of cyclin E expression was also observed after treatment by Ara-C and to a lesser degree by dFdC (not shown). The increase in fluorescence intensity, measured by flow cytometry, was confirmed by Western blotting to have been caused by a 3-fold increase in cyclin E protein expression (Fig. 9B). To investigate whether the overexpressed cyclin E protein was complexed with Cdk2, the proteins were precipitated by the anti-cyclin E antibody and the electrophoresed complex was Western blotted with an anti-Cdk2. Figure 9B shows that the level of Cdk2 was similar in control and treated cells, demonstrating that the overexpressed cyclin E was not complexed with Cdk2.
Perturbations of the cell cycle as well as the modification of cyclin E level induced by S23906-1 were also studied in the sensitive KB-3-1 andS23906-1 resistant KB/S23-500 cells. Interestingly, whereas a similar accumulation in S phase was induced by 2.5 μM S23906-1 in KB-3-1 cells and 5 μM S23906-1 in KB/S23-500 cells, no modification of the cyclin E protein level was detected for the KB/S23-500 resistant cell line exposed to the drug for 24 h (Fig.10). Higher concentrations (up to 20 μM) of S23906-1 failed to induce overexpression of cyclin E in resistant cells, which accumulated, however, at the G1-S transition (not shown)
Discussion
The acronycines have structural features that superficially suggest that they might possess classical DNA interacting properties (Fig. 1). However, an unusual spectrum of in vivo activity does not readily support this idea (Guilbaud et al., 2001). Indeed, previous experiments have shown that S23906-1 does not intercalate DNA (M.-H. David-Cordonnier, W. Laine, A. Lansiaux, A. P., J. A. H., and C. B., submitted), is devoid of effects on purified topoisomerases 1 and 2 and has no effects on tubulin polymerization (Léonce et al., 2000). The present study was undertaken to characterize the in vitro cytotoxic effects of this novel drug and to gain information that could explain its novel spectrum of activities. The unusual profile of cytotoxicity of S23906-1 (Fig. 2 and 3) and particularly its rapid onset of action and irreversibility suggest that S23906-1 accumulates and/or is strongly retained by tumor cells and that the lesions induced after only 1 h of exposure are not repaired.
We have shown previously that cytotoxic concentrations of the most potent acronycine derivatives in the current series induced an accumulation of L1210 cells in S phase of the cell cycle (Costes et al., 2000). Here we show that low concentrations of S23906-1 (0.1 to 1 μM) arrested HT29 cells in the G2+M phases, whereas higher concentrations induced an accumulation in S phase (2.5 and 5 μM) or at the G1-S boundary (10 μM). Kinetic experiments showed that 1 μM S23906-1 slowed the rate of progression through the S phase and induced an accumulation of cells in G2+M. The completion of the S phase was achievable, but the persistence of the lesions probably prevented cells from undergoing mitosis, thus stopping progression by a G2 checkpoint control. However, after removal of the compound, 20% of cells blocked in G2+M were able to divide further (Fig. 4). This partially reversible arrest may explain the incomplete inhibition of colony formation observed 14 days after the cells have been treated with 1 μM S23906-1 (Fig. 3). A higher concentration of 5 μM induced a totally irreversible arrest in S phase, which thus seemed to be implicated in the cytotoxic properties of S23906-1. These results suggest the possibility that different cellular targets were being affected, depending on the concentration ofS23906-1 or, alternatively, that a higher number of similar lesions induced different consequences. Such dual effects on the cell cycle were described for antitumor drugs that act through inhibition of DNA synthesis (Bhuyan et al., 1973). Because S23906-1 might affect the progress through the cell cycle in vitro via different mechanisms, depending on the concentration, it is not clear whether one or all of these mechanisms are responsible for the antitumor activity of S23906-1 in vivo. This requires further study of the pharmacokinetic profile of this drug with respect to peak concentrations and/or exposure time, as well as studies of the cell cycle perturbations induced in vivo in tumor cells.
Observations by microscopy revealed the presence of apoptotic cells after exposure to S23906-1, so induction of apoptosis was investigated by annexin-V labeling. A concentration-dependent increase of the number of apoptotic cells was observed, both in G2+M phases (1 μM) and S phase (5 μM) (Fig. 6). This suggests that the arrest in S or G2+M was required to activate the transduction pathway for apoptosis.
The changes in the cell cycle were associated with a profound inhibition of DNA synthesis by S23906-1, which was complete at 5 μM after only 4 h of incubation (Fig. 7). Interestingly, a similar profile was observed with cells treated by the structurally unrelated Ara-C and by dFdC (although to a lesser extent), two deoxycytidine analogs known to inhibit DNA synthesis and DNA polymerase α (Graham and Whitmore, 1970; Iwasaki et al., 1997). However, compared withS23906-1, the S phase arrest was completely reversed when Ara-C was removed from the medium (Gray et al., 1987), although dFdC induces an irreversible arrest followed by apoptosis (Chen et al., 2000). Clearly, the effects of S23906-1 were very different from the other standard cytotoxic agents investigated for their effects on DNA synthesis. It seems unlikely, considering the chemical structure of S23906-1, that the inhibition of DNA synthesis could be caused by incorporation of the molecule into DNA, as is the case for Ara-C and dFdC. More likely, it is possible that S23906-1 directly inhibits DNA polymerases or binds to DNA in a nonintercalative mode, thereby interfering with DNA chain elongation. These possibilities are currently under investigation. However, the in vivo pharmacological profile of this compound differs notably from existing DNA alkylating drugs, such as nitrosoureas or nitrogen mustards, suggesting a unique mode of interaction with DNA.
The irreversible inhibition of DNA synthesis and the effects on the cell cycle of S23906-1 might be expected to be associated with changes in the biochemistry of the cell cycle, reflected by activities of the cyclin-dependent kinases and the amounts and nature of the cyclins present. Given the different profile of S23906-1 on cell cycle progression and inhibition of DNA synthesis, compared with other agents, we were keen to determine its effects on the biochemistry of the cell cycle. A short incubation of HT29 cells with cytotoxic concentrations of S23906-1 was sufficient to induce an increase of cyclin E level (Fig. 9). Under the same conditions, no increases in the amount of cyclins D1, D2, D3, or A were detected.
Cyclin E is a critical regulator of the G1/S transition, and its overexpression, after genotoxic stress, such as γ-irradiation, has been reported to induce apoptosis of hematopoietic cells through activation of the caspase cascade (Mazumder et al., 2000). This proapoptotic effect was associated with an increased production of cyclin E/Cdk2 kinase activity (Gil-Gomez et al., 1998). Indeed, a dominant negative construct of Cdk2 prevented apoptosis induced by staurosporine and tumor necrosis factor α (Meikrantz and Schlegel, 1996), and it was suggested that radiation promoted an inappropriate entry into the cell cycle via the new synthesis of cyclin E (Mazumder et al., 2000). This effect was apparently not observed after radiation of a number of epithelial cell lines, whereas we observed an increase in cyclin E in such a cell line after treatment byS23906-1. Furthermore, we have not detected an increase of cyclin E-complexed to Cdk2 in S23906-1-treated cells (Fig. 9). Compared with radiation and other DNA reactive drugs, this suggests a different profile of activity for S23906-1. Taking into account that degradation of free cyclin E is required to control S phase progression (Singer et al., 1999), it can be postulated that the increase of cyclin E induced by S23906-1 resulted from an inhibition of the degradation of the free form of the protein. Experiments are currently in progress to determine the effects of S23906-1 on the members of cullin family proteins responsible for the cyclin E destruction machinery (Winston et al., 1999). Interestingly, cells made about 10-fold resistant to S23906-1, by stepwise exposure to the drug, still accumulated in S phase, but without any increase in cyclin E protein level (Fig. 10). This strongly supports the idea that cyclin E overexpression acts as an early signal for apoptosis induction by S23906-1. The relationship between the elevation of cyclin E and the onset of apoptosis induced by S23906-1 is the subject of further studies. In conclusion, S23906-1 is a cytotoxic agent with a novel mechanism of action and clear clinical potential on the basis of its pharmacological profile.
Acknowledgments
We thank Dr. N. Guilbaud for helpful discussions and Dr. L. Kraus-Berthier for critical reading of the manuscript.
Footnotes
- Received June 18, 2001.
- Accepted September 19, 2001.
Abbreviations
- S23906-1
- cis-1,2-diacetoxy-3,14-dihydro-3,3,14-trimethyl-6-methoxy-7H-benz[b]pyrano[3,2-d]acridin-7-one
- DMSO
- dimethyl sulfoxide
- Ara-C
- cytosine arabinoside
- dFdC
- gemcitabine (2′,2′-difluoro-2′-deoxycytidine)
- CDDP
- cisplatin
- MTX
- methotrexate
- FUra
- 5-fluorouracil
- BCNU
- carmustin
- FCS
- fetal calf serum
- MTT
- 3-(4,5 dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide
- PI
- propidium iodide
- FITC
- fluorescein isothiocyanate
- PBS
- phosphate-buffered saline
- BrdU
- bromodeoxyuridine
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}