


Abstract
Resistance to cisplatin (DDP) is often accompanied by impaired accumulation in mammalian cells. The mechanism of impaired DDP accumulation is unknown, but copper uptake is diminished as well. We investigated the ability of the copper transporter CTR1to control the accumulation of DDP in Saccharomyces cerevisiae. Parallel studies of copper and DDP cellular pharmacokinetics were carried out using an isogenic pair of wild-typeCTR1 and ctr1 knockout S. cerevisiae strains. Both copper and platinum accumulation increased linearly as a function of time and drug concentration in the parental cells. Deletion of CTR1 resulted in a 16-fold reduction in the uptake of copper and an 8-fold reduction in the uptake of DDP measured at 1 h. The CTR1-deficient cells accumulated 2.3-fold (p < 0.05) less platinum in their DNA and were 1.9-fold more resistant to the cytotoxic effect of DDP than the CTR1-replete cells. The kinetics of cellular copper accumulation were similar to those of DDP. Based on measurements of accumulation at 1 h, the K m for copper influx was 128.8 μM, and the V max was 169.5 ng/mg of protein/min; for DDP, the K m was 140.2 μM and the V max was 76.9 ng/mg of protein/min. DDP blocked the uptake of copper into the parental cells but notctr1-deficient cells. CTR1-deficient cells also demonstrated impaired accumulation of the DDP analogs carboplatin, oxaliplatin, and ZD0473 [cis-amminedichloro(2-methylpyridine) platinum (II)]. These results indicate that CTR1 function markedly influences the uptake of all of the clinically used platinum-containing drugs and suggest that this copper transporter may also transport DDP.
The effectiveness of cell killing by cisplatin (DDP) is generally acknowledged to be a function of how much drug gets into the cell, how much of it enters the nucleus and actually reacts with DNA, how tolerant the cell is of lesions in its DNA, and how effectively it removes these adducts (Andrews and Howell, 1990). Intracellular detoxification of DDP through mechanisms that involve binding to thiols may contribute to resistance (reviewed in Perez et al., 1993). Both defects in the ability of the cell to recognize adducts in DNA (reviewed in Fink et al., 1998) and enhanced repair of and tolerance to adducts (Johnson et al., 1997) have been identified as contributing to resistance in some cell types. However, impaired uptake of DDP is the most consistently identified characteristic of cells selected for DDP resistance both in vitro and in vivo (reviewed in Andrews and Howell, 1990; Gately and Howell, 1993).
The mechanism underlying impaired DDP accumulation in resistant cells is unknown; in fact, the mechanism by which DDP enters or exits cells remains poorly defined. DDP accumulates in cells relatively slowly compared to many other classes of anticancer agents, and earlier evidence suggested that at least one component of DDP uptake is mediated by a transport mechanism or channel (Andrews and Albright 1991; Andrews et al., 1991). In fact, the behavior of DDP is similar in many ways to that of transition metals such as copper. Both active transporter-mediated and passive processes contribute to the cellular uptake of DDP and copper (reviewed in Gately and Howell 1993; Pena et al., 1999), and DDP resistance is often accompanied by resistance to other metalloids (Tobey and Tesmer, 1985; Romach et al., 2000). Cross-resistance of cells to antimony (Chen et al., 1998), cadmium (Schilder et al., 1990; Naredi et al., 1994; Lee et al., 1995; Haga et al., 1997; Perego et al., 1997), zinc (Koropatnick and Pearson, 1990;Naredi et al., 1994), cobalt (Naredi et al., 1994), and copper (Nicholson et al., 1998) has been previously reported. A direct link between copper transport and DDP resistance has been identified byKomutsu et al. (2000), who found that cells molecularly engineered to express the copper efflux pump ATP7B become resistant to DDP. This finding has recently been confirmed for ovarian carcinoma cells in this laboratory (Katano et al., 2002c).
We have previously reported that human ovarian carcinoma cells selected for resistance to DDP are cross-resistant to copper (Katano et al., 2002b) and that cells selected for resistance to copper are cross-resistant to DDP (Safaei and Howell, 2001). The discovery that over-expression of the copper exporter ATP7B also mediates DDP resistance (Komatsu et al., 2000; Katano et al., 2002c) suggests that DDP may be sequestered and effluxed from the cell by pathways normally devoted to copper. These observations prompt the question of whether transporters involved in copper metabolism can also transport DDP. Because in most DDP-resistant cells there seems to be a defect in initial influx, the transporter responsible for the inward movement of copper across the plasma membrane is of particular interest. InS. cerevisiae and mammalian cells, the key influx transporter for copper is CTR1, a 190-amino acid protein with three transmembrane domains (Zhou and Gitschier 1997). In mammals, CTR1 mRNA is found in all tissues; the highest levels are in the liver and kidney (Zhou and Gitschier 1997; Lee et al., 2000). Several other homologous proteins, including CTR3 (Pena et al., 2000), CTR4, and CTR5 (Zhou and Thiele, 2001), may also play a role in copper uptake in yeast. In this study, we examined the connection between copper and DDP transport mechanisms using an isogenic pair of S. cerevisiae strains, a parental line and a subline in which the CTR1 gene had been deleted. We report here that the alterations in the cellular pharmacology of copper that accompany the loss of CTR1expression are paralleled by similar changes in the cellular pharmacology of DDP. These results indicate that DDP is transported into the cell by CTR1 or a mechanism regulated by CTR1.
Materials and Methods
Drugs and Reagents.
DDP was gift from Bristol-Myers Squibb (Princeton, NJ). The clinical formulation at a concentration of 3.33 mM was stored in the dark at room temperature. Carboplatin was purchased from Sigma (St. Louis, MO) and a stock solution was prepared at a concentration of 10 mM in water. ZD0473 was a gift from AstraZeneca Pharmaceuticals LP (Wilmington, DE) and a 10 mM stock solution was made up in 0.9% NaCl. Oxaliplatin was a gift from Sanofi Pharmaceuticals (Malvern, PA) and was dissolved in 0.9% NaCl solution at 10 mM. Cupric sulfate and other chemicals were obtained from Sigma and Fisher Scientific Co. (Tustin, CA). Protein concentration was measured using a kit from Bio-Rad Co. (Hercules, CA).
Yeast Strains and Cell Growth.
The S. cerevisiaestrains used in the study were obtained from the American Type Culture Collection (Manassas, VA). The parental BY4741 strain (ATCC 201388) contains a wild-type copy of the CTR1 gene, whereas its derivative strain BY4741-YPR124W (ATCC 4005539) has been molecularly engineered to delete the CTR1 coding sequences. Yeast cultures were seeded from single colonies grown on YPD agar plates. Growth of experimental cultures was initiated atA 660 nm = 0.05 or less; the cultures were allowed to grow to A 660 nm = 0.8 to 1.2 (log phase, 1.4–2.0 × 107 cells/ml) before use.
Cellular Accumulation of Copper, DDP, and DDP Analogs.
Cultures containing 10 ml of log-phase cells were harvested by centrifugation and resuspended in 10 ml of 30°C fresh YPD medium containing various test compounds at concentrations of 0 to 400 μM. After incubation for 1 h in at 30°C in a shaker at 200 rpm, cells were washed 3 times with ice-cold phosphate-buffered saline (PBS). The pellets were resuspended in 1 ml of PBS, an aliquot of 0.1 ml of the cell suspension was utilized for protein assay, and the remainder was digested in 70% nitric acid. Cell lysates were heated for 2 h at 65°C, diluted to 5% nitric acid and assayed for platinum and copper content on an inductively coupled plasma optical emission spectroscopy (ICP-OES) apparatus (Optima 3000 DV; PerkinElmer, Boston, MA) at the Analytical Facility at the Scripps Institute of Oceanography. V max andK m were determined via extrapolation to zero of the reciprocal plot of velocity versus substrate concentration using the Lineweaver-Burke equation.
Platinum Accumulation in DNA.
Cultures containing 10 ml of log-phase cells were treated with 50 μM DDP for 1 h. The cells were then washed three times with ice-cold PBS. A Wizard genomic DNA purification kit (Promega, Madison, WI) was used for isolation of DNA. Aliquots of the DNA were digested in 70% nitric acid at 65°C for 2 h and diluted to 5% nitric acid by adding appropriate volume of double distilled deionized water. Platinum in the hydrolysate was quantified by ICP-OES.
Cytotoxicity Assay.
Sensitivity to the cytotoxic effect of DDP was assessed using a colony formation assay. Cultures (1 ml) containing a total of 6 × 106 cells were exposed for 4 h to DDP at concentrations of 0.5, 1.0, 1.5, 2.0, and 2.5 mM, washed once in PBS, resuspended in YPD medium, diluted 1:4000, and plated onto 100-mm agar plates. After 2 days of growth at 30°C the number of colonies was counted manually. The IC50 was defined as the drug concentration that reduced the number of colony-forming units to 50% of the value in a control culture not exposed to drug. Each experiment was repeated three times with duplicate cultures for each drug concentration.
Statistics.
All the data were analyzed by use of a two-sided paired Student's t test with the assumption of unequal variance.
Results
Cellular Accumulation of Copper and DDP.
The cellular uptake of copper and platinum as a function of time during exposure of the parental CTR1 and the mutant ctr1 cells to 100 μM CuSO4 or DDP is shown in Fig.1. Accumulation of both copper and platinum was increased with time up to 1 h. The amount of cell-associated copper and platinum was markedly reduced in the ctr1 cells. In both strains, the uptake of copper was greater than that of platinum at all the time points tested. Fig.2 presents plots of the cellular accumulation of copper and platinum as a function of concentration after a 1-h exposure to copper or DDP. Both copper and platinum accumulation increased linearly as a function of concentration in theCTR1 and ctr1 cells. However, there was a substantial impairment of copper and platinum accumulation inctr1 cells. Based on the slope of the plot of uptake as a function of copper concentration, accumulation of copper in thectr1 cells was 16-fold less than in the CTR1cells. Likewise, accumulation of platinum during exposure to DDP was 8-fold less in the ctr1 than the CTR1 cells. Thus, CTR1 is important to the accumulation of both compounds, but the loss of CTR1 expression had a 2-fold larger effect on copper uptake than platinum accumulation.

Copper and platinum accumulation as a function of duration of exposure to 100 μM CuSO4 (▪) and DDP (♦). A, parental CTR1 S. cerevisiae strain BY4741; B,ctr1 BY4741-YPR124W mutant cells. Measurements were made on sequential aliquots withdrawn from the same culture.
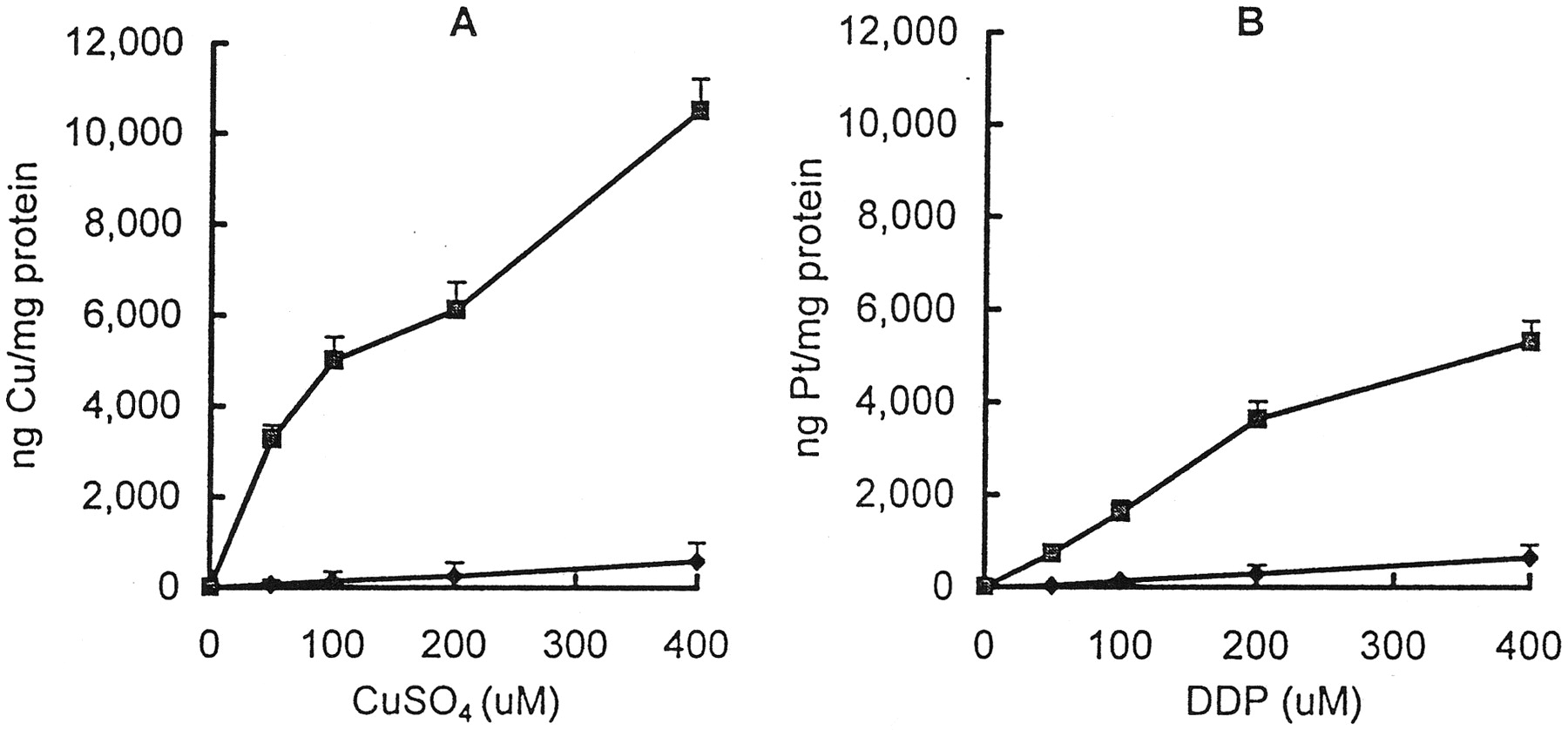
Copper (A) and platinum (B) accumulation in the whole cell at the end of a 1-h exposure as a function of CuSO4and DDP concentration in the parental CTR1 S. cerevisiaestrain BY4741 (▪) and its derivative ctr1 knockout strain BY4741-YPR124W (♦). Each data point presents the mean of three experiments, each performed with duplicate cultures.
True initial influx velocities could not be measured because of the relatively slow uptake rates and limitations on the sensitivity of the assays available. However, because uptake seemed to be linear with time once measurable levels were detectable, the 1-h accumulation data can be used to estimate the relative apparentK m andV max values for copper and DDP uptake. Figure 3A shows the plot of uptake velocity as a function of copper and platinum concentration, and Fig.3B shows the Lineweaver-Burke analysis of this data. For copper the estimated K m was 128.8 μM, and theV max was 169.5 ng/mg of protein/min. For DDP, the estimated K m was 140.2 μM and the V max was 76.9 ng/mg of protein/min. Thus, based on measurements made over a period of 1 h, although the uptake transport process in the CTR1 cells seemed to demonstrate similar affinity for the two agents, it had a 2-fold higher capacity to transport copper than DDP.
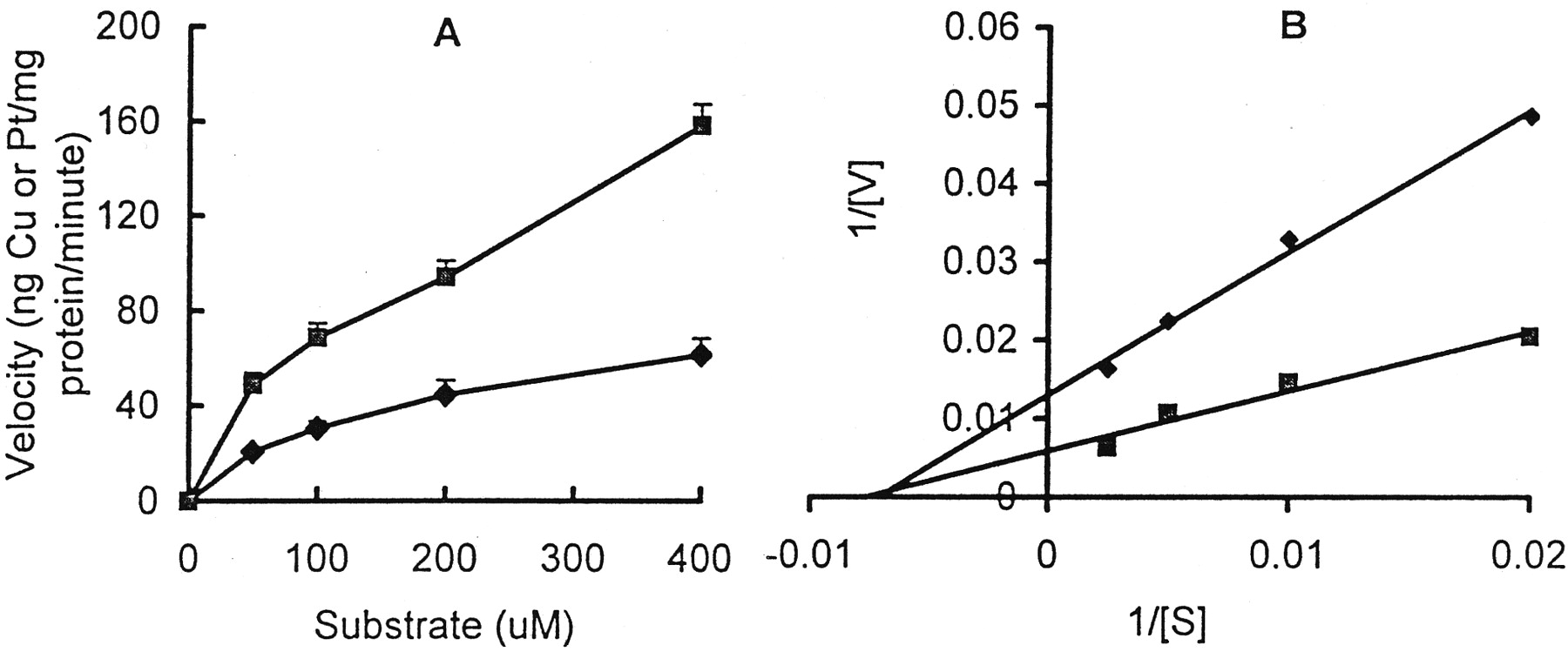
A, uptake velocity as a function of CuSO4(▪) or DDP (♦) concentration measured over the first 60 min of exposure in the wild-type CTR1 strain BY4741. B, a double reciprocal (Lineweaver-Burke) plot of the uptake velocity versus drug concentration. Each data point presents the mean of three experiments, each performed with duplicate cultures.
Accumulation of Platinum in the DNA of CTR1 versusctr1 Cells.
DDP has the potential to bind tightly to a large number of both intracellular and extracellular components, making it difficult to determine what fraction of the cell-associated platinum has actually entered the cell via a CTR1-dependent process. To establish that a CTR1-mediated process directly contributed to the amount of DDP reaching its primary intracellular target, the total amount of platinum per microgram of DNA was measured after a 1-h exposure to 50 μM DDP. As shown in Fig.4, the DNA-associated platinum was 2.3-fold lower (p < 0.05) in the ctr1 than in the parental CTR1 cells. This establishes that the majority of the DDP reacting with DNA was dependent on transport by CTR1, or a process linked to the expression of CTR1, for entry into the cell.

Platinum accumulation in DNA in CTR1BY4741 and ctr1 BY4741-YPR124W cells were exposed to 50 μM DDP for 1 h. Each bar represents the mean of three measurements made on duplicate cultures; vertical bars, S.E.M.
Cytotoxicity of DDP to CTR1 versusctr1 Cells.
The sensitivity of the CTR1and ctr1 cells to the cytotoxic effect of DDP was determined using colony-formation assays. Figure 5shows survival as a function of DDP concentration for the two strains. The IC50 for the CTR1 cells was 0.66 ± 0.05 μM (mean ± S.D.) whereas for thectr1 cells, it was 1.25 ± 0.15 μM. Thus, the CTR1-deficient cells were 1.9-fold resistant to DDP (p= 0.014).

Survival of CTR1 andctr1 cells as a function of DDP concentration. (▪),CTR1 cells; (♦), ctr1 cells. Each data point represents the mean of three independent experiments, each performed with duplicate cultures for each drug concentration. Vertical bars, S.E.M.
Effect of DDP on Copper Uptake.
The results reported above are consistent with the concept that copper and DDP share the same CTR1-dependent uptake mechanism. This was addressed more directly by examining the effect of increasing concentrations of DDP on the accumulation of copper in both the CTR1 and ctr1cells. The accumulation of copper during exposure to 100 μM CuSO4 for 1 h was measured in the presence of increasing concentrations of DDP. As shown in Fig.6A, DDP reduced the accumulation of copper in the CTR1 cells in a concentration-dependent manner; at 400 μM, DDP accumulation was reduced to 58 ± 10% (mean ± S.E.M.) of control. However, DDP had no effect on the accumulation of copper in the ctr1 cells (Fig. 6B). The differential effect observed in the CTR1 versusctr1 cells indicates that the effect of DDP was specific to the function of the CTR1 transporter.

Effect of DDP on copper accumulation. The parentalCTR1 BY4741 (A) and ctr1 BY4741-YPR124W (B) cells were treated with 100 μM CuSO4 in combination with increasing concentrations of DDP (0, 50, 100, 200, 400 μM) for 1 h. Each bar data point represents the mean of three measurements made on duplicate cultures; vertical bars, S.E.M.
Competition between Copper and DDP during Uptake.
To determine the nature of inhibitory interaction between copper and DDP during uptake into CTR1 cells, the accumulation of copper was measured at the end of a 1-h exposure to increasing concentrations of CuSO4 in the absence or presence of increasing concentrations of DDP. Figure 7 shows that, at all copper concentrations tested, as the DDP concentration increased, the copper accumulation decreased. Lineweaver-Burke analysis of the data yielded an apparent K i of 7 μM and the plots were most suggestive of a mixed-type interaction.

Interaction between copper and DDP during accumulation. The parental CTR1 BY4741 cells were treated with increasing concentration of CuSO4 in the presence of 0 to 400 μM DDP for 1 h. Each data point represents the mean of duplicate aliquots of the same culture.
Cellular Accumulation of DDP Analogs.
The effect ofCTR1 expression on the accumulation of other platinum-containing drugs in clinical use or in development was examined using the CTR1 and ctr1 cells. As shown in Fig. 8A, platinum accumulation increased linearly with the concentration for all the platinum drugs tested in the CTR1 cells. At any given concentration, platinum accumulation was greatest for ZD0473, followed in decreasing order by DDP, oxaliplatin, and carboplatin. Similar to its effect on DDP, the loss of CTR1 resulted in a substantial decrease in platinum uptake for each of these drugs as shown in Fig. 8B. The largest effect was observed for ZD0473 with an 89% reduction in platinum accumulation in the ctr1 cells compared with that in the CTR1cells at the highest concentration tested. These results indicate that, either directly or indirectly, CTR1 modulates the accumulation of all of the clinically relevant platinum-containing drugs despite the quite marked differences in their structures.

platinum accumulation following exposure of parentalCTR1 BY4741 (A) and ctr1 BY4741-YPR124W cells (B) to DDP or related analogs for 1 h. Each data point presents the mean of three experiments each performed with duplicate cultures.
Discussion
CTR1 was initially identified on the basis of its ability to mediate the high-affinity uptake of copper in yeast (Dancis et al., 1994). It was subsequently found to be expressed in all mammalian tissues and has been shown to be essential for copper accumulation in mice (Kuo et al., 2001; Lee et al., 2001). The results of the current study confirm the importance of CTR1 for accumulation of copper and indicate that CTR1 also markedly influences the cellular accumulation of DDP and other clinically important platinum-containing drugs inS. cerevisiae.
The cell-associated copper and DDP measured after a 1-h exposure to these agents represents the net contribution of nonspecific binding of the metalloids to the exterior of the cell, specific transporter-mediated entry of drug into the cell, any diffusion-mediated uptake that might occur, intracellular binding, and efflux. In the case of copper accumulation, the uptake into thectr1 cells was 16-fold less than into CTR1 cells across a wide spectrum of external copper concentrations. In the case of DDP, accumulation in the ctr1 cells was 8-fold less than in the CTR1 cells such that the effect of deletingCTR1 was approximately 2-fold less in magnitude for DDP than for copper. It is difficult to determine exactly what fraction of the platinum associated with the ctr1 cells represents drug nonspecifically bound to the exterior of the cell versus drug that has passed through the cell membrane. However, the importance of CTR1 to the ability of DDP to gain access to DNA was clearly demonstrated by the finding that loss of CTR1 function reduced DNA platinum content by 2.3-fold and decreased sensitivity to the cytotoxic effect of DDP by 1.9-fold. Similar linkage between DNA adduct formation and cytotoxicity is well established in other experimental systems (Strandberg et al., 1982).
Because it was not possible to make measurements of the true initial influx rate, the estimates of K m andV max presented here are based on net accumulation at 1 h. The observation that uptake was relatively linear with time for the first hour of accumulation and that the very extensive intracellular binding of both copper and DDP probably limited efflux over this period favor this approach. However, caution is required in interpreting this data, because the actual initial influx rate and the contribution of efflux has not been assessed. Substantial amounts of platinum became associated with the yeast cells even in the absence of CTR1 expression, and this may reflect the action of other transporters or, given the propensity of DDP to react with thiol-containing proteins, nonspecific binding. The fact that the drugs with the most and least rapid accumulation in the CTR1 cells were also those with the most and least rapid accumulation in thectr1 cells suggests the possible involvement of other transporters. In addition, data obtained with human cells suggest that DDP enters by several different mechanisms (Gately and Howell, 1993). Nevertheless, that copper and DDP seem to have similar affinities for the mechanism that transports them into the yeast cell suggests that the clear difference in the extent of accumulation is more likely attributable to differences in velocity of transport than affinity for the transporter(s).
Although CTR1 could alter DDP accumulation indirectly through effects on intracellular copper levels, several lines of evidence suggest that DDP is actually a substrate for CTR1. The external N-terminal domain of the protein contains 11 repeats of the MXXM sequence and histidine-rich sequences similar to those that have an established role in the binding of copper to other proteins. These are likely to be involved in the initial chelation of copper to CTR1 and its subsequent transfer into the channel formed by the transmembrane portions of the protein. DDP is also known to bind to both methionine and histidine in a variety of contexts (Djuran and Milinkovic, 2000, and references therein). Recent studies indicate that the bond linking DDP to the sulfur in methionine is labile enough to permit transfer of the DDP to histidine (van Boom et al., 1999; Djuran and Milinkovic, 2000), suggesting the feasibility of sequential chelation and transport steps similar to those of copper. Although the Cu(I) ion in water can exist in a tetrahedral structure, copper forms a square planar structure similar to that of DDP when coordinated by four amino groups (Theophanides and Anastassopoulou, 2002). Additional evidence supporting the concept that DDP is a substrate for CTR1 comes from the observation that DDP seems to be moved across cell membranes by other copper transporters as well. ATP7B is a copper transporter that sequesters copper from the cytoplasm into the trans-Golgi network, whence it is exported from the cell (Klomp et al., 1997). Wilson's disease is caused by mutations that disable the ability of ATP7B to export copper from the liver (Danks, 1995). Recent studies from this (Katano et al., 2002a,c) and other laboratories (Komatsu et al., 2000) indicate that overexpression of ATP7B renders cells resistant to both copper and DDP. Detailed studies of the cellular pharmacology of copper and DDP in cells selected for resistance to copper that also overexpress ATP7B demonstrate enhanced efflux of both compounds (Safaei et al., 2002).
DDP clearly blocked the uptake of copper, and this effect was specific to the CTR1 cells, indicating that it was mediated either by interaction with CTR1 itself, a target that impacted CTR1 function, or an effect of CTR1 on other transporters. The analysis based on Lineweaver-Burke plots did not clearly identify either a competitive or noncompetitive mechanism of interaction. There are multiple possible ways in which DDP could alter the CTR1-mediated uptake of copper. As noted above, the external domain of CTR1 is rich in histidines. DDP interacts with histidines in a variety of chemical environments, and may disturb the copper-trapping function of this domain. Alternatively, copper and DDP may truly compete for entrance into the channel putatively formed by CTR1; it would not be surprising to discover that both types of interactions occur. It interesting that another metalloid potentially capable of binding to CTR1, the lanthanide terbium, has been reported to modulate the cellular uptake of DDP in human cells and that it does so by binding to a membrane site within 10 Å of the site at which DDP binds (Canada and Paltoo, 1998).
One of the most interesting observations made in the current study is that CTR1 apparently mediates the cellular accumulation of platinum-containing drugs with quite a wide variety of different structures. The substrate specificity of CTR1 has not previously been well defined. The results of this study suggest that CTR1 might be quite promiscuous with respect to the substrates it can accommodate, a feature commonly found with other metalloid transporters [Naredi, 1995(and references therein); Romach et al., 2000]. Among the analogs tested, ZD0473 exhibited the greatest CTR1-dependent cellular accumulation. CTR1 is likely to play an important role in the absorption of copper from the gut, and it is noteworthy that among these analogs, ZD0473 has substantial oral bioavailability (Raynaud et al., 1998). Thus, screening for analogs with optimal CTR1 transport may permit identification of platinum compounds with even better absorption from the intestine.
Acknowledgments
We acknowledge the seminal information provided on the web by Dr. Ira Herskowitz (University of California, San Francisco;http://www.sacs.ucsf.edu/home/HerskowitzLab/) to the effect that CTR1 may play a role in DDP transport. We also thank Bristol-Myers Squibb for providing DDP.
Footnotes
- Received May 29, 2002.
- Accepted August 7, 2002.
-
The study was supported in part by National Institutes of Health grants CA78648 and CA95298. This work was conducted in part by the Clayton Foundation for Research–California Division. X.L. and S.B.H. are Clayton Foundation investigators.
Abbreviations
- DDP
- cisplatin
- ICP-OES
- inductively coupled plasma optical emission spectroscopy
- PBS
- phosphate-buffered saline
- YPD
- yeast extract (1%)/bacto-peptone (2%)/dextrose (2%)
- ZD0473
- cis-amminedichloro(2-methylpyridine) platinum (II)
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}