


Abstract
Previously we have shown that G protein-coupled receptor kinase (GRK) 6 plays a major role in the regulation of the human M3 muscarinic acetylcholine receptor (M3 mAChR) in the human neuroblastoma SH-SY5Y. However, 30-fold overexpression of the catalytically inactive, dominant-negative K215RGRK6 produced only a 50% suppression of M3 mAChR phosphorylation and desensitization. Here, we have attempted to determine whether other endogenous kinases play a role in the regulation of M3 mAChR signaling. In contrast to the clear attenuating effect of K215RGRK6 expression on M3 mAChR regulation, dominant-negative forms of GRKs (K220RGRK2, K220RGRK3, K215RGRK5) and casein kinase 1α (K46RCK1α) were without effect. In addition, inhibition of a variety of second-messenger-regulated kinases and the tyrosine kinase Src also had no effect upon agonist-stimulated M3 mAChR regulation. To investigate further the desensitization process we have followed changes in inositol 1,4,5-trisphosphate in single SHSY5Y cells using the pleckstrin homology domain of PLCδ1 tagged with green fluorescent protein (eGFP-PHPLCδ1). Stimulation of cells with approximate EC50 concentrations of agonist before and after a desensitizing period of agonist exposure resulted in a marked attenuation of the latter response. Altered GRK6 activity, through overexpression of wild-type GRK6 or K215RGRK6, enhanced or reduced the degree of M3 mAChR desensitization, respectively. Taken together, our data indicate that M3 mAChR desensitization is mediated by GRK6 in human SH-SY5Y cells, and we show that receptor desensitization of phospholipase C signaling can be monitored in 'real-time' in single, living cells.
Repeated or continuous exposure of G protein-coupled receptors to agonist stimulation often causes a progressive waning of the response, referred to as desensitization. In the majority of cases, the primary event underlying the desensitization process is thought to be phosphorylation of serine and threonine residues in the third intracellular loop and C-terminal tail of the receptor (Krupnick and Benovic, 1998; Pitcher et al., 1998; Ferguson, 2001). Agonist-mediated receptor phosphorylation can be mediated by second messenger-regulated kinases, such as cyclic AMP-dependent protein kinase (PKA) or protein kinase C (PKC) (Benovic et al., 1985; Clark et al., 1989; Pitcher et al., 1992), casein kinase 1α (CK1α; Tobin et al., 1997; Budd et al., 2000), or specific G protein-coupled receptor kinases (GRKs; Pitcher et al., 1998; Ferguson, 2001). Receptor phosphorylation is often closely followed by the recruitment of arrestin proteins, which bind and sterically inhibit receptor-G protein signaling and also initiate other regulatory functions, such as receptor internalization (Krupnick and Benovic 1998; Ferguson, 2001).
The human M3 muscarinic acetylcholine receptor (mAChR) possesses a large number of potential phospho-acceptor sites with more than 50 serine/threonine residues within the third intracellular loop alone. This region contains 1 PKA, 7 PKC, 1 Ca2+/camodulin kinase II, 14 CK1, 6 CK2 consensus phosphorylation sites, and two serine clusters (331SSS333 and 348SASS351) known to be phosphorylated by GRK2 (Wu et al., 2000), raising the possibility of regulation by many different receptor kinases. Indeed, there have been many previous reports that second messenger-regulated kinases (Willars et al., 1999; Bundey and Nahorski, 2001), CK1α (Tobin et al., 1997; Budd et al., 2000), and GRKs (DebBurman et al., 1995; Willets et al., 2001a) are able to phosphorylate M3 mAChRs.
However, receptor phosphorylation does not always lead to a subsequent reduction in receptor/G protein signaling. Overexpression of either GRK3 or GRK6 in SH-SY5Y neuroblastoma cells leads to enhanced agonist-mediated M3 mAChR phosphorylation (Willets et al., 2001a). Despite this, only GRK6-mediated M3 mAChR phosphorylation leads to reduction of receptor/G protein signaling via receptor/G protein uncoupling (Willets et al., 2001a). Furthermore, inhibition of endogenous GRK6 activity by expression of the dominant-negative K215RGRK6 blocked both agonist-mediated receptor phosphorylation and receptor/G protein uncoupling (Willets et al., 2002). It is interesting to note that despite recombinant expression of a 30-fold excess of K215RGRK6 M3 receptor phosphorylation and receptor/G protein uncoupling was reduced by only 50% (Willets et al., 2002). This suggests either that the large K215RGRK6 excess is insufficient entirely to inhibit the action of endogenous GRK6 or that other receptor kinases may be responsible for the remaining agonist-driven M3 mAChR phosphorylation.
SH-SY5Y cells endogenously express GRK2, GRK3, and CK1α, as well as GRK6 (Willets et al., 2002) and various PKC isoenzymes (Turner et al., 1994). In addition to the GRK2 phosphorylation sites already mapped (Wu et al., 2000), we have recently shown that GRK3 is able to phosphorylate the M3 mAChR in intact cells (Willets et al., 2001a). It is also established that CK1α is able to phosphorylate the M3 mAChR expressed in both HEK293 and CHO cells (Tobin et al., 1997; Budd et al., 2000). These data suggest that one or more other receptor kinases may be responsible for the remaining agonist-driven M3 mAChR phosphorylation seen when GRK6 activity is suppressed by dominant-negative K215RGRK6 expression.
In the present study, we have attempted to dissect which, if any, additional receptor kinases regulate M3 mAChR phosphorylation in the human neuroblastoma SH-SY5Y, through the introduction of dominant-negative GRK (Mundell et al., 1997; Willets at al., 2002) and CK1α (Budd et al., 2000) constructs. Furthermore, we have exploited 'biosensor' technology to monitor changes in InsP3 levels in single cells using an eGFP-PHPLCδ construct (Hirose et al., 1999; Nash et al., 2001a,b). This has allowed us to assess M3 mAChR desensitization in 'real-time' in live SH-SY5Y cells.
Materials and Methods
Creation of Stable, Dominant-Negative GRK-Transfected Cell Lines and Cell Culture. SH-SY5Y cells were cultured in 5% fetal calf serum and 5% newborn calf serum, penicillin (100 units/ml), streptomycin (100 μg/ml), and amphotericin B (2.5 μg/ml) (Invitrogen, Renfrew, UK). All cells were maintained at 37°C, under 5% CO2 in humidified conditions. Wild-type SH-SY5Y cells were transfected with either dominant-negative K220RGRK2 or K220RGRK3, cloned into pcDNA3 at BamHI/HindIII or BamHI/KpnI sites, respectively (original GRK constructs kindly provided by Dr. E. Kelly, University of Bristol, UK), using FuGENE 6 according to the manufacturer's instructions. After 48 h, G-418 (Geneticin; 300 μg/ml) was added to the cells. After 7 days, surviving colonies were selected for expansion into cell-lines.
Western Blotting. Expression of GRKs and dominant-negative GRKs was determined using standard Western blotting procedures. As described previously (Willets and Kelly, 2001b), cell lysates were subjected to electrophoretic separation and transfer to nitrocellulose. Wild-type and dominant-negative GRK expression was determined by addition of specific anti-rabbit polyclonal antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) against GRK2, GRK3, or GRK6. FLAG-tagged, dominant-negative K46RCK1α (Budd et al., 2000) was detected by using a specific, anti-mouse monoclonal antibody directed against the FLAG-tag (Budd et al., 2000) or a rabbit polyclonal anti-CK1α antibody (Budd et al., 2000). Protein expression was visualized by the addition of ECL-plus reagent (Amersham Biosciences, Little Chalfont, Buckinghamshire, UK) according to the manufacturer's instructions and exposed to Hyperfilm (Amersham Biosciences).
Determination of mAChR Number. The Kd and Bmax values were determined by N-methyl-[3H]scopolamine (Amersham Biosciences) saturation binding analysis of SH-SY5Y cell monolayers grown to confluence in 24-well plates as described previously (Willets et al., 2001a).
Creation of Adenoviral K46RCK1α. Adenoviral CK1α was created using the AdEasy Vector System following the manufacturer's instructions (Qbiogene, Carlsbad, CA). Briefly, K46RCK1α was cloned into the pShuttle-CMV (transfer) vector at the HindIII and XbaI sites. After cloning, the pShuttle vector and supercoiled adenoviral genome (pAdEasy-1 vector) were cotransformed into bacteria (BJ5183 strain) to allow recombination of both plasmids, under kanamycin selection. After screening and selection of recombinant plasmid by restriction enzyme analysis, DH5α cells were transformed, and plasmid DNA was prepared using standard techniques. Purified recombinant DNA was linearized by PacI digestion before transfection into QBI-293A cells to generate a recombinant adenovirus expressing the gene of interest under the cytomegalovirus promoter. After 12 to 15 days, cells were subjected to three freeze-thaw cycles to release the recombinant virus into the supernatant. The supernatant was used to further infect larger flasks of QBI-293A cells. Cells were harvested and the virus was extracted via four repeat freeze-thaw cycles and purified by density gradient centrifugation at 40,000g, 10°C, 24 h).
M3 mACh Receptor Phosphorylation. The effect of K220RGRK2, K220RGRK3, K215RGRK5, or K215RGRK6 on the phosphorylation of endogenously expressed M3 mACh receptors was assessed as described previously (Tobin and Nahorski, 1993; Willets et al., 2001a). Plasmid control, or cells overexpressing K220RGRK2, K220RGRK3, K215RGRK5, or K215RGRK6 were seeded into six-well culture plates; when confluent, they were loaded with [32P]orthophosphate (5 μCi/ml; Amersham Biosciences) in phosphate-free Krebs buffer (118.6 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 4.2 mM NaHCO3, 1.3 mM CaCl2, 10 mM HEPES, and 11.7 mM glucose), pH 7.4. After 60 min at 37°C, methacholine (MCh; 100 μM) was added to the cells for 3 min. Cells were then solubilized, and receptors were immunoprecipitated using a specific anti-human M3 mAChR antibody (Tobin and Nahorski 1993) and electrophoretically resolved as described previously (Willets et al., 2001a). Autoradiograms were documented and analyzed using the GeneGenius system and software (Syngene, Cambridge, UK). The effects on M3 mACh receptor phosphorylation of dominant-negative K46RCK1α delivered into cells by an adenovirus was also assessed 48 h after infection into either control or K215RGRK6 expressing cells.
Casein Phosphorylation. After 48-h transfection, wild-type CK1α or adenoviral dominant-negative K46RCK1α were immunoprecipitated from control (P3) cells, using either an anti-CK1α (Budd et al., 2000) or an anti-FLAG (M2; Sigma, UK) antibody, respectively. After immunoprecipitation, kinase activity was assessed as described previously (Willets et al., 2002).
Imaging Single Cell InsP3. Real-time visualization of changes in the subcellular distribution of eGFP-PHPLCδ was performed as described previously (Nash et al., 2001a,b). Briefly, images were taken using an Ultraview LCI confocal system (PerkinElmer Life Sciences, Boston, MA) connected to an Olympus IX-70 microscope with an oil immersion objective (100×). Cells were seeded onto 25-mm coverslips at about 50% confluence and transfected ∼2 h later with 1 μg of eGFP-PHPLCδ plasmid DNA using LipofectAMINE 2000 (Invitrogen), according to the manufacturer's instructions. After 48 h, cells were transferred to a custom-built chamber maintained at 37°C by a Peltier control unit. Cells were perfused with Krebs buffer (5 ml/min) using a Gilson Minipuls-2 pump. Confocal images were collected by excitation at 488 nm and light capture by a digital charge-coupled device camera. MCh was applied via the perfusion line for the times indicated in Figs. 6, 7, 8. Changes in cytosolic eGFP fluorescence are shown as ratios obtained by dividing the agonist-stimulated signal by the average prestimulation fluorescence, after subtraction of autofluorescence.

Measurement of changes in single cell InsP3 and receptor desensitization using the eGFP-PHPLCδ bioprobe. A, representative trace showing peak and plateau InsP3 response in P3 control cells caused by stimulation with 100 μM MCh for 8 min followed by atropine addition (1 μM) for 3 min. B, assessment of increases in cytosolic eGFP-PHPLCδ fluorescence as an index of InsP3 generation in response to 2-min MCh (100 μM) applications (as shown by the horizontal bars) interspersed by 5-min wash periods. Data are representative of four separate experiments.

Assessment of M3 mAChR desensitization using eGFP-PHPLCδ fluorescence measurements. A, representative trace showing the InsP3 responses to addition of MCh, at an approximate EC50 concentration (either 3 or 10 μM, depending on individual cells) applied for 2 min (R1) followed by a 5-min washout period; a desensitizing dose of MCh (100 μM) was then added for 3 min. After a 5-min washout period, a second application of the same EC50 MCh concentration was made for a further 2 min (R2). Receptor desensitization was determined as the reduction in peak InsP3 formation in R2 compared with R1. The images above the graph are from a representative experiment showing (from left to right) basal InsP3 levels, followed by peak fluorescence generated by subsequent addition of 3 μM (R1, for 2 min), 100 μM (for 3 min), and 3 μM (R2, for 2 min) MCh, respectively. B) Desensitization of the M3 mAChR-mediated InsP3 response in single vector-control (P3, ▪), GRK6 overexpressing cells (GRK6/24, □), or K215RGRK6-expressing (D1,  ) cells. Data are means ± S.E.M. (n = 5 to 10 separate experiments, per clone) for the percentage change in R2 relative to the R1 response. Receptor desensitization was significantly enhanced in cells overexpressing GRK6 and significantly reduced in K215RGRK6-expressing cells (*, p < 0.05; **, p < 0.01, two-way analysis of variance, followed by Student's t test).
) cells. Data are means ± S.E.M. (n = 5 to 10 separate experiments, per clone) for the percentage change in R2 relative to the R1 response. Receptor desensitization was significantly enhanced in cells overexpressing GRK6 and significantly reduced in K215RGRK6-expressing cells (*, p < 0.05; **, p < 0.01, two-way analysis of variance, followed by Student's t test).

Suppression of MCh-stimulated PLC activity by overexpression of K220RGRK2 and K220RGRK3. A, measurement of changes in single-cell InsP3 using the eGFP-PHPLCδ bioprobe in cells expressing K220RGRK2, K220RGRK3, or empty vector (P3). Representative traces are shown for P3 control, K220RGRK2 expressing (clone 9), or K220RGRK3 expressing (clone 6) cells stimulated with MCh (100 μM) for 60 s. The bar above the graph indicates the MCh (100 μM) application period. B, cumulative data showing inhibition of peak InsP3 accumulation. C, time-courses of InsP3 mass accumulation in P3 control (▪), K220RGRK2-expressing (clone 9, •), or K220RGRK3-expressing (clone 6, □) after stimulation with MCh (100 μM). D, MCh-stimulated concentration-response curves in P3 (control), K220RGRK2 expressing (clone 9, ▪), and K220RGRK3 expressing (clone 6, □) cells. For data shown in C and D, InsP3 accumulation was determined as described under Materials and Methods. Data are shown as means ± S.E.M. for four experiments, each performed in duplicate.
Measurement of InsP3 Mass. Plasmid control or cells expressing K220RGRK2 or K220RGRK3 were seeded into 24-well plates. When confluent, cells were washed twice with Krebs buffer, pH 7.4, before addition of MCh. For time-course experiments, MCh (100 μM) was added for up to 5 min; for concentration-response curves, increasing concentrations of MCh (10-8 to 10-4 M) were added for 3 min. Incubations were terminated by rapid aspiration and addition of 0.5 M trichloroacetic acid. Extracts were neutralized and InsP3 determined as described previously (Challiss et al., 1988).
Cyclic AMP Measurements. Confluent monolayers of SH-SY5Y cells stably expressing the plasmid control (P3) or overexpressing GRK6 (6/24) or the dominant-negative GRK6 protein (D1) were incubated with either the A2 adenosine receptor agonist CGS21680 (100 μM) or vehicle for 15 min. After this time, the medium over the cells was carefully aspirated, and cells were washed four times with 1 ml of KHB. The indicated concentration of CGS21680 (1-100 μM) was added 5 min after initiating the washing procedure and after 3-min incubations terminated by aspiration and addition of ice-cold 0.5 M trichloroacetic acid. Samples were neutralized and cyclic AMP concentrations determined exactly as described previously (Brown et al., 1971).
Data and Statistical Analysis. All data were analyzed using one or two-way analysis of variance (Excel 5.0; Microsoft, Redmond, WA). Significance was accepted when p < 0.05.
Results
Creation of Stable, Dominant-Negative GRK-Expressing Cell Lines. Transfection of wild-type SH-SY5Y cells with either K220RGRK2 or K220RGRK3 produced several G-418-resistant colonies that were expanded into cell lines. Clones that overexpressed either K220RGRK2 or K220RGRK3 were chosen for further study and were matched with previously characterized plasmid-transfected control, K215RGRK5, and K215RGRK6 cell lines (Willets et al., 2002) with respect to receptor expression based on N-methyl-[3H]scopolamine binding (Table 1). M3 mACh receptor characteristics (Bmax and KD values) in each clone were not found to change with passage (data not shown). Estimation of the level of dominant-negative GRK was determined by quantification of Western blots (Fig. 1). K220RGRK2 expression in clones 2 and 9 was 22- and 32-fold greater and K22ORGRK3 expression in clones 6 and 10 was 30- and 15-fold greater compared with GRK2 and GRK3 expression levels in vector-transfected (P1/P3) control cells. As described previously, levels of K225RGRK6 were ∼30-fold greater than endogenously expressed GRK6 in SH-SY5Y cells (Willets et al., 2002). The clones used were also examined to determine CK1α expression using a polyclonal anti-CK1α antibody. CK1α expression was similar in all clones examined (Fig. 1D).
Whole cell [3H]NMS saturation binding to determine M3 mAChR expression in plasmid control (P3), K220RGRK2, K220RGRK3, K215RGRK5, and K215RGRK6 clones The Bmax and KD values were determined using nonlinear regression analysis and curve-fitting programs of GraphPad Prism 3. The data are expressed as the means of specific binding ± S.E.M. for three to five separate experiments.

Determination of (dominant-negative) GRK and CK1α expression in stably transfected SH-SY5Y cells. Whole-cell lysates were subjected to SDS-polyacrylamide gel electrophoresis followed by Western transfer and immunoblotting with polyclonal antibodies recognizing GRK2 (A), GRK3 (B), GRK6 (C), or CK1α (D). In all cases, lanes were loaded with 40 μg of protein: lane 1, P3 (plasmid control); lane 2, K220RGRK2 (clone 2); lane 3, K215RGRK6 (clone D1); lane 4, K220RGRK3 (clone 6); lane 5, P1 (plasmid control); lane 6, K220RGRK2 (clone 9); lane 7, K220RGRK3 (clone 10); lane 8, GRK6 clone 24; lane 9, K215RGRK5 (clone 2). Blots shown are representative of experiments performed on at least three separate occasions with similar results.
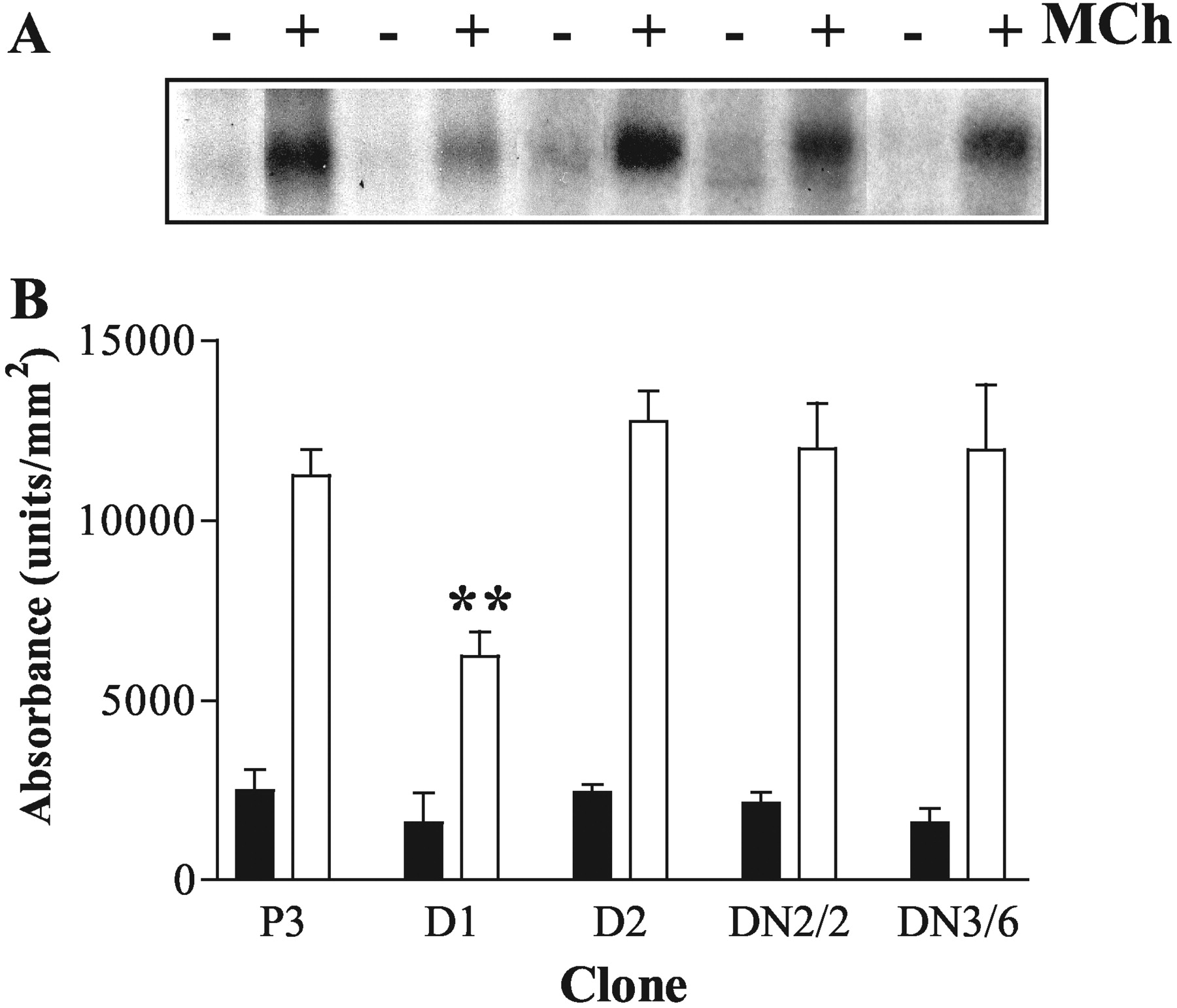
Effects of Dominant-Negative GRKs on Agonist-Stimulated M3 mAChR Phosphorylation. After 3 min of stimulation with MCh (100 μM), a robust M3 mAChR phosphorylation was detected in plasmid control cells (Fig. 2). As observed previously (Willets et al., 2002), agonist-driven M3 mAChR phosphorylation was inhibited (by ∼50%) in cell-lines overexpressing K215RGRK6 (D1; Fig. 2). The residual increase in receptor phosphorylation in K215RGRK6-expressing cells suggests that other kinases may also phosphorylate the M3 mACh receptor in an agonist-dependent manner. In addition to GRK6, the SH-SY5Y cell line endogenously expresses GRKs 2 and 3 (Fig. 1), and we have previously shown that GRK3 can attenuate M3 mAChR signaling (Willets et al., 2001a). Therefore, we investigated whether K220RGRK2 or K220RGRK3 in this cell line could affect MCh-stimulated M3 mAChR phosphorylation. Neither K220RGRK2 (clone GRK2DN/2) nor K220RGRK3 (clone GRK3DN/6) had any effect on agonist-stimulated receptor phosphorylation (Fig. 2). Furthermore, and as shown previously (Willets et al., 2002), K215RGRK5 overexpression (clone D2; Fig. 2) had no effect on MCh-stimulated M3 mAChR phosphorylation, probably because of the relative or absolute lack of GRK5 expression in SH-SY5Y cells. Densitometric analysis confirmed that basal phosphorylation of M3 mAChR was unaffected by expression of any of the dominant-negative GRKs, and only K215RGRK6 significantly (p < 0.01) inhibited agonist-stimulated M3 mAChR phosphorylation (Fig. 2B). Similar data were obtained using another set of SH-SY5Y clones expressing K220RGRK2 (clone 9), K220RGRK3 (clone 10), or K215RGRK6 (clone D5) (data not shown).
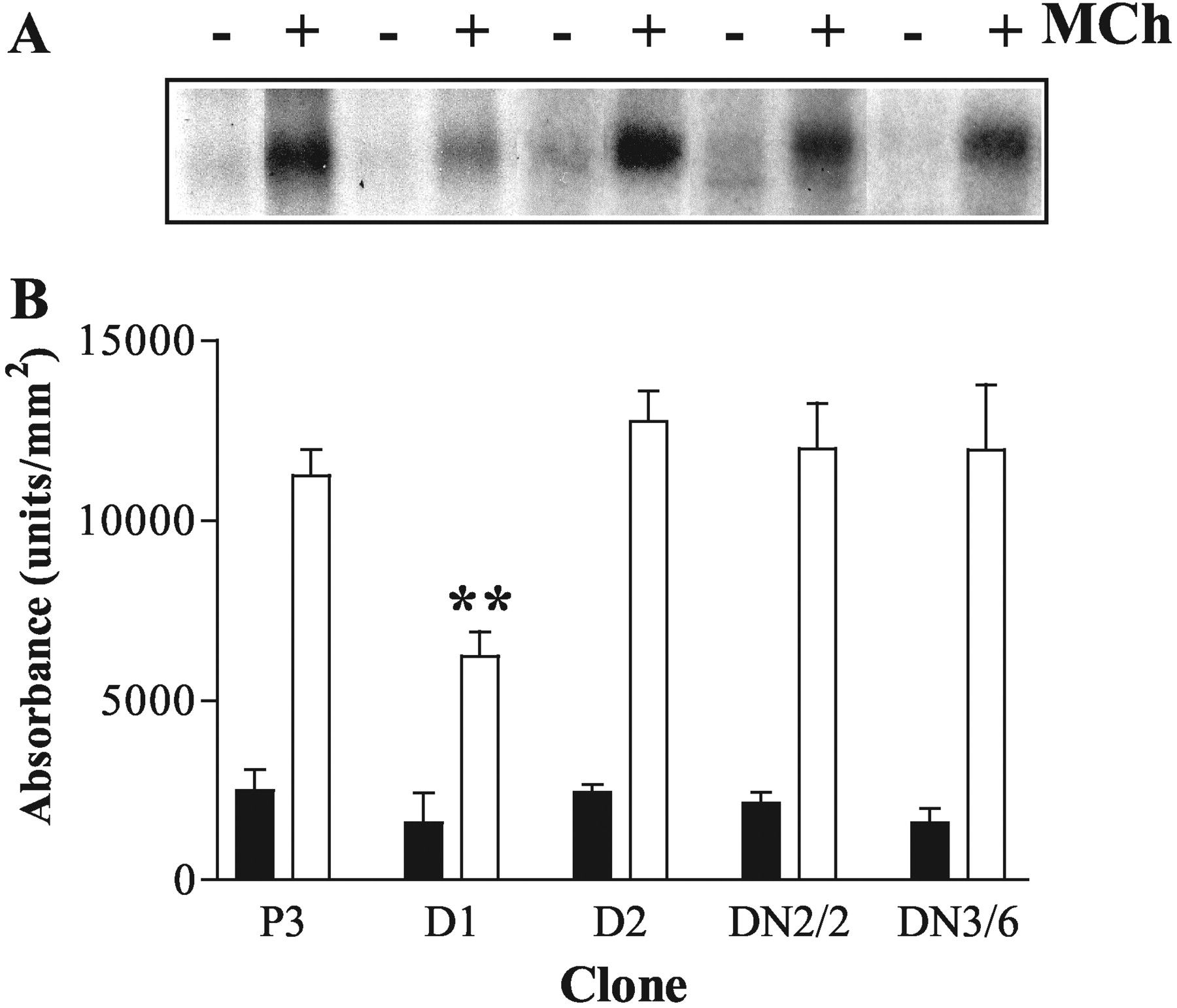
Effects of stable, dominant-negative GRK expression on agonist-stimulated M3 mACh receptor phosphorylation in SH-SY5Y cells. Confluent plasmid control, and cells expressing K220RGRK2, K220RGRK3, K215RGRK5, or K215RGRK6, were loaded with [32P]orthophosphate (5 μCi/ml), incubated, and processed for visualization of M3 mAChR phosphorylation as described under Materials and Methods. A, representative autoradiogram of M3 mAChR phosphorylation in P3 (lanes 1 and 2), D1 (K215RGRK6-expressing; lanes 3 and 4), D2 (K215RGRK5-expressing; lanes 5 and 6), K220RGRK2-expressing (clone 2; lanes 7 and 8), or K220RGRK3-expressing (clone 6, lanes 9 and 10) cell lines. B, densitometric analysis of agonist-stimulated M3 mAChR phosphorylation. M3 mAChR phosphorylation is shown as basal (▪), and after MCh stimulation (100 μM, 3 min, □). Data are expressed as the means ± S.E.M. of three separate experiments. Expression of K215RGRK6 significantly inhibited MCh-stimulated M3 mAChR phosphorylation (**, p < 0.01) compared with plasmid controls and cell lines expressing other dominant-negative GRKs.
Effects of Protein Kinase C on M3 mAChR Phosphorylation. Agonist stimulation of M3 mACh receptors leads to activation of Ca2+ and/or diacylglycerol-stimulated PKC isoenzymes, which may feedback to phosphorylate the receptor. We examined the effect of down-regulating PKC isoenzymes by PDBu (1 μM; 24 h) treatment on agonist-stimulated M3 mAChR phosphorylation. In control cells (P3), short-term treatment with PDBu (1 μM; 3 min) stimulated an increase of ∼2-fold in M3 mAChR phosphorylation that was not observed after 24-h PDBu pretreatment (Fig. 3A). However, long-term PDBu pretreatment had no effect on agonist-stimulated M3 mAChR receptor phosphorylation (Fig. 3A). Furthermore, the 50% inhibition of M3 mAChR phosphorylation seen in K215RGRK6-overexpressing (D1) SHSY5Y cells was not further affected by long-term PDBu pretreatment (Fig. 3A). In addition, the broad spectrum PKC inhibitor Ro 31-8220 had no effect on agonist-stimulated M3 mAChR phosphorylation in SH-SY5Y (P3) cells at a concentration (10 μM) that completely blocked PDBu-induced receptor phosphorylation (Fig. 3B).

Effect of PKC down-regulation or inhibition on agonist-stimulated M3 mAChR phosphorylation. A, plasmid-control (P3; □) or K215RGRK6-expressing (D1; ▪) cell lines were pretreated with PDBu (1 μM) or vehicle for 24 h before loading with [32P]orthophosphate, incubation, and processing for visualization of M3 mAChR phosphorylation as described under Materials and Methods. Basal, no MCh; MCh (100 μM; 3 min); Basal + PDBu, basal after PDBu pretreatment; MCh + PDBu, MCh (100 μM for 3 min after PDBu pretreatment); PDBu, short-term PDBu (1 μM, 3 min); PDBuPT, PDBu, (1 μM, 3 min) after PDBu pretreatment. Expression of K215RGRK6 significantly inhibited MCh-stimulated M3 mAChR phosphorylation compared with plasmid control cells in both vehicle and PDBu pretreated cells; **, p < 0.01. B, effect of the PKC inhibitor Ro 31-8220 (10 μM) on agonist-stimulated M3 mAChR phosphorylation was determined in control (P3) cells. Cells were either pre-incubated with vehicle (□) or Ro 31-8220 (▪) for 15 min before MCh stimulation for 3 min. PDBu (1 μM) was also added for 3 min to stimulate M3 mAChR phosphorylation in the presence or absence of Ro 31-8220. All data are shown as means ± S.E.M. for four separate experiments.
Effect of K46RCK1α on M3 mACh Receptor Phosphorylation. SH-SY5Y cells express CK1α, which has been shown to phosphorylate recombinant M3 mAChRs expressed in HEK293 and CHO cells (Tobin et al., 1997; Budd et al., 2000). To assess whether CK1α phosphorylates endogenously expressed M3 mAChR in SH-SY5Y cells, we infected control (P3) and K215RGRK6-expressing (D1) cells with an adenoviral dominant-negative CK1α construct (K46RCK1α). Adenovirus-infected SH-SY5Y cells seemed healthy and expressed ∼20-fold higher levels of K46RCK1α compared with endogenous CK1α (Fig. 4C). To determine whether the adenoviral K46RCK1α is devoid of kinase activity, K46RCK1α was immunoprecipitated from SH-SY5Y cell lysates (48 h after infection) with the commercial anti-FLAG (M2) antibody. Wild-type CK1α was also immunoprecipitated from noninfected SH-SY5Y cells using a specific anti-CK1α antibody (Budd et al., 2000). Kinase activity was assessed by incubation with dephosphorylated α-casein, in the presence of [γ-32P]ATP. Robust α-casein phosphorylation was detected in the presence of wild-type CK1α but not in the presence of K46RCK1α (Fig. 4B). Infection of either control (P3) or K215RGRK6-expressing SH-SY5Y cell monolayers with K46RCK1α had no effect on either basal or MCh (100 μM, 3 min) stimulated M3 mAChR phosphorylation (Fig. 4A).
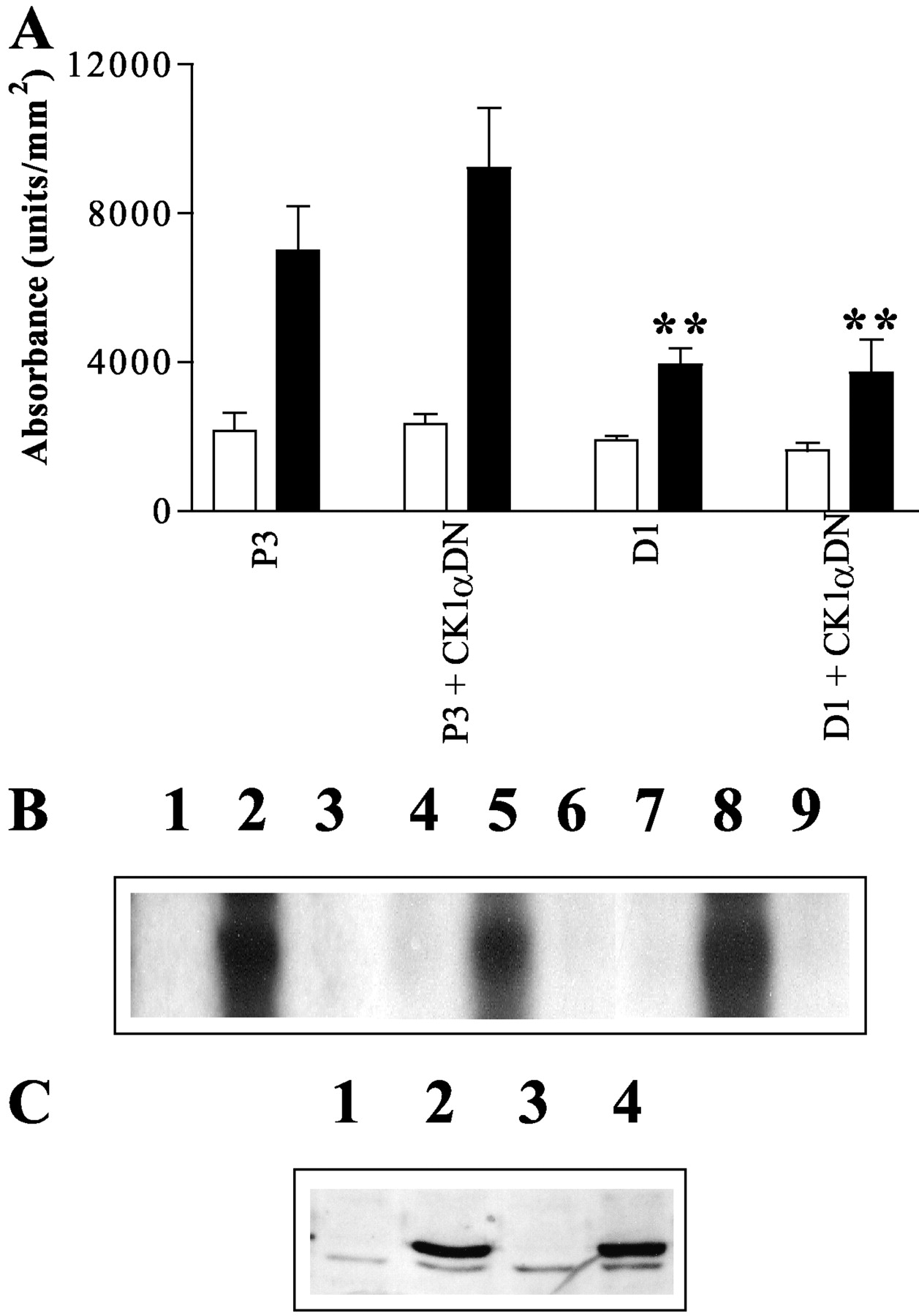
Inhibition of MCh-stimulated M3 mAChR phosphorylation by dominant-negative K215RGRK6 but not K46RCK1α. A, control (P3) or K215RGRK6-expressing (D1) cells at ∼50% confluence were infected with adenovirus encoding K46RCK1α. After 48 h, cells were [32P]orthophosphate-loaded, incubated with agonist, and processed for assessment of M3 mAChR phosphorylation as described under Materials and Methods. Autoradiograms were quantified and results expressed as means ± S.E.M. for four separate experiments. M3 mAChR phosphorylation is shown for basal conditions (□) and for MCh-stimulation (100 μM, 3 min; ▪). Expression of K215RGRK6, but not K46RCK1α (CK1αDN), significantly inhibited MCh-stimulated M3 mAChR phosphorylation (**, p < 0.01) compared with plasmid controls. B, the K46RCK1α protein is devoid of catalytic activity. Control (P3) cells were infected with K46RCK1α adenovirus for 48 h before immunoprecipitation of K46RCK1α using the specific monoclonal M2 anti-FLAG antibody (lane 3). Wild-type CK1α was immunoprecipitated from nontransfected P3 cells, using a specific polyclonal anti-CK1α antibody (lane 2). Immunoprecipitates were added to dephosphorylated α-casein and [γ-32P]ATP (10 min, 37°C) and α-casein phosphorylation was determined by autoradiography. In addition, the catalytic activities of the dominant-negative K220RGRK2 and K215RGRK6 were also analyzed. HEK293 cells were transfected with either GFP-tagged wild-type GRK2 or GRK6, or GFP-tagged K220RGRK2 or K215RGRK6 for 48 h before immunoprecipitation with a specific anti-GFP antibody. Lane 1 shows α-casein phosphorylation after immunoprecipitation in the absence of primary antibody. Lanes 4 and 7 show α-casein phosphorylation after immunoprecipitation in the absence of primary antibody. Robust casein phosphorylation was observed after immunoprecipitation of GFP-tagged wild-type GRK2 (lane 5) and GRK6 (lane 8). No phosphorylation was detected in the presence of GFP-tagged K220RGRK2 (lane 6) or K215RGRK6 (lane 9). C, Western blot detection of adenoviral K46RCK1α. Cell lysates (40 μg per lane) were subjected to SDS-polyacrylamide gel electrophoresis followed by Western transfer and immunoblotting with the anti-CK1a antibody. Lane 1, noninfected P3 cells; lane 2, P3 cells infected with K46RCK1α adenovirus; lane 3, noninfected D1 cells; D1 cells infected with K46RCK1α adenovirus. For B and C, the blots shown are representative of three or four separate experiments, respectively.
Effects of Other Kinases on Agonist-Stimulated M3 mAChR Phosphorylation. A range of inhibitors or activators was tested to assess their influence on agonist-stimulated M3 mAChR phosphorylation in SH-SY5Y cells. These included use of the Src-family tyrosine kinase inhibitor PP1 (5 μM); possible cyclic AMP/protein kinase A involvement was assessed by direct activation of adenylyl cyclase by forskolin (10 μM) or addition of the PKA inhibitor myristoylated-PKA inhibitor 12-22-amide (20 μM); and assessment of Ca2+-calmodulin-dependent protein kinase involvement was made using KN62 (5 μM). Of the pharmacological agents used, only KN62 had any effect, producing a small inhibition (18 ± 5%; mean ± S.E.M. for n = 4 experiments) of MCh-stimulated M3 mAChR phosphorylation.
Possible Involvement of GRK6 in the Regulation of other Endogenous G Protein-Coupled Receptors. To assess whether altered GRK6 activity in SH-SY5Y cells affects other endogenously expressed G protein-coupled receptors, agonist-stimulated desensitization of adenosine A2 receptors was examined. Pretreatment of confluent P3 control cells with the adenosine A2 agonist CGS21680 (100 μM) for 15 min caused a 52 ± 7% decrease in maximal cyclic AMP response (Emax) and only a slight shift in the CGS21680 EC50 value (Fig. 5). The effect of pretreatment with CGS21680 was similar in both GRK6-overexpressing (D6/24) and K215RGRK6-expressing (D1) cell-lines, causing 51 ± 5 and 47 ± 4% decreases in Emax, respectively. Comparative studies between the three cell lines using either shorter preincubation times (5 min) or lower concentrations of CGS21680 also failed to reveal any differences (data not shown). Thus, adenosine A2 receptor desensitization seems to occur independently of GRK6 in SH-SY5Y cells.

Effects of stable or dominant-negative expression of GRK6 on A2 adenosine receptor responses in SH-SY5Y cell-lines. Confluent monolayers of vector control (P3; □, ▪), D1 (K215RGRK6-expressing; ○, •), and GRK6/24 (GRK6-overexpressing; ▿, ▾) cell lines were incubated in KHB containing 300 μM 3-isobutyl-1-methylxanthine for ∼30 min before addition of 100 μM CGS21680 (closed symbols) or vehicle (open symbols) for 15 min. Cell monolayers were carefully washed over a 5-min period and then rechallenged with the indicated concentrations of CGS21680 for 3 min. Incubations were terminated and neutralized, and cyclic AMP levels were determined as described under Materials and Methods. Data are expressed as a percentage of the maximal response observed in P3 cells prestimulated with vehicle (basal, 9.1 ± 1.6; +100 μM CGS21680, 94.4 ± 6.9 pmol/mg protein) and represent means ± S.E.M. for three experiments, each performed in duplicate.
`Real-Time' Assessment of M3 mAChR Desensitization in Single SH-SY5Y Cells. The development of fluorescent bioprobes, such as the PH domain of PLCδ1, provides a means to measure agonist-stimulated InsP3 generation in single cells in real time (Nash et al., 2001a,2001b). This approach can be adapted to follow the time course of receptor desensitization in single SH-SY5Y cells. In initial experiments, control (P3) cells were transfected with eGFP-PHPLCδ (1 μg) for 48 h, and eGFP-PHPLCδ translocation to the cytoplasm was detected upon stimulation of the M3 mAChR. MCh (100 μM) produced a rapid peak followed by a plateau phase, which was rapidly reversed by the addition of the mAChR antagonist atropine (1 μM) (Fig. 6A). In an attempt to assess M3 mAChR desensitization at the level of the InsP3 response, 2-min additions of MCh (100 μM) were applied with 5-min intervening wash periods. A 5-min wash period was chosen because within this period, cytosolic eGFP-PHPLCδ fluorescence intensity returns to prestimulation levels, and previous work has shown that phosphatidylinositol-4,5-bisphosphate levels are fully restored in this cell type (Willars et al., 1998). However, using this protocol, we observed only a gradual waning of the M3 mAChR-stimulated peak InsP3 response (Fig. 6B), and it proved difficult to observe reproducible differences in P3, GRK6-overexpressing (GRK6/24), or K215RGRK6-expressing cells (data not shown).
A different protocol was then adopted that involved a comparison of the eGFP-PHPLCδ translocation response with a submaximal agonist concentration before and after a 'desensitizing' agonist treatment (100 μM MCh, 3 min) (see Fig. 7A). In SH-SY5Y cell population studies, the EC50 for MCh-stimulated InsP3 accumulation is ∼5 μM (Martin et al., 1999). However, we found that EC50 values varied between individual SH-SY5Y cells, which caused us to use either 3 or 10 μM MCh as an approximate EC50 concentration depending on the cell studied. Thus, 3 (or 10) μM MCh was applied for 2 min and, after a 5-min washout, the desensitizing concentration of MCh (100 μM) was added for 3 min followed by a further 5-min washout. A second application of either 3 (or 10) μM was then made for a further 2 min (see Fig. 7A). In control (P3) cells, comparison of the second peak InsP3 (R2) response evoked by an EC50 concentration of MCh compared with the initial peak (R1) showed a clear reduction (desensitization) of 48 ± 4% and 64 ± 16% at 3 and 10 μM MCh, respectively (Fig. 7B). Using this protocol, we showed that overexpression of GRK6 significantly increased the extent of receptor desensitization, whereas K215RGRK6-expressing cells showed a significant reduction in the desensitization (Fig. 7B). These data confirm and extend our previous findings in populations of SH-SY5Y cells (Willets et al., 2001a, 2002).
Despite its ability to enhance M3 mAChR phosphorylation and inhibit PLC signaling when overexpressed in SH-SY5Y cells, GRK3 does not enhance M3 mAChR/Gαq/11 uncoupling (Willets et al., 2001a). These data suggest that GRK3 inhibits PLC signaling independently of its ability to phosphorylate the M3 mAChR. We examined whether either dominant-negative GRK2 or GRK3 was able to inhibit M3 mAChR-stimulated PLC signaling in SH-SY5Y cells in real time. Overexpression of K220RGRK2 or K220RGRK3 suppressed maximal MCh (100 μM, 60 s)-stimulated InsP3 production by 75% when measuring translocation of eGFP-PHPLCδ (Fig. 8, A and B) and was as effective as overexpression of wild-type GRK2 (data not shown). A similar inhibition of PLC activity was observed by analysis of InsP3 mass. Overexpression of K220RGRK2 or K220RGRK3 decreased peak InsP3 formation to maximal MCh stimulation and reduced plateau InsP3 levels by ∼50% (Fig. 8C), whereas MCh concentration-response curves (at 3 min) exhibited suppressed maximal InsP3 responses and right-shifted curves (Fig. 8D).
Atropine Reversal of MCh-Stimulated M3 mAChR Phosphorylation. Measurement of receptor desensitization in single cells using eGFP-PHPLCδ required a 5-min wash period between individual agonist applications to minimize the potential effects of Ca2+ and/or phosphatidylinositol-4,5-bisphosphate depletion on MCh-stimulated PLC signaling. However, the wash period after the desensitizing dose of MCh (100 μM, 3 min) may allow dephosphorylation of the M3 mAChR to occur via the action of protein phosphatases. In an attempt to gauge the effect of the washing period on M3 mAChR phosphorylation, [32P]orthophosphate-loaded P3 cells were stimulated with MCh (100 μM, 3 min), and then the mACh receptor antagonist atropine was added for 3, 5, or 10 min. Examination of 32P-labeled M3 mAChRs after atropine addition showed that 44 ± 5% had been lost after 5 min (Fig. 9).
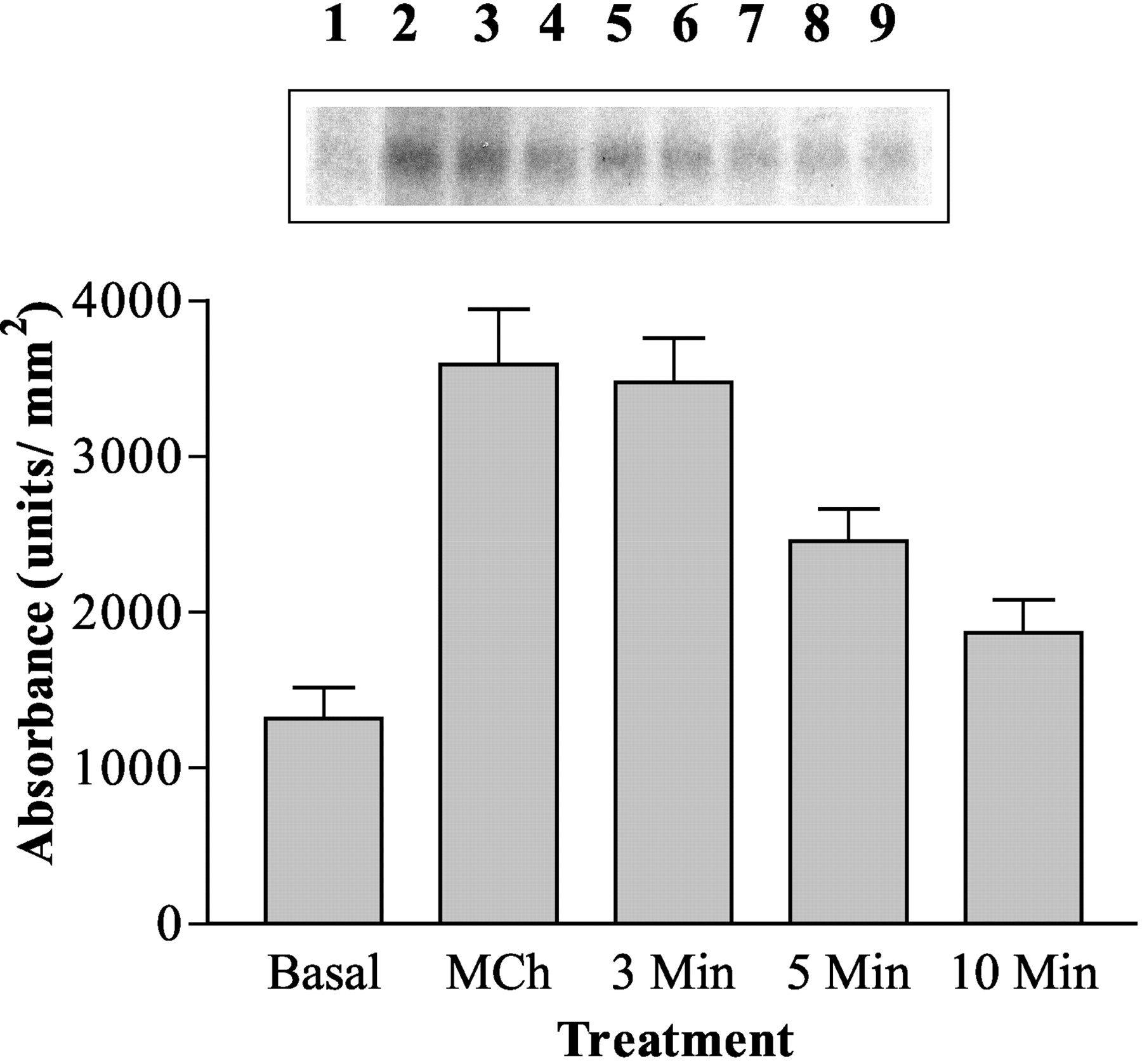
Reversal of MCh-stimulated M3 mAChR phosphorylation after atropine addition. Confluent P3 cells were loaded with [32P]orthophosphate as described under Materials and Methods. Cells were challenged with MCh (100 μM) for 3 min (lanes 2 and 3) before addition of atropine (1 μM) for increasing time periods (3 min, lanes 4 and 5; 5 min, lanes 6 and 7; or 10 min, lanes 8 and 9, respectively). Lane 1 shows receptor phosphorylation after addition of vehicle alone (basal). After agonist/antagonist challenge, cells were processed as described under Materials and Methods. The upper shows a representative autoradiogram of M3 mAChR phosphorylation under the different conditions. In the graph, data are shown for densitometrically analyzed autoradiograms and are expressed as means ± S.E.M. for single or duplicate samples from four separate experiments.
Discussion
In previous studies, we have shown the important role that GRK6 plays in the regulation of endogenous human M3 mAChRs expressed in SH-SY5Y neuroblastoma cells (Willets et al., 2001a; 2002). These studies also highlighted the phosphorylation-independent regulation by GRK3, possibly exerted via the N-terminal regulator of G protein signaling (RGS) domain associating with Gαq (Carman et al., 1999; Sallese et al., 2000). In these studies, suppression of the PLC activity was monitored biochemically after continuous stimulation of the M3 mAChR. In the present study, we have used a single cell imaging approach to assess InsP3 generation by the translocation of the pleckstrin homology domain of PLCδ (eGFP-PHPLCδ) (Hirose et al., 1999; Nash et al., 2001a,b). This approach has allowed us not only to follow signaling in real time but also to assess a prestimulation-mediated desensitization. Using such a protocol, we have previously shown that application of maximal MCh concentration for 3 min will induce ∼50% M3 mAChR-Gαq uncoupling when assessed by guanosine 5′-O-(3-[35S]thio)triphosphate binding in subsequently prepared membranes of SH-SY5Y cells (Willets et al., 2002). However, when used to assess M3 mAChR desensitization in single cells using eGFP-PHPLCδ, this protocol results in little desensitization even after multiple applications. This almost certainly reflects the significant receptor reserve that exists between activated Gαq and PLC activation, because when submaximal agonist concentrations were used to assess the responsiveness of pretreated cells, a highly significant suppression was detected. Crucially, manipulations of the GRK6-mediated M3 mAChR phosphorylation by overexpression of GRK6 or the catalytically inactive K215RGRK6 was accompanied by enhanced or reduced desensitization, respectively.
The inability to produce a more complete desensitization could reflect the experimental limitations of these imaging experiments in which an extensive (5 min) washout of pre-exposed agonist is required to assess subsequent receptor responsiveness. Removal of agonist activation of the M3 mAChR by atropine leads to significant dephosphorylation of the receptor by 5 min, implying that reversal by active protein phosphatases may limit the extent of desensitization under these conditions.
Another conundrum approached in these studies is that despite a 30-fold overexpression of K215RGRK6, it was difficult to observe suppression of agonist-mediated receptor phosphorylation by more than 50%. These data suggest two possibilities. First, 30-fold overexpression of K215RGRK6 is not sufficient for total inhibition of endogenous GRK6 activity. Alternatively, the residual agonist-mediated M3 mAChR phosphorylation is undertaken by other protein kinases. Indeed, SH-SY5Y cells contain many different kinases, which have the potential to phosphorylate the M3 mAChR in an agonist-dependent manner. In the present study, we have attempted to dissect any involvement of endogenous kinases other than GRK6 in M3 mAChR phosphorylation.
Because SH-SY5Y cells endogenously express both GRK2 and -3, either kinase may well phosphorylate the M3 mAChR on agonist stimulation. However, inhibition of GRK2 and GRK3 activity by expression of the dominant-negative K220RGRK2 or K220RGRK3 proteins failed to inhibit agonist-mediated M3 mAChR phosphorylation. These findings were surprising because other researchers and we have implicated GRK2 (Wu et al., 2000) and GRK3 (Willets et al., 2001a) in the regulation of M3 mAChR phosphorylation. Indeed, overexpression of wild-type GRK3 enhanced receptor phosphorylation within the same cell background (Willets et al., 2001a). Furthermore, studies using purified GRK2 have shown enhanced phosphorylation of M3 mAChRs (DebBurman et al., 1995). Moreover, Wu et al. (2000) have mapped the regions of the M3 mAChR third intracellular loop phosphorylated by GRK2. However, it is difficult to directly compare in vitro studies with those conducted in intact cells. In general, cellular models overexpressing both receptors and GRKs lead to an enhanced receptor phosphorylation, independent of the receptor examined (Diviani et al., 1996; Oppermann et al., 1996; Freedman et al., 1997; Blaukat et al., 2001; Willets et al., 2001a). It is also noteworthy that under in vitro conditions, GRK2 and GRK3 are equally proficient in phosphorylating the human M3 mAChR, whereas GRK6 seems ineffective (DebBurman et al., 1995).
Increasing numbers of studies are beginning to show that GRK selectivity for receptor regulation is revealed only through the inhibition of endogenous GRK activities toward endogenously expressed receptors (Mundell et al., 1997; Willets et al., 1999; Aiyar et al., 2000; Fong et al., 2002; Shetzline et al., 2002; Ghadessy et al., 2003). Indeed, despite the fact that overexpression of wild-type GRK 3 and GRK6 enhanced M3 mAChR phosphorylation (Willets et al., 2001a), in the present study, inhibition of endogenous GRK3 and GRK6 shows that only the latter mediates agonist-stimulated phosphorylation. Despite their inability to inhibit agonist-mediated M3 mAChR phosphorylation, overexpression of K220RGRK2 or K220RGRK3 suppressed agonist-stimulated PLC activity. Our data concur with a number of reports suggesting that GRK2 and GRK3 are able to inhibit agonist-stimulated PLC activity through a phosphorylation-independent mechanism (Carman et al., 1999; Sallese et al., 2000), probably via binding of the GRK N-terminal RGS-like domain to Gαq/11-GTP. These findings highlight the potential problems associated with the use of dominant-negative GRKs when examining PLC-coupled receptor desensitization and also suggest a different mechanistic basis for GRK2 and GRK3 regulation of Gαq/11-coupled receptors.
The lack of involvement of GRKs other than GRK6 in the regulation of M3 mAChRs in SH-SY5Y cells leaves open the possibility of regulation by other protein kinases. Previous studies from these laboratories have reported that CK1α enhances human M3 mAChR agonist-mediated phosphorylation when coexpressed in CHO cells (Tobin et al., 1997). Furthermore, K46RCK1α was able to inhibit agonist-stimulated M3 mAChR phosphorylation when coexpressed in HEK293 or CHO cells (Budd et al., 2000). However, our data show that expression by adenoviral infection of a catalytically inactive K46RCK1α surprisingly failed to inhibit agonist-mediated M3 mAChR phosphorylation in SH-SY5Y cells. In addition, no further inhibition of M3 mAChR above that seen with K215RGRK6 expression alone was observed upon K46RCK1α coexpression. The inability of K46RCK1α to inhibit M3 mAChR phosphorylation in SH-SY5Y cells may be related to cell background or experimental design and may again highlight the differential regulation of overexpressed and endogenous M3 mAChR populations.
In agreement with our previous findings, we report that the M3 mAChR is a substrate for PKC (Willars et al., 1999; Bundey and Nahorski, 2001). However, inhibition of PKC activity, or down-regulation by long-term pretreatment with phorbol ester, did not alter agonist-mediated M3 mAChR phosphorylation. It is also interesting that inhibition of PKC fails to prevent M3 mAChR-G protein uncoupling in SHSY5Y cells after agonist pretreatment (Bundey and Nahorski, 2001). These data suggest that although the human M3 mAChR is a substrate for PKC, PKC plays no discernible role in M3 mAChR regulation in SH-SY5Y cells. In addition, it seems that because GRK6-mediated M3 mAChR phosphorylation is unaffected by inhibition of PKC, PKC is unable to regulate GRK6 activity, unlike GRK2 or GRK5 (Chuang et al., 1995; Pronin and Benovic, 1997).
Further manipulation of second messenger-regulated kinases indicated that PKA also has no role in M3 mAChR regulation alone. PKA has recently been identified as a regulator of GRK2 activity via direct phosphorylation of Ser-685, which enhances Gβγ subunit binding and subsequently increases GRK2 membrane association (Cong et al., 2001); however, because inhibition of PKA has no effect upon M3 mAChR phosphorylation, it seems unlikely that PKA regulates GRK6 in a manner similar to that seen with GRK2 (Cong et al., 2001). The nonreceptor tyrosine kinase Src has been implicated in β2 adrenergic receptor regulation and desensitization (Fan et al., 2001) via the creation of an SH2 domain after agonist-mediated Tyr-350 phosphorylation, promoting Src binding and subsequent GRK2 recruitment and receptor phosphorylation. These authors reported that inhibition of Src abolished GRK2 phosphorylation and β2 adrenergic receptor desensitization. In SH-SY5Y cells, agonist-mediated M3 mAChR phosphorylation was unaffected by Src inhibition, which suggests either that the M3 mAChR is not a substrate for Src phosphorylation or that GRK6 activity is not regulated by Src phosphorylation.
Our data suggest that in SH-SY5Y cells, agonist-mediated phosphorylation and regulation of the M3 mAChR is undertaken by endogenous GRK6. However, it is unclear whether this action of GRK6 is specific for M3 mAChR receptors. In the present study, we have also examined the potential role of GRK6 to regulate another endogenously expressed GPCR in this neuroblastoma cell line. Endogenously expressed adenosine A2 receptors were unaffected by manipulations of GRK6 activity, and it is likely that this Gs-coupled receptor is regulated by alternate GRKs. Indeed, it has previously been shown that adenosine A2 receptors are regulated exclusively by GRK2 in NG108-15 cells (Mundell et al., 1997; Willets et al., 1999). Taken together, these data suggest that GRK6 seems to regulate selectively M3 mAChR receptor phosphorylation and desensitization in SH-SY5Y cells.
Overall, our data suggest that GRK6 is the predominant or only kinase that regulates the phosphorylation and desensitization of the M3 mAChR endogenously expressed in SHSY5Y cells. The inability to totally suppress agonist-stimulated receptor phosphorylation through overexpression of K215RGRK6 suggests the involvement of alternative kinases, although we have been unable to implicate GRK2, GRK3, GRK5, PKC, or CK1α in this process. Alternatively, it may be impossible to express enough of the exogenous kinase-dead mutants within the putative micro-domains in which endogenous receptor signaling/regulation processes may be concentrated. Currently, we are continuing to investigate regulation of PLC signaling in single cells including primary neurons. Gene silencing by means of antisense or RNA interference should identify relevant GRKs in the physiological regulation of muscarinic receptors.
Acknowledgments
We thank Dr. Andrew B. Tobin and John E. McDonald for providing the M3 mAChR and CK1α antisera, and adenoviral K46RCK1α, and Dr. Eamonn Kelly (Department of Pharmacology, University of Bristol, UK) for providing the dominant-negative GRK mutants. In addition, we are grateful to Andrew Tobin for many helpful discussions and his constructive criticism of this manuscript.
Footnotes
-
This work was supported by the Wellcome Trust Program Grant 062495/Z/00.
-
ABBREVIATIONS: PKA, protein kinase A; PKC, protein kinase C; CK, casein kinase; GRK, G protein-coupled receptor kinase; mAChR, muscarinic acetylcholine receptor; PLC, phospholipase C; HEK, human embryonic kidney; CHO, Chinese hamster ovary; MCh, methacholine; eGFP-PHPLCδ, enhanced green fluorescent protein tagged pleckstrin homology domain of phospholipase δ1; InsP3, inositol 1,4,5-trisphosphate; CGS21680, 2-[4-](2-carboxyethyl)phenyl]ethylamino]-5′-N-ethylcarboxamidoadenosine; PDBu, phorbol 12,13-dibutyrate; KN62, 1-(N,O-bis-[5-isoquinolinylsulfonyl]-N-methyl-l-tyrosyl)-4-phenylpiperazine; RGS, regulator of G protein signaling.
- Received April 15, 2003.
- Accepted July 28, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}