


Abstract
Cannabinoids have recently been shown to induce the expression of the cyclooxygenase-2 (COX-2) isoenzyme in H4 human neuroglioma cells. Using this cell line, the present study investigates the contribution of the second messenger ceramide to this signaling pathway. Incubation of cells with the endocannabinoid analog R(+)-methanandamide (R(+)-MA) was associated with an increase of intracellular ceramide levels. Enhancement of ceramide formation by R(+)-MA was abolished by fumonisin B1, a ceramide synthase inhibitor, whereas inhibitors of neutral sphingomyelinase (spiroepoxide, glutathione) and serine palmitoyltransferase (l-cycloserine, ISP-1) were inactive in this respect. R(+)-MA caused a biphasic activation of the p38 and p42/44 mitogen-activated protein kinases (MAPKs), with phosphorylation peaks occurring after 15-min and 4- to 8-h treatments, respectively. Inhibition of ceramide synthesis with fumonisin B1 was associated with a suppression of R(+)-MA-induced delayed phosphorylations of p38 and p42/44 MAPKs and subsequent COX-2 expression. The involvement of ceramide in COX-2 expression was corroborated by findings showing that C2-ceramide and neutral sphingomyelinase from Bacillus cereus caused concentration-dependent increases of COX-2 expression that were suppressed in the presence of 4-(4-fluorophenyl)-2-(4-methylsulfonylphenyl)-5-(4-pyridyl)imidazol (SB203580, a p38 MAPK inhibitor) or 2′-amino-3′-methoxyflavone (PD98059, a p42/44 MAPK activation inhibitor). In contrast, dihydro-C2-ceramide being used as a negative control did not induce MAPK phosphorylation and COX-2 expression. Collectively, our results demonstrate that R(+)-MA induces COX-2 expression in human neuroglioma cells via synthesis of ceramide and subsequent activation of p38 and p42/44 MAPK pathways. Induction of COX-2 expression via ceramide represents a hitherto unknown mechanism by which cannabinoids mediate biological effects within the central nervous system.
The cyclooxygenase (COX) enzyme catalyzes the first step in the metabolism of arachidonic acid to prostaglandins (PGs) and thromboxanes. The enzyme is bifunctional with fatty acid COX activity (catalyzing the reaction from arachidonic acid to PGG2) and PG hydroperoxidase activity (catalyzing the reaction from PGG2 to PGH2). In the early 1990s COX was demonstrated to exist as two genetically distinct isoforms. COX-1 is constitutively expressed as a “housekeeping” enzyme in most tissues and mediates homeostatic functions such as cytoprotection of the stomach and regulation of platelet aggregation. In contrast, COX-2, which is encoded by an immediate-early gene, can be up-regulated by various pro-inflammatory agents, including bacterial lipopolysaccharide, cytokines, and mitogens. However, although COX-2 was initially regarded as a source of only pathological prostanoids, recent studies have indicated that this isoenzyme mediates a variety of physiological responses (for review, see Hinz and Brune, 2002).
Over the past years, a relationship between cannabinoids and PGs has been established by several lines of evidence. Accordingly, various actions of cannabinoids within the central nervous system, including hippocampal neuronal death (Chan et al., 1998), dilation of cerebral arterioles (Ellis et al., 1995), psychoactive and behavioral effects (Burstein et al., 1989; Yamaguchi et al., 2001), and reduction of intraocular pressure (Green et al., 2001) have been associated with an increased production of PGs. Nonetheless, the mechanism of action responsible for enhanced PG production attributed to cannabinoid compounds has been elusive. A recent study from our laboratory has shown that cannabinoids induce the expression of COX-2 via a cannabinoid receptor-independent pathway (Ramer et al., 2001). However, the signaling events involved in the cannabinoid-mediated induction of COX-2 expression remain to be established. Recent investigations indicate that several actions of cannabinoids, including inhibition of glioma cell growth (Galve-Roperh et al., 2000), effects on energy metabolism (Guzman and Sanchez, 1999), and stimulation of ketogenesis (Blazquez et al., 1999) and glucose metabolism (Sanchez et al., 1998) are signaled through accumulation of the lipid second messenger ceramide. Ceramide is a membrane sphingolipid that can be generated by ceramide synthase, a key enzyme involved in de novo sphingolipid biosynthesis and in the reacylation of free sphingoid bases derived from sphingolipid turnover (Wang et al., 1991). Alternatively, ceramide can be formed as a result of sphingomyelin hydrolysis by sphingomyelinases (SMases) (Hannun, 1994). Besides a stimulatory action on the expression of a variety of proinflammatory mediators, ceramide has previously been reported to induce COX-2 expression (Subbaramaiah et al., 1998; Newton et al., 2000) and to activate mitogen-activated protein kinases (MAPKs) (Reunanen et al., 1998, Subbaramaiah et al., 1998), which have been implicated as upstream targets regulating COX-2 expression.
In the present study, we examined the involvement of the ceramide pathway in the induction of p38 and p42/44 MAPK phosphorylation and COX-2 expression by the endocannabinoid analog R(+)-methanandamide (R(+)-MA), in H4 human neuroglioma cells. Our results show that ceramide synthesized upon the action of ceramide synthase confers activation of MAPKs and increased COX-2 expression by R(+)-MA. Induction of ceramide production and COX-2 expression constitutes an as-yet-unknown mechanism by which cannabinoids may exert their diverse biological actions within the central nervous system.
Materials and Methods
Materials. AM-251, AM-630, anandamide, arachidonyl trifluoromethylketone (AACOCF3), and the neutral sphingomyelinase (nSMase) spiroepoxide inhibitor [N-[1R)-1-(hydroxymethyl)-2-oxo-2-[(8-oxo-1-oxaspiro[2.5]octa-4.6-dien-5-yl)amino]ethyl]decane-amid] were purchased from Alexis Deutschland GmbH (Grünberg, Germany). R(+)-MA, C2-ceramide, dihydro-C2-ceramide, C16-ceramide, fumonisin B1, PD98059, and SB203580 were purchased from Calbiochem (Bad Soden, Germany). Capsazepine, l-cycloserine, glutathione (reduced and oxidized forms), myriocin from Mycelia sterilia (ISP-1), and nSMase from Bacillus cereus were obtained from Sigma (Deisenhofen, Germany). WIN-55,212-2 was purchased from Biotrend (Köln, Germany). Dulbecco's modified Eagle's medium with 4 mM l-glutamine and 4.5 g/l glucose was from Cambrex Bio Science Verviers S.p.r.l. (Verviers, Belgium). Fetal calf serum and penicillin-streptomycin were obtained from PAN Biotech (Aidenbach, Germany) and Invitrogen (Karlsruhe, Germany), respectively.
Cell Culture. H4 human neuroglioma cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. The cells were grown in a humidified incubator at 37°C and 5% CO2. All incubations were performed in serum-free medium.
Quantitative RT-PCR Analysis. H4 neuroglioma cells were grown to confluence in 24-well plates. After incubation of cells with the respective test compounds or vehicle for the indicated times, supernatants were removed and cells were lysed for subsequent RNA isolation. Total RNA was isolated using the RNeasy total RNA Kit (Qiagen, Hilden, Germany). β-Actin-(internal standard) and COX-2 mRNA levels were determined by quantitative real-time RT-PCR. Briefly, this method uses the 5′→3′ exonuclease activity of the Thermus aquaticus polymerase to cleave a probe during PCR. A probe consists of an oligonucleotide coupled with a reporter dye (6-carboxyfluorescein) at the 5′-end of the probe and a quencher dye (6-carboxytetramethylrhodamine) at an internal thymidine. After cleavage of the probe, reporter and quencher dye become separated, resulting in an increased fluorescence of the reporter. Accumulation of PCR products was detected directly by monitoring the increase in fluorescence of the reporter dye using the integrated thermocycler and fluorescence detector ABI Prism 7700 Sequence Detector (Applied Biosystems, Darmstadt, Germany). Quantification of mRNA was performed by determining the threshold cycle (CT), which is defined as the cycle at which the 6-carboxyfluorescein fluorescence exceeds 10 times the S.D. of the mean baseline emission for cycles 3 to 10. COX-2 mRNA levels were normalized to β-actin according to the following formula: CT (COX-2) - CT (β-actin) = ΔCT. Subsequently, COX-2 mRNA levels were calculated using the ΔΔCT method: ΔCT (test compound) - ΔCT (vehicle) = ΔΔCT (test compound). The relative mRNA level for the respective test compound was calculated as 2-ΔΔCT × 100%. RT-PCR reaction was performed using the One Step RT-PCR kit (QIAGEN, Hilden, Germany). RNA samples were amplified using specific primers for human β-actin and COX-2 (TIB MOLBIOL, Berlin, Germany) as described previously (Ramer et al., 2001).
Western Blot Analysis. Cells grown to confluence in 10-cm dishes were incubated with test substance or vehicle for the indicated times. Afterward, H4 neuroglioma cells were washed, harvested, and pelleted by centrifugation. Cells were than lysed in solubilization buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% (v/v) Triton X-100, 10% (v/v) glycerol, 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, and 10 μg/ml aprotinin), homogenized by sonication, and centrifuged at 10,000g for 5 min. Supernatants were used for Western blot analysis. Proteins were separated on a 10% SDS-polyacrylamide gel. After transfer to nitro-cellulose and blocking of the membranes with 5% milk powder, blots were probed with specific antibodies raised to p38 MAPK, phosphop38 MAPK, p42/44 MAPK, or phospho-p42/44 MAPK (New England BioLabs GmbH, Frankfurt, Germany). Subsequently, membranes were probed with horseradish peroxidase-conjugated anti-rabbit IgG (New England BioLabs). Antibody binding was visualized by enhanced chemiluminescence (ECL) Western blotting detection reagents (Amersham Biosciences, Freiburg, Germany). For determination of cannabinoid and vanilloid receptors membrane fractions of proteins were used. Cells were lysed in solubilization buffer (250 mM sucrose, 10 mM EGTA, 2 mM EDTA, 20 mM Tris-HCl, pH 7.5, 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, and 10 μg/ml aprotinin), homogenized by sonication, and centrifuged at 13,000g for 20 min. Supernatants were centrifuged at 40,000g for 1 h. Pellets were resuspended in solubilization buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% (v/v) Triton X-100, 10% (v/v) glycerol, 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, and 10 μg/ml aprotinin). The blots were probed with antibodies raised to the CB1-(BD Biosciences GmbH, Heidelberg, Germany), CB2-(Calbiochem, Bad Soden, Germany), or VR1 receptor (Chemicon International, Temecula, CA).
Determination of PGE2 and Glutathione. Cells grown to confluence in 24-well plates were incubated with test substances or its vehicles for the indicated times. PGE2 concentrations in cell culture supernatants and intracellular levels of glutathione were determined using commercially available enzyme immunoassay kits (Cayman, Ann Arbor, MI).
Quantification of Intracellular C16-Ceramide Levels. Cells grown to confluence in 10-cm dishes were incubated with test substances or its vehicles for the indicated times. Cells were washed and harvested with ice-cold PBS and afterward pelleted by centrifugation. Pellets were resuspended in chloroform and methanol (1:2, v/v) containing internal standard (41.6 pg/ml dihydro-C2-ceramide). Extraction of lipids was performed according to the method of Bligh and Dyer (1959). Intracellular ceramide was determined by liquid chromatography mass spectrometry. The high-performance liquid chromatography system consisted of a PU-1585 pump and an AS-1550 auto injector (Jasco, Groβ-Umstadt, Germany). Masses were acquired on a Finnigan MAT LCQ ion trap spectrometer equipped with an atmospheric pressure chemical ionization interface (Thermoquest, Egelsbach, Germany) and connected to a PC running the standard software Navigator (version 1.2). High-performance liquid chromatography was carried out isocratically at ambient temperature using a Nucleosil C8 guard column (120-5, 11 × 2 mm; Macherey-Nagel, Düren, Germany) and an eluent comprising methanol and water (90:10, v/v) and 0.2% formic acid, at a flow rate of 200 μl/min. The injection volume was 20 μl. The run time was 5 min. The outlet was coupled to the mass spectrometer's atmospheric pressure chemical ionization source. The vaporizer temperature was set to 325°C, and N2 was applied as sheath and auxiliary gas at flow rates of 40 and 10 (arbitrary units), respectively. The heated capillary was maintained at 120°C. Mass analysis was performed at unit resolution in the positive ion mode with the corona discharge current set to 10 μA, the potential of tube lens to 50 V, and the potential of capillary to 26 V. The ion trap was operated in the tandem mass spectroscopy mode and the transition of C16-ceramide [parent m/z 538.2, collision-induced dissociation energy 38%, isolation width 3 Da, product m/z 520.3] and dihydro-C2-ceramide [parent m/z 344.3, collision-induced dissociation energy 35%, isolation width 2 Da, product m/z 326.2] were followed by full-scan tandem mass spectroscopy. Cellular lipid extracts were resuspended in 1 ml of methanol just before mass analysis. Standards were analyzed at concentrations ranging from 7.2 nM to 1 μM for intraday reproducibility of the method. The peak areas of C16-ceramide and dihydro-C2-ceramide were determined. Dihydro-C2-ceramide was used as internal standard to quantify the efficiency of lipid extraction. Ratios of peak areas (C16-ceramide/dihydro-C2-ceramide) were normalized to protein amounts.
Statistics. Comparisons between groups were performed with Student's two-tailed t test.
Results
Time Course of R(+)-MA-Induced COX-2 mRNA Expression and PGE2 Synthesis. Incubation of H4 neuroglioma cells for 0.25 to 24 h with R(+)-MA (10 μM) led to a continuous increase of COX-2 mRNA during the first 12 h up to a 3.2-fold induction over vehicle (Fig. 1A). Induction of PGE2 synthesis by R(+)-MA became evident between 8 and 12 h after stimulation and reached an induction of approximately 6.8-fold 24 h after stimulation (Fig. 1B).

Time course of R(+)-MA-induced COX-2 mRNA expression (A) and PGE2 synthesis (B) by H4 human neuroglioma cells. Cells were incubated with R(+)-MA (10 μM) or its vehicle for the indicated times. Percentage of control represents comparison with vehicle-treated cells (100%) in the absence of test substance. Values are means ± S.E.M. of n = 3 experiments. *, P < 0.05; ***, P < 0.001, versus corresponding vehicle control (Student's t test).
Time Course and Characteristics of R(+)-MA-Induced Ceramide Formation. Looking for second messengers conferring COX-2 expression by R(+)-MA, cell lysates of H4 cells were assayed for C16-ceramide using liquid chromatography-mass spectrometry technique. As demonstrated in Fig. 2A, cellular ceramide levels significantly increased within the first 2 h of exposure to R(+)-MA and reached a 4.9-fold induction over baseline after 8 h. Thereafter, ceramide levels declined, returning to baseline after a 24-h incubation of cells with R(+)-MA.

Time course of R(+)-MA-induced C16-ceramide formation by H4 human neuroglioma cells (A) and influence of the ceramide synthase inhibitor fumonisin B1 (FB1) and the nSMase spiroepoxide inhibitor (Spiro) on R(+)-MA-induced C16-ceramide formation (B). Cells were incubated with R(+)-MA (10 μM) or its vehicle for the indicated times (A) or for 4 h (B) in the presence or absence of fumonisin B1 (50 μM; 1-h preincubation) and nSMase spiroepoxide inhibitor (10 μM; 1.5-h preincubation). Percentage of control represents comparison with vehicle-treated cells (100%) in the absence of test substance. Values are means ± S.E.M. of n = 3 experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; versus corresponding vehicle control or indicated group (Student's t test).
To examine the mechanism of ceramide generation upon treatment of cells with R(+)-MA, the nSMase and ceramide synthase were focused on as possible targets of enzymatic action. The question of whether R(+)-MA acted through nSMase to induce ceramide formation was examined using the nSMase inhibitor glutathione and a selective inhibitor of nSMase, referred to as nSMase spiroepoxide inhibitor. Experiments performed to assess the impact of de novo ceramide synthesis were performed using the potent and selective inhibitor of ceramide synthase fumonisin B1 as well as the inhibitors of serine palmitoyltransferase (SPT), l-cycloserine, and ISP-1. Whereas complete suppression of R(+)MA-stimulated ceramide formation was observed in the presence of fumonisin B1 (Fig. 2B), glutathione (Table 1), the nSMase spiroepoxide inhibitor (Fig. 2B), as well as l-cycloserine and ISP-1 (Table 1), were virtually inactive in this respect. l-Cycloserine left R(+)-MA-induced ceramide formation unaltered even when a 5-fold higher concentration (i.e., 5 mM) of this inhibitor was used (data not shown). Fumonisin B1 did not alter basal ceramide levels (data not shown).
Influence of inhibitors of nSMase [glutathione in its reduced (GSH) and oxidized forms (GSSH)], cPLA2 (AACOCF3), MAPKs (SB203580, PD98059), and SPT (L-cycloserine, ISP-1) on R(+)-MA—induced C16-ceramide formation
Cells were incubated with R(+)-MA (10 μM) or its vehicle for 4 h in the presence of the test substances (1 h preincubation). Values are comparisons with the vehicle-treated cells (100%) in the absence of test substance. Values are means ± S.E.M. of n = 3 to 4 experiments.
In previous investigations, decreases of intracellular glutathione levels (Liu and Hannun, 1997) or increased liberation of arachidonic acid (Jayadev et al., 1994) have been implicated in activation of nSMase activity. Thus, for further confirmation that nSMase is not involved in the observed ceramide accumulation, intracellular levels of glutathione were assayed over a 4-h time course after stimulation with R(+)-MA. However, there was no significant change of glutathione levels (data not shown). Likewise, inhibition of cytosolic phospholipase A2 (cPLA2) by AACOCF3 left ceramide synthesis unaltered (Table 1).
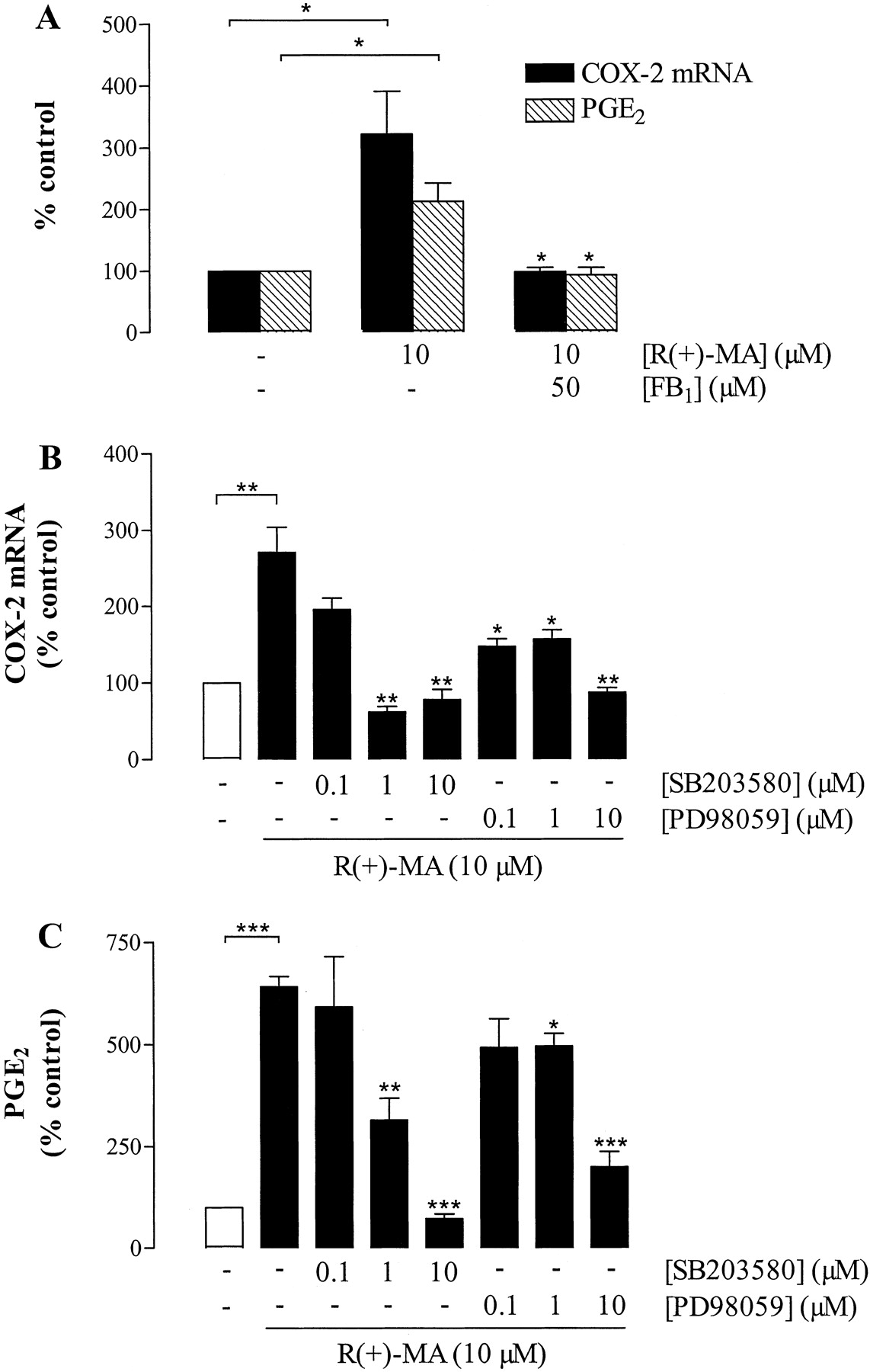
To investigate whether R(+)-MA-mediated C16-ceramide formation was a downstream event of p38 and p42/44 MAPK activation, cells were incubated with R(+)-MA in the presence of SB203580, an inactivator of p38 MAPK, and PD98059, an inhibitor of p42/44 MAPK activation. However, analysis of ceramide showed that MAPK activation was not necessary for the observed increase of ceramide (Table 1).
Time Course of R(+)-MA-Induced p38 and p42/44 MAPK Phosphorylation. According to previously published short-term experiments that were performed over a 45-min incubation period (Ramer et al., 2001), R(+)-MA causes phosphorylation of p38 and p42/44 MAPKs. However, analysis of a broader time frame in the present study revealed that R(+)-MA stimulation leads not only to an early but also to a delayed activation of both kinases, with maximum phosphorylation 4 h after stimulation (Fig. 3, A and B). Early and delayed peaks of MAPK phosphorylation referring to the respective vehicle controls are shown in Fig. 3C. As internal standards, the blots have been incubated with antibodies binding to the unphosphorylated forms of p38 and p42/44 MAPK.

Time course of R(+)-MA-induced phosphorylation of p38 (A) and p42/44 MAPKs (B). Cells were incubated with R(+)-MA (10 μM) or its vehicle for the indicated times. Phosphorylation peaks at 15 and 240 min after stimulation with R(+)-MA were compared with the respective vehicle control (C). Activation of p38 and p42/44 MAPKs was analyzed by Western blotting using phospho-p38 MAPK and phospho-p42/44 MAPK antibodies, and antibodies against the nonphosphorylated forms as internal standards, respectively. Results are representative of three experiments with similar results.
Effect of Fumonisin B1 on R(+)-MA-Induced p38 and p42/44 MAPK Phosphorylation, COX-2 Expression, and PGE2 Synthesis. To examine whether synthesis of ceramide by R(+)-MA leads, in turn, to activation of MAPKs, the influence of fumonisin B1 on R(+)-MA-induced phosphorylation of p38 and p42/44 MAPKs was investigated. Blocking of ceramide synthesis with fumonisin B1 inhibited R(+)-MA-induced phosphorylation of p38 and p42/44 MAPKs occurring after a 4-h incubation with the cannabinoid (Fig. 4). In contrast, first peak phosphorylation of both MAPKs (15 min) remained unaltered in the presence of fumonisin B1 (Fig. 4).

Effect of the ceramide synthase inhibitor fumonisin B1 on R(+)MA-induced phosphorylation of p38 and p42/44 MAPKs. Cells were incubated with R(+)-MA (10 μM) or its vehicle for 15 min or 4 h. Fumonisin B1 (50 μM) was added 1 h before stimulation with R(+)-MA. Activation of p38 and p42/44 MAPKs was analyzed by Western blotting using phosphop38 MAPK and phospho-p42/44 MAPK antibodies and antibodies against the nonphosphorylated forms as internal standards, respectively. Results are representative of three experiments with similar results.
In further experiments, the impact of ceramide synthesis by R(+)-MA on COX-2 mRNA expression and PGE2 release was determined. R(+)-MA-induced COX-2 mRNA expression and subsequent PGE2 formation were totally abolished by fumonisin B1 (Fig. 5A). In contrast, inhibitors of nSMase (spiroepoxide, glutathione), cPLA2 (AACOCF3), or SPT (l-cycloserine) left effects of R(+)-MA virtually unaltered (data not shown). Fumonisin B1 did not interfere with basal COX-2 expression and PGE2 synthesis (data not shown).

Effect of inhibitors of ceramide synthase (fumonisin B1), p38 MAPK (SB203580), and p42/44 MAPK activation (PD98059) on R(+)MA-induced COX-2 mRNA expression and PGE2 release by H4 human neuroglioma cells. Cells were incubated with R(+)-MA (10 μM) or its vehicle for 4 h (COX-2 mRNA) or 24 h (PGE2 formation). Fumonisin B1 (50 μM) was added 1 h before R(+)-MA, whereas SB203580 (0.1-10 μM) and PD98059 (0.1-10 μM) were added 1 h after R(+)-MA. Percentage of control represents comparison with the vehicle-treated cells (100%) in the absence of test substance. Values are means ± S.E.M. of n = 3 experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; versus sole R(+)-MA treatment, unless otherwise indicated (Student's t test).
Effect of p38 and p42/44 MAPK Inhibitors on R(+)MA-Induced COX-2 Expression and PGE2 Synthesis. To examine whether the delayed phosphorylations of p38 and p42/44 MAPK confer COX-2 expression by R(+)-MA, SB203580 and PD98059 were added to H4 cells 1 h after the start of the 4-h (COX-2 mRNA) or 24-h (PGE2 determination) incubation with R(+)-MA. Figure 5 shows that both R(+)MA-induced COX-2 expression and PGE2 synthesis were suppressed when cells were treated with SB203580 and PD98059 under these experimental conditions. Significant inhibition of both COX-2 expression and PGE2 release were observed at inhibitor concentrations as low as 1 μM. In the absence of R(+)-MA, neither SB203580 nor PD98059 significantly altered basal COX-2 expression and PGE2 release (data not shown).
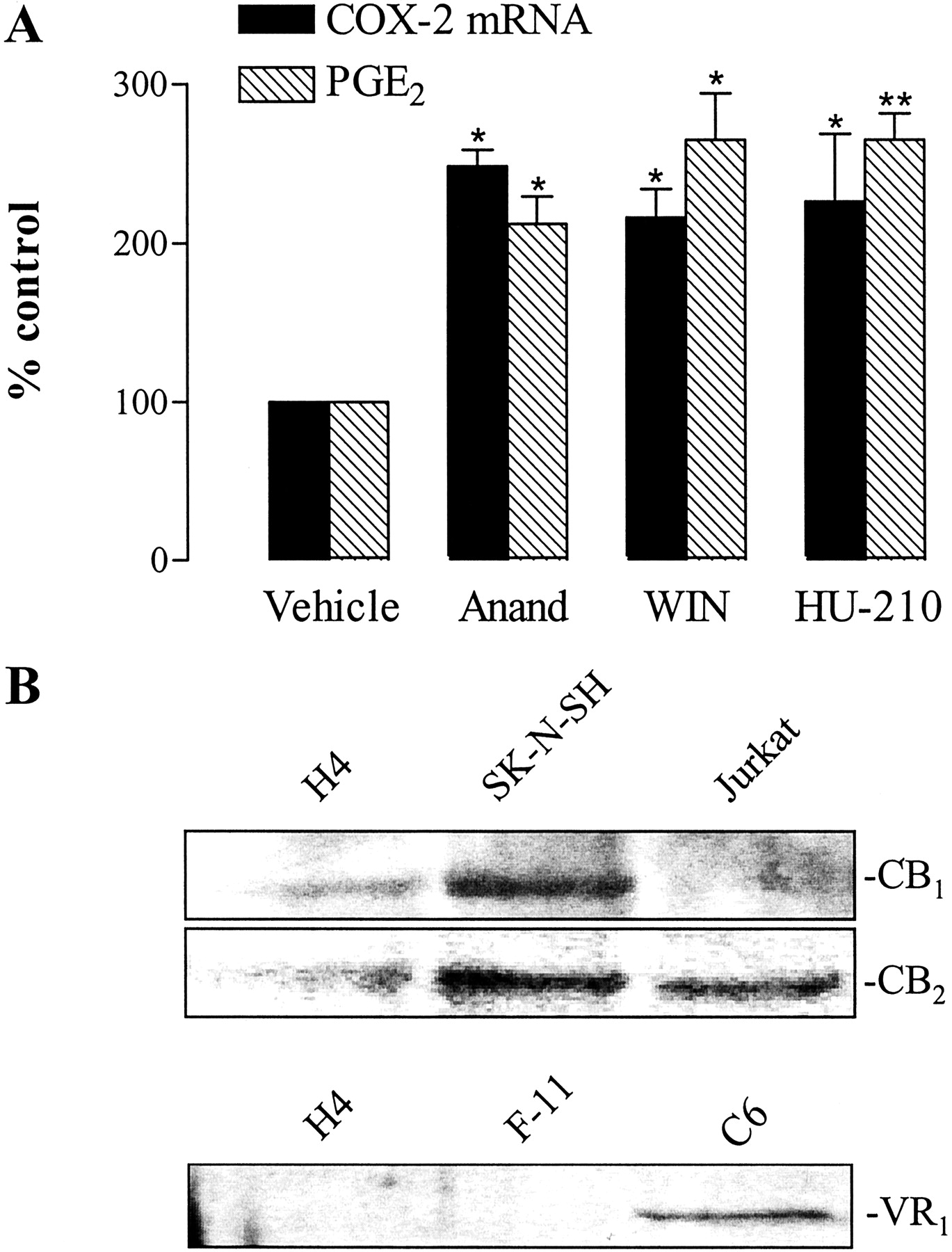
Evaluation of the Involvement of CB1-, CB2-, and VR1 Receptors in COX-2 Induction by R(+)-MA. To further study the mechanism of R(+)-MA action, we next investigated whether COX-2 expression by H4 human neuroglioma cells was also induced by cannabinoids structurally similar (anandamide) or not similar (WIN-55,212-2, HU-210) to R(+)-MA. In the presence of these substances, significant inductions of COX-2 expression and PGE2 synthesis were observed at threshold concentrations of 10 μM (Fig. 6A).

Effect of different cannabinoids on COX-2 expression and PGE2 synthesis by H4 cells (A) and expression of CB1-, CB2-, and VR1 receptors in H4 human neuroglioma cells (B). A, cells were incubated with anandamide (10 μM), WIN-55,212-2 (10 μM), HU-210 (10 μM), or its vehicle for 4 (COX-2 mRNA) or 24 h (PGE2). Percentage of control represents comparison with the vehicle-treated cells (100%) in the absence of test substance. Values are means ± S.E.M. of n = 3 experiments. *, P < 0.05; **, P < 0.01; versus corresponding vehicle control (Student's t test). B, expression of receptors was analyzed by Western blotting using specific antibodies raised to the CB1-, CB2-, or VR1 receptor. Lysates from C6 cells (glial tumor, rat), F-11 cells (hybridoma of mouse neuroblastoma and rat dorsal root ganglion cells), Jurkat cells (acute T cell leukemia, human), and SK-N-SH cells (neuroblastoma, human) were included for comparison.
To ascertain a possible role of CB and VR1 receptors in the stimulatory action of R(+)-MA on COX-2 expression, R(+)MA-treated cells were preincubated with the CB1 receptor antagonist AM-251, the CB2 receptor antagonist AM-630, or the VR1 antagonist capsazepine. However, none of the three substances, tested alone or in combination, suppressed R(+)MA-induced COX-2 expression (Table 2). Treatment of cells with the antagonists alone without R(+)-MA caused no significant change in COX-2 expression by H4 cells (data not shown).
Influence of CB1-, CB2-, and VR1 receptor antagonists on R(+)-MA—induced COX-2 expression
Cells were incubated with R(+)-MA (10 μM) or its vehicle for 4 h. AM-251 (1 μM; selective CB1 antagonist), AM-630 (1 μM; selective CB2 antagonist), and capsazepine (Caps; 1 μM; VR1 antagonist) were added to the cells 1 h before R(+)-MA. Values represent comparison with the vehicle-treated cells (100%) in the absence of test substance. Values are means ± S.E.M. of n = 3 experiments.
The expression profile of cannabinoid and vanilloid receptors in H4 cells compared with other cell lines was examined using Western blotting. As shown in Fig. 6B, H4 cells were found to express relatively low amounts of both CB1- and CB2 receptor subtypes compared with other cell lines, where a profound expression of both CB receptors (SK-N-SH cells) or CB2 receptor (Jurkat cells) was detected. In the case of VR1 receptors, we were unable to detect this protein in H4 and F-11 cells, whereas a substantial amount of this receptor was found in rat C6 cells.
Time Course of C2-Ceramide-Induced p38 and p42/44 MAPK Phosphorylation. To further confirm whether ceramide confers induction of MAPK phosphorylation, cells were incubated with C2-ceramide, a cell-permeable, short-chain ceramide analog. As negative controls, cells were stimulated with the respective vehicle control and dihydro-C2-ceramide, an inactive analog of C2-ceramide. After treatment of cells with C2-ceramide, maximum phosphorylation of MAPKs was observed after 4 (p38) and 8 h (p42/44), respectively (Fig. 7, A and B). At these time points, vehicle- and dihydro-C2-ceramide-treated cells exhibited no significant phosphorylation of p38 and p42/44 MAPK (Fig. 7C) compared with C2-ceramide-treated H4 cells.

Time course of C2-ceramide-induced phosphorylation of p38 (A) and p42/44 MAPKs (B). Cells were incubated with C2-ceramide (10 μM) or its vehicle for the indicated times. Phosphorylation peaks at 240 and 480 min after stimulation with C2-ceramide were compared with dihydro-C2-ceramide and the respective vehicle control (C). Activation of p38 and p42/44 MAPKs was analyzed by Western blotting using phospho-p38 MAPK and phospho-p42/44 MAPK antibodies, and antibodies against the nonphosphorylated forms as internal standards, respectively. Results are representative of three experiments with similar results.
Effect of C2-Ceramide on COX-2 Expression and PGE2 Synthesis by H4 Neuroglioma Cells. Incubation of cells with C2-ceramide led to a concentration-dependent increase in COX-2 expression and subsequent PGE2 formation (Fig. 8A). In contrast, dihydro-C2-ceramide left COX-2 expression and PGE2 synthesis virtually unaltered (Fig. 8A). To confirm a causal link between the phosphorylation of p38 and p42/44 MAPK and C2-ceramide-elicited induction of COX-2 expression, cells were preincubated with SB203580 and PD98059. Both compounds concentration-dependently inhibited the induction of COX-2 at the mRNA (Fig. 8B) and PGE2 (Fig. 8C) levels.

Effect of C2-ceramide and dihydro-C2-ceramide on COX-2 mRNA expression and PGE2 release by H4 human neuroglioma cells (A) and influence of SB203580 and PD98059 on C2-ceramide-induced COX-2 mRNA expression (B) and PGE2 release (C). Cells were incubated with C2-ceramide, dihydro-C2-ceramide, or its vehicle for 4 (COX-2 mRNA) or 24 h (PGE2 formation). SB203580 (0.1-10 μM) and PD98059 (0.1-10 μM) were added to the cells 1 h before C2-ceramide. Percentage of control represents comparison with the vehicle-treated cells (100%) in the absence of test substance. Values are means ± S.E.M. of n = 3 experiments. A, *, P < 0.05; **, P < 0.01; ***, P < 0.001; versus vehicle control (Student's t test). B and C, *, P < 0.05; **, P < 0.01; versus sole C2-ceramide treatment, unless otherwise indicated (Student's t test).
Effect of nSMase from B. cereus on COX-2 Expression and PGE2 Synthesis by H4 Neuroglioma Cells. The stimulatory effect of ceramide on COX-2 expression was confirmed by inducing ceramide formation with nSMase from B. cereus. In our hands, nSMase at 100 mU/ml increased intra-cellular basal levels of C16-ceramide up to 10-fold after a 1-h incubation of H4 cells (data not shown). COX-2 mRNA expression, as well as subsequent PGE2 production, were upregulated by nSMase in a concentration-dependent manner, reaching an induction of almost 4- or 5-fold at 100 mU/ml, respectively (Fig. 9A). As for C2-ceramide, inhibition of p38 MAPK and p42/44 MAPK activation attenuated the induction of COX-2 mRNA (Fig. 9B) and PGE2 formation (Fig. 9C) by nSMase in a concentration-dependent manner.

Effect of nSMase from B. cereus on COX-2 mRNA expression and PGE2 release by H4 human neuroglioma cells (A) and influence of SB203580 and PD98059 on nSMase-induced COX-2 mRNA expression (B) and PGE2 release (C). Cells were incubated with nSMase or its vehicle for 4 (COX-2 mRNA) or 24 h (PGE2 formation). SB203580 (0.1-10 μM) and PD98059 (0.1-10 μM) were added to the cells 1 h before nSMase. Percentage of control represents comparison with the vehicle-treated cells (100%) in the absence of test substance. Values are means ± S.E.M. of n = 3 experiments. A, *, P < 0.05; **, P < 0.01; versus vehicle control (Student's t test). B and C, *, P < 0.05; **, P < 0.01; ***, P < 0.001; versus sole SMase treatment, unless otherwise indicated (Student's t test).
Discussion
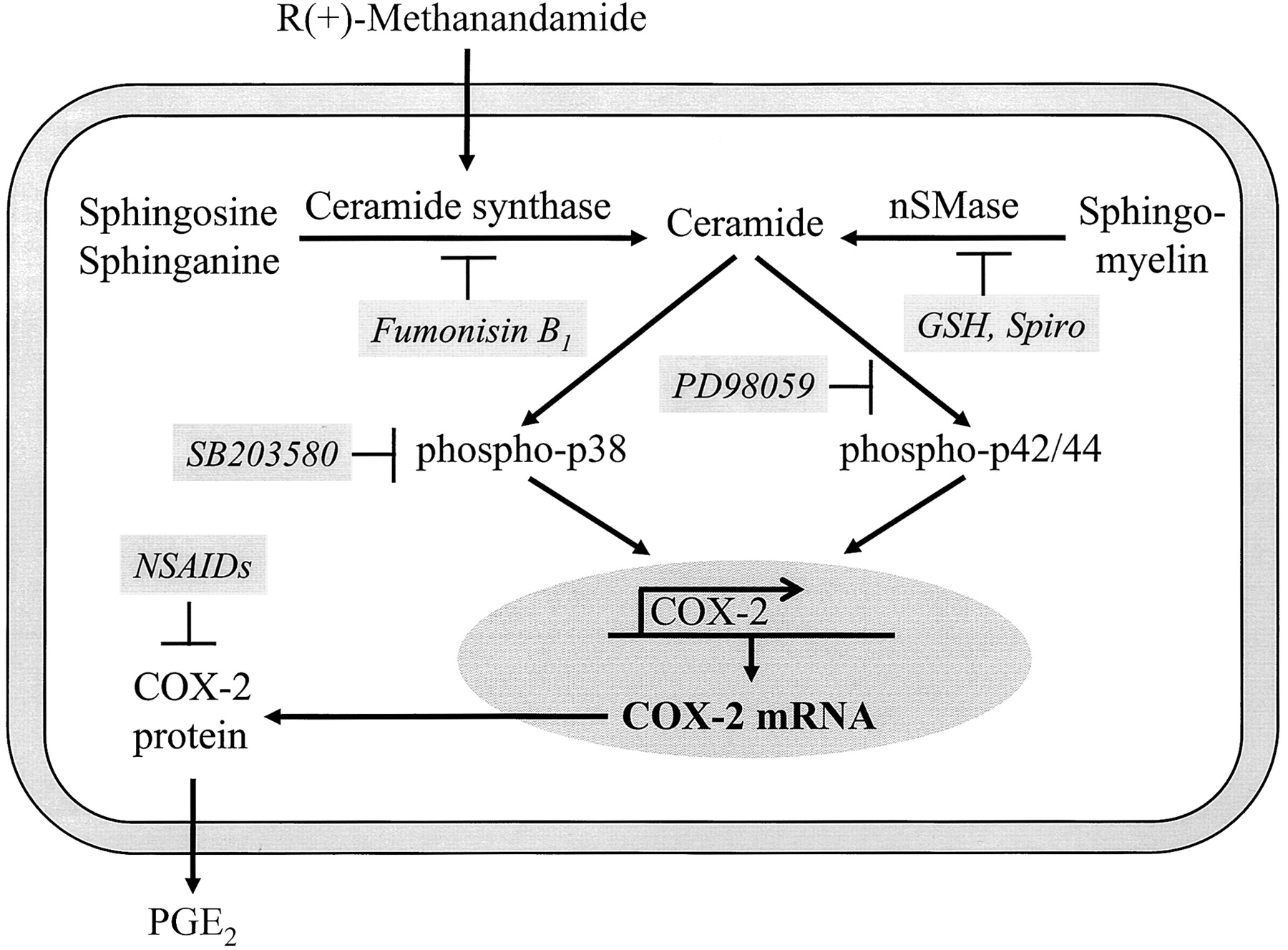
Induction of COX-2 expression has recently been shown to confer enhanced formation of PGs by cannabinoids in human neuroglioma cells (Ramer et al., 2001). In the current work, we demonstrated that the lipid second messenger ceramide is involved in R(+)-MA-induced phosphorylation of p38 and p42/44 MAPKs and subsequent up-regulation of COX-2 expression in these cells (Fig. 10).

Schematic illustrating the proposed mechanism by which R(+)-MA enhances COX-2 expression in H4 human neuroglioma cells. R(+)-MA increases synthesis of ceramide via the enzyme ceramide synthase, which in turn leads to phosphorylation of MAPKs, COX-2 expression, and PGE2 synthesis.
Ceramide can be generated through different pathways such as hydrolysis of sphingomyelin by SMases (Hannun, 1994), de novo, or via reacylation of free sphingoid bases (Wang et al., 1991). The mechanisms thought to be involved in SMase activation include depletion of intracellular glutathione (Liu and Hannun, 1997) and liberation of arachidonic acid (Jayadev et al., 1994). However, several lines of evidence suggest that the cannabinoid-induced ceramide synthesis in H4 cells does not involve a nSMase-mediated pathway. First, R(+)-MA did not decrease intracellular glutathione, and exogenous addition of nSMase inhibitors, including reduced and oxidized glutathione (Liu and Hannun, 1997) and spiroepoxide, an irreversible and selective nSMase inhibitor (Arenz and Giannis, 2000), left R(+)-MA-induced ceramide formation virtually unaltered. Second, the cPLA2 inhibitor AACOCF3 failed to interfere with this response. Third, induction of ceramide synthesis was delayed, whereas ceramide generation depending on SMases has been reported to occur within minutes (Kolesnik and Krönke, 1998). Rather, our data suggest that R(+)-MA-induced ceramide formation is mediated via the enzyme ceramide synthase. Accordingly, induction of ceramide formation by R(+)-MA was prevented by the mycotoxin fumonisin B1, a structural sphingolipid analog that blocks the acylation step in ceramide synthesis by competing with sphinganine and sphingosine (Wang et al., 1991). Surprisingly, inhibition of another enzyme involved in de novo ceramide synthesis, SPT (Perry, 2002), left R(+)MA-induced ceramide synthesis unaltered. In a recent study, cannabinoids have been shown to induce the activity of this enzyme in glioma cells (Gomez del Pulgar et al., 2002). Although the reason for our differential findings with inhibitors of ceramide synthase (fumonisin B1) and SPT activity (l-cycloserine, ISP-1) requires further investigation, the dual inhibitory action of fumonisin B1 within the cascade of ceramide synthesis may provide a possible explanation concerning this matter. Apart from inhibiting N-acylation of sphinganine, the enzymatic step, which occurs downstream to the action of SPT, fumonisin B1 also blocks the reacylation of sphingosine released by the turnover of complex sphingolipids (Wang et al., 1991). Thus, it is tempting to speculate that the latter reaction might play the more important role in R(+)-MA-induced ceramide accumulation. Interestingly, similar results have been published by Jacobsson et al. (2001), who showed an inhibitory effect of fumonisin B1, but not of l-cycloserine, on the antiproliferative action of anandamide on rat C6 glioma cells. Fumonisin B1 has also been suggested to inhibit sphingomyelin breakdown (Tonnetti et al., 1999). However, given the negative results with several nSMase inhibitors, we can exclude an involvement of this enzyme in the inhibitory effect of fumonisin B1 in our system.
Activation of p38 and p42/44 MAPKs has recently been shown to occur upon treatment of H4 cells with R(+)-MA within a 15-min exposure (Ramer et al., 2001). Analysis of a broader time frame in the present study revealed a biphasic phosphorylation of both enzymes by R(+)-MA, with a rapid transient phosphorylation after 15 min and a delayed phosphorylation after 4 h of stimulation. Fumonisin B1 suppressed only the late-phase activation of p38 and p42/44 MAPK phosphorylation, suggesting that the delayed induction was mediated by endogenous ceramide. This is corroborated by time course experiments showing that specifically late phase phosphorylation of MAPKs coincided with significant increases in intracellular levels of C16-ceramide. Further experiments indicated that specifically delayed, ceramide-dependent phosphorylations of p38 and p42/44 MAPK confer COX-2 expression and subsequent PGE2 synthesis by R(+)-MA. Consistent with its inhibitory effect on the activation of MAPKs, fumonisin B1 potently blocked R(+)-MA-induced COX-2 expression and PGE2 synthesis in H4 cells. Apart from the biphasic p42/44 MAPK activation shown for R(+)-MA in the present study, a similar regulation has previously been reported for growth factors, angiotensin II, nitric oxide, and reactive oxygen species generators (Meloche et al., 1992; Callsen et al., 1998; York et al., 1998; Hannken et al., 2000; Jin et al., 2000; Fukuzawa et al., 2002). In the case of p38 MAPK, a biphasic phosphorylation has been observed in response to angiotensin II and endothelin (Hannken et al., 2000; Ohanian et al., 2001).
Additional experiments addressed the role of cannabinoid and vanilloid receptors in R(+)-MA-induced COX-2 expression. Recently, occupation of both CB1 and CB2 receptors was implicated in the cytotoxic effect of cannabinoids on C6 rat glioma cells (Galve-Roperh et al., 2000). In another study, the antiproliferative effect of cannabinoids was associated with a combined activation of cannabinoid and vanilloid receptors (Jacobsson et al., 2001). In H4 cells, Western blot analysis revealed the presence of CB1 and CB2 receptors, whereas VR1 receptors were not detectable. However, neither selective antagonists of CB1 (AM-251) or CB2 receptors (AM-630) nor the VR1 receptor antagonist capsazepine affected R(+)MA-induced COX-2 expression. Moreover, and in line with a cannabinoid-receptor-independent event, the COX-2-inducing effects of several cannabinoids did not correlate with their different receptor binding profiles, with HU-210 and WIN-55,212-2 displaying higher affinities to CB1 and CB2 receptors than R(+)-MA and anandamide. Accordingly, all four cannabinoids showed a “threshold”-like profile in inducing COX-2 expression, with significant stimulations confined to micromolar concentrations. Collectively, these data support previous observations from our laboratory (Ramer et al., 2001) that alternative receptor-independent signaling pathways are involved in COX-2 expression by R(+)-MA. A possible mode of action of the lipophilic R(+)-MA may lie in alterations of membrane fluidity, which in turn may lead to activation of enzymes conferring ceramide synthesis and MAPK activation. In a recent study, cholesterol-rich membrane lipid rafts have been proposed to confer cannabinoidand vanilloid-receptor-independent toxic effects of anandamide on rat pheochromocytoma cells, possibly via endocannabinoid membrane transporter or carrier proteins (Sarker and Maruyama, 2003).
The involvement of ceramide in COX-2 expression was further substantiated by findings demonstrating inductions of COX-2 expression and PGE2 synthesis by the cell-permeable short-chain ceramide analog, C2-ceramide, and by nSMase from B. cereus. Inhibitor experiments confirmed the involvement of both p38 and p42/44 MAPKs within this process. In contrast to R(+)-MA, C2-ceramide caused a monophasic activation of p38 and p42/44 MAPKs, with the respective peaks coinciding with the induction of COX-2 expression. The stimulatory effect of C2-ceramide was specific in that dihydro-C2-ceramide did not induce MAPK phosphorylation, COX-2 expression, or PGE2 production. Overall, the inductive action of C2-ceramide is in good agreement with previous studies showing that exogenous ceramide induces COX-2 expression (Subbaramaiah et al., 1998; Newton et al., 2000) and MAPKs (Reunanen et al., 1998, Subbaramaiah et al., 1998) in human mammary epithelial cells, human pulmonary A549 cells, and human skin fibroblasts.
Other investigations that addressed the role of ceramide as a second messenger of cannabinoids have shown a biphasic ceramide response. Whereas the rapid transient peak of ceramide induced by cannabinoids is linked to activation of SMase and has been implicated in the regulation of metabolic functions (Blazquez et al., 1999; Guzman and Sanchez, 1999), other actions of cannabinoids, including inhibition of glioma cell growth (Galve-Roperh et al., 2000), are associated with sustained ceramide generation. In the latter study, accumulation of de novo synthesized ceramide was observed after a latency of days. However, rapid increases (within minutes) in ceramide synthase activity have been reported in hydrogen peroxide-challenged renal tubular epithelial cells (Ueda et al., 2001) and in heat shock-stimulated lymphocytes (Jenkins et al., 2002), suggesting that latency and duration of de novo synthesized ceramide strongly depends on experimental conditions.
There is a current renaissance in the study of the potential clinical use of cannabinoids. One possibility is their use as therapeutic agents for the management of malignant brain tumors (for review, see Guzman et al., 2001). In this context, the apoptotic effect of cannabinoids in glial cells has been shown to rely on sustained ceramide accumulation (Galve-Roperh et al., 2000). The induction of COX-2 expression by cannabinoids raises several questions in this regard. Because COX-2 has been implicated in inhibition of apoptosis (Tsujii and DuBois, 1995), it cannot be ruled out that cannabinoids may diminish their pro-apoptotic action by virtue of their capacity to induce COX-2 expression. On the other hand, recent studies suggest that induction of COX-2 does not necessarily confer resistance to apoptosis but rather may sensitize these cells to apoptotic death (Bagetta et al., 1998; Corasaniti et al., 2000; Na and Surh, 2002). Ongoing studies in our laboratory focus on the functional consequence of a cannabinoid/ceramide-induced COX-2 in terms of viability and apoptosis of neuroglioma cells.
In summary, we have demonstrated that a ceramide-dependent pathway confers p38 and p42/44 MAPK activation and subsequent COX-2 expression by the endocannabinoid derivative, R(+)-MA, in human neuroglioma cells. The involvement of ceramide in this process defines a novel mechanism by which cannabinoids may mediate PG-dependent effects within the central nervous system.
Footnotes
-
This study was supported by the Deutsche Forschungsgemeinschaft (HI 813/1-1 and SFB 539, BI.6).
-
ABBREVIATIONS: COX, cyclooxygenase; PG, prostaglandin; Smase, sphingomyelinase; C2-ceramide, d-erythro-sphingosine; dihydro-C2-ceramide, dihydro-N-acetyl-sphingosine; C16-ceramide, N-palmitoyl-sphingosine; MAPK, mitogen-activated protein kinase; R(+)-MA, R(+)-methanandamide (R-(+)-arachidonyl-1′-hydroxy-2′-propylamide); nSMase, neutral sphingomyelinase; AM-251, [N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide]; AM-630, [(6-iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl) (4-methoxyphenyl)methanone]; PD98059, 2′-amino-3′-methoxyflavone; SB203580, 4-(4-fluorophenyl)-2-(4-methylsulfonylphenyl)-5-(4-pyridyl)imidazol; RT, reverse transcriptase; PCR, polymerase chain reaction; SPT, serine palmitoyltransferase; ISP-1, myriocin from Mycelia sterilia; AACOCF3, arachidonyl trifluoromethylketone; cPLA2, cytosolic phospholipase A2; WIN-55,212-2, (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone; HU-210, (6aR)-trans-3-(1,1-dimethylheptyl)-6a,7,10,10a-tetrahydro-1-hydroxy-6,6-dimethyl-6H-dibenzo[bd]pyran-9-methanol.
- Received April 11, 2003.
- Accepted August 11, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}