


Abstract
G protein-coupled receptors form a ternary complex of ligand, receptor, and G protein heterotrimer (LRG) during signal transduction from the outside to the inside of a cell. Our goal was to develop a homogeneous, small-volume, bead-based approach compatible with high-throughput flow cytometry that would allow evaluation of G protein coupled receptor molecular assemblies. Dextran beads were derivatized to carry chelated nickel to bind hexahistidine-tagged green fluorescent protein (GFP) and hexahistidine-tagged G proteins. Ternary complexes were assembled on these beads using fluorescent ligand with wild-type receptor or a receptor-Giα2 fusion protein, and with a nonfluorescent ligand and receptor-GFP fusion protein. Streptavidin-coated polystyrene beads used biotinylated anti-FLAG antibodies to bind FLAG-tagged G proteins for ternary complex assembly. Validation was achieved by showing time and concentration dependence of ternary complex formation. Affinity measurements of ligand for receptor on particles, of the ligand-receptor complex for G protein on the particles, and receptor-Giα2 fusion protein for Gβγ, were consistent with comparable assemblies in detergent suspension. Performance was assessed in applications representing the potential of these assemblies for ternary complex mechanisms. We showed the relationship for a family of ligands between LR and LRG affinity and characterized the affinity of both the wild-type and GFP fusion receptors with G protein. We also showed the potential of kinetic measurements to allow observation of individual steps of GTP-induced ternary complex disassembly and discriminated a fast step caused by RG disassembly compared with the slower step of Gαβγ disassembly.
GPCRs interact with extracellular stimuli, such as photons, hormones, neurotransmitters, and odorants (Gilman, 1995). These stimuli cause conformational changes in the receptor, leading to binding of intracellular G protein heterotrimers, each with one copy of a guanyl nucleotide binding α subunit and a βγ dimer (Neer, 1995). After stimulation, the α subunit binds GTP, which promotes dissociation of the α subunit from the βγ dimer, exposing new surfaces to cytoplasmic effectors, such as adenylyl cyclase and phospholipase C. The human genome contains ∼600 GPCR genes, 27 α, 5 β, and 13 γ (Venter et al., 2001), with smaller numbers of these G proteins (17, 5, and 12, respectively) found to date. With such large numbers, determining how productively any given GPCR couples to a particular αβγ heterotrimer is daunting (1020 αβγ combinations alone). The assembly of a high agonist-affinity complex is a good criterion of productive partners (Gilman, 1987).
The formyl peptide receptor (FPR) responds to the presence of N-formyl methionine-containing peptides resulting from bacterial and mitochondrial protein synthesis, as well as other hydrophobic peptides (Gao et al., 1994). This receptor has served as a model for signal transduction in phagocytic cells and for inflammatory and autoimmune diseases (Prossnitz and Ye, 1997). The receptor has been cloned and overexpressed in tissue culture cells, solubilized, and assembled with a formyl peptide ligand and G protein to form a high agonist-affinity ternary complex in solution (Bennett et al., 2001b).
The soluble receptor reconstitutes with ligand, G proteins, and arrestin in a manner that is sensitive to receptor phosphorylation and mutations in both the receptor and G proteins. The assembly can be measured in real-time with fluorescent ligands, and the assemblies are consistent with cellular colocalizations observed by fluorescence confocal microscopy (Bennett et al., 2001a,b; Key et al., 2001).
Whereas ternary complex assemblies have been the subject of experimental investigation and mathematical modeling over several decades (Kent et al., 1980), the tools to examine the affinities and kinetics of individual steps in complex formation, disassembly, activation, and termination have only been accessible in a limited way (Christopoulos and Kenakin, 2002). For rhodopsin, it has been possible to measure complex assembly and disassembly through the spectroscopic signature of the metarhodopsin II-transducin complex (Mitchell et al., 2001). GPCRs also activate transmembrane channels in the subsecond time frame (Mark et al., 2000), where ternary complex dynamics can be inferred from measurements of ion currents. Such measurements have given a Gt (transducin) activation rate of ∼120 s-1 (Leskov et al., 2000), probably unique to the visual transduction system, and a Gq activation rate of 2 s-1 (Mukhopadhyay and Ross, 1999). Both surface plasmon resonance (Rebois et al., 2002) and flow cytometry (Nolan and Sklar, 1998) could be general tools for measuring individual rate constants.
Because GPCRs are prominent targets in drug discovery, we developed generic assembly capabilities for GPCRs using a homogeneous approach in which a flow cytometer can distinguish fluorescent molecules associated with a particle from those free in solution around the particle (Sklar et al., 2002). Based on solubilization in dodecyl maltoside, we showed that an epitope-tagged receptor could be associated with particles and analyzed by flow cytometry using a fluorescent ligand to detect the assembled complex (Sklar et al., 2000). We envisioned adapting such assemblies for analysis of ternary complex formation involving both signal transduction and termination partners and that these approaches would be compatible with high-throughput flow cytometry (Kuckuck et al., 2001). This initial approach suffered from two important limitations. First, we failed in our efforts to detect signaling assemblies when the receptor was anchored to the particles. Second, detection of the assembly required a fluorescent ligand to detect receptor affinity changes induced by subsequent receptor assemblies.
Both of these problems have now been addressed. We report here the formation of high-affinity complexes of the FPR with ligands on beads that have been coated with epitope-tagged G protein subunits. Ternary complexes have been assembled using three different receptor constructs (wild type, FPR-Giα fusion, and FPR-GFP fusion), two types of epitope-tagged G proteins, two α and β subunits, and two types of beads. The affinities of ligand for receptor, ligand-receptor complex for G protein, and α for βγ were estimated for the detergent-solubilized receptor. We evaluated LR and LRG formation for a family of ligands, measured the rate at which different receptor forms dissociate from G proteins, and used the beads as a sensor for RG assembly to evaluate the affinity of wild-type and GFP fusion receptors for G protein.
Materials and Methods
Reagents and Cell Culture. The cloning of the FPR (Boulay et al., 1990) and its expression in U937 cells (Kew et al., 1997) have been described. Plasticware was from VWR (West Chester, PA), and chemicals and reagents were from Sigma (St. Louis, MO) unless otherwise noted. The cells were grown in tissue culture treated flasks (Corning Inc., Corning, NY) in RPMI 1640 medium (Hyclone, Logan,UT) with 10% fetal bovine serum (Hyclone), 2 mM glutamine, 10 mM HEPES, 10 units/ml penicillin, and 2 μg/ml streptomycin. The cultures were grown at 37°C with 5% CO2 and passaged from subconfluent cultures every 3 to 4 days by reseeding at 2 × 105 cells/ml. The cells were expanded for membrane preparations in 1-liter baffled Pyrex spinner flasks by seeding at 2 × 105 cells/ml, equilibrated with 5% CO2, then sealed and incubated at 37°C, with stirring. The cells were harvested when the density reached 106 cells/ml. Receptor expression level decreased with passage, so freshly thawed cells were incubated with 10 nM fMLFK-FITC and sorted for the highest 5% expression to maintain 200,000 to 500,000 receptors/cell as needed, then frozen in aliquots for future use.
Generation of FPR-Giα2 and FPR-GFP Fusion Constructs. The human FPR (containing an EcoRI site and a NotI site embedded within the 5′ and 3′ primers, respectively) and rat Giα2 (containing a NotI site and an EcoRI site embedded within the 5′ and 3′ primers, respectively) were amplified by standard polymerase chain reaction protocols using Platinum TaqDNA polymerase (PerkinElmer Life Sciences, Boston, MA). The digested polymerase chain reaction products were ligated into EcoRI-digested and phosphatase-treated pSFFV.Neo and screened for orientation of the insert. Appropriate clones were confirmed by dideoxy sequence analysis. The final fusion protein contained three alanine residues between the last amino acid of the FPR and the first amino acid of the Giα2 protein reading frame. A similar strategy was used to construct a plasmid to produce the FPR-GFP fusion protein using HindIII, NotI, and Xba, which again had three alanine residues between the last amino acid of the FPR and the first amino acid of the GFP (optimized for fluorescence using standard fluorescein filter sets) (enhanced GFP; BD Biosciences Clontech, Palo Alto, CA).
Membrane Preparation by Nitrogen Cavitation. The procedure was performed at 4°C. Cells were harvested by centrifugation at 450g for 5 min and resuspended in cavitation buffer (10 mM HEPES, pH 7.3, 100 mM KCl, 3 mM NaCl2, 3.5 mM MgCl2, and 1× protease inhibitor cocktail 1 (Calbiochem, San Diego, CA) at a density of 107 cells/ml. This cell suspension was placed in a nitrogen bomb and pressurized to 450 psi for 20 min, after which the suspension was slowly released into a sample tube. Unbroken cells and nuclei were removed by centrifugation at 1000g for 5 min. The membranes in the supernatant were pelleted by centrifugation twice at 135,000g for 30 min, resuspended in buffer (25 mM HEPES, pH 7.5, and 200 mM sucrose), and then separated into aliquots at 108 cell equivalents in 0.5 ml and stored at -80°C.
Solubilization of the FPR. An aliquot of membrane was thawed, 700 μl of buffer A (30 mM HEPES, pH 7.5, 100 mM KCl, 20 mM NaCl, and 1 mM MgCl2) was added, and the membranes were removed from the sucrose by centrifugation in a microcentrifuge for 15 min. The supernatant was removed, the pellet was resuspended in 220 μl of buffer A by 10 passes back and forth through a 25-G needle, 25 μl of 10% dodecyl maltoside, and 2.5 μlof100× protease inhibitor cocktail were added, and the suspension was gently mixed for 2 h at 7°C. The unsolubilized material was removed by centrifugation as above for 15 min, giving a supernatant of solubilized FPR at 4 × 108 cell equivalents/ml (∼5 mg/ml protein), and used within 6 h. Solubilization was essentially 100%, so that 150,000 receptors/cell resulted in about 100 nM soluble FPR using this procedure. For FPR-GFP (RF) preparations, 2 μl of 10-4 M ligand (fMLFFGGK) was added to 200 μl of the preparation when desired to give quantitative conversion of RF to LRF. For FPR, LF (fMLFF-FITC) was added when desired at greater than the concentration of receptor to ensure nearly quantitative conversion of R to LFR while keeping the concentration of free ligand low to minimize nonspecific binding to the beads. The solublilized receptor preparation retained >90% activity after freezing at -80°C.
Use of Formyl Peptides. LF (fMLFK-FITC) was obtained from Bachem (King of Prussia, PA). Typically, 1 mg was dissolved in 10 ml of methanol, and 30 μl of the solution at about 0.1 mM was diluted in 3 ml of buffer A with 0.1 mg/ml BSA to obtain the absorbance at 495 nm. The concentration of LF was calculated using an extinction coefficient of 76,000 M-1cm-1. Aliquots of the methanol solution were transferred into microcentrifuge tubes to give 10-8 mol of LF, and dried in a Speedvac (Thermo Savant, Holbrook, NY). These aliquots were stored at -20°C, dissolved in 10 μl of DMSO to give 10-3 M LF, then diluted at least 100-fold in buffer A with 1 mg/ml BSA to give 10-5 M LF.
L (fMLFFGGK) was synthesized by Commonwealth Biotechnologies, Inc. (Richmond, VA). Dry peptide (8.2 mg) was dissolved in a final volume of 1 ml of acetic acid/water: 100 μl of acetic acid dissolved the powder, then 900 μl of 50% acetic acid was added. This was diluted 100-fold into buffer A, which was brought back to pH 7.5 with NaOH, giving 10-4 M L. It was divided into 1-ml and 10 μl-aliquots, stored at -20°C, and thawed fresh each day.
Synthesis of Dextran Chelate Nickel (DCNi) Beads. Superdex peptide beads, a crosslinked agarose/dextran matrix with an exclusion limit of 7000 Da and an average size of 13 μm, were removed from a packed column purchased from Amersham Biosciences (Piscataway, NJ). (Superdex 30 Prep Grade beads, average size 34 μm, are also compatible with flow cytometric analysis.) The beads were activated with a water-soluble bis-epoxide (Sundberg and Porath, 1974) and then coupled to a chelator that contained an amino group (Hochuli et al., 1987). Twelve milliliters of a 50% slurry of beads was reduced to a wet cake by vacuum filtration using a 60-ml coarse sintered glass funnel, and then washed three times with 50 ml of water to remove the ethanol in which the beads were supplied. The wet cake was transferred to a 25-ml Erlenmeyer flask, the funnel was rinsed with 5 ml of water, and this rinse was added to the flask. One milliliter of 5 M NaOH, 10 mg of NaBH4, and 5 ml of 1,4-butanediol diglycidyl ether (Sigma) were then added, and the flask was rotated to keep the beads in suspension for 8 h at 37°C; some bubbling occurred in the first hour. The beads were washed by vacuum filtration twice with water, twice with phosphate-buffered saline, twice with water again, then stored for up to 1 week at 4°C or for 2 months dried at 4°C. One settled volume of these epoxy-activated beads was coupled with 1 volume of the chelator Na,Na-bis(carboxymethyl)-l-lysine (Fluka) in 0.2 M Na2CO3, pH 11, adjusting the pH again after addition to the beads; we used 2.5, 25, and 250 mM chelator in three different reactions to obtain different substitution levels on the beads. The coupling proceeded at 22°C overnight with gentle mixing to keep the beads in suspension. The beads were washed as above and then treated with 10 volumes of 0.1 M NiCl2 for 1 min in column or batch mode; the two most highly substituted batches became visibly blue/green, whereas the lightly substituted batch remained white. The beads were rinsed with water and phosphate-buffered saline. Atomic absorption analysis of the three samples showed the content of Ni to be 1.5, 16, and 30 mM for the settled beads: substitution seemed proportional to the concentration of amino compound up to 25 mM in the reaction, then began to saturate.
Coating DCNi beads with H6-Tagged G Proteins. N-terminal hexahistidine-tagged γ2 subunit (H6γ2) cDNA was created by standard recombinant DNA techniques. β1H6γ2 dimer was produced by coexpression of β1 subunit and H6γ2 subunit in Sf9 insect cells, and the dimer was purified essentially as described previously (Kozasa and Gilman, 1995) using a Ni2+ chelate column followed by a Mono S column (Amersham Biosciences). The β1H6γ2 preparation was 46 μM, and 14 μl was incubated with 15 μl of 42 μM αi3 subunit (Calbiochem) and 44 μl of G buffer (0.1% dodecyl maltoside, 30 mM HEPES, pH 7.5, 100 mM KCl, 20 mM NaCl, 1 mM MgCl2, and 1 mM dithiothreitol) for 5 min on ice to allow G protein heterotrimer to form, then frozen in 2-μl (17 pmol) aliquots. After thawing, 2.5 μl of a 50% slurry of DCNi beads (2.5 × 108 beads/ml) was added, and the volume was brought to 100 μl with G buffer. The beads were kept suspended with rotation at 7°C for 1 h, then pelleted by centrifugation and brought to 50 μl with G buffer. This gave 1.2 × 107 beads/ml, nominally coated with 18 × 106 G protein αβH6γ per bead, with an unknown amount left on the beads in an active orientation (for comparison, assuming that random fall results in about 50% coverage, one expects about 7 × 106 BSA molecules per 13-μm sphere); 2 μl of bead suspension was used per 10 μl assay, consuming about 0.7 pmol of Gαβγ per assay on 24,000 beads. The beads retained more than 90% of their binding activity after freezing at -80°C. When the FPR-αi2 fusion protein was used for an assembly, only β1H6γ2 dimer was used to coat the DCNi beads as above.
Standard LRG Assembly Assay. The standard 10-μl assay consisted of 2 μl of water or 10-4 M GTPγS, 6 μl of soluble receptor preparation with or without ligand, and 2 μl of beads prepared as above, with 0.7 pmol of G protein used per assay and an unknown fraction left on the beads in the proper orientation. For FPR and R-αi2 assemblies, LF (fMLFK-FITC) was added in excess of receptor to ensure that essentially all the receptor was bound. For FPR-GFP assembly, 10-6 M L (fMLFFGGK) was added to the receptor preparation to ensure that essentially all the receptor was bound. Each mixture was mixed by pipetting to ensure a uniform starting suspension of the beads in plates containing 96 V-wells (Costar), and then mixed at low speed on a vortex mixer at 4 to 7°C, the temperature variation of our cold-room during use, for 2 h. The 10-μl assays were individually brought to 200 μl with 0.1% dodecyl maltoside in buffer A in 12 × 75-mm tubes for flow cytometric measurement of fluorescence of the beads. LRG assembly was defined as the difference between fluorescence without GTPγS and that with GTPγS. All determinations were done in duplicate. For assays with the FPR-αi2 chimera, a low amount of GTP was present (see Fig. 4B).

Calibrated LFRG assembly with the R-αi2 fusion protein. A, schematic diagram of the assembly, including LF, R-αi2 fusion protein, and Gβγ protein coated beads. Without ligand, the receptor does not bind to the beads. B, the standard assembly included 24 nM R-αi2, 40 nM LF, and 24,000 G protein-coated beads in 10 μl, which was mixed for 2 h at 4 to 7°C, then diluted to 200 μl for flow cytometric determination of bead fluorescence as described under Materials and Methods. Results for the standard assembly are shown in column 3, whereas variations in how the bead was coated and changes in the buffer are indicated beside the graph.
Flow Cytometry Analysis and Calibration. Flow cytometry was carried out using FACScan cytometers (BD Biosciences, San Jose, CA), obtaining 3000 gated events (see Fig. 2A for a typical gate of the DCNi beads) for a sample to obtain a mean channel fluorescence (MCF). These numbers were converted to the mean equivalent of soluble fluorophores on a bead using calibrated beads (Bangs Laboratories, Fishers, IN). The number of fluorescent ligands on a bead was determined by multiplying the mean equivalent of soluble fluorophores by 1.22 to reflect the smaller fluorescence of conjugated fluorescein compared with free fluorescein (it takes 122 conjugated fluorescein molecules to give the fluorescence intensity of 100 free fluorescein molecules) (Buranda et al., 2001). Beads with calibrated numbers of GFP molecule equivalents are available from BD Biosciences for GFP determinations in flow cytometric experiments.

Characterization of dextran chelate nickel (DCNi) beads by flow cytometry. A, dot plot of forward scatter versus side scatter. B, histogram of DCNi fluorescence with 0 or 10 nM hexahistidine-tagged green fluorescent protein (H6-GFP). C, time course of DCNi binding to 10 nM H6-GFP in the presence of 10 mM EDTA (▾), absence of EDTA (▴), and with 10 mM EDTA added after 30 min (□). D, DCNi binding to H6-GFP through five washes over 2 h (▴) and with 10 mM EDTA added after 2 h (□). Beads were kept in suspension with moderate mixing at 0 to 4°C, and 200-μl aliquots were removed for flow cytometric measurement of bead fluorescence. E, membranes from cells expressing N-terminal hexahistidine-tagged FPR (N-H6:FPR) or C-terminal hexahistidine-tagged FPR (FPR:CH6) were solubilized as described under Materials and Methods, giving 18.5 nM FPR:C-H6 or 10 nM N-H6:FPR, and incubated with 50,000 DCNi beads in 200 μlof 0.1% dodecyl maltoside in buffer A with moderate mixing for 7 h in duplicate. The beads were washed by centrifugation and resuspended in fresh buffer, incubated with various concentrations of LF as shown in the graph for 30 min, and then the bead fluorescence was measured by flow cytometry. F, the dissociation of LF from beads, which were coated with FPR: C-H6 as in E, was monitored using a custom built rapid mixer, described under Materials and Methods. The initial bead fluorescence was measured for 20 s, then anti-FITC antibody was mixed with the beads and the decease in bead fluorescence was measured for another 100 s. The line shown is a best fit to a plateau, followed by an exponential decrease to the bottom.
Coating Streptavidin-Coated Beads with Biotinylated Anti-FLAG Antibody and FLAG-Tagged G Proteins. Twenty microliters of 6.2-μm diameter streptavidin-coated polystyrene beads at 4 × 107 beads/ml (Spherotech Inc., Libertyville, IL) were mixed with 20 μl of 1 mg/ml biotinylated anti-FLAG antibody (Sigma) for 2 h at 4°C and then washed three times in buffer to give ∼9 × 106 FLAG-FITC binding sites per bead at 4000 beads/μl (Buranda et al., 2001). G protein γ2-H6-FLAG subunits were coexpressed with β4 subunits in Sf9 cells, which were extracted as described previously (McIntire et al., 2001). The extract was loaded on a 3-ml FLAG column (Sigma) and eluted with FLAG peptide according to the manufacturer's instructions. The eluate was immobilized on a 4-ml nickel column, washed with increasing concentrations of salt and detergent, then eluted with imidazole. The eluate was immobilized on a 15Q column (Amersham Biosciences), eluted with salt, concentrated with a Centricon 30, formed into aliquots, and stored at -80°C. This βγ preparation was combined with equimolar αi3 (Calbiochem) as above. Fifty microliters of the beads were mixed with 1 μl of 3.4 μM αi3β4γ2-FLAG-H6 for 1 h, spun, and resuspended in 40 μl of buffer to give beads nominally coated with 9 × 106 G protein αβγ per bead; 2 μl of this suspension was used per assay, ∼0.17 pmol per assay, on 10,000 beads. These beads are smaller than the DCNi beads and easier to keep in suspension.
Kinetic LRG Disassembly. LRG was assembled according to one of the three methods above, depending on the receptor type. The 10-μl assay was brought to 200 μl as usual at the flow cytometer, and an initial fluorescence was recorded for 20 s; then the tube was removed, 2 μl of 0.01 M GTPγS or 6 μM anti-FITC antibody was added at 25 s, and the tube was put back on the flow cytometer for dynamic measurement of fluorescence. A 2 × 5-mm stir bar (Bel-Art; Pequannock, NJ) was driven by a magnetic stirrer brought near the tube to keep the beads in suspension. The time course data were converted to ASCII format using the FCSQuery program (developed by Bruce Edwards and available upon request), which puts the raw data into bins of the desired time period, calculates an MCF for each bin, and outputs the results to an Excel file. Dissociation curves of this series of MCF values were analyzed using Prism (GraphPad Software, San Diego, CA).
Spectrofluorometric Analysis of Soluble Complexes. Fluorescence was measured with an SLM 8000 spectrofluorometer (SLM Instruments, Inc., Rochester, NY) using the photon counting mode. The sample holder was fitted with a cylindrical cuvette adapter, which allowed the use of 200-μl samples in 7 × 45-mm cylindrical cuvettes (Sienco, Wheat Ridge, CO), stirred with 2 × 5-mm stir bars (Bel-Art, Pequannock, NJ). Excitation was at 490 nm, and stray light was reduced with a 490 ± 10 nm filter (Spectra-Physics, Franklin, MA). Emission was monitored using a 520 ± 10 nm filter (Spectra-Physics) and a 500-nm long-pass filter (Kopp, Pittsburgh, PA). Additions to samples during kinetic measurements were made through an injection port on the top of the sample holder with 10-μl glass syringes (Hamilton, Reno, NV). For each concentration of fluorescent ligand used, a sample of solubilized proteins from membranes containing receptor and membranes without receptor were measured, typically 5 μl of a ∼60 nM R preparation to give 3 nM R, as used in Fig. 1.

Soluble receptor determination. Fluorescent ligand (LF) was incubated in the presence of an unknown amount of solubilized formyl peptide receptor in 200-μl aliquots in a spectrofluorometer with stirring as described under Materials and Methods. Bound and free ligand were discriminated with the use of an anti-FITC antibody, which rapidly quenched the free LF. Concentrations of free LF and receptor-bound LF were obtained for each total concentration of LF. These paired values were plotted to obtain the dissociation constant of the LF for the receptor, and the concentration of the receptor.
Results
Soluble FPR Assay. We have previously demonstrated the presence of LR and LRG complexes in solution using a fluorometric assay in which FPR are quantitatively solubilized (Sklar et al., 2000; Bennett et al., 2001a,b; Key et al., 2001). The detection of LR in a spectrofluorometer was accomplished with a fluoresceinated ligand, fMLFK-FITC (LF), and an anti-FITC antibody that quenched the fluorescence of the FITC on the ligand about 91% when it was bound. The dissociation half-time for LF was 14 s at room temperature. Here, we extend this to a quantitative assay by performing the analysis with the fluorescent ligand used at multiple initial concentrations, resulting in a series of bound and free determinations. Figure 1 shows a plot of the data, in the form of a ligand-binding curve, from which one can obtain a Kd of 4.8 nM, and a Bmax of 3.3 nM FPR. The dissociation rate, its insensitivity to guanine nucleotide (not shown), and Kd were consistent with the LR but not the LRG form of the receptor.
Soluble Receptor Display on Beads. In previous studies (Sklar et al., 2000), we used commercial porous silica particles intended for protein purification (QIAGEN, Valencia, CA). Although they could bind several million receptors on an average size particle, the particles were heterogeneous in size, appeared to break under gentle stirring as monitored by flow cytometry light scatter patterns, and settled rapidly in aqueous media. We therefore prepared a hydrophilic particle, DCNi, as described under Materials and Methods. Preliminary experiments were performed with purified hexahistidine-tagged enhanced green fluorescent protein (H6-GFP; generously supplied by John Nolan) (Lauer and Nolan, 2002) to determine the maximum number of binding sites for a hexahistidine-tagged protein that was unencumbered by competition against other proteins or detergent, that had defined fluorescence, and that was more readily available to other researchers than our particular constructs. This H6-GFP was found to have a molar fluorescence, or quantum yield, in solution of 60% compared with our standard fluoresceinated formyl peptide ligand, fMLFK-FITC.
DCNi beads of the lowest level of substitution were suspended in phosphate-buffered saline at 50,000 beads/ml at 4°C, with or without 10 mM EDTA. Figure 2A is a dot plot of these beads' forward scatter versus side scatter, which vary slightly more than those of a cell population. Figure 2B displays a histogram of unstained and stained beads. These two plots display the same overall shapes for these beads using any fluorescent material that we have used. The kinetic data of Fig. 2C show that in the absence of EDTA, addition of 10 nM H6-GFP resulted in maximal bead fluorescence after about 20 min and displayed about 5 × 106 fluors per bead by comparison with standardized fluorescent microspheres. A portion of these beads was brought to 10 mM EDTA at 30 min, and the H6-GFP on the beads was reduced by 80% after 30 min. The stable binding of this platform is demonstrated in Fig. 2D, in which the H6-GFP remained on the beads for five washes over 2 h, after which the H6-GFP was displaced by 10 mM EDTA as before. Similar rates of displacement by EDTA and stability of bound fluorescence were obtained for the hexahistidine-tagged FPRs (data not shown). It is well known that nickel chelate beads bind proteins without hexahistidine tags, and the presence of other protein or detergent (1 mg/ml BSA or 0.1% Tween 20, respectively) reduced the binding of H6-GFP by 90% (data not shown), giving about 500,000 binding sites under these conditions.
As described previously (Sklar et al., 2000), several million hexahistidine-tagged receptors could bind to a porous silica nickel chelate bead in an LR form with a Kd similar to that of the soluble receptor in detergent. DCNi beads were able to bind about 400,000 formyl peptide receptors with a C-terminal hexahistidine tag (FPR:C-H6) in a crude membrane homogenate, as detected by LF (Fig. 2E). The amount of receptor bound was a function of the position of the tag, with the FPR:C-H6 consistently binding more than the FPR with an N-terminal hexahistidine tag (N-H6:FPR). Although the concentration FPR:C-H6 was 1.9-fold greater than N-H6:FPR in this experiment, the FPR:C-H6 displayed 5-fold greater binding of N-H6:FPR. The binding of receptor in this complex mixture of solubilized proteins was very slow, and continued to increase even after the 7-h data shown in Fig. 2E (data not shown). The Kd for ligand binding was estimated to be 8 nM, and the ligand dissociation rate on beads was similar to the rate in detergent solution, with a 14-s half-time of dissociation (Fig. 2F). There seems to be a systematic deviation of the fitted line from the data points, which affects only about 15% of the total fluorescence and could be caused by receptor bound in different orientations. Although the receptor seemed to behave normally, addition of heterotrimeric G did not alter the ligand Kd or dissociation rate using the N-H6: FPR, which was expected to have a free binding site for G protein while bound to the beads. We therefore took the route to ternary complex assembly described below.
Detection of LFRG Complexes on Beads. Because structural analysis and functional studies suggested that the amino terminus of the γ subunit could be modified without interfering with ternary complex assembly (McIntire et al., 2001), we used purified, epitope-tagged G-proteins to coat the particles.
To prove the concept of assembly on beads, fluorescent ligand, LF, was used to form LFRG on the beads (Fig. 3A). G protein-coated beads (G beads) were prepared and washed as described under Materials and Methods, giving beads coated with αi3β1H6γ2. (18 × 106 αβγ were provided per bead, with ≤500,000 binding sites in proper orientation.) Evidence that fluorescence on the beads was caused by LFRG included the requirement that LF, R, and G were all necessary for fluorescence over nonspecific, background fluorescence. As shown in Fig. 3B, column 1, uncoated beads gave a background binding equivalent to about 9000 fluorophores. The binding doubled when βγ was on the beads and tripled when αβγ was on the beads. We interpret these data to indicate that the αβγ beads, having the highest fluorescence, had everything necessary for LFRG formation, whereas the βγ beads, probably with endogenous αi supplied in the crude solubilized membrane preparation of FPR, gave an intermediate, weaker signal. The addition of GTPγS, which should dissociate α from βγ and from R, resulted in only background fluorescence (similar to unlabeled beads) both with βγ and αβγ beads, as expected. This observation rules out binding of LFR to βγ alone and indicates that an α subunit, either exogenous or in the receptor preparation, is required. Use of an irrelevant fluorescent peptide-organic molecule chimera, specific for the α4 integrin (Chigaev et al., 2001), instead of a fluorescent formyl peptide, also showed only nonspecific binding. Substitution of parental cell extracts that contained no receptor showed increased binding, which was attributed to the fact that free ligand was higher in the absence of FPR, which binds the majority of the total ligand; a high concentration of the free ligand alone gives a nonspecific signal of this magnitude (data not shown). Thus, LF, R, and G were all necessary for the specific fluorescent signal, defined as column 3 minus column 4. Under more nearly optimal conditions, we have observed total fluorescence to background levels as high as 4:1 in this assembly with 30,000 ternary complexes per particle.

Calibrated LFRG assembly with the wild-type FPR. A, schematic diagram of the assembly, including LF, R, and G protein-coated beads. Without the fluorescent ligand, the receptor does not bind to the beads. B, the standard LRG assembly (column 3) included 60 nM R, 75 nM LF, and 24,000 G protein-coated beads in 10 μl, which was mixed for 2 h at 4 to 7°C, then diluted to 200 μl for flow cytometric determination of bead fluorescence as described under Materials and Methods. Results for the standard assembly are shown in column 3, whereas variations in how the bead was coated and changes in soluble components, are indicated beside the graph. 'Wrong L' refers to a peptide-organic molecule chimera which binds specifically to the α4 integrin (Chigaev et al., 2001), and 'No R' refers to the addition of membrane extracts from untransfected U937 cells.
Detection of LFR-αGβγ Complexes on Beads. An FPR-αi2 fusion protein was generated as described under Materials and Methods and solubilized. With this construct (Fig. 4A), we anticipated that endogenous βγ in the solubilized fusion protein preparation might bind to FPR-αi2 to form LRG complex in solution (Shi et al., 2003) and prevent the FPR-αi2 from binding the βγ on the beads. Therefore, we examined the ability of GTP to promote the dissociation of FPR-αi2 from endogenous βγ, and as the GTP was hydrolyzed, the ability of more FPR-αi2 to bind the βγ beads. βγ beads (24,000) were mixed with 24 nM FPR-αi2 and 40 nM LF as in the standard protocol, with GTP as indicated, in Fig. 4B. Uncoated beads, and beads coated with βγ but incubated in the presence of GTPγS, gave background binding equivalent to about 5000 fluors. Assembly in the absence of GTP showed about 12,000 fluors, whereas assemblies in the presence of 0.1 to 10 μM GTP all showed up to 20,000 fluors. Assemblies conducted in the presence of yet higher amounts of GTP showed less bead fluorescence than assembly with no GTP, consistent with excess GTP remaining after the incubation. 1 μM GTP was optimal for the highest binding on the beads and the highest specific signal, defined as column 6 minus column 2. Additional experiments with regulator of G protein signaling suggested that GTP consumption played a role in ternary complex assembly, the effect being a shift in the dose-response curve in LRG formed (data not shown); this could be used for a qualitative test for the presence of regulator of G protein signaling activity but would be difficult to use quantitatively, because the hydrolysis of GTP and binding of R-αi2 to the beads take place simultaneously. The best total fluorescence to background ratio (column 6 compared with column 2) was 2.7:1, similar to that observed for the wild-type receptor above.
Detection of LRFG Complexes on Beads. The third assembly used a fusion protein of FPR with enhanced green fluorescent protein (Fig. 5A; FPR-GFP or RF). The fusion protein was expressed and solubilized as described under Materials and Methods. This receptor bound to the beads in a manner consistent with LRFG formation. In Fig. 5B, background binding of this receptor, with saturating amounts of the nonfluorescent ligand fMLFFGGK, to uncoated beads gave a background binding equivalent to about 5000 fluors, binding to βγ-coated beads to about 30,000 fluors, and binding to αβγ-coated beads to about 60,000 fluors. The assembly on the βγ beads was probably a result of the endogenous αi subunit from the solubilized receptor preparation, because in the presence of GTPγS, the signal was virtually the same as background. The control experiment with no receptor in the assembly reaction was not carried out, because our GFP (without receptor) had a hexahistidine tag on it. The control experiment without ligand gave nearly the same signal as the nonspecific binding. The best total binding to background ratio (column 3 compared with column 4) was 4.9:1, slightly better than above. Thus, three FPR variants were used to demonstrate the formation of an LRG complex on beads. At least tens of thousands of each of the ternary complexes could be formed on the beads.
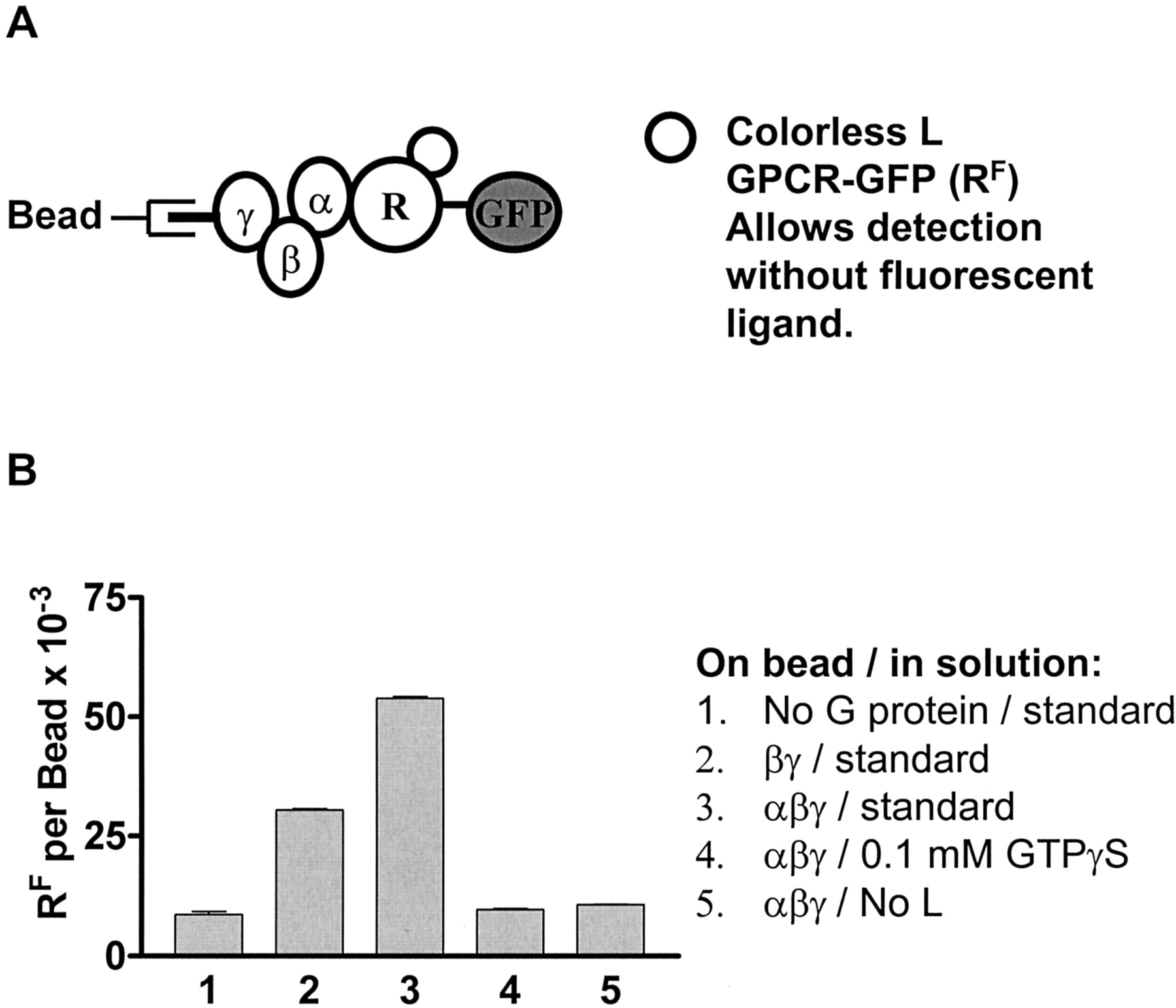
Calibrated LRG assembly with the FPR-GFP fusion protein (RF). A, schematic diagram of the assembly, including L, RF, and G protein-coated beads. Without the ligand, the receptor does not bind the beads. B, the standard LRG assembly included 200 nM RF, 600 nM L, and 24,000 G protein-coated beads in 10 μl, which was mixed for 2 h at 4 to 7°C, then diluted to 200 μl for flow cytometric determination of bead fluorescence as described under Materials and Methods. Results for the standard assembly are shown in column 3, whereas variations in how the bead was coated and changes in the buffer are indicated beside the graph.
Kinetics and Concentration Dependences of the Standard LRFG Assembly. The availability of three receptor forms provided an opportunity to evaluate the affinity of individual steps of the ternary complex model (L to R, LR to G, and α to βγ). To accomplish this task, we first determined the assembly time course for LRFG assembly (schematic of Fig. 5A), which revealed a half-time of 13 min and a calculated association of ∼30,000 LRFG complexes/bead (Fig. 6A). Other experiments showing that maximum assembly was achieved in one to 3 h led us to choose 2 h as the standard time of assembly, which is therefore near equilibrium.

The effects of time and ternary complex partner concentrations on LRFG assembly. The standard assembly for LRFG was performed and bead fluorescence was measured as described in the legend of Fig. 5, with variations in each. A, the time of assembly LRFG was varied. B, the amount of Gαβγ used to coat the beads was varied for LRFG assembly. C, the concentration of ligand used was varied for LRFG assembly. D, the concentration of FPR-GFP used was varied for LRFG assembly.
In Fig. 6B, the amount of G protein incubated with the beads in the standard coating procedure was varied; the line shown is a fit to a simple binding curve, giving half saturation of the beads at 0.35 pmol of G protein applied per assembly assay, corresponding to about 9 × 106 αβγ provided per bead, and a Bmax of 50,000 LRFG per bead. We believe that this curve reflects bead saturation rather than an EC50 for LRG assembly, which is described in Fig. 7B. Our standard protocol thus resulted in 67% saturation of beads with respect to G protein.

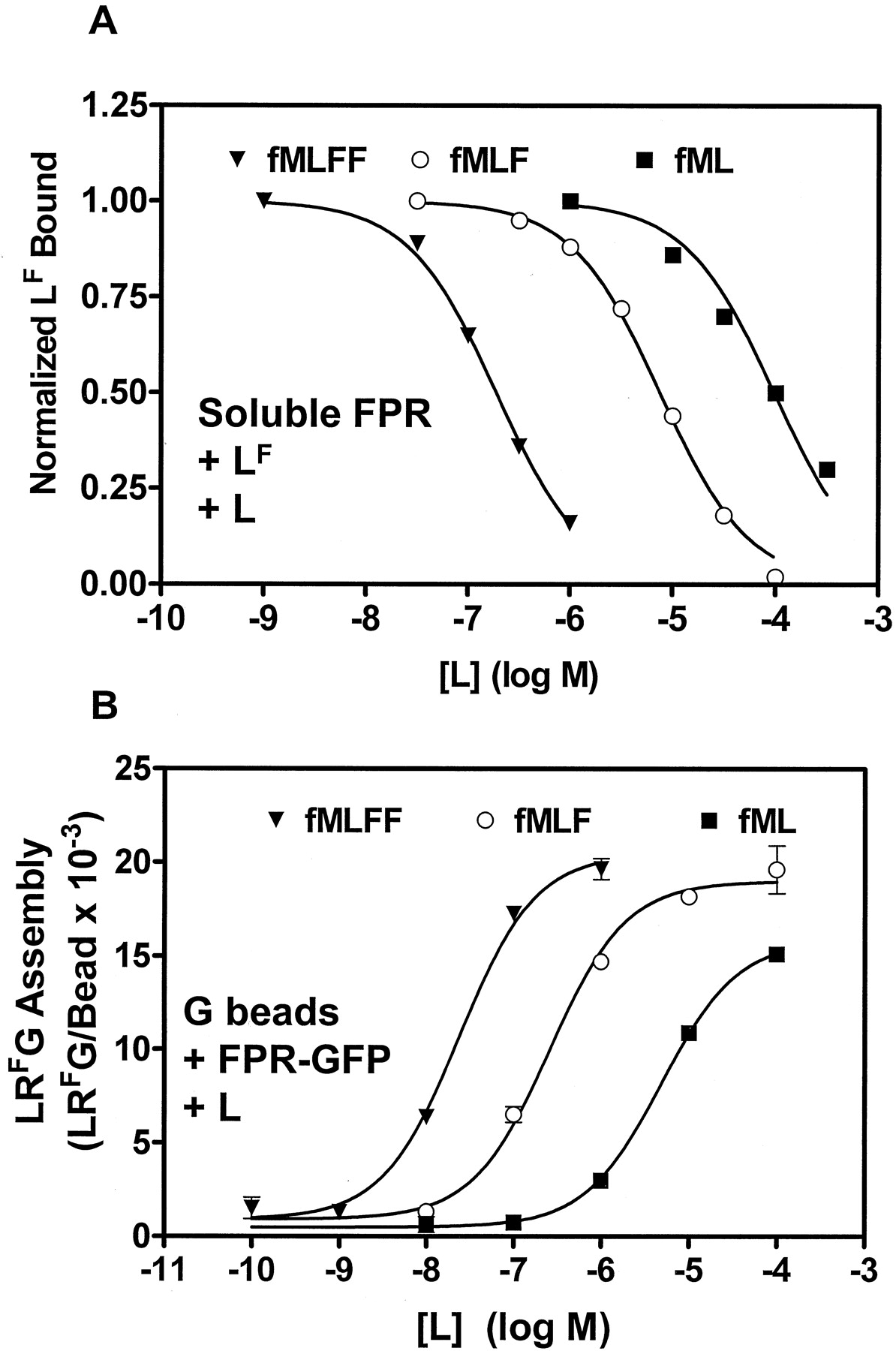
Determination of interaction constants for nonfluorescent ligands. ▾, fMLFF; ○, fMLF; ▪, fML. A, determination of the dissociation constants of nonfluorescent ligands for R by competition with LF in the spectrofluorometer. Receptor (5 nM) and LF (3 nM) were incubated at 22°C for two min, then anti-FITC antibody was added and the resulting trace of fluorescence was analyzed as described previously (Bennett et al., 2001b) to give a maximal amount of LF bound. Increasing amounts of nonfluorescent ligand were added to a fresh sample of R for 2 min, then LF was added for another 2 min to compete for the R, after which anti-FITC antibody was added to determine the LF bound for each amount of nonfluorescent ligand. Although limited by solubility of the ligands, the data were analyzed to give a bottom, or nonspecific binding value, for each data set, shown as zero. IC50 values were calculated from these curves, then initial Ki values were calculated for each ligand by the method of Cheng and Prusoff, using the known [LF], [R], and the Kd of LF for R, using Prism (GraphPad Software). Because the program did not account for free ligand or free receptor depletion, a second program was used to calculate an accurate Ki, accounting for free LF, bound LF, free L, bound L, free R, Kd for LF (4.8 nM), and the Ki for each L. This was done by varying the value of Ki until the calculated values of LF bound for the data set were closest to the experimental values. B, determination of the EC50 for nonfluorescent ligands for LRFG formation. The standard LRG assembly assay was conducted with 30 nM RF and increasing amounts of each ligand as shown. EC50 values were obtained from analysis of the curves.
In Fig. 6C, the concentration of L was varied, and the results again followed a simple binding curve, with half-maximal LRFG assembly at 115 nM L (half of the RF concentration). Because depletion of RF was required for the assay, the affinity of L for RF was not revealed. Our standard assembly, with ligand concentration at least 20% higher than the receptor concentration, gave near saturation with respect to LRG assembly.
In Fig. 6D, the concentration of RF was varied with saturating L. The binding was nearly linear over the accessible concentration range of receptor. It was not possible to calculate a Kd, other than that it must be >200 nM, or a Bmax from these data. We have inserted a theoretical line for a fit to a Kd of 1 μM, which was obtained from solution measurement for LR to G (Bennett et al., 2001b), only to show that the present data are not in disagreement with earlier work and to emphasize that these measurements were made in a system displaying a low affinity component. The standard assembly at 200 nM RF was thus saturating for time and ligand, 67% saturating for G protein, nearly linear with RF, and gave about 30,000 LRFG ternary complexes per bead.
An analogous experiment was performed for the LFR-αGβγ assembly (schematic of Fig. 4A), which examined the affinity of the α to βγ interaction using βγ on the beads, the FPRGαi2 fusion protein, and excess fluorescent ligand (data not shown). The apparent Kd of the R-α to βγ assembly was 26 nM in a range observed previously in other detergent solutions with fluorescent subunits alone on beads, 3 to 50 nM (Sarvazyan et al., 1998). The earlier studies showed a 30-fold difference when performed with different detergents, and because there was no experiment including dodecyl maltoside, no direct comparison can be made. These results do not reveal a contribution of the R to Gβγ interaction.
Comparison of LR and LRG Affinities for a Family of Nonfluorescent Ligands. The complexes on particles offered the ability to examine additional features of ternary complex assembly. First, the Ki values of a series of unlabeled ligands for the FPR were determined by spectrofluorometry (Fig. 7A) and compared with LRG assembly (Fig. 7B). Competitive binding experiments were conducted in which increasing amounts of L were allowed to compete with a fixed amount of fluorescent ligand and soluble receptor. Insolubility limited the highest concentrations of ligands, but Prism software gave IC50 values and both maximum and minimum values of bound ligand for each data set and calculated preliminary Ki values using the Cheng-Prusoff approximation. A second program was used to fit the fraction of maximal LF bound to a single site competition model in which the concentrations of free LF, bound LF, free R, bound L, and free L were calculated using the Kd for LF, 4.8 nM. The Ki was varied for each nonfluorescent L until a consistent fit was obtained, resulting in Ki values about 40% of the IC50 values. Using at least three experiments of the form shown in Fig. 7A, the averages of the Ki values for fMLFF, fMLF, and fML were 5.6 × 10-8, 2.6 × 10-6, and 3.7 × 10-5 M, respectively.
The EC50 values of the ligands for LRG formation were determined using the standard LRFG assembly, as shown in Fig. 7B. Assemblies were conducted in which increasing amounts of each nonfluorescent ligand were added to the standard assembly with 30 nM RF, and the experiment was repeated at 7 nM RF for fMLFF so as to minimize ligand depletion (data not shown). The EC50 values for fMLFF, fMLF, and fML were 1.3 × 10-8, 2.6 × 10-7, and 4.9 × 10-6 M, respectively. The ratios of Kd (for LR) to EC50 (for LRG formation) for fMLFF, fMLF, and fML were 4.3, 10, and 7.6, respectively. These data suggest that in this system, LRG assembly is a function of occupancy. The calculated maximal assembly of fML was 78 ± 3% of the LRG assembly of the longer peptides. Although obtained near the limit of solubility of fML, the data suggest the possibility of partial agonism at the LRG assembly step in signal transduction, consistent with partial agonism for the dipeptide fMF for oxidant production in cells (Sklar et al., 1985).
The presence of the GFP on the tail of the FPR could perturb its ability to interact with G proteins. With 0.4 nM receptor in the 10-μl assay (100,000 receptors per bead, and 24,000 beads), the bead assembly can be used as a sensor in an assembly with tens of nanomolar ligand and receptor. We measured the competition between G protein on beads and soluble G protein in LRG assemblies in an attempt to measure the Kd of LR for G. For FPR with excess LF, the bead-borne LRG assembly was decreased by 50% with 300 to 400 nM soluble G protein, whereas for FPR-GFP with excess L, the bead-borne LRG assembly was decreased by 50% with 150 to 350 nM soluble G protein (data not shown). These results indicate that the GFP moiety does not inhibit the G protein interaction with FPR and that the Kd of this interaction is in the 100 to 400 nanomolar range, similar to the 1 μM value obtained in solution with αi3 and bovine brain βγ (Bennett et al., 2001b).
Real-Time Dissociation Kinetics by Flow Cytometry. To assess a kinetic and mechanistic potential, we examined ternary complex disassembly with wild-type receptor and LF. LRG complexes generally display a higher affinity for L than do LR complexes alone (Gilman, 1987), and we have observed a slower dissociation rate of ligand from LRG complexes than from LR complexes of FPR in detergent (Bennett et al., 2001a,b; Key et al., 2001). We anticipated ternary complex disassembly after GTPγS addition with a half-time <<14 s, the half-time associated with LR dissociation in solution and on beads (Fig. 2F).
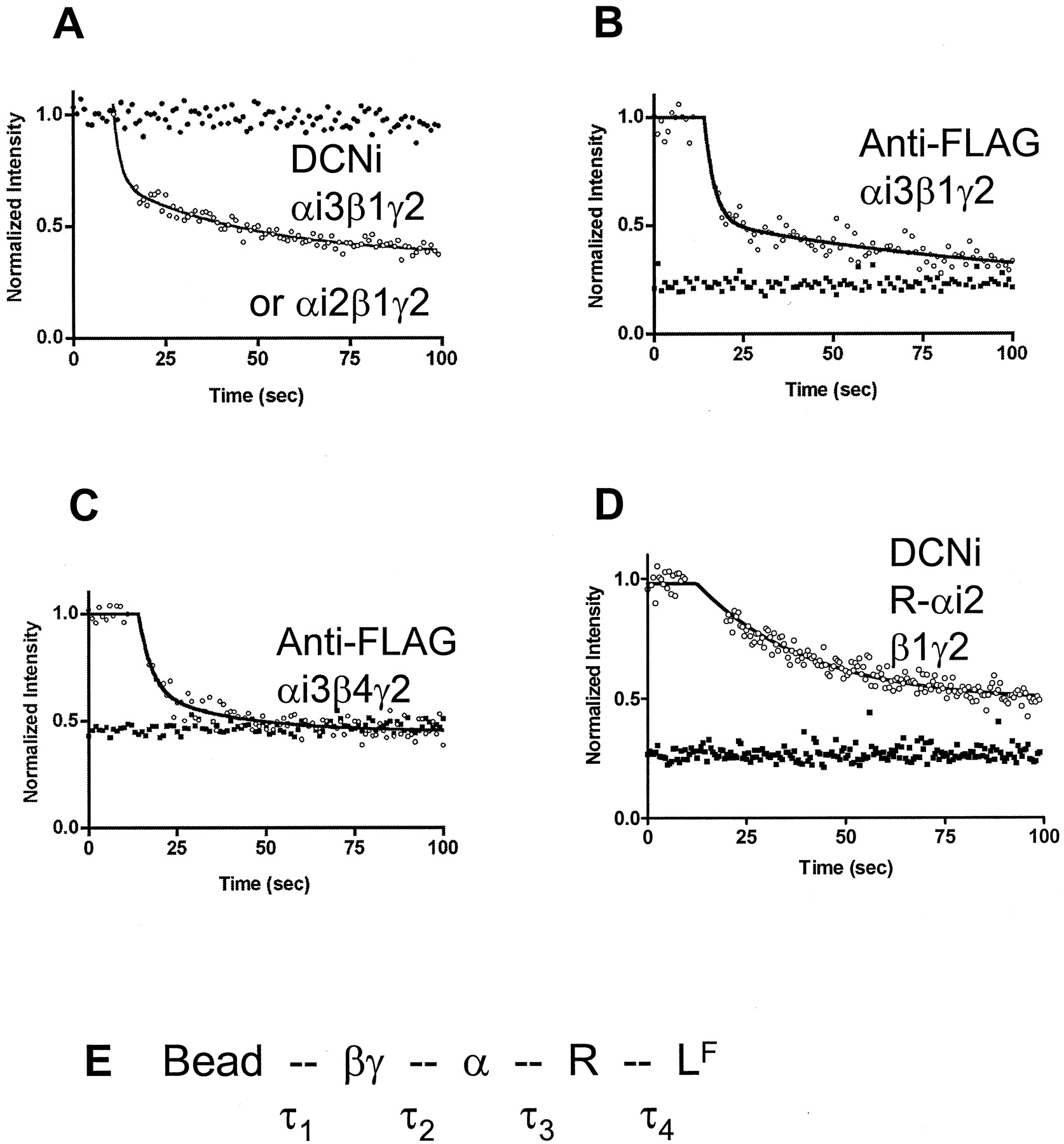
The dissociation of LRG complexes was followed by flow cytometry using manual addition of GTPγS to the bead suspensions (Fig. 8). The time of manual addition and mixing of the GTPγS was determined by the elapsed time indicator on the cytometer and is accurate to about 1 s. Fig. 8A shows results using LFRG on DCNi beads. The half-time for loss of fluorescence (LF) from the particles in the absence of nucleotide was much greater than 100 s, corresponding to LFRG. With addition of saturating GTPγS, a fast component was observed, with a half-time of <5 s, or faster than LR dissociation, using either αi3 (shown) or αi2 (not shown). To evaluate the possibility that nonspecific interactions were contributing to the kinetics, we assembled complexes using streptavidin beads coated with biotin labeled anti-FLAG antibody then coated with αi3+βγ-FLAG complexes as described under Materials and Methods. Studies were performed with two different β subunits, β1 and β4, which have both been shown to complex efficiently with receptors in αi1 complexes (Lim et al., 2001). In assemblies here with αi3 and LF, guanine nucleotide induced dissociation with β1γ2 and β4γ2. As in earlier studies using different α subunits (Bennett et al., 2001b), the reconstitution of the formyl peptide receptor into ternary complex was more efficient with αi3 than αi2 (data not shown). The half-times faster than LR dissociation could potentially result from either RG dissociation, αβγ dissociation, or both. We probed this possibility using the FPR-αi2 fusion protein assembly, in which there can be no RG dissociation (Fig. 8D). This fusion protein has been characterized extensively (Shi et al., 2003) where we found that the ligand dissociation rates for LR (∼14 s) and LRG (>100 s) were indistinguishable from the wild-type receptor. The fact that the bead assembly using this fusion protein (Fig. 8D) dissociated only as rapidly as LR, suggests that αβγ dissociation is no faster than LR and therefore that RG disassembly is the fast step for the wild-type receptor (Fig. 8E). These results are consistent with measurements of slow αβγ dissociation using biotinylated subunits and aluminum-magnesium-fluoride complex as the GTP analog (Sarvazyan et al., 1998).

LFRG disassembly with GTPγS. A, wild-type FPR was used in the standard assembly to form LFRG on DCNi beads; then the samples were diluted to 200 μl for kinetic flow cytometric measurement of bead fluorescence. Samples were applied to the cytometer for determination of initial bead fluorescence, then manually removed for addition of the GTPγS, after which the samples were returned to the cytometer for measurement of bead fluorescence (note the 5-s gap). •, no GTPγS addition. The initial ○ have been averaged over 10 s, normalized to 1.0, and are shown as a single point for clarity at the point of GTPγS addition, with the best fit to a two-exponential decay shown as a line. B and C, 6-μm streptavidin-coated polystyrene beads were coated with biotinylated anti-FLAG antibodies as described under Materials and Methods. The beads were then incubated with Gαi3β1γ2H6-FLAG (B) or Gαi3β4γ2H6-FLAG (C) in which the γ2 subunit was tagged with H6 and FLAG epitopes, as described under Materials and Methods. Standard LFRG assembly assays were conducted with (▪) or without (○) 0.1 mM GTPγS as indicated on the graphs, after which the 10-μl assays were diluted to 200 μl, and the bead fluorescence was measured by flow cytometry, as above. B required two exponential decays to obtain a good fit, whereas C shows a line fit to a single exponential decay. D, LRG was assembled with R-αi2- and βγ-coated DCNi beads and diluted to 200 μl for kinetic flow cytometric analysis. GTPγS was added manually, as above. The line represents a single exponential decay. E, schematic of the bead-based LFRG assembly, with noncovalent bonds represented as dotted lines between the components. Each noncovalent bond has been assigned a half-time for dissociation, which would result in decreasing bead fluorescence if the bond were broken. τ1 is long. τ4 is ∼14 s for the LR form of the receptor and >100 s for the LRG form. When GTPγS is present, the observed half-time of less than 5 s must therefore reside in the bond between α and βγ or between α and R (τ2 or τ3). When the bond between α and R is covalent (D), there is no fast dissociation, indicating that the α to R bond (τ3) represents the fast kinetic component in this system.
Discussion
Reconstitution of high-affinity binding of agonist to receptor with the addition of G proteins in detergent solutions is a well studied method of proving selectivity for the appropriate G proteins (Freissmuth et al., 1991). This report demonstrates the formation of LRG complexes on beads with three FPR variants. It extends work in which reconstitution of soluble receptors with signal transduction partners has been a valuable adjunct to cell physiology and confocal microscopy (Bennett et al., 2001a). Achieving ternary complex formation in detergent on particles involved evaluating several types of beads, attachment schemes with several epitope tags, and several approaches to tether ternary complexes that failed. These included using hexahistidine-tagged receptors on DCNi beads (Fig. 2E) and biotinylated ligand on streptavidin beads (not shown). With hexahistidine-tagged receptors, the problem was nonspecific binding of G proteins to the particles. The biotinylated ligands that recognized soluble receptors in suspension did not capture those receptors on beads.
Assembly and Detection of Ternary Complexes on Beads. The wild-type receptor (R) used in the LFRG assembly provides a direct comparison to the assembly of LFRG in solution (Bennett et al., 2001a,b; Key et al., 2001). The conditions (60 nM R, 75 nM LF, Kd = 5 nM) ensured nearly quantitative conversion of R to LFR, with 15 nM LF free to interact with the beads nonspecifically. The unavailability of fluorescent ligands for other GPCRs is a barrier to transferring this technology. Although the receptor-Gαi2 (R-αi2) construct allows high-affinity complex assembly with the α subunit available at no additional cost, it still uses a fluorescent ligand for detection and is not applicable to other receptors. The receptor-GFP fusion protein (RF) allows quantification of the receptor and obviates the development of a fluoresceinated ligand for every GPCR. It is the obvious construct for high-throughput drug-discovery applications. A triple fusion protein incorporating receptor, Gα subunit, and GFP (Bevan et al., 1999) would allow high-affinity assemblies to be generalized to other receptors.
Affinities of the Components of the Complexes. Previous work with the solubilized FPR (Sklar et al., 2000; Bennett et al., 2001a,b; Key et al., 2001) enabled an analysis of the affinities of LR and LRG. The assembly of LRG in detergent solution took place in a 10-μl volume, with high concentrations of all components, as did assembly onto G protein-coated beads. The 2-h time for assembly in solution was similar to assembly on the bead. Because of the high receptor concentration, ligand depletion at concentrations of ligand similar to that of receptor prevented direct analysis of L affinity with the LRFG complex. Because the receptor concentration was <500 nM, we were unable to unequivocally determine the affinity of LRF for G by varying the concentration of receptor, but the data were consistent with Kd ∼1 μM, similar to the solution value (Bennett et al., 2001a,b; Key et al., 2001); when varying the soluble G protein concentration in competition with the bead-borne G, the FPR and FPR-GFP both exhibited a Ki of 200 to 400 nM. The α to βγ affinity in the presence of LR, 26 nM, was based on the binding of R-αi2 to βγ. Although this is similar to the affinity observed in other detergents with fluorescent subunits alone on beads, 3 to 50 nM (Sarvazyan et al., 1998), we do not resolve R to βγ contributions.
Applications. The three forms of receptor used in this study allowed assemblies to be probed in novel ways. We used FPR-GFP to study ternary complex for a family of ligands, the FPR-αi2 for exploration of kinetic disassembly mechanism and αβγ affinity, and the wild-type receptor and FPR-GFP on beads as sensors for receptor availability in solution. For a set of agonists, the affinities of LR and LRG varied essentially in parallel over 3 orders of magnitude with a hint that a partial agonist might be reflected in fractional LRG assembly (Fig. 7). We have also simultaneously discriminated among antagonists, full agonists, and partial agonists in a format compatible with high throughput (P. C. Simons, S. Biggs, A. Waller, T. D. Foutz, D. F. Cimino, Q. Guo, R. R. Neubig, W.-J. Tang, E. Prossnitz, and L. A. Sklar, submitted for publication).
The assembly and disassembly kinetics of complexes on particles can provide insight into the ternary complex activation. Figure 2 shows dissociation of LFC-H6:FPR on DCNi beads (half-time was ∼14 s in solution and on beads). The dissociation of LFRαβγ on beads was far slower but enhanced by the binding of GTPγS to include a half-time faster than that observed for LR (Fig. 8). The combination of sensitivity of LRG and insensitivity of LR to nucleotide, the Kd values, and the kinetics indicate that we can observe both binary and ternary complexes on beads as well as in solution.
The wild-type ternary complex (LFRαβγ) dissociation was characterized in Fig. 8. During cell activation, the dissociation of R from α, or of α from βγ, could occur in a time frame much faster than LR dissociation. Either of these mechanisms would account for loss of fluorescence from the bead at a rate greater than dissociation of LF from R. Because nonspecific interactions between proteins and DCNi beads could stabilize assembly and slow disassembly, the measurements were repeated with streptavidin beads, biotinylated anti-FLAG antibody, and FLAG-tagged βγ dimer. In all cases (two types of beads, two β subunits) there was a fast component of dissociation that seemed, as expected, to be faster than dissociation of LF from R. These results are consistent with activation faster than ligand release. The dissociation, being faster than LR dissociation, could have been accounted for by dissociation of RG or of αβγ. Experiments with FPR-αi2 showed dissociations no faster than LR, suggesting that RG rather than αβγ dissociation is the fast step.
Beads. The bead display of LRG complexes seems to be general, and the nickel-to-hexahistidine bond seems stable. LRG formation occurs for three forms of FPR (wild-type, receptor-Gα, and receptor-GFP), two epitope tags (hexahistidine and FLAG), two Gα subunits (αi2, αi3), and two Gβ subunits (β1 and β4). We have also demonstrated ternary complex formation with a β2-adrenergic receptor-GFP fusion protein (data not shown). Other types of molecular assemblies are amenable to this technology.
Nonspecific binding with nickel-chelate beads is a potential problem. For purified hexahistidine-tagged GFP, total binding was several million sites per bead, of which ∼90% could be blocked by 0.1% bovine serum albumin, leaving ≤500,000 sites. About 400,000 epitope-tagged receptors from a crude mixture of solubilized proteins bound per particle. For optimal ternary complex formation, about 100,000 G protein sites per particle were accessible. We hypothesize that the purified H6-GFP binding (Fig. 2) reflects all possible modes of binding (hexahistidine-tag dependent and independent) and is within an order of magnitude or less of covering the surface. On the other hand, specific binding of the nonHis-tagged receptor plus ligand to G beads represent hexahistidine-tagged G proteins that are displayed in correct orientation on the surface, ∼100,000. We anticipate that most of the G protein, between the 100,000 displayed correctly and the total number of binding sites (∼5 × 106), was bound nonspecifically and with improper orientation. The use of anti-FLAG beads (Buranda et al., 2001) avoids this problem.
Screening and Proteomics. The receptor-GFP fusion protein should adapt to high throughput screening, especially when coupled to HyperCyt, which delivers beads to a flow cytometer from multiwell plates at rates up to 100 samples per minute (Kuckuck et al., 2001). Particle-based screening is compatible with a search for ligands for both known and orphan receptors (Stadel et al., 1997), agonists promoting assembly on particles, and antagonists inhibiting them. Proteomic applications could be based on bead arrays (Nolan and Sklar, 1998): in this situation, color-coded particles would display individual αβγ combinations, one combination per color code. Specific subunit interactions could be assessed as a GFP-receptor binds to a subset of the combinations. Commercial hardware and software are already available for decoding the results of soluble, multiplex cytometric arrays (Lund-Johansen et al., 2000). Our standard coating of the nickel beads uses 0.7 pmol of Gαβγ per assay to obtain a 3:1 ratio of total signal to nonspecific signal, whereas the anti-FLAG beads use 0.17 pmol of Gαβγ per assay to obtain the same 3:1 ratio, using tens of thousands of beads. Smaller volumes and fewer beads would produce a more efficient screening process.
Footnotes
-
This work was supported by National Institutes of Health Bioengineering Consortium grant GM60799/EB00264 (L.A.S.), grants HL46417, and GM39561 (R.R.N.) and National Science Foundation grant MCB-9907611 (T.B.).
-
ABBREVIATIONS: FPR, formyl peptide receptor; GPCR, G protein-coupled receptor; H6-GFP, hexahistidine-tagged GFP; GFP, green fluorescent protein; R, receptor; L, ligand; G, G protein or heterotrimeric regulatory G protein; LF, fluorescent ligand; LR, ligand-receptor complex; LRG, ligand-receptor-G-protein complex; RF, FPR-GFP fusion protein; RG, receptor-G protein complex; RF, fluorescent receptor; FITC, fluorescein isothiocyanate; fMLFK, formyl-methionyl-leucyl-phenylalanyl-lysine; BSA, bovine serum albumin; DCNi beads, dextran chelate nickel beads; GTPγS, guanosine 5′-O-(3-thio)triphosphate; MCF, mean channel fluorescence; α, Gα subunit; αβγ, Gαβγ heterotrimer; β, Gβ subunit; βγ, Gβγ dimer; γ, Gγ subunit.
- Received April 8, 2003.
- Accepted August 11, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}