


Abstract
The biochemical and pharmacological characteristics in human proinflammatory cells of BRL 50481 [3-(N,N-dimethylsulfonamido)-4-methyl-nitrobenzene], a novel and selective inhibitor of phosphodiesterase (PDE) 7, are described. BRL 50481 inhibited the activity of hrPDE7A1 expressed in baculovirus-infected Spodoptera frugiperda 9 cells in a competitive manner (Ki value of 180 nM) and was 416 and 1884 times less potent against PDE3 and 38 and 238 times less potent against PDE4 at a substrate concentration of 1 μM and 50 nM cAMP, respectively. Western blotting identified HSPDE7A1 but not HSPDE7A2 in three human cell types that are implicated in the pathogenesis of chronic obstructive lung disease, namely, CD8+ T-lymphocytes, monocytes, and lung macrophages. BRL 50481 had no effect on the proliferation of CD8+ T-lymphocytes and only marginally (∼2-11%) reduced the generation of tumor necrosis factor (TNF)α from blood monocytes and lung macrophages. However, in the presence of BRL 50481 the inhibitory effect of rolipram was enhanced on all three cell types. The expression of HSPDE7A1 was increased in a time-dependent manner in monocytes that were “aged” in culture medium. Under this condition, BRL 50481 now inhibited TNFα generation in a concentration-dependent manner. In aged monocytes, rolipram, Org 9935 (a PDE3 inhibitor), and prostaglandin E2 inhibited TNFα generation in a concentration-dependent manner and interacted additively with BRL 50481. BRL 50481 is the first fully documented PDE7 inhibitor that has acceptable selectivity for in vitro studies. Furthermore, although BRL 50481 had only a modest inhibitory effect per se on the proinflammatory cells studied, it acted at least additively with other cAMP-elevating drugs, especially when HSPDE7A1 was up-regulated.
Phosphodiesterase (PDE) 4 is the most extensively studied PDE (Houslay et al., 1998; Conti and Jin, 1999; Houslay and Adams, 2003). Enzymes within this family are found in most proinflammatory and immune cells where they are important regulators of cAMP metabolism (Giembycz, 1992; Torphy, 1998). Moreover, PDE4 inhibitors abrogate inflammation in animal models of respiratory diseases, which has led to the view that PDE4 may represent a target amenable to therapeutic intervention with small molecule inhibitors (Torphy, 1998). Although the use of PDE4 inhibitors for the treatment of airways inflammatory diseases is based on a conceptually robust hypothesis, the clinical efficacy of the current generation of compounds is compromised by dose-limiting side effects (Giembycz, 2000) of which nausea and vomiting are the most common and worrisome (Torphy et al., 1999; Giembycz, 2001, 2002). These adverse effects represent an extension of the pharmacology of PDE4 inhibitors (Duplantier et al., 1996), and improving their therapeutic ratio has proved a major challenge that is still ongoing.
Several strategies have been considered to dissociate the beneficial from detrimental effects of PDE4 inhibitors (Torphy, 1998; Giembycz, 2000) with some degree of success (Torphy et al., 1999; Giembycz, 2001). However, compounds with an optimal pharmacophore still have not been described. An alternative approach is to target other cAMP PDE families that are expressed in proinflammatory and immune cells in the hope that therapeutic activity can be retained with a reduced side effect profile. One such candidate is PDE7, which was first isolated at the gene level in 1993 from a human glioblastoma cDNA library and expressed in a cAMP-deficient strain of the yeast Saccharomyces cerevisiae (Michaeli et al., 1993). PDE7A encodes a cAMP-specific PDE that is insensitive to cGMP and inhibitors of PDE3 and PDE4 and has an amino acid sequence distinct from other cAMP PDEs (Michaeli et al., 1993). Two PDE7 genes (PDE7A and PDE7B) have been identified in humans (Michaeli et al., 1993; Gardner et al., 2000; Hetman et al., 2000; Sasaki et al., 2000). Transcription of PDE7A can give rise to three isoenzymes (PDE7A1, PDE7A2, and PDE7A3) as a consequence of alternative mRNA splicing (Han et al., 1997; Glavas et al., 2001). In contrast, PDE7B exists as a single isoenzyme in humans with approximately 70% sequence similarity to, and distinct kinetic properties from, PDE7A (Gardner et al., 2000; Hetman et al., 2000; Sasaki et al., 2000).
We have reported recently that PDE7A1 mRNA and protein are distributed ubiquitously across human proinflammatory and immune cells (Smith et al., 2003). On the other hand, PDE7A2, which is generated by 5′-splicing and differs from PDE7A1 at its N terminus (Bloom and Beavo, 1996; Han et al., 1997), was never detected at the protein level, despite unequivocal identification of its mRNA (Smith et al., 2003). The distribution of PDE7A3 is largely unknown, but it has been found in human T-lymphocytes (Glavas et al., 2001) and may also be present in many PDE7A1-expressing cells as both transcripts are probably regulated by the same promoter (Torras-Llort and Azorin, 2003). In contrast, PDE7B is abundant in the brain, liver, heart, thyroid glands, and skeletal muscles, but it is not found in leukocytes (Gardner et al., 2000). Together, the distribution of PDE7A1 across human proinflammatory and immune cells mirrors the expression pattern of PDE4 and raises the possibility that it could be exploited to therapeutic advantage to suppress chronic inflammation that characterizes several airway diseases, including asthma and COPD. This hypothesis is supported by the finding that anti-CD3/anti-CD28-driven interleukin (IL)-2 production by, and proliferation of, human naive peripheral blood-derived T-lymphocytes are associated with induction of PDE7A1 and that delivery to these cells of antisense oligonucleotides directed against PDE7A prevented these responses (Li et al., 1999; Glavas et al., 2001). Despite these data, the functional role of PDE7A is still largely unexplored. Here, we report the discovery of a sulfonamide PDE7 inhibitor, BRL 50481 (Fig. 1) and describe its pharmacological activity on proinflammatory responses in human blood monocytes, lung macrophages, and CD8+ T-lymphocytes that are believed to contribute to the inflammation that is a characteristic feature of COPD.

Structure of BRL 50481.
Materials and Methods
Patients providing blood samples gave written informed consent. The Ethics Committee of the Royal Brompton and Harefield National Health Service Trust and National Heart and Lung Institute approved this study.
Purification of Leukocytes. Blood was collected from normal healthy individuals by antecubital venepuncture into acid citrate dextrose (160 mM disodium citrate and 11 mM glucose, pH 7.4), and leukocytes were purified as described below.
T-Lymphocytes. CD8+ T-lymphocytes were purified from the peripheral blood mononuclear cell fraction, which was obtained from density gradient centrifugation of anti-coagulated blood on Ficoll-Hypaque (Amersham Biosciences UK, Ltd., Little Chalfont, Buckinghamshire, UK). Blood was diluted 1:1 with Hanks' balanced salt solution, layered onto Ficoll-Hypaque (1.077 g/ml) and centrifuged (400g; 20 min) at room temperature. Peripheral blood mononuclear cells were harvested from the plasma/Ficoll-Hypaque interface, washed twice in Hanks' balanced salt solution, and further purified by negative immunoselection using the MACS system (Miltenyi Biotech, Bisley, Surrey, UK) according to the manufacturer's instructions to obtain highly purified leukocytes. The cocktail used to isolate the CD8+ T-lymphocytes included antibodies against CD4, CD11b, CD16, CD19, CD36, and CD56.
Monocytes. Monocytes were isolated from peripheral blood, centrifuged on discontinuous Percoll gradients, and purified either by adherence to tissue culture plastic (Seldon et al., 1995) or by negative immunoselection using the Miltenyi MACS system (Miltenyi Biotech). Cells were then cultured in RPMI 1640 medium for the times indicated in the text for TNFα and Western blotting experiments.
Lung Macrophages. Macroscopically normal lung tissue, obtained from patients undergoing surgical resection for carcinoma, was lavaged with RPMI 1640 medium containing 5 mM EDTA, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2.5 μg/ml amphotericin. The cells were washed, resuspended in 2 ml of phosphate-buffered saline, and layered on top of a discontinuous Percoll density gradient [65%/25% (v/v)]. After centrifugation (20 min; 20°C; 500g), the macrophage-enriched fraction was collected at the 65% (v/v) and 25% (v/v) Percoll interface, and the cells were washed twice in phosphate-buffered saline and resuspended in RPMI 1640 medium supplemented with 10% (v/v) fetal calf serum, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2.5 μg/ml amphotericin. After 2 h of incubation in 24-well cell culture plates, the nonadherent cells were removed and fresh medium was added. The adherent purified macrophages were incubated overnight, and the medium was changed the next day before beginning the experiment.
Assessment of CD8+ T-Lymphocyte Proliferation. Isolated CD8+ T-lymphocytes were resuspended at 5 × 105 cells/ml in supplemented RPMI 1640 medium. Unless stated otherwise, 100 μl of the T cell suspension was added to each well of a 96-well plate containing IL-15 as indicated. Cells were incubated in a final volume of 200 μl at 37°C under an atmosphere of 5% CO2 in air for 60 h. At this time 1 μCi of [3H]thymidine in 20 μl of RPMI 1640 medium was added to each well for a further 18 h. In general, inhibitors were diluted in supplemented RPMI-1640 and added to the cells at the start of the incubation 30 min before addition of mitogen and were present throughout the experiment. At 72 h, cells were harvested (Packard Filtermate 196 harvester) onto Packard GF/C 96-well filter plates prewetted with distilled water, washed with excess water, rinsed with 70% (v/v) ethanol, and allowed to air dry. Packard Microscint-20 (50 μl) was then added to each well, the plates shaken for approximately 5 min, and radioactivity incorporated into cellular DNA was counted using a TopCount scintillation counter (PerkinElmer Life and Analytical Sciences, Boston, MA). Proliferation determined using this method was confirmed by manual counting.
Measurement of TNFα. Monocytes and macrophages were pretreated for 30 min with PDE inhibitor(s) or 5 min with PGE2 before being exposed to LPS (3 ng/ml) for 24 h and were present throughout the experiment. The medium was removed and assayed for TNFα by ELISA (human matched pair antibodies, R & D Systems Europe, Abingdon, UK) according to the manufacturer's instructions. The detection limit of this assay is 16 pg/ml.
Measurement of cAMP. MOLT-4 cells (American Type Culture Collection, Manassas, VA) in 96-well plates were treated for 30 min with PDE inhibitor as indicated in the text. The cAMP content was then determined by an immunospecific ELISA. Results are expressed as a percentage of the response effected by 100 μM IBMX.
Measurement of PDE Activity. PDE assays were conducted at room temperature and initiated by the addition of partially purified enzyme to a reaction buffer (100 μl of total volume) containing (final concentration) 50 mM Tris-HCl, pH 7.5, 8.3 mM MgCl2, and 1 μM or 50 nM of either cAMP or cGMP (supplemented with 50,000 dpm/pmol [3H]cAMP or [3H]cGMP, respectively) with or without added inhibitors. The assay was allowed to proceed for 1 h after which the reaction was terminated by the addition of 100 μl of 0.1 M TES, pH 8.0, containing 25 mM EDTA and 100 μM [14C]5′-AMP/5′-GMP (specific activity ∼300 dpm nmol/100 μl of TES “stop” solution) to estimate recovery. The sample was applied to a column containing dry (unwashed) neutral alumina, and unhydrolyzed substrate was eluted with 2 ml plus 10 ml of 0.1 M TES, pH 8.0, and discarded. The 5′-nucleotide products were eluted with 2 ml of 2 M NaOH into 15 ml of Gold Ultima XR liquid scintillation cocktail, and the radioactivity was counted by liquid scintillation spectrometry. The protein concentration in the cytosolic and particulate fractions was adjusted such that less than 20% of the substrate was hydrolyzed and cyclic nucleotide hydrolysis was linear for at least 60 min.
Determination of Kinetics Constants. Km and Vmax were determined by varying the amount of unlabeled cAMP in the reaction cocktail in the presence of a fixed concentration of cyclic nucleotide tracer. Appropriate corrections were made for the changes in specific activity of the substrate. Ki values were derived by the method of Dixon using substrate and inhibitor concentrations that spanned the Km and estimated Ki.
Western Immunoblot Analysis. Cells (3 × 106) were lysed in ice-cold buffer [10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% (v/v) Nonidet P-40, 0.25% (w/v) sodium deoxycholate, 0.1% (w/v) SDS, and 0.25% (v/v) Triton X-100] supplemented with phenylmethylsulfonyl fluoride (0.1 mg/ml), leupeptin (10 μg/ml), aprotinin (25 μg/ml), pepstatin (10 μg/ml), sodium orthovanadate (10 μg/ml), sodium fluoride (100 μg/ml), and sodium pyrophosphate (200 μg/ml) and centrifuged (12,000g; 10 min) to remove insoluble material. The lysates were diluted (5:1) in sample buffer [62.5 mM Tris, 10% (v/v) glycerol, 1% (w/v) SDS, 1% (v/v) β-mercaptoethanol, and 0.01% (w/v) bromphenol blue, pH 6.8] and boiled for 5 min. Denatured proteins were subsequently separated by electrophoresis upon 4 to 12% (w/v) gradient SDS polyacrylamide vertical gels and transferred to Hy-bond ECL membranes (Amersham Biosciences UK, Ltd.) in 50 mM Tris base, pH 8.3, 192 mM glycine, and 20% (v/v) methanol. Nonspecific binding sites were blocked by immersing the membranes in nonfat milk [5% (w/v) in TBS/Tween 20] for 1 h at room temperature. PDE7A expression was detected using a rabbit anti-PDE7A antibody, which is specific for a 15-amino acid sequence at the C-terminal end of HSPDE7A1 and HSPDE7A2. Primary labeling was performed at room temperature using 1 μg/ml of the rabbit anti-PDE7A in nonfat milk 5% (w/v) in TBS/Tween 20 for 1 h at room temperature. After washing in TBS/Tween 20, the membranes were incubated for 60 min with a peroxidase-conjugated goat anti-rabbit antibody (DakoCytomation California Inc., Carpinteria, CA), washed again, and the antibody-labeled proteins were visualized by enhanced chemiluminescence (Amersham Biosciences UK, Ltd.).
Construction of a NFκB Plasmid. An NFκB-dependent reporter, 6NFκBtk.luc, was constructed containing three tandem repeats of the sequence 5′-AGC TTA CAA GGG ACT TTC CGC TGGGGA CTT TCC AGG GA-3′, which harbors two copies of the NFκB binding site (underlined) upstream of a minimal thymidine kinase promoter (-105 to +51) driving a luciferase gene. Neomycin resistance was conferred by ligating a HincII (blunted)/PvuI fragment from pMC1neoPoly(A) (Stratagene, Cambridge, UK) into the PvuI site of 6NFκBtk.luc downstream of the luciferase gene, producing 6NFκBtk.luc.neo.
Stable Transfection and Luciferase Assay. Subconfluent (60%) A549 cells (American Type Culture Collection) in T-75 flasks were washed and incubated in medium containing 8 μg of plasmid and Tfx50 (Promega, Southampton, UK) for 2 h. Cells were washed again, grown in fresh medium for a further 16 h, and then seeded in T-175 flasks containing 500 μg/ml G-418 (Invitrogen, Paisley, UK). In the continued presence of G-418, foci (∼1000) of stable transfected cells developed after approximately 14 days of culture. To create a heterogeneous cell population with regard to integration site, multiple clones were harvested and used in experiments over eight additional passages while being maintained in medium containing 500 μg/ml G-418. After being incubated overnight in serum-free medium in the absence of G-418 selection, cells in 23-well plates were stimulated with IL-1β as indicated in the text and figure legends, and the effect of PDE inhibitors on NFκB-dependent transcription was determined. Luciferase activity was measured using a commercially available Luciferase Assay System according to the manufacturer's instructions (Promega).
Cell Viability. At the end of each experiment, cell viability was determined colorimetrically by measuring the reduction of the tetrazolium salt 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium to formazan by mitochondrial dehydrogenases.
Drugs and Analytical Reagents. PGE2, rolipram, and human recombinant IL-15 were obtained from Sigma-Aldrich (Poole, Dorset, UK), Calbiochem (Merck Biosciences, Nottingham, UK) and Peprotech (Rocky Hill, NJ) respectively. [3H]Thymidine, [3H]cAMP, [3H]cGMP, [14C]5′-AMP, and [14C]5′-GMP were from Amersham Biosciences UK, Ltd. Human recombinant PDE7A1, hrPDE4A4, siguazodan, and BRL 50481 [3-(N,N-dimethylsulfonamido)-4-methyl-nitrobenzene] were made “in-house” at GlaxoSmithKline. PDE1B, PDE1C, PDE2, PDE3, and PDE5 were purified from canine trachea as described previously (Torphy and Cieslinski, 1990). Org 9935 was donated by Organon Laboratories (Newhouse, Lanarkshire, UK). The PDE7A antibody was provided by Celltech Chiroscience (Cambridge, UK). All other reagents were of the highest commercial grade available.
Data and Statistical Analyses. Data points, and values in the text and figure legends, represent the mean ± S.E. mean of n independent determinations using cells from different donors. Concentration-response curves were analyzed by least-squares, nonlinear iterative regression with the Prism curve-fitting program (GraphPad Software Inc., San Diego, CA), and ECX and ICX values were subsequently interpolated from curves of best fit. Where appropriate log-transformed data were analyzed statistically using Student's paired t test or by one-way analysis of variance/Newman-Keuls multiple comparison test. The null hypothesis was rejected when P < 0.05.
Results
Kinetic Characteristics of hrPDE7A1 and Sensitivity to BRL 50481. The hydrolysis of cAMP by hrPDE7A1 expressed in baculovirus-infected Spodoptera frugiperda (Sf9) cells followed simple Michaelis-Menten kinetics from which a Km value of 9.4 ± 0.7 nM and a Vmax value of 74.5 ± 1.8 μmol/min/mg of protein were derived (Fig. 2). At substrate concentrations of 50 nM and 1 μM, BRL 50481 inhibited the hydrolysis of cAMP by hrPDE7A1 with IC50 values of 0.26 and 2.4 μM, respectively, whereas siguazodan (PDE3 inhibitor) and rolipram (PDE4 inhibitor) were >400-fold less active (Table 1). BRL 50481 was a selective inhibitor of hrPDE7A1 being 416 and 1884 times less potent against PDE3 and 38 and 238 times less potent against hrPDE4A4 at 1 μM and 50 nM cAMP, respectively (Table 1). Likewise, BRL 50481 failed to significantly inhibit PDE1B, PDE1C, PDE2, and PDE5 at concentrations below 100 μM (Table 1). IBMX also inhibited hrPDE7A1 but was nonselective, yielding similar IC50 values for the inhibition of PDE3 and PDE4 (Table 1). Kinetic analysis demonstrated that BRL 50481 was a competitive inhibitor (with respect to substrate) of hrPDE7A1 (Fig. 3a) with a Ki value, derived from secondary (Dixon) plots, of 180 ± 10 nM (Fig. 3b).
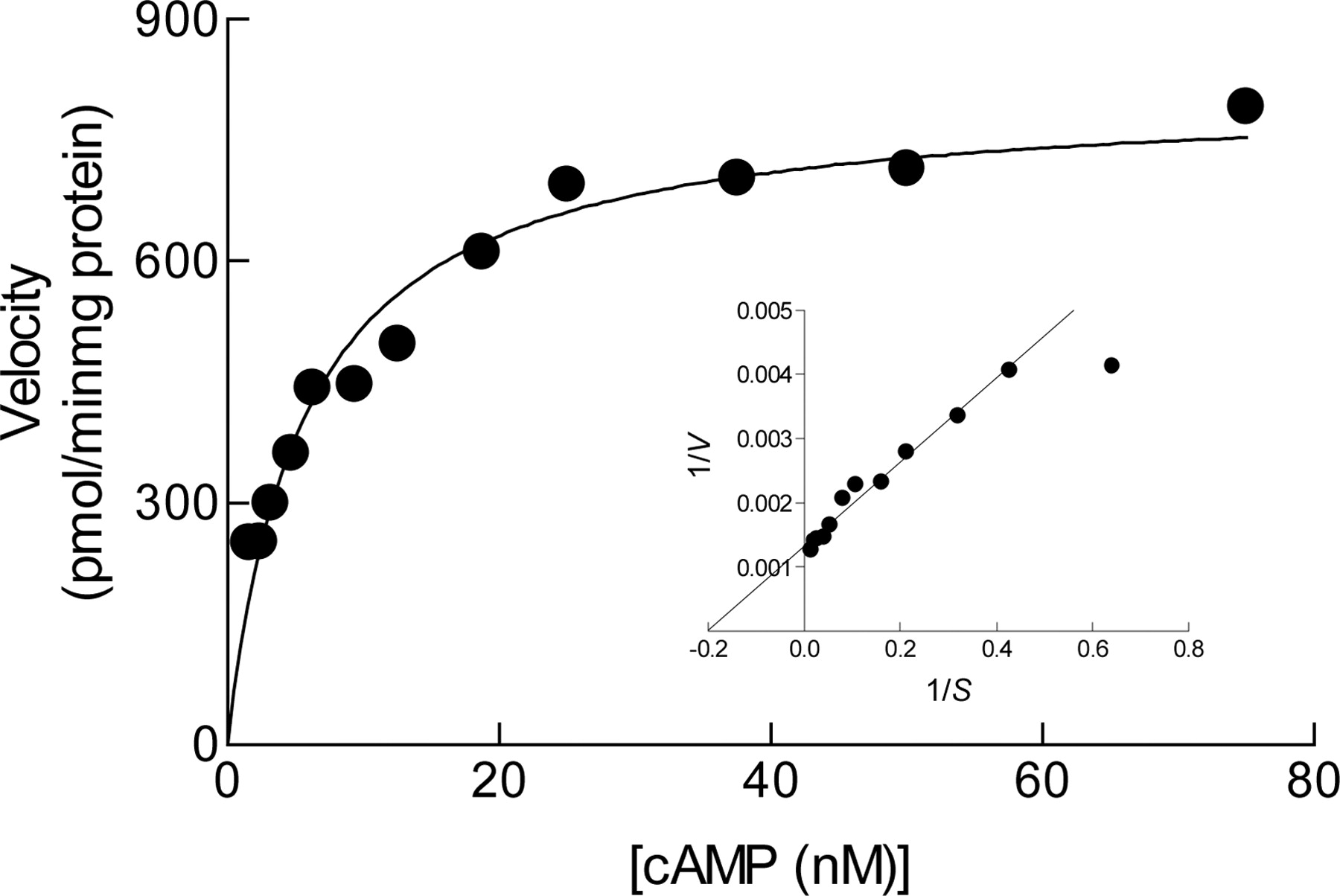
Kinetics of cAMP hydrolysis catalyzed by hrPDE7A1. Lineweaver-Burke plots of the same data are shown in the inset and are typical of five independent determinations.
PDE isoenzyme selectivity of BRL 50481

Kinetic analysis of the inhibition by BRL 50481 of hrPDE7A1. In a, the Lineweaver-Burk plots of cAMP hydrolysis catalyzed by hrPDE7A1 in the absence and presence of BRL 50481 (50-600 nM) are shown. In b, a Dixon plot is illustrated from which a Ki value of 185 nM was obtained in this experiment. These data are typical of three independent determinations.
Identification of PDE7A1. Western blotting was used to establish PDE7A isoform expression in human monocytes, tissue macrophages, and CD8+ T-lymphocytes. For this purpose, a rabbit polyclonal antibody raised against the 15-amino acid sequence (FELNSQLLPQENRLS) at the extreme C terminus of PDE7A, which is common to both PDE7A1 and PDE7A2, was used. In these experiments, the T cell line HUT-78 or U-937 monocytic cells were used as positive controls (Bloom and Beavo, 1996; Smith et al., 2003). As shown in Fig. 4, the anti-PDE7A antibody labeled a peptide in CD8+ T-lymphocytes, monocytes, and macrophages that migrated as a 57-kDa band on SDS polyacrylamide gels. On a protein basis, CD8+ T cells and macrophages expressed similar amounts of PDE7A relative to monocytes where expression was very weak. In each cell type, these immunoreactive proteins were identical in size to PDE7A1 expressed by HUT-78 cells and of a molecular mass consistent with hrPDE7A1 expressed in Sf9 insect cells (Bloom and Beavo, 1996). PDE7A2 (50-kDa protein) was never detected in any of the three cell types studied.

Identification of PDE7A1 in human proinflammatory cells by Western analysis. Highly purified cells were lysed, insoluble proteins were removed, and 20 μg of soluble extract was denatured and subjected to electrophoresis on 4 to 12% (w/v) SDS polyacrylamide gels. Proteins were transferred onto nitrocellulose and incubated with a rabbit anti-PDE7A antibody. The nitrocellulose was subsequently incubated with a peroxidase-conjugated goat anti-rabbit Ig antibody and labeled proteins were detected by enhanced chemiluminescence. The gels are representative of five independent experiments. See Materials and Methods for further details. 1, macrophage; 2, monocyte; 3, HUT-78; and 4, CD8+ T-lymphocyte.
Effect of BRL 50481 on the cAMP Content in the MOLT-4 T Cell Line. Rolipram (100 nM-100 μM) increased in a concentration-dependent manner the cAMP content of MOLT-4 T cells with a pEC50 value of 5.31 ± 0.05 (Fig. 5). At the maximally effective concentration (30 μM), the increase in cAMP mass amounted to 18.1 ± 1.9% of the response elicited by IBMX (100 μM). BRL 50481 also increased the cAMP content (19.1 ± 6.2% of IBMX response at 300 μM) but was considerably less potent (EC50 >> 100 μM). When the effect of BRL 50481 (10-300 μM) on cAMP mass was examined in the presence of a submaximal concentration of rolipram (10 μM; ∼EC80), synergy was observed such that the mean BRL 50481 concentration-response curve was displaced upwards of a magnitude significantly greater than the sum of the cAMP response effected by rolipram and BRL 50481 alone (Fig. 5). Thus, at the highest concentration of BRL 50481 used, the cAMP content amounted to 57.6 ± 3.8% of the IBMX maximum (Fig. 5).

Effect of BRL 50481 on the cAMP content of MOLT-4 T cells. Cells were treated for 30 min with BRL 50481 (10-300 μM), rolipram (100 nM-100 μM), and rolipram (10 μM) in the presence of BRL 50481 (10-300 μM). The cAMP content was then determined by immunospecific ELISA and expressed as a percentage of the response produced by IBMX (100 μM). Each data point represents the mean ± S.E. of four independent determinations.
Effect of BRL 50481 on Human CD8+ T-Lymphocyte Proliferation. Exposure of CD8+ T-lymphocytes to the T cell mitogen IL-15 (Grabstein et al., 1994) increased the amount of [3H]thymidine incorporated in to DNA in a concentration-dependent manner with a pEC50 value (grams per milliliter) of 7.99 ± 0.02 (Fig. 6a). Pretreatment (30 min) of CD8+ T-lymphocytes with rolipram (10 μM) suppressed proliferation by 39.0 ± 4.8%, which was associated with a small (2-fold) but significant (P < 0.05) decrease in the potency of the mitogen (pEC50 value of 7.74 ± 0.3). In contrast, BRL 50481 (30 μM) failed to suppress proliferation by itself (data not shown) but significantly potentiated the effect of rolipram. Thus, [3H]thymidine incorporation was reduced by 67.0 ± 3.6% without a further reduction in the potency of IL-15 (pEC50 value of 7.63 ± 0.02; Fig. 6a).

Effect of BRL 50481 on IL-15-induced proliferation of human CD8+ T-lymphocytes. Cells in 96-well plates were pretreated for 30 min with PDE inhibitors or vehicle. IL-15 was added, and the cells were incubated at 37°C for 60 h under a 5% CO2 atmosphere and then for a further 18 h in the presence of [3H]thymidine (1 μCi). At 72 h, cells were harvested on to GF/C filters, which were washed and counted. In a, the effect of rolipram (10 μM) and rolipram and BRL 50481 (30 μM) in combination is shown on IL-15 (1-400 ng/ml)-induced [3H]thymidine uptake. b shows the antimitogenic action of rolipram (300 nM-10 μM) in the absence and presence of BRL 50481 (30 μM) on IL-15 (100 ng/ml)-induced [3H]thymidine uptake. Each data point represents the mean ± S.E. of four independent determinations using cells from different donors. Note that BRL 50481 (30 μM) did not suppress IL-15-induced proliferation (data not shown). See Materials and Methods for further details. *, P < 0.05, significant inhibition of proliferation. **, P < 0.05, significant inhibition of proliferation over the effect of rolipram alone.
Pretreatment of CD8+ T-lymphocytes with rolipram (30 nM-10 μM) prevented IL-15 (100 ng/ml; ∼EC95)-induced proliferation in a concentration-dependent manner with a pIC50 value (molar) of 5.56 ± 0.11 (Fig. 6b). At the highest concentration (10 μM) of rolipram tested, the cellular incorporation of [3H]thymidine was reduced by 60.0 ± 4.3% (Fig. 5b). BRL 50481 (30 μM) had no effect on IL-15-induced proliferation but augmented the inhibitory effect of rolipram. Thus, the potency of rolipram was increased 4-fold (pIC50 value of 6.17 ± 0.06; P < 0.05) and the maximum inhibition of proliferation was enhanced to 77.1 ± 2.5% (Fig. 6b).
Effect of BRL 50481 on TNFα Release from Human Monocytes. The amount of TNFα released spontaneously from human monocytes after they had been cultured for 24 h in RPMI 1640 medium was low or below the detection limit of the ELISA. However, treatment of these cells with LPS (3 ng/ml; ∼EC90 in freshly isolated cells) resulted in appreciable TNFα production that amounted to 233 ± 32 pg/ml.
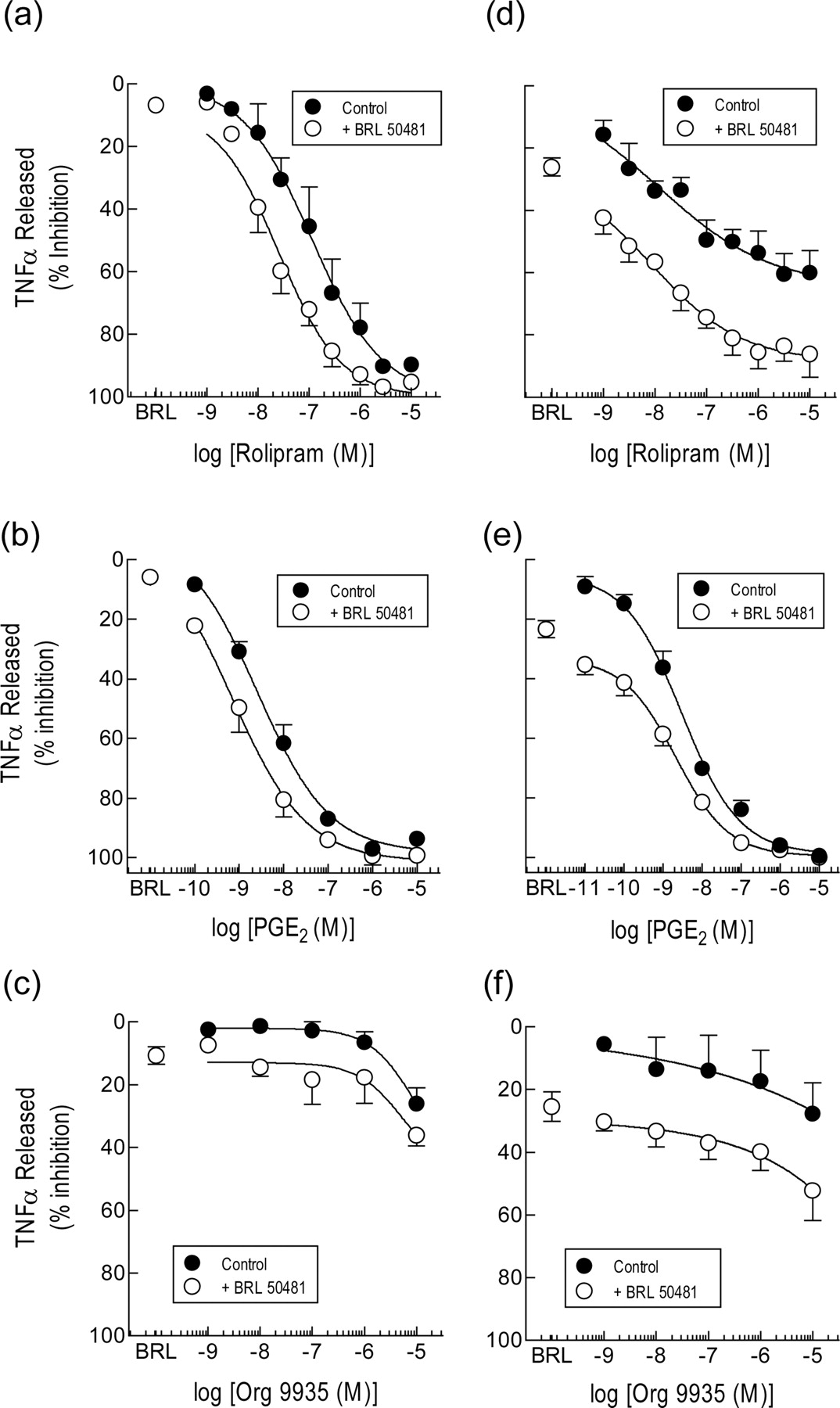
Pretreatment (30 min) of monocytes with rolipram inhibited LPS-induced TNFα release in a concentration-dependent manner with a pEC50 value (molar) of 7.06 ± 0.29 and maximum inhibition of 94.2 ± 2.0% (Fig. 7a). Pretreatment (30 min) of human monocytes with BRL 50481 had, by itself, a negligible (∼2-10%) inhibitory effect on TNFα output at all concentrations tested. However, BRL 50481 (30 μM) displaced 5-fold to the left and in a parallel manner the rolipram concentration-response curve that described the inhibition of TNFα release, which was statistically significant (pEC50 value of 7.76 ± 0.17; P < 0.05; Fig. 7a).

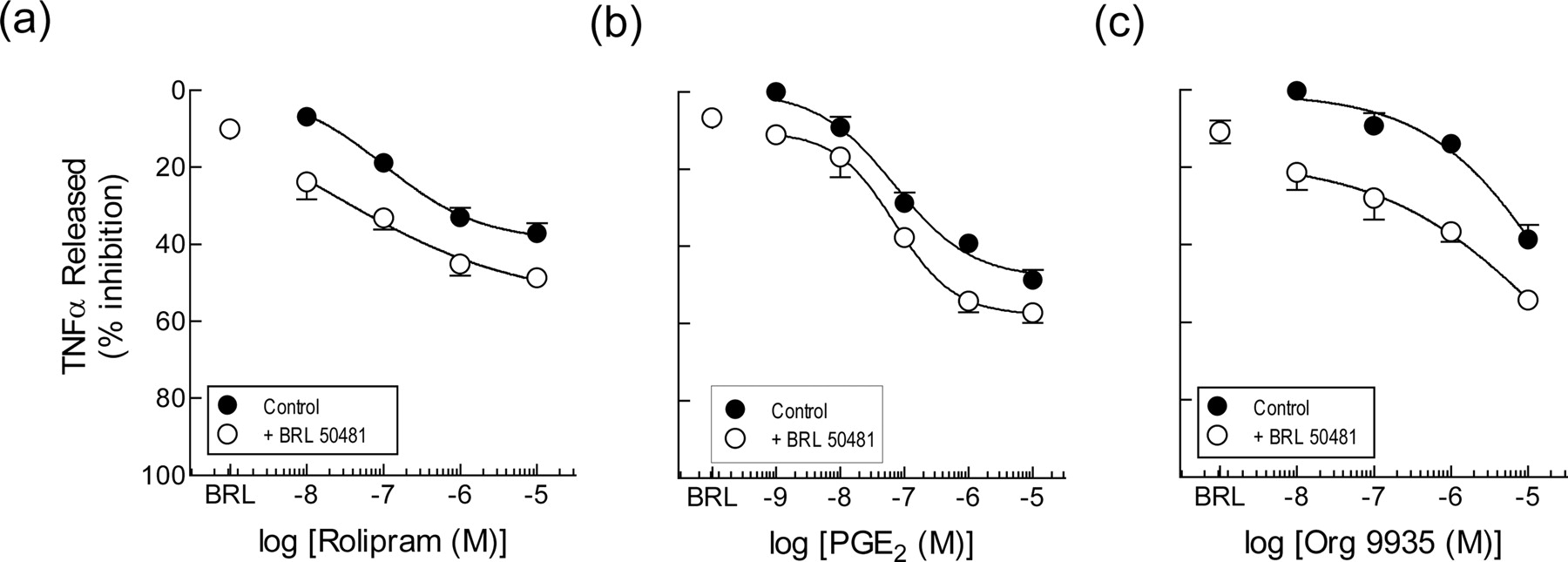
Effect of BRL 50481 on the inhibition by rolipram, PGE2 and Org 9935 of TNFα release from human monocytes. Freshly isolated cells (a, b, and c) or cells aged in culture for 24 h (d, e, and f) were pretreated (30 min) with rolipram (1 nM-10 μM), PGE2 (0.1 nM-10 μM), or Org 9935 (1 nM-10 μM) in the absence and presence of BRL 50481 (30 μM). LPS (3 ng/ml) was then added, and the TNFα elaborated into the culture medium was determined 24 h later by ELISA. Each data point is the mean ± S.E. of five and four independent determinations from different donors for fresh and aged cells, respectively. See Materials and Methods for further details.
BRL 50481 also potentiated the inhibitory effect of PGE2 on LPS-induced TNFα release (Fig. 7b). Indeed, the mean PGE2 concentration-response curve was displaced to the left in an apparently parallel fashion by BRL 50481 (30 μM) increasing the prostanoid's potency approximately 4-fold (pEC50 values: PGE2 = 8.37 ± 0.14; PGE2 + BRL 50481 = 9.02 ± 0.16; P < 0.05). In contrast, neither Org 9935 per se nor Org 9935 in the presence of BRL 50481 (30 μM) modified TNFα release, accept at high concentrations where isoenzyme selectivity may be compromised (Shahid et al., 1991) (Fig. 7c).
Effect of BRL 50481 on TNFα Release from Human Tissue Macrophages. Fig. 8, a and b, shows the inhibitory effect of rolipram and PGE2 on TNFα release from LPS-stimulated human tissue macrophages. Both drugs suppressed TNFα release in a concentration-dependent manner but were less effective (maximum inhibition <50%) and potent compared with their activity on human blood monocytes (cf. Fig. 7, a and b). The PDE3 inhibitor Org 9935, which was essentially inactive on human monocytes, also incompletely suppressed TNFα release from tissue macrophages at concentrations (≤10 μM) of the compound where isoenzyme selectivity is preserved (Fig. 8c).

Effect of BRL 50481 on the inhibition by rolipram, Org 9935, and PGE2 of TNFα release from human lung macrophages. Freshly isolated cells were pretreated (30 min) with BRL 50481 (30 μM) by itself, or BRL 50481 in the absence and presence of rolipram (10 nM-10 μM; a), PGE2 (1 nM-10 μM; b), or Org 9935 (10 nM-10 μM; c). LPS (3 ng/ml) was then added, and the amount of TNFα elaborated in to the culture medium was determined 24 h later by immunospecific ELISA. Each data point is the mean ± S.E. of six, four, and five independent determinations from different donors for rolipram, Org 9935, and PGE2, respectively. See Materials and Methods for further details.
In contrast, BRL 50481 (30 μM) was a very weak inhibitor of TNFα output (4-11% in the experiments shown in Fig. 8). However, when BRL 50481 (30 μM) was combined with rolipram, Org 9935, or PGE2, an additive (or possibly synergistic) effect was seen where the maximum inhibition of TNFα output and potency of rolipram and Org 9935 were significantly increased (Fig. 8).
Effect of PDE Inhibitors on κB-Dependent Transcription. The effect of BRL 50481, rolipram, and Org 9935 on κB-dependent transcription was assessed in A549 cells stably expressing a reporter plasmid, 6NFκBtk.luc.neo, that features two copies of the NFκB binding site upstream of a minimal thymidine kinase promoter driving a luciferase gene. IL-1β (1 ng/ml) increased luciferase expression 19.0 ± 3.5-fold (n = 4) over baseline. Pretreatment of cells with rolipram and Org 9935 inhibited the induction of the luciferase gene in a concentration-dependent manner with pIC30 values (molar) of 5.34 ± 0.13 and 5.97 ± 0.18, respectively (Fig. 9). At the highest concentration where selectivity for PDE3 and PDE4 is preserved, Org 9935 and rolipram repressed the luciferase gene by 52.1 ± 7.2 and 51.9 ± 1.9%, respectively (Fig. 9). In contrast, BRL 50481 had no significant effect by itself on κB-dependent transcription (5.6 ± 1.9% inhibition at 30 μM) and failed to enhance the effect of rolipram (maximum inhibition, 52.9 ± 2.7%; pIC30 value of 5.33 ± 0.12).

Effect of PDE inhibitors on IL-1β-induced activation of a κB-dependent reporter construct. A549 cells were stably transfected with a reporter plasmid, 6NFκBtk.luc.neo, featuring two copies of the NFκB binding site upstream of a minimal thymidine kinase promoter driving a luciferase gene. Cells were pretreated (30 min) with Org 9935, rolipram, BRL 50481 (30 μM), or a combination of rolipram and BRL 50481, exposed to IL-1β (1 ng/ml), and luciferase activity was measured 8 h later. Each data point is the mean ± S.E. four independent determinations. See Materials and Methods for further details.
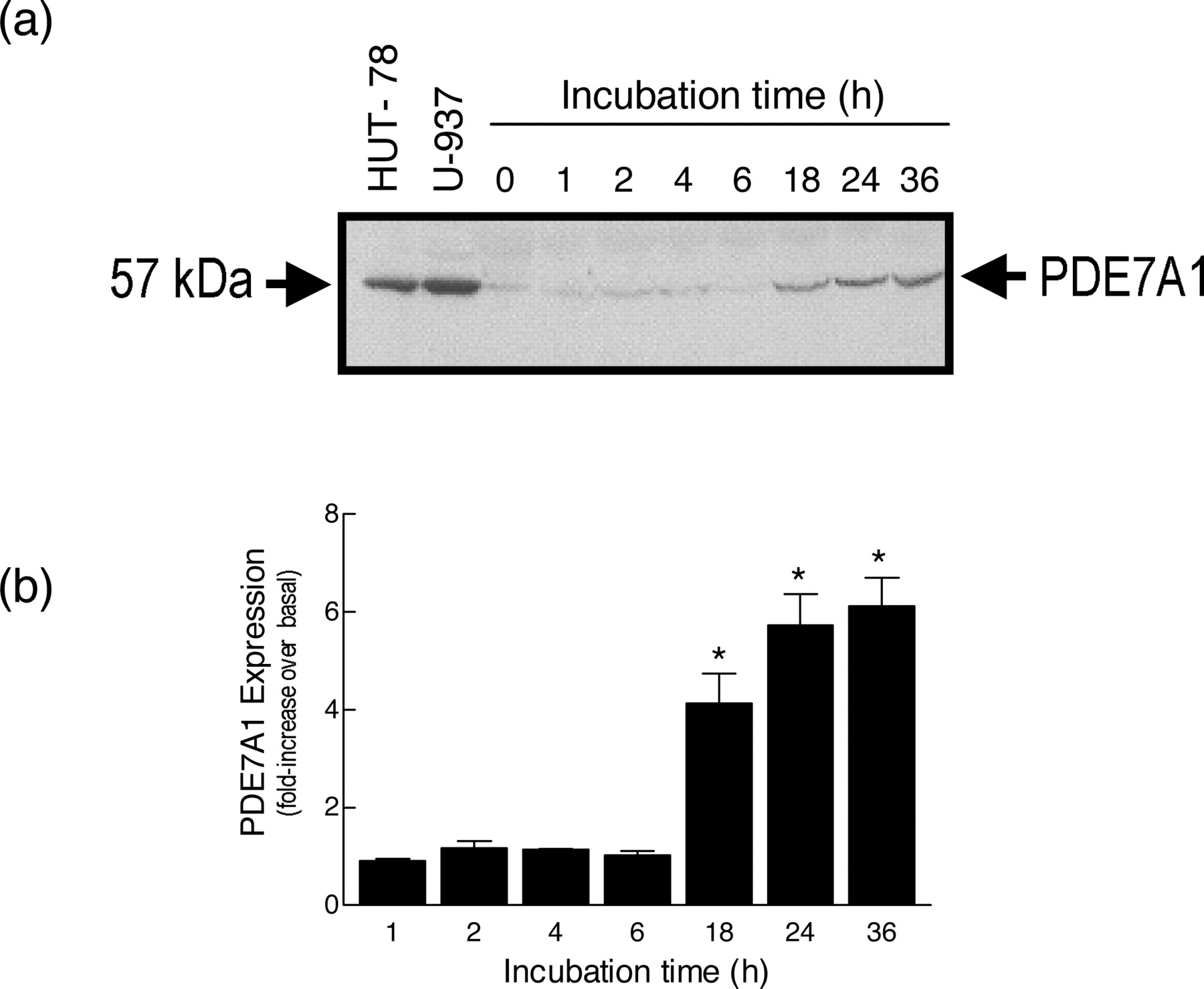
Effect of “Ageing” Monocytes on the Expression of PDE7A1 and Sensitivity to BRL 50481. Culture of monocytes in RPMI 1640 medium for 36 h was associated, after an approximate 6-h lag, with a time-dependent increase in the expression of PDE7A1 compared with freshly isolated cells (Fig. 10). Maximal up-regulation (5.7 ± 0.6-fold) of PDE7A1 was seen after 24 h of culture, and this remained stable for the duration of the experiment. BRL 50481 suppressed, in a concentration-dependent manner, LPS-induced TNFα release in monocytes in which PDE7A1 was induced (21.7 ± 1.6% inhibition at 30 μM at the 12-h time point; Fig. 11a). Kinetic studies showed that the maximal inhibition of TNFα output effected by BRL 50481 was reached by 12 h and was steady at all further time points examined (Fig. 11b). In contrast, whereas the ability of rolipram to reduce LPS-induced TNFα release was preserved in aged monocytes, the characteristics of the inhibition now resembled those seen in human tissue macrophages (cf. Figures 7d and 8a). Thus, rolipram was 11-fold more potent in the aged (12-h) population of cells (pEC50 value of 7.86 ± 0.53) than in macrophages (pEC50 value of 6.82 ± 0.36), whereas the maximum inhibition achieved was similar (∼52 versus. 41%) and considerably less than that effected in freshly purified monocytes (>95%; Fig. 7a). As shown in Fig. 11b, culture of monocytes beyond 12 h was not associated with any further reduction in the maximum inhibitory effect of rolipram, and, accordingly, all further experiments were performed at this time.

Effect of ageing monocytes on the expression of HSPDE7A1. Cells (3 × 106) were cultured for 1 to 36 h in RPMI 1640 medium, lysed, and the soluble extract was denatured and subjected to electrophoresis on 4 to 12% (w/v) SDS-polyacrylamide gels. Proteins were transferred onto nitrocellulose and incubated with a rabbit anti-PDE7A antibody. The nitrocellulose was subsequently incubated with a peroxidase-conjugated goat anti-rabbit Ig antibody, and labeled proteins were detected by enhanced chemiluminescence and shown as fold-increase over expression at the “0” time point. a and b show a representative gel and a bar chart of the mean ± S.E. of four determinations using cells from different donors. HUT-78 and U-937 cells were used as positive controls. See Materials and Methods for further details. *, P < 0.05, significant induction of PDE7A1 in aged versus freshly harvested cells.

Effect of BRL 50481 and rolipram on LPS-induced TNFα generation from human aged monocytes. Cells were aged in culture for up to 48 h, treated (30 min) with either BRL 50481 or rolipram as indicated, exposed to LPS (3 ng/ml), and the TNFα elaborated into the culture medium was determined 24 h later by immunospecific ELISA. a shows the inhibitory effect of BRL 50481 in monocytes aged for 24 h relative to freshly isolated cells. b shows the inhibitory effect of a fixed concentration of rolipram (10 μM) and BRL 50481 (30 μM) on LPS-induced TNFα release in fresh and aged monocytes. Each data point is the mean ± S.E. of five independent determinations using cells from different donors. See Materials and Methods for further details. *, P < 0.05, significantly greater inhibition of TNFα release effected by BRL 50481 in aged versus freshly isolated monocytes. **, P < 0.05, significant change in maximal TNFα output compared with release at the “0” time point.
In the presence of BRL 50481 (30 μM), which in this set of experiments suppressed the elaboration of TNFα by 26.2 ± 5.9%, the rolipram concentration-response curve in aged monocytes was displaced downwards by an amount that was purely additive (Fig. 7d). Thus, the 86% inhibition of TNFα output produced by BRL 50481 (30 μM) and rolipram (10 μM) in combination amounted to the sum of the individual effect of BRL 50481 (26.2%) and rolipram (60.1% at 10 μM). Therefore, this was reflected as equivalence in the potency of rolipram (pEC50 value of 7.97 ± 0.62).
On aged human monocytes, the PDE3 inhibitor Org 9935 evoked a very modest inhibitory effect on the elaboration of TNFα (mean pEC25 of 5.13), which was in keeping with its weak of activity on freshly isolated cells (cf. Figure 7, c and f). In contrast, BRL 50481 (30 μM) suppressed TNFα output by 25.5 ± 4.7% and displaced downwards the Org 9935 concentration-response curve in an additive manner (Fig. 7f). In this respect, the data were reminiscent of those obtained with rolipram (Fig. 7d). Thus, the 52% inhibition of TNFα release produced by BRL 50481 (30 μM) and Org 9935 (10 μM) in combination amounted to the sum of the individual effect of BRL 50481 (25.5%) and Org 9935 (27.7% at 10 μM). Therefore, this was reflected as equivalence in the potency of Org 9935 (mean pIC25 value of 5.26).
Consistent with the rolipram and Org 9935 results described above, BRL 50481 interacted with PGE2 in, at most, an additive manner (Fig. 7e). However, because PGE2 abolished TNFα output, additivity was seen only at low agonist concentrations as the asymptotes of the PGE2 and PGE2/BRL 50481 concentration-response curves converged at 100% inhibition (Fig. 7e). Nevertheless, the pEC50 value of PGE2 in the presence of the PDE7A inhibitor was 8.71 ± 0.17, which was not significantly different from the potency of PGE2 alone (pEC50 value of 8.98 ± 0.27; P > 0.05).
Discussion
Phosphodiesterase 4 inhibitors provide a promising new approach for alleviating the chronic inflammation that characterizes COPD (Torphy et al., 1999; Giembycz, 2001; Giembycz, 2002). However, despite some evidence of clinical efficacy (Compton et al., 2001; Gamble et al., 2003), this class of compound is compromised by dose-limiting side effects that are proving difficult to overcome (Giembycz, 2000). In theory, an alternative way of evoking anti-inflammatory activity in the lung and airways is to target, with small molecule inhibitors, other cAMP PDE families that share a similar pulmonary cellular distribution to PDE4 in the hope that an improved therapeutic ratio will be realized. Of the novel cAMP PDEs thus far identified, PDE7A is a prime candidate because it is expressed in essentially all proinflammatory and immune cells (Smith et al., 2003) and may modulate human T cell function (Li et al., 1999). As an initial step to testing this hypothesis, we describe here the discovery and pharmacology in human CD8+ T-lymphocytes, monocytes, and macrophages of BRL 50481, a selective PDE7 inhibitor.
At the protein level, human proinflammatory and immune cells express PDE7A1 but not PDE7A2 (Smith et al., 2003). Therefore, the inhibition by BRL 50481 of hrPDE7A1 (engineered in Sf9 cells) was characterized, and its selectivity was assessed against hrPDE4A4 and PDEs 1 to 5 purified from canine trachealis. Enzyme kinetic analyses established that BRL 50481 was a competitive inhibitor (with respect to substrate) of hrPDE7A1 with a Ki value in the high nanomolar range. At a substrate concentration of 50 nM, where the IC50 value of cAMP PDE inhibitors closely approximates to the Ki value (Lee et al., 2002), BRL 50481 was at least 200-fold selective for hrPDE7A1 over all other PDEs examined (Table 1). BRL 50481 was not tested against PDE7A2, PDE7A3, or PDE7B in the present study. However, because the three PDE7A splice variants differ only at their extreme amino and carboxy termini, it is likely that they will all be inhibited with similar potency (Giembycz and Smith, 2004). Likewise, BRL 50481 is a catalytic site inhibitor and comparable activity would be expected against PDE7B in the same way that rolipram does not readily discriminate between PDE4 isoenzymes.
Few compounds have been reported with inhibitory activity against PDE7. Martinez et al. (2000) have described a series of benzothieno and benzothiadiazine dioxides that inhibit hrPDE7A expressed in yeast. However, none of the compounds tested showed statistically significant selectivity for PDE7A over other cAMP PDE isoenzymes. Nevertheless, a three-dimensional quantitative structure-activity relationship study using comparative molecular field analysis of 19 compounds within these two series has allowed the design, in silico, of inhibitors with theoretically enhanced activity toward PDE7A (Castro et al., 2001). More recently, data on ICOS corporation's IC242 was disclosed. This compound has an IC50 against PDE7A of 370 nM (at 32 nM cAMP) and has a similar isoenzyme selectivity profile to BRL 50481 (Lee et al., 2002). Several series of compounds from Pfizer and Bristol-Myers Squibb with PDE7 inhibitory activity have also been reported (Lorthiois et al., 2004; Pitts et al., 2004; Vergne et al., 2004). One of these, BMS-586353, is a potent (IC50 value of 8 nM), bioavailable inhibitor of PDE7 with excellent selectivity relative to other PDE isoenzyme families (3722-, 6277-, 1250-, 1231-, and 553-fold less potent against PDE1, PDE3, PDE4, PDE5, and PDE6, respectively; Yang et al., 2003). Finally, chemists from Celltech have found that 8-bromo-9-substituted derivatives of guanine are selective inhibitors of PDE7A (Barnes et al., 2001). In particular, incorporation of a bromo-substituted tetralin ring at position 9 of the guanine template results in a compound with an IC50 value for HUT-78 PDE7A of 1.3 μM with weak activity against PDE3 and PDE4 (14 and 10% inhibition at 10 μM, respectively). Thus, several selective PDE7 inhibitors have now been described, including BRL 50481, which are suitable for in vitro pharmacological testing (see below).
Despite the unequivocal identification of PDE7A1 in CD8+ T cells, BRL 50481 had no effect on IL-15-driven proliferation, even though the cAMP content in MOLT-4 T cells was significantly elevated over the basal level. This was an unexpected finding given that Beavo and colleagues have reported that IL-2 production by, and proliferation of, human blood T-lymphocytes evoked by anti-CD3 and anti-CD28 antibodies is associated with induction of PDE7A1 and prevented by antisense oligonucleotides directed against PDE7A (Li et al., 1999; Glavas et al., 2001). Nevertheless, our data are consistent with results obtained in a more recent investigation where proliferation, and T helper 1 and T helper 2 cytokine production evoked by ligation of CD3/CD28 was preserved in T-lymphocytes taken from PDE7A knockout mice and from wild-type animals treated with BMS-586353 (Yang et al., 2003). It is not clear why these results fail to corroborate data reported by Li et al. (1999), but based on current evidence we suggest that it is unlikely to be species related or to a redundant mechanism in mice that compensates for the deficiency in PDE7A. However, two additional possibilities may account for the discrepancy. In the present study, CD8+ T-lymphocytes were isolated from other leukocytes by negative immunoselection using a mixture of antibodies against CD4, CD11b, CD16, CD19, CD36, and CD56. In the experiments described by Li et al. (1999), antibodies against CD25 and HLA-DR were also used, which will remove all activated and proliferating T cells. Thus, it is possible that naive T-lymphocytes are regulated differently by PDE7A than their activated and proliferating counterparts. On the other hand, the use of naked antisense oligonucleotides, as used by Li et al. (1999), may not have targeted specifically the mRNA of interest or, alternatively, evoked toxic effects that were sequence nonspecific (Stein, 2001).
Consistent with other reports, rolipram suppressed the cellular uptake of [3H]thymidine into CD8+ T-lymphocytes, providing further support that PDE4 is a major regulator of T cell growth and division (Averill et al., 1988; Robicsek et al., 1991; Giembycz et al., 1996; Staples et al., 2001). In contrast, BRL 50481 was inert at all concentrations tested. To determine whether inhibition of PDE7A could enhance the antimitogenic activity of a PDE4 inhibitor, rolipram concentration-response curves were constructed in the absence and presence of a fixed concentration (30 μM) BRL 50481 where selectivity for PDE7 in intact cells is preserved. As shown in Fig. 6, BRL 50481 significantly enhanced the antimitogenic activity of rolipram in a manner that was reminiscent of the behavior of PDE3 inhibitors in human CD8+ T-lymphocytes (Giembycz et al., 1996). Moreover, in MOLT-4 T cells BRL 50481 (30 μM) produced only a very modest increment in cAMP mass compared with rolipram. However, when used in combination BRL 50481 and rolipram acted synergistically increasing cAMP to a level that was significantly greater than that elicited by a maximally effective concentration of rolipram. Thus, PDE7A regulates proliferation and the cAMP content of human CD8+ T cells provided PDE4 is inhibited concomitantly.
On human monocytes and lung macrophages, BRL 50481 was also inactive or, at most, poorly active (2-11% inhibition) in suppressing LPS-induced TNFα release but interacted with rolipram, PGE2, and Org 9935 in at least an additive manner. This profile of activity closely resembles the behavior of BRL 50481 on human T-lymphocytes and demonstrates that inhibition of PDE7A can negatively regulate the proinflammatory activity of cells from the monocytes/macrophage lineage when PDE4 is inhibited.
Many cAMP-elevating agents are known to inhibit the induction of a distinct set of NFκB-regulated genes, including Tnf. To determine whether BRL 50481 also shares this mechanism, a NFκB reporter plasmid, 6NFκBtk.luc.neo, was stably transfected into A549 cells and the effect of PDE inhibitors on IL-1β-induced κB-dependent transcription assessed. Consistent with previous reports (Ollivier et al., 1996; Jimenez et al., 2001; Takahashi et al., 2002), rolipram and the PDE3 inhibitor, Org 9935, effectively repressed luciferase gene expression. In contrast, BRL 50481 was inactive and also failed to enhance the inhibitory effect of rolipram (compare TNFα output). How cAMP blocks κB-dependent transcription has not been fully elucidated and controversy still prevails. In human monocytes, we have found previously that rolipram has no effect on IL-1β-induced IκB degradation or on NFκB/DNA binding (K. K. Meja and M. A. Giembycz, unpublished observations). These data are consistent with the results of comparable studies with human umbilical vein endothelial cells and Jurkat T-lymphocytes and may be explained by the recent demonstration that cAMP blocks κB-dependent transcription in susceptible cells by modifying, directly or indirectly, the transactivation domain of the p65 subunit of NFκB independently of the PKA recognition site at Ser276 (Ollivier et al., 1996; Takahashi et al., 2002). However, blockade of NFκB may not be the primary mechanism of action through which cAMP-elevating drugs suppress TNFα release from monocytes. Indeed, IL-1β-driven, κB-dependent luciferase expression in A549 cells was relatively insensitive (∼20% inhibition) to rolipram at a concentration (3 μM) that reduced IL-1β-induced TNFα output from monocytes by >90%. Furthermore, Org 9935 effectively inhibited the reporter construct at concentrations where TNFα release was unaffected. Although these comparisons are made across two different cells type, these results may, nevertheless, account for the lack of effect of BRL 50481 on IL-1β-stimulated luciferase expression.
Culture of human monocytes in RPMI 1640 medium for 36 h resulted in an ∼6-fold up-regulation of PDE7A1 and conferred functional sensitivity to BRL 50481 such that LPS-induced TNFα release was now significantly inhibited. Moreover, in monocytes where PDE7A1 was up-regulated, the ability of rolipram, PGE2, and Org 9935 to prevent TNFα release was enhanced by BRL 50481 in a purely additive manner. Previous studies have found that ageing monocytes is also associated with a down-regulation of PDE4 activity that is primarily attributable to PDE4D family members (Gantner et al., 1997; Shepherd et al., 2004) and that this enzyme remodeling accounts for the reduction in the maximum inhibition of TNFα output produced by rolipram (Gantner et al., 1997). Therefore, collectively, these data demonstrate that with respect to the regulation of TNFα release, the increased expression of PDE7A1 compensates, at least in part, for the down-regulation of PDE4. In theory, this could be exploited to therapeutic advantage in inflammatory diseases where cells of the monocyte/macrophages lineage play a pathogenic role.
It is important to emphasize that the ability of BRL 50481 to suppress TNFα output at the concentration (i.e., 30 μM) used in the combination experiments was not caused by the inhibition of PDE4 because the rolipram and rolipram plus BRL 50481 concentration-response curves did not converge at high concentrations. Together, these data imply that PDE7A can regulate the responsiveness of monocytes and possibility other proinflammatory and immune cells under circumstances when PDE7A is highly expressed, such as conditions of chronic inflammation. In this respect, many cytokines relevant to the pathogenesis of airway inflammatory diseases signal, in part, through a protein kinase C-dependent mechanism (Kontny et al., 2000), and Torras-Llort and Azorin (2003) have reported that the human PDE7A1 promoter is activated by the phorbol ester phorbol 12-myristate 13-acetate.
In conclusion, the results of the present investigation demonstrate that BRL 50481 is a PDE7 inhibitor with nanomolar potency and is sufficiently selective against other PDE isoenzyme families for in vitro pharmacological studies. BRL 50481 was poorly active in suppressing human T cell proliferation and TNFα release from monocytes and macrophages but, nevertheless, acted in at least an additive manner with the PDE4 inhibitor rolipram. These findings suggest that hybrid PDE4/PDE7 inhibitors may be more efficacious and display a superior therapeutic index than a PDE4 inhibitor alone. This is an important consideration as the two most clinically advanced PDE4 inhibitors in trials of COPD are compromised by dose-limiting side-effects (see Introduction). In this respect, it noteworthy that several pharmaceutical companies have filed patents claiming for dual PDE4/PDE7 inhibitors with potential anti-inflammatory activity (Pitts et al., 2002; Hatzelmann et al., 2004). Finally, up-regulation of PDE7A1 in monocytes conferred increased sensitivity to BRL 50481, indicating that inhibitors of this isoenzyme family per se could be effective in inflammatory indications where HSPDE7A is induced.
Acknowledgments
We thank Peter Goldstraw (Department of Cardiothoracic Surgery, Royal Brompton Hospital) for generously providing human lung specimens for macrophage isolation and Lisa Cambridge for assistance with the NFκB reporter studies.
Footnotes
-
This study was funded by GlaxoSmithKline (Stevenage, UK). M.A.G. is an Alberta Heritage Foundation Senior Medical Scholar. M.A.G. and R.N. are funded by the Canadian Institutes of Health Research.
-
ABBREVIATIONS: PDE, phosphodiesterase; hrPDE, human recombinant phosphodiesterase; COPD, chronic obstructive pulmonary disease; IL, interleukin; TNF, tumor necrosis factor; LPS, lipopolysaccharide; ELISA, enzyme-linked immunosorbent assay; IBMX, 3-isobutyl-1-methylxanthine; TBS, Tris-buffered saline; NFκB, nuclear factor κB; PG, prostaglandin; Org 9935, 4,5-dihydro-6-(5,6-dimethoxybenzo[b]thien-2-yl-5-methyl-3(2H)-pyridazinone.
- Received May 5, 2004.
- Accepted September 14, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}