


Abstract
Interactions between the histone deacetylase (HDAC) inhibitors suberanoylanilide hydroxamic acid (SAHA) and sodium butyrate (SB) and the heat shock protein (Hsp) 90 antagonist 17-allylamino 17-demethoxygeldanamycin (17-AAG) have been examined in Bcr-Abl+ human leukemia cells (K562 and LAMA84), including those sensitive and resistant to STI571 (imatinib mesylate). Cotreatment with 17-AAG and SAHA or SB synergistically induced mitochondrial dysfunction (cytochrome c and apoptosis-inducing factor release), caspase-3 and -8 activation, apoptosis, and growth inhibition. Similar effects were observed in LAMA84 cells and K562 cells resistant to STI571, as well as in CD34+ cells isolated from the bone marrows of three patients with chronic myelogenous leukemia. These events were associated with increased binding of Bcr-Abl, Raf-1, and Akt to Hsp70, and inactivation of extracellular signal-regulated kinase 1/2 and Akt. In addition, 17-AAG/SAHA abrogated the DNA binding and the transcriptional activities of signal transducer and activator of transcription (STAT) 5 in K562 cells, including those ectopically expressing a constitutively active STAT5A construct. Cotreatment with 17-AAG and SAHA also induced down-regulation of Mcl-1, Bcl-xL, and B-Raf; up-regulation of Bak; cleavage of 14-3-3 proteins; and a profound conformational change in Bax accompanied by translocation to the membrane fraction. Moreover, ectopic expression of Bcl-2 attenuated cell death induced by this regimen, implicating mitochondrial injury in the lethality observed. Together, these findings raise the possibility that combining HDAC inhibitors with the Hsp90 antagonist 17-AAG may represent a novel strategy against Bcr-Abl+ leukemias, including those resistant to STI571.
17-Allylamino 17-demethoxygeldanamycin (17-AAG), a derivative of the ansamycin antibiotic geldanamycin, has shown promising preclinical activity against a variety of tumor cell types and is currently undergoing phase II clinical evaluation (Dunn, 2002). 17-AAG antineoplastic activity has been attributed to binding to the ATP/ADP binding pocket of the heat shock protein (Hsp) 90, thereby inhibiting its function as a molecular chaperone. Hsp90 plays a key role in the intracellular trafficking and maturation of diverse cell signaling proteins, including those involved in cell survival such as Bcr-Abl, Raf-1, ErbB2, and Akt. Inhibition of Hsp90 interferes with the folding of client proteins, thereby targeting them for ubiquitination and subsequent proteasomal degradation (Nimmanapalli et al., 2001).
Histone deacetylase (HDAC) inhibitors represent a structurally diverse group of compounds that inhibit the deacetylation of histones, permitting chromatin scaffolding to assume a more relaxed state. Recent evidence suggests that these agents may also enhance acetylation of nonhistone proteins (Marks et al., 2003). After treatment with HDAC inhibitors, various genes are activated (e.g., p21CIP1) or repressed (e.g., ErbB2) (Marks et al., 2003). Although the mechanisms determining which genes are activated or down-regulated by HDAC inhibitors remain unclear, it is likely that transcription factors play an important role in this phenomenon. For example, the signal transducer and activator of transcription (STAT) 6 seems to bind to its target promoters and enhance transcription only when histones are acetylated (Shankaranarayanan et al., 2001). Activity of the transcription factor Sp1 is also enhanced after histone acetylation (Park et al., 2003). On the other hand, hyperacetylation of nonhistone proteins may disrupt certain signaling molecules, leading to diminished expression of their target genes (Nimmanapalli et al., 2003a). Several HDAC inhibitors are currently undergoing clinical evaluation, including butyrate, MS-275, and depsipeptide (Marks et al., 2003).
Chronic myelogenous leukemia (CML) is characterized by the Philadelphia chromosome, formed by the reciprocal translocation of chromosomes 9 and 22, resulting in a constitutively activated fusion oncoprotein (Bcr-Abl) (Faderl et al., 1999). The Bcr-Abl protein has tyrosine kinase activity and signals downstream to multiple survival pathways, including STATs, Akt, extracellular signal-regulated kinase (ERK), and nuclear factor-κB, among others (Van Etten, 2004). In addition to providing hematopoietic cells with a survival advantage (Horita et al., 2000), the Bcr-Abl oncoprotein renders leukemic cells particularly resistant to conventional cytotoxic agents (Bedi et al., 1995). Treatment of CML has recently been revolutionized by the introduction of STI571 (imatinib mesylate), a Bcr-Abl kinase inhibitor that effectively induces cell death in Bcr-Abl+ cells (Druker et al., 1996). However, the emergence or preexistence of imatinib resistance, through either gene amplification or mutation (von Bubnoff et al., 2003), has prompted the search for additional therapeutic approaches. In this regard, strategies combining STI571 with other novel antisignaling agents, including PD184352 (Yu et al., 2002b), histone deacetylase inhibitors (Yu et al., 2003a), flavopiridol (Yu et al., 2002a), and arsenic trioxide (Porosnicu et al., 2001), seem promising.
Several lines of evidence suggest that both 17-AAG and HDAC inhibitors may play a useful role in the treatment of CML and related Bcr-Abl+ hematologic malignancies. For example, 17-AAG has been shown to induce Bcr-Abl down-regulation and may preferentially induce degradation of mutant protein (Gorre et al., 2002). Furthermore, HDAC inhibitors, including SAHA, and more recently, NVP-LAQ824, have been shown to potentiate the lethal effects of imatinib mesylate in Bcr-Abl+ cells in association with Bcr-Abl down-regulation (Nimmanapalli et al., 2003a; Yu et al., 2003a). Very recently, we have observed that 17-AAG and HDAC inhibitors interact in a highly synergistic manner to induce apoptosis in multiple human Bcr-Abl- leukemia cells (Rahmani et al., 2003a). In light of these findings, and in view of the documented resistance of Bcr-Abl+ cells to more conventional cytotoxic agents (Bedi et al., 1995), the notion that combined exposure of Bcr-Abl+ cells to these agents might lead to enhanced apoptosis seemed worthy of investigation. Here, we report that cotreatment with the HDAC inhibitors SAHA or sodium butyrate and 17-AAG potently induces apoptosis in Bcr-Abl+ leukemic cells, including those sensitive and resistant to imatinib mesylate. These events are associated with multiple perturbations in signaling pathways, including pronounced down-regulation of Bcr-Abl protein expression, marked inhibition of the DNA binding and the transcriptional activities of STAT5, and inactivation of Akt and Raf/MEK/ERK axis, accompanied by a profound Bax conformational change and translocation to the mitochondria. Together, these findings suggest that combined treatment with HDAC inhibitors and Hsp90 antagonists such as 17-AAG may represent a novel therapeutic approach in Bcr-Abl+ leukemias.
Materials and Methods
Cells. The human leukemia K562 cells and LAMA84 cells (German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany) were cultured as reported previously (Rahmani et al., 2003b). STI571-resistant K562 and LAMA84 cells, designated K562-STI-R and LAMA-STI-R, respectively, were generated by culturing cells in progressively higher concentrations of STI571 as we have described previously (Yu et al., 2002a, 2003b). These cells, which exhibit an STI571 IC50 value ∼15-fold greater than parental cells, were maintained in 1 μM STI571 and washed free of drug 2 days before experimentation. K562 cells ectopically overexpressing constitutively active STAT5 were obtained by stable transfection of K562 cells using electroporation as described previously (Rahmani et al., 2003b) with a plasmid encoding for a constitutively active form of STAT5 (FLAG-tagged pMX-STAT5A-N642H) (Ariyoshi et al., 2000) or the empty vector pMX-neo.
Bone marrow cells were obtained with informed consent from three patients with CML and from two patients with nonmyeloid hematologic disorders (e.g., iron deficiency and thrombocytopenia). These studies have been sanctioned by the Investigational Review Board of Virginia Commonwealth University/Medical College of Virginia. Mononuclear cells were isolated by Ficoll-Hypaque density gradient separation as described previously in detail (Yu et al., 2003b). Mononuclear cells from patients with CML were enriched for CD34+ cells using a Miltenyi microbead separation system (Miltenyi BioTech, Auburn, CA) according to the manufacturer's protocol.
Reagents. 17-AAG was provided by the Cancer Treatment and Evaluation Program, National Cancer Institute (Bethesda, MD). SAHA was purchased from Alexis Corporation (San Diego, CA). Sodium butyrate was obtained from Calbiochem (San Diego, CA). All reagents were formulated as recommended by their suppliers.
Assessment of Apoptosis. Apoptosis was routinely assessed by annexin V-fluorescein isothiocyanate staining and in some experiments by morphological assessment of Wright Giemsa-stained cytospin preparations as described previously (Rahmani et al., 2002).
Cell Growth and Viability. Cell growth and viability were assessed using the [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt] (MTS) compound. In brief, after cell exposure to indicated drugs for various intervals, cells were seeded in 96-well plates (100 μl/well) in the presence of 20 μl of MTS solution (Promega, Madison, WI). Cells were cultured for an additional 4 h, after which absorbance, reflecting reduction of MTS by viable cells, was determined at 490 nm using a microplate reader. Values were expressed as a percentage relative to those obtained in untreated controls.
Electrophoretic Mobility Shift Assay (EMSA). EMSA was performed on nuclear extracts as described previously (Rahmani et al., 2001) using double-stranded oligonucleotides corresponding to STAT5 binding site (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). For supershift analysis, specific antibodies directed against STAT5A (Santa Cruz Biotechnology, Inc.) or anti-FLAG M2 (Sigma-Aldrich, St. Louis, MO) were used.
Transient Transfection and Luciferase Reporter Gene Assays. Transient transfections were performed by electroporation using the Amaxa nucleofector (Koeln, Germany) as recommended by the manufacturer. High transfection efficiency was obtained routinely in these cells [i.e., 70 to 80% of cells were green fluorescent protein-positive when cells were transfected with an enhanced green fluorescent protein plasmid (Amaxa)]. K562 cells were transfected with Bcl-2 and pUSEamp vectors (Upstate Biotechnology, Lake Placid, NY), incubated for 24 h, and then treated as indicated in the results. To determine the transcriptional activity of STAT5, K562 cell were cotransfected with pSTAT5-luc plasmid encoding for firefly luciferase (Panomics, Redwood City, CA) and pRL-TK-luc plasmid encoding for Renilla reniformis luciferase. Cells were incubated for 6 h and then treated with indicated agents for an additional 20 h, after which activity of firefly and R. reniformis luciferase was measured using the Dual-Luciferase reporter assay system (Promega). Values of firefly luciferase activity were normalized to those obtained for R. reniformis luciferase activity.
Immunoprecipitation and Immunoblotting. For immunoprecipitation, cells were lysed in buffer containing 20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, antiproteases, and 1% Triton X-100, after which 500 μg of protein lysates was subjected to immunoprecipitation using indicated antibodies (c-abl, Raf-1, and Akt). Immunoblotting was performed using the immunoprecipitates or the whole cells lysates as described previously in detail (Rahmani et al., 2002). The primary antibodies used in this study were cytochrome c, caspase-3, polyclonal Bax, and Mcl-1 (BD Biosciences PharMingen, San Diego, CA); Bcl-2 (Upstate Biotechnology); caspase-8 (Alexis); Bid, poly(ADP-ribose) polymerase (PARP), phospho-Akt (Ser473), Lyn, Phospho-Lyn (Tyr507), Akt, p38, Bcl-xL, and XIAP (Cell Signaling Technology Inc., Beverly, MA); total and phospho-ERK1/2 (p44/42 Tyr204), Bim, 14--3, c-Src, c-Abl, Raf-1, B-raf, hsp70, hsp90, and AIF (Santa Cruz Biotechnology, Inc.); FLAG-M2 (Sigma-Aldrich); and Bak and α-tubulin (Calbiochem).
Bax Conformational Change. Cells were lysed in buffer containing 150 mM NaCl, 10 mM HEPES, pH 7.4, anti-proteases, and 1% CHAPS or 1% Triton X-100 (Murphy et al., 2000). Then, 500 μg of protein lysates was subjected to immunoprecipitation using anti-Bax monoclonal antibody 6A7 (Sigma-Aldrich) that recognizes only Bax protein that has undergone conformational change (Murphy et al., 2000). Immunoprecipitates were then subjected to Western blot analysis with anti-Bax polyclonal antibody (BD Biosciences PharMingen).
Subcellular Fractionation. K562 cells (4 × 106) were lysed using digitonin buffer (Rahmani et al., 2002), after which cytosolic and membrane fractions were separated by centrifugation, solubilized in Laemmli buffer, and boiled for 5 min. Proteins were analyzed by Western blot to evaluate cytochrome c release into the cytosol and Bax translocation into the membrane fraction. The purity of cytosolic and membrane fractions was routinely confirmed by Western blot using antibody against cytochrome c oxidase subunit IV (Cox4), which resides strictly in the inner mitochondrial membrane. Bax detected in the membrane fraction is presumed to represent mitochondrial Bax.
Statistical Analysis. The significance of differences between experimental conditions was determined using Student's t test for unpaired observations. Analysis of synergism was performed by median dose effect analysis using commercially available software (Calcusyn; Biosoft, Ferguson, MO) (Chou and Talalay, 1984).
Results
Cotreatment with SAHA or SB and 17-AAG Results in a Striking Increase in Growth Arrest, Mitochondrial Injury, and Apoptosis in K562 Cells. To characterize interactions between 17-AAG and the histone deacetylase inhibitor SAHA in K562 cells, dose-response studies were performed (Fig. 1). Logarithmically growing K562 cells were exposed to various concentrations of SAHA and 17-AAG alone or in combination for 24 h, after which apoptosis was monitored by annexin V staining. As shown in Fig. 1, A and B, 17-AAG and SAHA were minimally toxic when administered alone at concentrations as high as 2.5 and 3 μM, respectively. However, exposure of cells to 2 μM SAHA in combination with 250 nM 17-AAG resulted in a clear increase in apoptosis (∼25%), and at 2.5 μM 17-AAG, the majority of cells was apoptotic (70%). Likewise, when cells were exposed to 1.5 μM 17-AAG and increasing concentration of SAHA, a significant increase in apoptosis was observed at 0.5 μM SAHA, and the majority of cells was apoptotic at SAHA concentration ≥2 μM (Fig. 1B).

Cotreatment with SAHA and 17-AAG results in a striking increase in apoptosis and decrease in cell growth in K562 cells in a time- and dose-dependent manner. A, K562 cells were exposed for 24 h to the designated concentration of 17-AAG alone (empty squares) or in conjunction with 2 μM SAHA (solid squares), after which the percentage of apoptotic cells was determined by annexin V analysis as described under Materials and Methods. B, K562 cells were exposed to the designated concentration of SAHA alone (empty squares) or in combination with 1.5 μM 17-AAG (solid squares), after which apoptosis was determined by annexin V analysis. C, cells were exposed to 17-AAG (1.5 μM) ± 2 μM SAHA for the indicated interval, after which the percentage of apoptotic cells was determined by annexin V analysis. D, median dose effect analysis of apoptosis induction by SAHA and 17-AAG. K562 cells were exposed to varying concentrations of SAHA and 17-AAG at a fixed ratio (1:0.75) for 24 h, after which apoptosis was monitored by annexin V/propidium iodide analysis. Combination index (CI) values were determined in relation to the fractional effect using a commercially available software program as described under Materials and Methods. CI values <1.0 correspond to a synergistic interaction. E, K562 cells were exposed to 2 mM SB and 1.5 μM 17-AAG alone or in combination for 24 h, after which the percentage of apoptotic cells was determined by annexin V analysis. F, K562 cells were exposed to 2 μM SAHA or 2 mM SB alone or in combination with 1.5 μM 17-AAG for the indicated time, after which cell growth was evaluated using MTS assay as described under Materials and Methods. For all studies, values represent the means for three separate experiments performed in triplicate ± S.D.
A time-course analysis of 17-AAG/SAHA-induced apoptosis was conducted using the annexin V assay. As shown in Fig. 1C, whereas 1.5 μM 17-AAG or 2 μM SAHA administered individually was minimally toxic over a 48-h treatment interval, combined treatment resulted in an increase in apoptosis (e.g., 30%) by 16 h and a very substantial increase in lethality after 24 h (∼60%).
Median dose effect analysis of apoptosis induction by SAHA and 17-AAG administered at a fixed ratio (1:0.75) yielded combination index values considerably less than 1.0, corresponding to a highly synergistic interaction (Fig. 1D).
Attempts also were made to extend these findings to another HDAC inhibitor. As shown in Fig. 1F, a 24-h exposure to 2 mM sodium butyrate (SB) was minimally toxic to K562 cells. However, when this agent was combined with 1.5 μM 17-AAG, which was essentially nontoxic by itself, a pronounced increase in lethality was observed. Finally, MTS assays demonstrated that cotreatment with 17-AAG and either SAHA or SB for 72 h dramatically reduced the number of surviving cells relative to the effects of the agents administered individually (Fig. 1F).
Consistent with the previous results, Western blot analysis (Fig. 2A) revealed that cotreatment with SAHA and 17-AAG (24 h) resulted in a marked increase in release of cytochrome c and AIF into the cytosolic fraction. In contrast, SAHA alone was ineffective in triggering cytochrome c and AIF release, whereas 17-AAG alone exerted only modest effects. Combined treatment also resulted in a pronounced increase in procaspase-3 and -8 processing and PARP degradation. Together, these findings indicate that cotreatment with the HDAC inhibitors SAHA or SB and 17-AAG results in a striking increase in growth arrest, mitochondrial injury, and apoptosis in Bcr-Abl+ K562 cells.

Combined exposure to 17-AAG and SAHA results in a marked increase in apoptosis and mitochondrial injury in K562 and LAMA84 cells sensitive and resistant to STI571 as well as in primary CD34+ cells. A, cells were treated with SAHA (2 μM) and 17-AAG (1.5 μM) alone or in combination for 24 h, after which whole cell lysates were obtained, the proteins separated by SDS-polyacrylamide gel electrophoresis, and Western blot analysis used to monitor expression of procaspase-3, caspase-8, and PARP. On the other hand, mitochondria-free cytosolic fractions were obtained as described under Materials and Methods, and subjected to Western blot analysis to monitor release of cytochrome c and AIF. The blots were subsequently reprobed with anti-tubulin (Tub) antibodies to document equivalent loading and transfer. The result of a representative study is shown; two additional experiments yielded equivalent results. B, LAMA84 cells were exposed to 1.5 μM SAHA and 1 μM 17-AAG alone or in combination for 24 h, after which the percentage of apoptotic cells was determined by annexin V analysis as described under Materials and Methods. Thereafter, protein lysates were prepared and subjected to Western blot analysis using procaspase-3 and PARP antibodies (inset). C, K562-STI-R and LAMA-STI-R were exposed to SAHA (2 and 1 μM, respectively) and 17-AAG (1.5 and 0.5 μM, respectively) alone or in combination for 24 h, after which the extent of apoptosis was determined as in A. For B and C, values represent the means for three separate experiments ± S.D. D, CD34+ cells were isolated as described under Materials and Methods from the bone marrow of a patient newly diagnosed with CML in chronic phase (patient 1) and a patient with CML in blast crisis (patient 2) or accelerated phase (patient 3), both of whom had previously received STI571. CD34+ cells were exposed to 2 μM SAHA ± 1.5 μM 17-AAG for 24 h, after which the extent of apoptosis was determined by morphological analysis of Wright Giemsa-stained cytospin preparations. Values represent the mean percentages of apoptotic cells in 10 to 15 randomly selected fields encompassing a total of >1000 cells. E, normal bone marrow mononuclear cells were isolated as described under Materials and Methods and exposed to 2 μM SAHA ± 1.5 μM 17-AAG for 24 h, after which the extent of apoptosis was determined as in D.
Cotreatment with 17-AAG and SAHA Results in Enhanced Lethality in Other Bcr-Abl+ Leukemia Cell Types, Including Those Resistant to STI571, as Well as in Primary CD34+ CML Cells. To determine whether these events could be extended to include other Bcr-Abl+ cells, parallel studies were performed in LAMA84 cells. As shown in Fig. 2B, cotreatment with 1 μM 17-AAG and 1.5 μM SAHA resulted in a marked increase in caspase-3 activation, PARP cleavage (inset), and annexin V positivity, whereas individual treatment was ineffective. Similar results were obtained when 1 μM 17-AAG was combined with 1.5 mM SB (data not shown).
To determine whether cotreatment with 17-AAG and SAHA also triggered cell death in Bcr-Abl+ cells refractory to STI571, parallel studies were conducted in K562 and LAMA84 cells made resistant to this agent by subculturing in progressively higher concentration of STI571 (K562-STI-R and LAMA-STI-R) as we have described previously (Yu et al., 2002b, 2003a). As shown in Fig. 2C, exposure of K562-STI-R cells for 24 h to SAHA (2 μM) alone had no effect, and 1.5 μM 17-AAG increased annexin V positivity over controls by ∼10%. However, combined treatment, although somewhat less toxic than in parental K562 cells, induced apoptosis in approximately 40% of cells. Equivalent results were obtained in cells exposed to 17-AAG and SB (2 mM) (data not shown). Similarly in LAMA-STI-R cells, SAHA (1 μM) had no significant effect; whereas 17-AAG (0.5 μM) induced apoptosis in approximately 38% of cells. However combined treatment with SAHA and 17-AAG triggered apoptosis in the majority of cells (70%).
It is significant that, cotreatment with 17-AAG (1.5 μM) and SAHA (2 μM) resulted in a marked increase in cell death in CD34+ leukemia cells isolated from the bone marrow of a newly diagnosed patient with chronic phase Philadelphia chromosome-positive CML (Fig. 2D, patient 1). As observed in Bcr-Abl+ leukemic cell lines, SAHA or 17-AAG administered individually exerted only marginal effects. A clear increase in apoptosis was also observed with combined treatment in CD34+ cells obtained from a CML patients in blast crisis (patient 2) and accelerated phase (patient 3), both of whom had previously received STI571. In contrast, SAHA and 17-AAG, either alone or in combination, exerted minimal toxicity toward normal bone marrow mononuclear cells isolated from two subjects (Fig. 2E). Thus, cotreatment with 17-AAG and HDAC inhibitors leads to a marked increase in lethality in several human Bcr-Abl+ leukemia cell types, including those resistant to STI571, but exerts relatively little toxicity toward normal bone marrow mononuclear cells.
Effects of 17-AAG/SAHA Cotreatment on the Expression of Bcl-2 Family Members. Because Bcl-2 family proteins play a critical role in regulating apoptosis (Chao and Korsmeyer, 1998), Western blot analysis was used to monitor expression of these proteins in K562 cells treated for 24 h with 17-AAG (1.5 μM) and SAHA (2 μM) alone or in combination (Fig. 3A). Although SAHA alone modestly decreased Bcl-xL expression, exposure of cells to 17-AAG ± SAHA for intervals ≥8 h resulted in a pronounced reduction in levels of this protein. Furthermore, treatment of cells with SAHA alone resulted in a clear increase in expression of Mcl-1 after 8 h of treatment, whereas coexposure to 17-AAG led to a marked reduction in Mcl-1 protein levels. In addition, an increase in expression of the proapoptotic protein Bak was observed in K562 exposed to SAHA and 17-AAG alone and in combination for ≥16 h. Increased Bak expression and diminished Mcl-1 and Bcl-xL expression were also observed in LAMA84 cells exposed to 17-AAG ± SAHA (Fig. 3D). In addition, cells exposed to both 17-AAG and SAHA for 24 h exhibited a clear decrease in Bid protein levels, reflecting cleavage/activation, whereas no major changes were noted in the levels of expression of Bax, Bim, or XIAP proteins with any treatment (Fig. 3B). Finally, the pan-caspase inhibitor N-benzyloxycarbonyl-VAD fluoromethyl ketone failed to block the decrease of Bcl-xL and the increase in Bak expression and minimally reversed the decrease in Mcl-1 and Bid expression (Fig. 3C), indicating that perturbations in expression of these proteins in SAHA/17-AAG cells cannot be solely attributed to caspase activation. Thus, combined exposure of Bcr-Abl+ leukemia cells to 17-AAG and SAHA was associated with increased expression of Bak, down-regulation of Bcl-xL and Mcl-1, and activation of Bid.

Effects of 17-AAG and SAHA on Bcl-2 family members, XIAP, and 14-3-3 protein expression. Bax conformational change and distribution. A to C, K562 cells were exposed to SAHA (2 μM) and 17-AAG (1.5 μM) alone or in combination for the indicated time (A) or 24 h (B and C) in the presence or absence of N-benzyloxycarbonyl-VAD (25 μM), after which protein lysates were prepared and subjected to Western blot analysis as described under Materials and Methods using indicated antibodies. D, LAMA84 cells were exposed to SAHA (1.5 μM) and 17-AAG (1 μM) alone or in combination for 24 h, after which protein lysates were prepared and subjected to Western blot analysis. E, K562 cells were exposed to SAHA (2 μM) and 17-AAG (1.5 μM) alone or in combination for 24 h, after which cells were lysed in buffer containing 1% CHAPS or 1% Triton X-100 as a positive control, after which conformationally changed Bax protein was immunoprecipitated using anti-Bax 6A7 antibody and subjected to Western blot analysis using polyclonal Bax antibody. Otherwise, cells were lysed, and proteins were extracted from membrane, cytosolic, and total fractions as described under Materials and Methods and subjected to Western blot analysis using polyclonal Bax and 14-3-3 antibodies. F, K562 cells were transfected with a Bcl-2 vector or the empty vector pUSEamp, cultured for 24 h, and subsequently treated with SAHA (2 μM) and 17-AAG (1.5 μM) for 24 h, after which the extend of apoptosis was determined using annexin V analysis. Values represent the means for three independent experiments ± S.D. Asterisk (*) represents significantly lower than values obtained for empty vector pUSEamp cells (P < 0.05). In all Western blots, each lane was loaded with 25 μg of protein; blots (total and cytosolic fractions) were subsequently reprobed with antibodies directed against α-tubulin (Tub) to control for equal loading and transfer of proteins. In each case, at least two additional experiments yielded equivalent results.
Cotreatment of K562 Cells with SAHA and 17-AAG Results in a Pronounced Bax Conformational Change and Translocation to the Membrane (Mitochondrial) Fraction. In nonapoptotic cells, Bax protein is primarily distributed in the cytosol. During apoptosis induced by various agents, including 17-AAG, Bax protein undergoes a conformational change, accompanied by translocation and integration into the mitochondrial membrane (Murphy et al., 2000; Nimmanapalli et al., 2003b), thereby facilitating the release of cytochrome c, AIF, and Smac/DIABLO (Rosse et al., 1998; Bidere et al., 2003; Yamaguchi et al., 2003). To investigate the effects of 17-AAG and SAHA alone and in combination on Bax conformational changes in K562 cells, Bax was immunoprecipitated from cells lysed in CHAPS buffer using a monoclonal anti-Bax antibody (6A7) and subjected to immunoblotting using a polyclonal anti-Bax antibody. As shown in Fig. 3E, both SAHA (2 μM) as well as 17-AAG (1.5 μM) administered alone modestly induced a conformational change in Bax, and this process was associated with a moderate translocation of Bax into the membrane (mitochondrial) fraction, accompanied by depletion of the cytosolic fraction. However, combined treatment with SAHA and 17-AAG resulted in a very marked Bax conformational change, along with a striking translocation of Bax to the mitochondrial membrane. Consistent with these results, combined treatment induced cleavage of the 14-3-3 protein, an event previously associated with Bax conformational change (Nomura et al., 2003), whereas no cleavage was noted in cells exposed to the agents administered individually (Fig. 3E). To investigate the functional role that perturbation of Bcl-2 family members might play in SAHA/17-AAG-mediated cell death, K562 cells were transiently transfected with a Bcl-2 plasmid. As shown in Fig. 3F (top), robust expression of Bcl-2 was observed in cells transfected with the Bcl-2 vector in the absence as well as in the presence of SAHA and 17-AAG, whereas no Bcl-2 protein was detected in cells transfected with the empty vector (pUSEamp). It is significant that cotreatment with SAHA and 17-AAG induced lethality was significantly attenuated in Bcl-2-overexpressing cells (Fig. 3F, bottom). Together, these findings indicate that combined exposure of Bcr-Abl+ leukemia cells to 17-AAG and SAHA results in a pronounced increase in Bax conformational change and mitochondrial translocation, events previously associated with mitochondrial release of proapoptotic proteins into the cytosol (Rosse et al., 1998; Bidere et al., 2003; Yamaguchi et al., 2003). Finally, ectopic expression of Bcl-2 attenuated cell death induced by this regimen, supporting the notion that proapoptotic actions of SAHA/17-AAG involve perturbations in Bcl-2 family members implicated in regulation of mitochondrial integrity.
Exposure to 17-AAG and SAHA Diminishes Bcr-Abl Expression and Abrogates STAT5 DNA Binding and Transcriptional Activities in K562 Cells. In view of evidence that signaling molecules such as Bcr-Abl, STAT5, CrkL, and Src family members play major roles in the survival of Bcr-Abl+ leukemia cells (Druker et al., 1996; de Groot et al., 1999; Sillaber et al., 2000; Klejman et al., 2002; Wilson et al., 2002), the effects of 17-AAG and SAHA, alone and in combination, were examined with respect to the expression and activation of these proteins. Consistent with previous reports (Nimmanapalli et al., 2001; Gorre et al., 2002) Western blot analysis demonstrated that exposure of K562 cells to 1.5 μM 17-AAG resulted in a marked decrease in Bcr-Abl protein expression after 8-h exposure (Fig. 4A). Moreover, combined treatment (for ≥16 h) with 17-AAG and SAHA led to the virtual loss of Bcr/Abl expression. Levels of total or phosphorylated Lyn, a Src protein member constitutively active in Bcr-Abl cells (Wilson et al., 2002), sharply declined in K562 cells treated with 17-AAG alone or in combination with SAHA for intervals ≥8 h. On the other hand, no major changes were observed in Hck, or CrkL protein expression after individual drug treatment, whereas slight declines in STAT5 and c-Src protein level were noted in SAHA/17-AAG-treated cells (Fig. 4B). In separate studies, down-regulation of the latter proteins was attenuated by the caspase inhibitor N-tert-butoxycarbonyl-D-fluoromethyl ketone (25 μM; data not shown), suggesting that these events were secondary to caspase activation. Similar expression patterns, e.g., down-regulation of Bcr/Abl and phospho-Lyn were observed in LAMA84 cells exposed to 17-AAG alone and in combination with SAHA for 24 h (Fig. 4C).
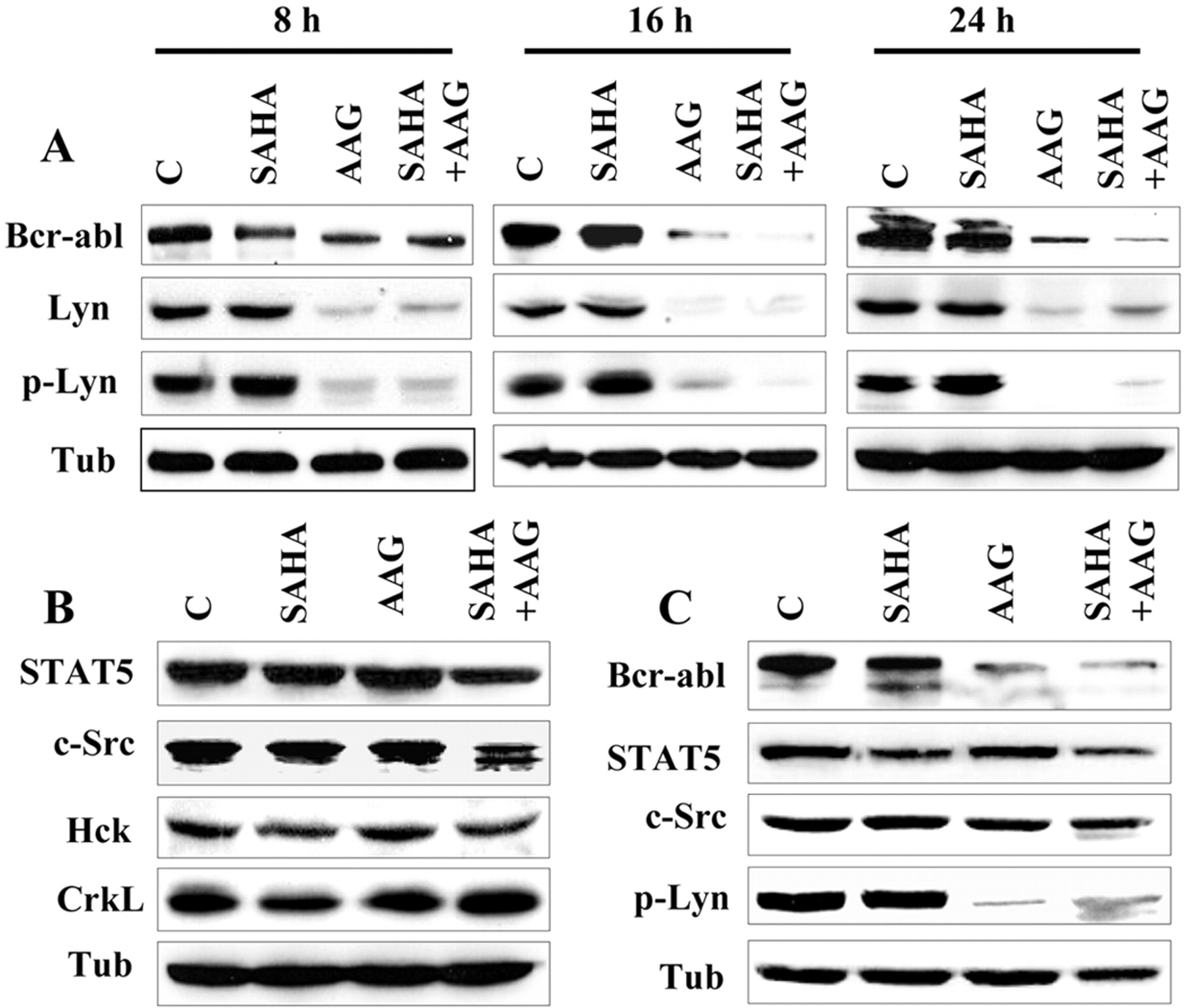
Exposure to 17-AAG and SAHA diminishes Bcr-Abl and Lyn expression in K562 and LAMA84 cells. K562 (A and B) and LAMA84 (C) cells were exposed to SAHA (2 and 1.5 μM, respectively) and 17-AAG (1.5 and 1 μM, respectively) alone or in combination for the indicated intervals (A) or for 24 h (B and C), after which protein lysates were prepared and subjected to Western blot analysis as described under Materials and Methods using specific antibodies for the indicated proteins. Each lane was loaded with 25 μg of protein; blots were subsequently reprobed with antibodies were directed against α-tubulin (Tub) to control for equal loading and transfer of proteins.
The effects of 17-AAG and SAHA, alone and in combination, were then examined in relation to DNA binding and transcriptional activities of STAT5. EMSA analysis revealed that exposure of K562 cells to SAHA and 17-AAG alone for 20 h resulted in a reduction in STAT5 DNA binding activity (Fig. 5A). However, combined treatment with these agents essentially abolished STAT5 DNA binding activity. Luciferase reporter gene assay revealed that 17-AAG and SAHA individually reduced but together largely abrogated STAT5 transactivation activity (Fig. 5B). Together, these findings indicate that combined exposure of K562 cells to 17-AAG and SAHA results in extensive down-regulation of Bcr-Abl and a very pronounced loss of STAT5 activity.
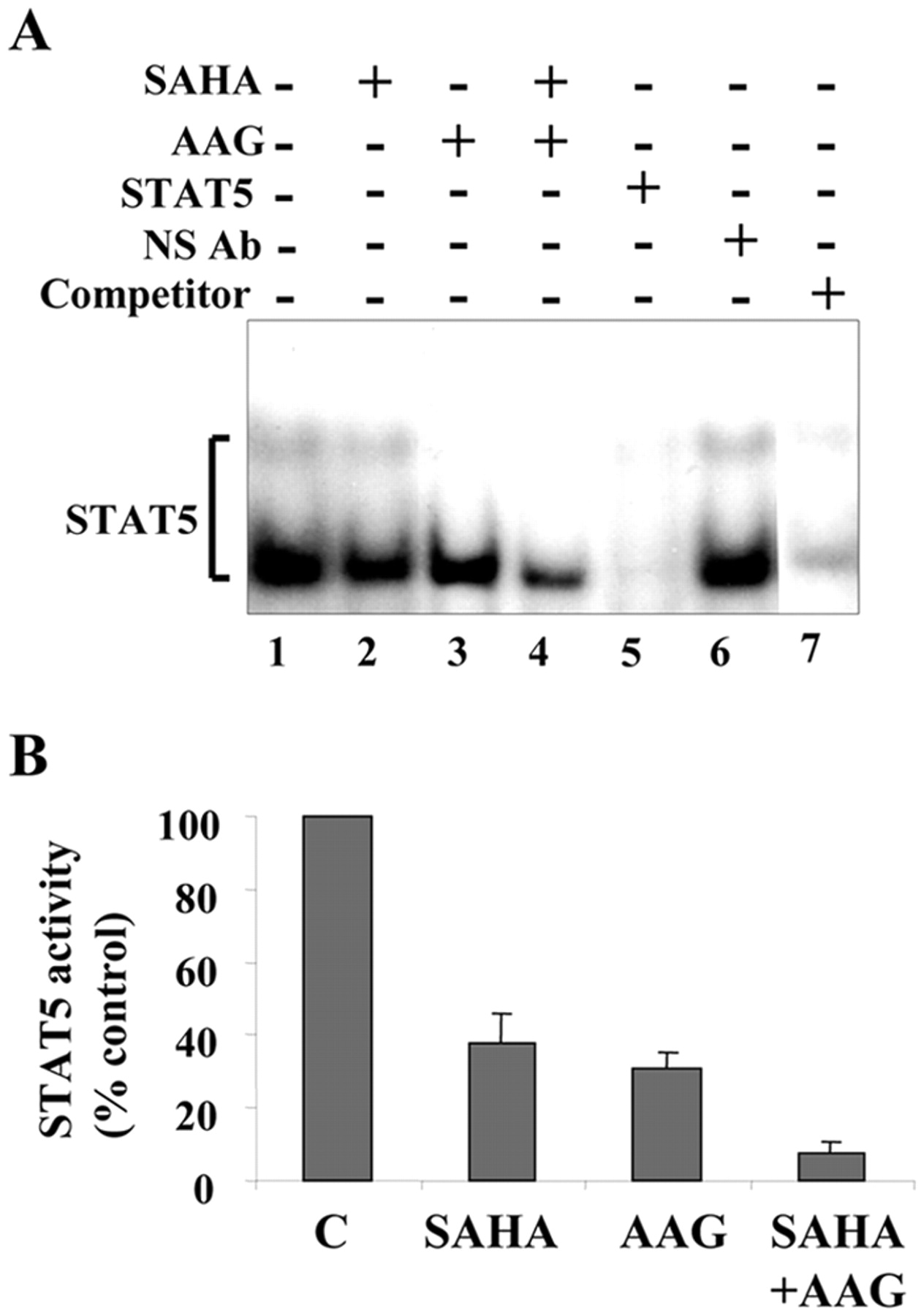
Exposure to 17-AAG and SAHA abrogates STAT5 activity in K562 cells. A, K562 cells were untreated (lane 1) or treated with SAHA (2 μM, lane 2), 17-AAG (1.5 μM, lane 3), or the combination (lane 4) for 20 h. At the end of this interval, nuclear extracts were prepared and subjected to EMSA analysis to evaluate DNA binding activity of STAT5. Nuclear extracts prepared from untreated cells were incubated 30 min with STAT5A specific antibody (lane 5), a nonspecific antibody (NS Ab, lane 6), or 100-fold excess of unlabeled oligonucleotides (competitor, lane 7) before addition of the labeled oligonucleotides. Two additional studies yielded equivalent results. B, K562 cell were cotransfected with pSTAT5-luc and pRL-TK-luc plasmids, left in the incubator for 6 h, and then treated with indicated agents for an additional 20 h after which, activity of firefly and R. reniformis luciferase was measured using the Dual-Luciferase reporter assay system. Values for firefly luciferase activity were normalized to those obtained for R. reniformis luciferase activity and expressed relative to the control. The average of three independent experiments ± S.D. is shown.
Combined Treatment with 17-AAG and SAHA Reverses Enhanced STAT5 Activation in K562 Cells Expressing a Constitutively Active STAT5 Mutant (N642H). In view of evidence of the critical role that STAT5 plays in the survival of Bcr-Abl+ cells, attempts were made to determine whether the pronounced STAT5 inactivation observed plays a functional role in 17-AAG/SAHA-induced apoptosis. A construct encoding the constitutively active form of STAT5A (STAT5A-N642H), which is associated with high DNA binding and transactivation activities (Ariyoshi et al., 2000), was used to generate K562 cells displaying constitutive activation of STAT5A. As shown in Fig. 6A, two separate clones (cl-4 and cl-18) exhibited expression of STAT5A-N642-FLAG (top) and increased constitutive STAT5 DNA binding activity (bottom). Reporter assays confirmed the pronounced increase in STAT5 activity in mutant cells (Fig. 6A, right). It is interesting that cotreatment with 17-AAG and SAHA abrogated the pronounced increase in STAT5 DNA binding (Fig. 6B, left) and transactivation activity, reflected by reporter gene assays (Fig. 6B, right) in transfectant cells. Thus, constitutive activation of STAT5 failed to prevent the dramatic reduction in DNA binding and transcriptional activities of STAT5 triggered by 17-AAG/SAHA treatment. Consistent with the persistent inhibition of STAT5 activity, SAHA/17-AAG-induced apoptosis was not attenuated in either constitutively active STAT5-expressing cell line (P > 0.05 for cl-4 and cl-18; Fig. 6C).

Exposure to 17-AAG and SAHA inhibits STAT5 activity in STAT5A N642H-expressing cells. A, proteins were extracted from two clones (cl-4 and cl-18) of K562 cells expressing STAT5A-N642H-FLAG or the empty vector (pMX-neo) and subjected to Western blot analysis using Anti-FLAG M2 antibody (top). Otherwise, nuclear proteins were extracted from these cells, and EMSA was performed to evaluate DNA binding activity of STAT5 (bottom). In parallel, STAT5A-N642H (cl-18) and pMX-neo cells were cotransfected with pSTAT5-luc and pRL-TK-luc plasmids and then cultured for 24 h after which firefly and R. reniformis luciferase activities were determined using the Dual-Luciferase reporter assay system as described under Materials and Methods (right). Three separate experiments yielded similar results. The data shown represent the means ± S.D. of one representative experiment performed in triplicate. B, STAT5A-N642H (cl-18) and pMX-neo cells were coexposed to SAHA (2 μM) and 17-AAG (1.5 μM) for 20 h, after which nuclear proteins were prepared and subjected to EMSA analysis (left). Otherwise, cells were cotransfected with pSTAT5-luc and pRL-TK-luc plasmids and coexposed to SAHA (2 μM) and 17-AAG (1.5 μM) for 20 h, after which firefly and R. reniformis luciferase activities were determined (right). Firefly luciferase activities were normalized to R. reniformis luciferase activities and expressed as a percentage relative to those obtained in untreated controls. The results shown represent the means ± S.D. of three separate experiments performed in triplicate. For A and B (left), specificity of STAT5 complexes was demonstrated by a supershift using anti-FLAG and anti-STAT5A antibodies as well as by competition with 100-fold excess of cold probe (competitor) on nuclear extracts from untreated cells (cl-18). The supershifted band is indicated by an arrow. C, K562 cells expressing STAT5A-N642H (cl-4 and cl-18) and the empty vector (pMX-neo) were exposed to SAHA (2 μM) and 17-AAG (1.5 μM) for 24 h, after which the extent of apoptosis was determined by annexin V analysis. Values represent the means for three separate experiments ± S.D. Asterisk (*) indicates not significantly different from values obtained for empty vector pMX-neo cells (P > 0.05).
Coexposure of K562 Cells to 17-AAG and SAHA Results in Disruption of the Raf/MEK/ERK and Akt Pathways. Interactions between SAHA and 17-AAG were then examined with respect to effects on mitogen-activated protein kinase and Akt signaling pathways (Fig. 7). Consistent with previous reports (Nimmanapalli et al., 2003b), exposure of K562 cells to 17-AAG for 8 or 24 h resulted in a substantial decrease in Raf-1 and a small decrease in B-raf protein expression as well as a decline in ERK phosphorylation, whereas protein levels of ERK remained unperturbed. Exposure of cells to SAHA alone induced no changes in expression of B-Raf, or ERK, but clear reductions in ERK phosphorylation. However, down-regulation/inactivation of Raf-1 and ERK were slightly more pronounced when 17-AAG was combined with 2 μM SAHA, whereas B-Raf expression was substantially reduced.
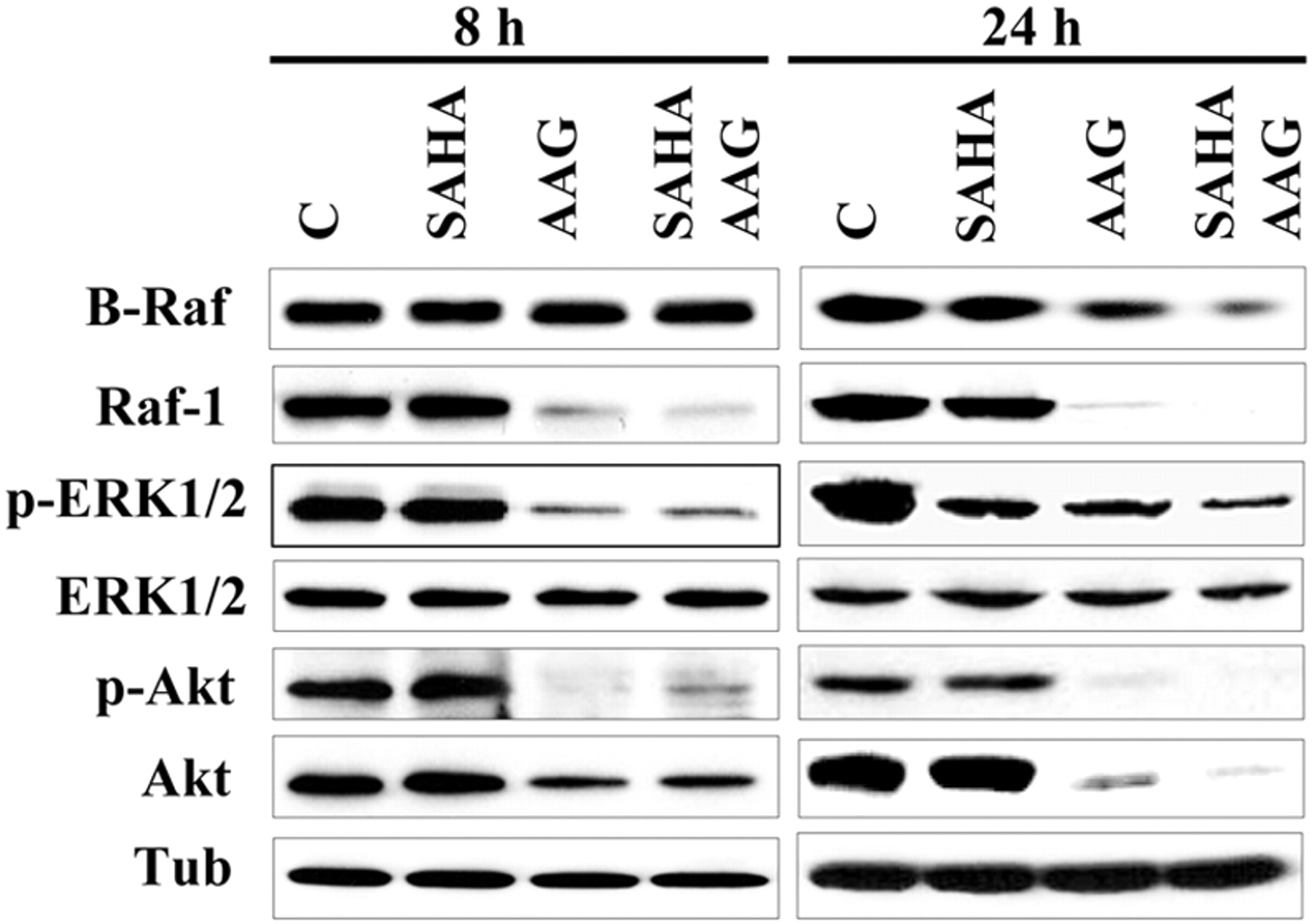
Effect of SAHA and 17-AAG on expression/activation of MAP kinases and Akt in K562 cells. K562 cells were exposed to SAHA (2 μM) and 17-AAG (1.5 μM) alone or in combination for 8 to 24 h, after which protein lysates were prepared and subjected to Western blot analysis as described under Materials and Methods using indicated antibodies. Each lane was loaded with 25 μg of protein; blots were subsequently reprobed with antibody directed against α-tubulin to control for equal loading and transfer of proteins. Two additional experiments yielded similar results.
In addition to these changes, a profound decline in Akt protein levels as well as diminished Akt and GSK3α/β (data not shown) phosphorylation was observed in cells exposed to 17-AAG ± SAHA. Thus, cotreatment with SAHA and 17-AAG was associated with pronounced inactivation of the cytoprotective Raf/MEK/ERK and Akt pathways.
Finally, exposure to 17-AAG ± SAHA for 20 h induced a marked accumulation of Hsp70, whereas Hsp90 protein levels remained unperturbed (Fig. 8A). In addition, exposure to 17-AAG had no major effect on SAHA-induced acetylation of histones H3 and H4. Furthermore, immunoprecipitation experiments followed by immunoblotting (Fig. 8B) revealed that exposure of K562 to 17-AAG alone resulted in a very modest decrease in Bcr-Abl associating with Hsp90, whereas combined treatment with 17-AAG and SAHA essentially eliminated Bcr-Abl associating with Hsp90 and increased levels of Hsp70-associated protein (Fig. 8B). Likewise, marked increases in Hsp70-associated fractions of Raf-1 and Akt were observed in cells treated with 17-AAG alone or in combination with SAHA (Fig. 8, C and D). These observations are consistent with previous reports suggesting that agents acting at the level of Hsp90 (e.g., Hsp90 antagonists or histone deacetylase inhibitors) promote the association of client proteins with an unstable Hsp70 multichaperone protein complex, resulting in proteasomal degradation (Nimmanapalli et al., 2001).

Effect of SAHA and 17-AAG on association of Hsp70 and Hsp90 with Bcr-Abl, Raf-1, and Akt. A, K562 cells were exposed to SAHA (2 μM) and 17-AAG (1.5 μM) alone or in combination for 20 h, after which protein lysates were prepared and subjected to Western blot analysis using antibodies against Hsp90, Hsp70, acetylated histones H3 (Ac-H3), and H4 (Ac-H4). B to D, K562 cells were treated as in A, and protein lysates were prepared and subjected to immunoprecipitation as described under Materials and Methods using antibodies against c-abl (B), Raf-1 (C), and Akt (D). The immunoprecipites were then subjected to Western blot analysis using antibodies against Hsp90 and Hsp70, as well as Bcr-Abl, Raf-1, and Akt (bottom). The results shown are representative of three separate experiments.
Discussion
Results of the present study indicate that cotreatment with the Hsp90 antagonist 17-AAG and clinically relevant HDAC inhibitors results in a striking increase in mitochondrial injury, caspase activation, and apoptosis in Bcr-Abl+ human leukemia cells. These events are associated with Bcr-Abl down-regulation; multiple perturbations in Bcl-2 family member proteins, particularly induction of Bax conformational change; and disruption of diverse signaling/cell cycle regulatory pathways, including those related to STAT5, Raf/MEK/ERK, and Akt.
Translocation and integration of cytoplasmic Bax into the mitochondrial membrane represent critical steps in activation of the mitochondrial apoptotic pathway in multiple systems (Yamaguchi et al., 2003). Moreover, a conformational change in Bax, resulting in exposure of the NH2 and COOH termini, is required for release of proapoptotic mitochondrial proteins (Murphy et al., 2000). The present results demonstrate that 17-AAG induces a conformational change in Bax as well as its translocation to the mitochondria, consistent with recent findings in Bcr-Abl- HL-60 cells (Nimmanapalli et al., 2003b). It is interesting that SAHA, administered alone, also triggered a conformational change in Bax, and potentiated its mitochondrial translocation. To the best of our knowledge, promotion of Bax conformational change by HDAC inhibitors has not been reported previously, although the HDAC inhibitor apicidin has been shown to induce Bax translocation to the mitochondria (Kwon et al., 2002). It is significant that combined treatment of K562 cells with 17-AAG and SAHA resulted in a considerably greater Bax conformational change and more extensive mitochondrial translocation of Bax, implicating these events in antileukemic synergism. Nomura et al. (2003) reported that 14-3-3 proteins physically interact with Bax and inhibit its conformational change and mitochondrial translocation independently of Bax phosphorylation status but partially dependent upon caspase activation. Here, cleavage of 14-3-3 proteins in cells exposed to combined (but not individual) treatment with 17-AAG/SAHA was observed. Together, these observations raise the possibility that cleavage of 14-3-3 proteins in 17-AAG/SAHA-treated cells may promote Bax conformational change and mitochondrial translocation, culminating in apoptosis.
Multiple perturbations in other Bcl-2 family member proteins were observed in 17-AAG/SAHA-treated K562 cells that might have contributed to enhanced cell death. It should be noted that some of these perturbations were observed in cells treated with agents individually i.e., down-regulation of Mcl-1 and Bcl-xL in 17-AAG-treated cells, whereas other perturbations were more pronounced after combined drug treatment, i.e., up-regulation of Bak and cleavage of Bid. Each of these events has been associated with apoptosis induction and may contribute to Bax conformational change and mitochondrial translocation (Chao and Korsmeyer, 1998). Such findings are consistent with the notion that enhanced activity of SAHA/17-AAG against Bcr-Abl+ leukemic cells involves, at least in part, alterations in the expression of Bcl-2 family members responsible for maintaining mitochondrial integrity. This notion is strongly supported by the finding that ectopic expression of Bcl-2, which is known to oppose the actions of proapoptotic multidomain proteins such as Bax at the mitochondrial level (Chao and Korsmeyer, 1998), significantly attenuated the lethality of this combination. In this context, it is noteworthy that enhanced expression of Bcl-xL in Bcr-Abl+ cells has been shown to proceed through a STAT5-dependent mechanism (Horita et al., 2000).
Constitutive activation of transcriptional factors such as STATs has been observed in diverse malignant cell types, including leukemia, lymphoma, and multiple myeloma, among others (Weber-Nordt et al., 1996; Catlett-Falcone et al., 1999). Furthermore, inhibition of STAT5 by dominant-negative STAT5 reverses the transformed phenotype and induces apoptosis in human Bcr-Abl+ leukemia K562 cells (de Groot et al., 1999), documenting the essential role of STAT5 in cell survival in this disorder. The present results indicate that both the Hsp90 antagonist 17-AAG as well as the HDAC inhibitor SAHA result in declines in both the DNA binding and transcriptional activities of STAT5. It is significant that combination of these agents essentially abrogated STAT5 activity. It is noteworthy that the combination of 17-AAG and SAHA markedly reduced Bcr-Abl protein levels, although 17-AAG by itself and, to a lesser extent, SAHA was active in this regard. This combination also induced a marked decrease in phosphorylated Lyn and a caspase-dependent decline in c-Src protein levels. Because Bcr-Abl as well as Src proteins regulate STAT5 activity in CML cells (Klejman et al., 2002), the possibility that abrogation of STAT5 DNA binding activity results, at least in part, from down-regulation and/or inactivation of the upstream kinases Bcr-Abl, Lyn, and c-Src cannot be excluded.
It is interesting to note that 17-AAG/SAHA-induced cells death was not attenuated in cells expressing a constitutively active STAT5A mutant; moreover, 17-AAG administered alone and particularly in combination with SAHA continued to inhibit DNA binding activity as well as transcriptional activities in these cells. Thus, expression of a constitutively active STAT5 mutant was unable to overcome inactivation of the STAT5 pathway in cells exposed to the 17-AAG/SAHA regimen, nor was it able to protect cells from the lethal effects of this combination. One possible explanation for this phenomenon is that 17-AAG/SAHA may down-regulate STAT5 through activation of phosphatases, or, alternatively, activation of STAT repressors such as SOCS or PIAS (Levy and Darnell, Jr., 2002). In any case, although the present findings involving constitutively active STAT5 mutants do not provide direct evidence that inactivation of STAT5 contributes to 17-AAG/SAHA lethality, they are entirely consistent with this notion.
In addition to down-regulation of Bcr-Abl and Lyn, expression/activation of the antiapoptotic proteins Raf-1 and Akt were also attenuated in cells exposed to 17-AAG ± HDACIs. The finding that 17-AAG potentiated binding of client proteins (e.g., Bcr-Abl, Raf-1, and Akt) to Hsp70 is in agreement with previous reports (Nimmanapalli et al., 2001). In particular, such studies suggest that Hsp90 antagonists promote the association of client proteins with unstable multiprotein complexes involving Hsp70, resulting in proteasomal degradation. In addition, the present studies indicate, for the first time, that B-raf, which is a more potent activator of MEK1/2 than Raf-1 (Pritchard et al., 1995), is also down-regulated by 17-AAG in Bcr-Abl+ cells. The role, if any, that B-raf down-regulation plays in synergistic interactions between 17-AAG and HDAC inhibitors remains to be determined. In view of recent evidence that HDAC inhibitors interfere with Hsp90 function (Nimmanapalli et al., 2003a), it is tempting to postulate that HDAC inhibitors potentiate the ability of 17-AAG to disrupt Hsp90 chaperone capacity. However, it should be noted that cotreatment with HDAC inhibitors and 17-AAG resulted in only modest further reductions in expression of certain client proteins (e.g., Bcr-Abl and B-raf) and essentially no further reductions in others (e.g., Akt). For these reasons, it is likely that factors other than or in addition to disruption of Hsp90 function play major roles in synergism between 17-AAG and HDAC inhibitors in Bcr-Abl+ cells.
In summary, the present study indicate that cotreatment with 17-AAG and the HDAC inhibitors SAHA or SB induces a marked increase in mitochondrial injury and apoptosis in Bcr-Abl+ leukemia cells, events associated with down-regulation of Bcr-Abl, Lyn, Raf-1, B-Raf, Akt, Mcl-1, and Bcl-xL; up-regulation of Bak; abrogation of STAT5 activity; increased Bid cleavage; and a profound conformational change in and mitochondrial translocation of Bax. Although the introduction of imatinib mesylate into the clinic has revolutionized the treatment of CML, the development or preexistence of drug resistance because of Bcr-Abl amplification or mutation represents a continuing challenge (von Bubnoff et al., 2003). In this regard, evidence has surfaced that both 17-AAG and HDAC inhibitors such as SAHA may be active, either alone or in combination, against imatinib mesylate-resistant Bcr-Abl+ cells (Gorre et al., 2002; Yu et al., 2003a). Therefore, the finding that 17-AAG and SAHA interact synergistically to induce apoptosis, at least in certain imatinib mesylate-resistant cells, including primary CD34+ cells from CML patients, has potential therapeutic implications for this disease.
Footnotes
-
This work was supported by National Institutes of Health grants CA93738, CA63753, and CA100866, Leukemia and Lymphoma Society of America award 6045-03, and Department of Defense award DAMD-17-03-1-0209.
-
ABBREVIATIONS: 17-AAG, 17-allylamino 17-demethoxygeldanamycin; Hsp, heat shock protein; HDAC, histone deacetylase inhibitor; STAT, signal transducer and activator of transcription; CML, chronic myelogenous leukemia; ERK, extracellular signal-regulated kinase; SAHA, suberanoylanilide hydroxamic acid; MEK, mitogen-activated kinase kinase; MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt; EMSA, electrophoretic mobility shift assay; PARP, poly(ADP-ribose) polymerase; AIF, apoptosis-inducing factor; CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]propanesulfonate; SB, sodium butyrate; MS-275, N-(2-aminophenyl)-4-[N-(pyridine-3-ylmethoxycarbonyl)aminomethyl]benzamide; PD184352, 2-[2-chloro-4-iodo-phenilamino]-N-cyclopropylmethoxy-3,4-difluoro-benzamide.
- Received October 4, 2004.
- Accepted December 28, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}