


Abstract
The CB1 cannabinoid receptor has been shown to play important physiological roles in the central nervous system, as well as peripherally, and is a target for development of therapeutic medications. To gain insight on the ligand binding site(s) and structural features of activation, we designed and synthesized (-)-7′-isothiocyanato-11-hydroxy-1′,1′-dimethylheptylhexahydrocannabinol (AM841), a classical cannabinoid affinity label that incorporates an isothiocyanate substituent as an electrophilic reactive group capable of interacting irreversibly with a suitably located and properly oriented nucleophilic amino acid residue at or near the binding site. To obtain evidence for the site of covalent attachment of AM841, C6.47, identified in part by interactive ligand docking, was mutated to serine, alanine, and leucine to reduce or eliminate the nucleophilic character. Wild-type (WT) and mutant CB1 receptors were evaluated for their abilities to recognize a series of cannabinergic ligands. Each bound comparably to WT, excluding C6.47L, which displayed a reduced affinity for 3H-labeled (1R,3R,4R)-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-4-(3-hydroxypropyl)cyclohexan-1-ol (CP55940), AM841, 11-hydroxy-1′,1′-dimethylheptylhexahydrocannabinol (AM4056), and (-)-7′-bromo-11-hydroxy-1′,1′-dimethylheptylhexahydrocannabinol (AM4043) and an improvement in affinity for (-)-trans-Δ9-tetrahydrocannabinol (Δ9-THC). The affinity of 3H-labeled [2,3-dihydro-5-methyl-3-[(4-morpholinyl)methyl]pyrrolo-[1,2,3-de]-1,4-benzoxazin-6-yl](naphthyl)methanone (WIN55212-2) was unchanged across all mutants. It is noteworthy that AM841 was shown to bind irreversibly to WT CB1 but exhibited no covalent attachment with the mutants and behaved as an agonist suggesting irreversible attachment to C6.47 maintains CB1 in its active state. The evidence presented identifies C6.47 as the site of covalent bond formation with AM841 and combined with the binding data fully supports the molecular modeling. These studies present the first report of tandem applications of affinity labeling, site-directed mutagenesis, and interactive ligand docking for CB1.
The CB1 and CB2 cannabinoid receptors are relatively new members in the G-protein-coupled receptor (GPCR) superfamily. They have been shown to play important physiological roles and represent targets for development of therapeutic medications. From a pharmacological standpoint, agonist activation of both receptors results in the release of Gαi-proteins, causing a concomitant reduction in intracellular cAMP. The CB1 receptor (Fig. 1) is very densely distributed throughout the central nervous system and in various tissues in the periphery, whereas CB2 is present on cells of immune origin. Activation of CB1 has been shown to modulate D2-dopamine receptor activity presynaptically and inhibits excitotoxic glutamate release, among a host of other effects, including generalized neuroprotection (Howlett et al., 2002).
Cannabinoid receptors are activated by endocannabinoids as well as other cannabinergic ligands and are classified as belonging to family 1A GPCRs along with the adrenergic and dopaminergic receptors as well as Rhodopsin. Ligands capable of associating with cannabinoid receptors (for a recent review, see Palmer et al., 2002) include classical cannabinoids, commonly represented by the major psychoactive constituent of marijuana, Δ9-THC; nonclassical cannabinoids, with CP55940 as a leading example; aminoalkylindoles such as WIN55212-2 (Fig. 2); and the endogenous cannabinergics anandamide and 2-arachidonyl glycerol, which are biosynthesized from arachidonic acid. Despite the obvious structural dissimilarity of these ligands, each is an agonist capable of activating CB1, whereas the diarylpyrazoles, such as SR141716A, represent a key class of CB1-selective antagonists/inverse agonists. Understanding the precise molecular interactions of these molecules with their receptor sites of action can enhance rational design of novel ligands with improved receptor subtype selectivity, fewer undesirable side effects, and enhanced potency and efficacy, leading to potentially attractive therapeutic agents that produce their effects by modulating functionality of the cannabinergic system.
Characterization of GPCR ligand binding domains is a major goal in the design of improved drug molecules in that nearly 60% of all approved and marketed medications target GPCRs as their site of action (Muller, 2000). To accomplish this, the evaluation of small molecule-receptor target SAR is frequently coupled with site-directed mutagenesis to provide three-dimensional information on ligand recognition. The recent publication of the 2.8-Å crystal structure determination of bovine Rhodopsin has validated many of the existing models of GPCR structure previously based on fragment-derived or lower-resolution biophysical studies (Farrens et al., 1996; Palczewski et al., 2000; Yeagle et al., 2000). Moreover, the crystallographic study has hastened the process of identifying potential recognition domains by facilitating the construction and refinement of homology-based, computationally derived, three-dimensional models to allow interactive ligand docking. As a consequence, recognition domains for many GPCRs have begun to be elucidated.

Helical-net representation of the human CB1 receptor. The amino acid mutated in this investigation, C6.47(355), is indicated in bold. Representative locants in respective TMHs, identified by arrowheads, are given using the convention established by Ballesteros and Weinstein (1995) and are accompanied by their sequence positions as shown. For simplicity, the first 105 amino acids from the N termini and the final 42 amino acids from the C termini have been omitted.
We have reported the development of cannabinergic covalent probes and their potential application in GPCR binding site structural studies (Charalambous et al., 1992; Guo et al., 1994; Morse et al., 1995; Picone et al., 2002). The work described here clearly supports the role of the CWXP motif, which is found in Rhodopsin as well as other family 1A GPCRs, located in TMH6 of the CB1 receptor and its involvement in ligand recognition and binding. Our results, based on site-directed mutagenesis, identify the cysteine residue C6.47(355) within this key amino acid tetrad as the site of covalent attachment of AM841, an electrophilic classical cannabinoid affinity label designed to interact selectively with a nucleophilic amino acid in the ligand binding domain. This covalent ligand-receptor interaction is abolished when either C6.47(355) is mutated to weaker or non-nucleophilic residues or the electrophilic isothiocyanate group on AM841 is exchanged with nonelectrophilic substituents. AM841 was also shown, upon covalent attachment, to behave as a highly potent CB1 agonist. Interactive ligand docking in a computer-derived model of the CB1 receptor is fully consistent with the observed radioligand binding SAR and functional data.
Within the spectrum of mutations designed and evaluated in this SAR study, we present evidence for the molecular basis by which the characteristic C-3 alkyl side chains of differing lengths, possessed by natural and synthetic classical cannabinoids, interact with CB1. This study represents the first report of the tandem application of affinity labeling, site-directed mutagenesis, and interactive ligand docking involving elucidation of recognition and activation elements for CB1 and suggests that this conserved CWXP motif may have a similar role in a number of other family IA GPCRs.
Materials and Methods
Materials. Chemicals and reagents were obtained from Sigma (St. Louis, MO) at the highest purity/grade possible unless otherwise noted. AM841, AM4043, and AM4056 were synthesized in our laboratory; the details of the methods used will be published elsewhere. Regisil was obtained from Regis Technologies (Downers Grove, IL). All glassware and plasticware were treated with Regisil before use to minimize the adherence of lipophilic cannabinoids to their surfaces. pRC/CMV-hCB1 was a gift from Dr. T. I. Bonner (National Institute of Mental Health, Bethesda, MD). Oligonucleotide primers were synthesized by Sigma-Genosys (St. Louis, MO). QuikChange site-directed mutagenesis kit and XL-1 Blue supercompetent Escherichia coli were obtained from Stratagene (La Jolla, CA). LipofectAMINE 2000 and Thermoscript RT-PCR kit were obtained from Invitrogen (Carlsbad, CA). RNeasy and Midi-Prep kits were obtained from QIAGEN (Valencia, CA). CHO-K1 cells were from the American Type Culture Collection (Manassass, VA). Detergent-compatible protein assay kit was from Bio-Rad (Hercules, CA). [3H]CP55940 (110–180 Ci/mmol) and [3H]WIN55212-2 (43–50 Ci/mmol) were from PerkinElmer Life and Analytical Sciences (Boston, MA). CP55940 and Δ9-THC were provided by the National Institute on Drug Abuse drug supply program.
Amino Acid Numbering Scheme. This report uses the numbering scheme proposed by Ballesteros and Weinstein (1995). The most highly conserved amino acid in a particular transmembrane helix (TMH) is assigned a locant of 0.50. This number is preceded by the helix number and followed in parentheses by the sequence number. All other amino acids in a given helix are assigned a locant relative to that most conserved residue. For example, the most conserved residue in TMH6 of hCB1 is Pro358, which is accordingly referred to as P6.50(358). The residue of interest in the current mutation study is C6.47(355) which corresponds to the residue three amino acids N-terminal to P6.50(358).
Site-Directed Mutagenesis, Transfection, and Cell Culture. Mutations to pRC/CMV-hCB1 were introduced using the QuikChange site-directed mutagenesis kit as described by Tao et al. (1999). Mutagenic oligonucleotide primers were annealed and extended during 16 cycles of thermocycling with Pfu DNA polymerase. After an initial melting for 30 s at 95°C, each cycle consisted of 30 s at 95°C, 60 s of annealing at 55°C, and 14 min of extension at 68°C. Primers employed to make the C6.47(355) mutations were: C6.47(355)S: forward, 5′-G GTG GTG TTG ATC ATC TCC TGG GGC CCT CTG CTT GCA ATC ATG G-3′; reverse, 5′-C CAT GAT TGC AAG CAG AGG GCC CCA GGA GAT GAT CAA CAC CAC C-3′; C6.47(355)A: forward, 5′-G GTG GTG TTG ATC ATC GCC TGG GGC CCT CTG CTT GCA ATC ATG G-3′; reverse, 5′-C CAT GAT TGC AAG CAG AGG GCC CCA GGC GAT GAT CAA CAC CAC C-3′; C6.47(355)L: forward, 5′-G GTG GTG TTG ATC ATC TTA TGG GGC CCT CTG CTT GCA ATC ATG G-3′; reverse, 5′-C CAT GAT TGC AAG CAG AGG GCC CCA TAA GAT GAT CAA CAC CAC C-3′ [the bold and underlined sequences refer to base changes (deviating from WT)]. After thermocycling, the product was treated with DpnI and then used to transform XL-1 Blue supercompetent E. coli. Plasmid DNA was isolated using the QIAGEN Midi-Prep Kit. To ensure that only the desired mutations had been produced, plasmid DNA was sequenced at the UCONN Biotechnology Center using SP6 and T7 primers, as well as the RPP-For-hCB1 primer (described below). Near the point of mutation, both top and bottom strand DNA sequences were obtained and verified. With DNA sequence integrity verified, purified mutagenic plasmid DNA was used to transfect CHO-K1 cells.
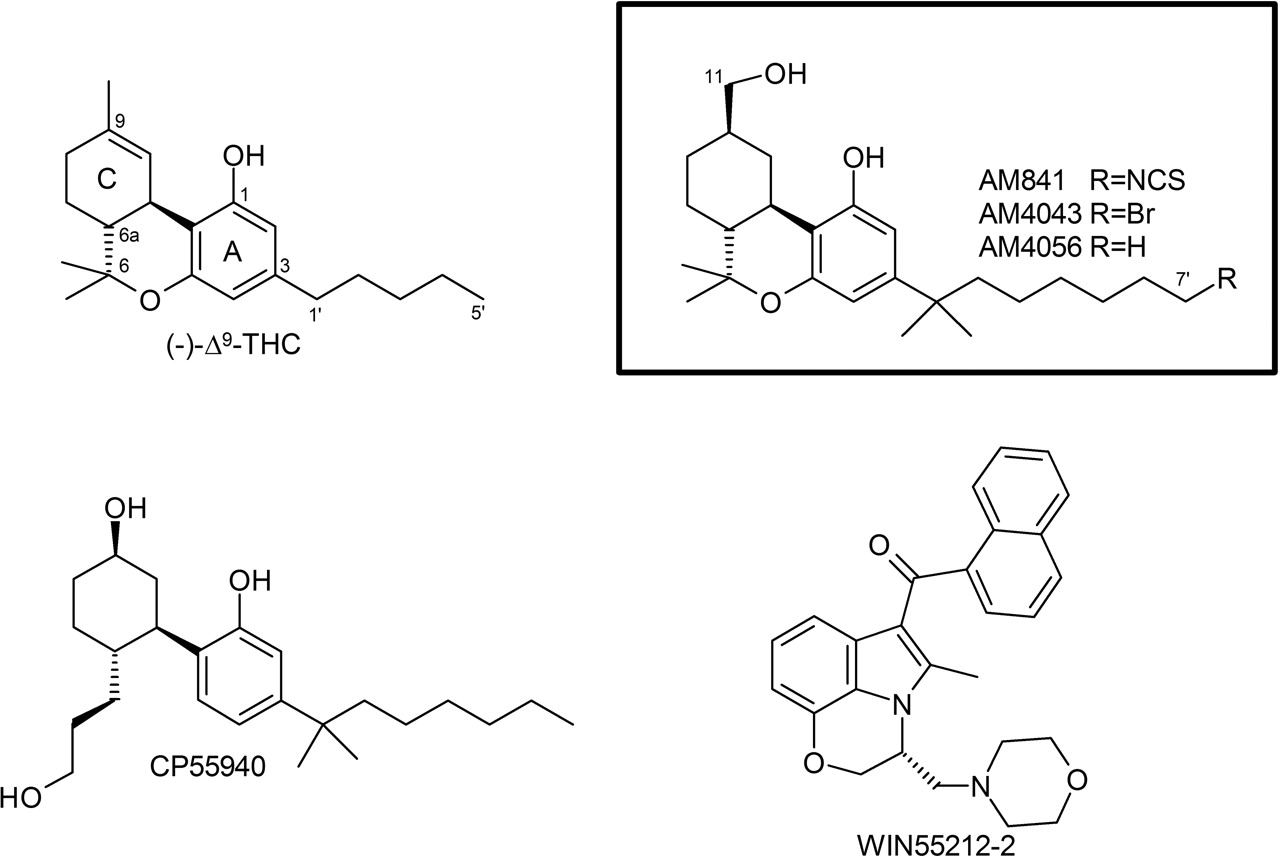
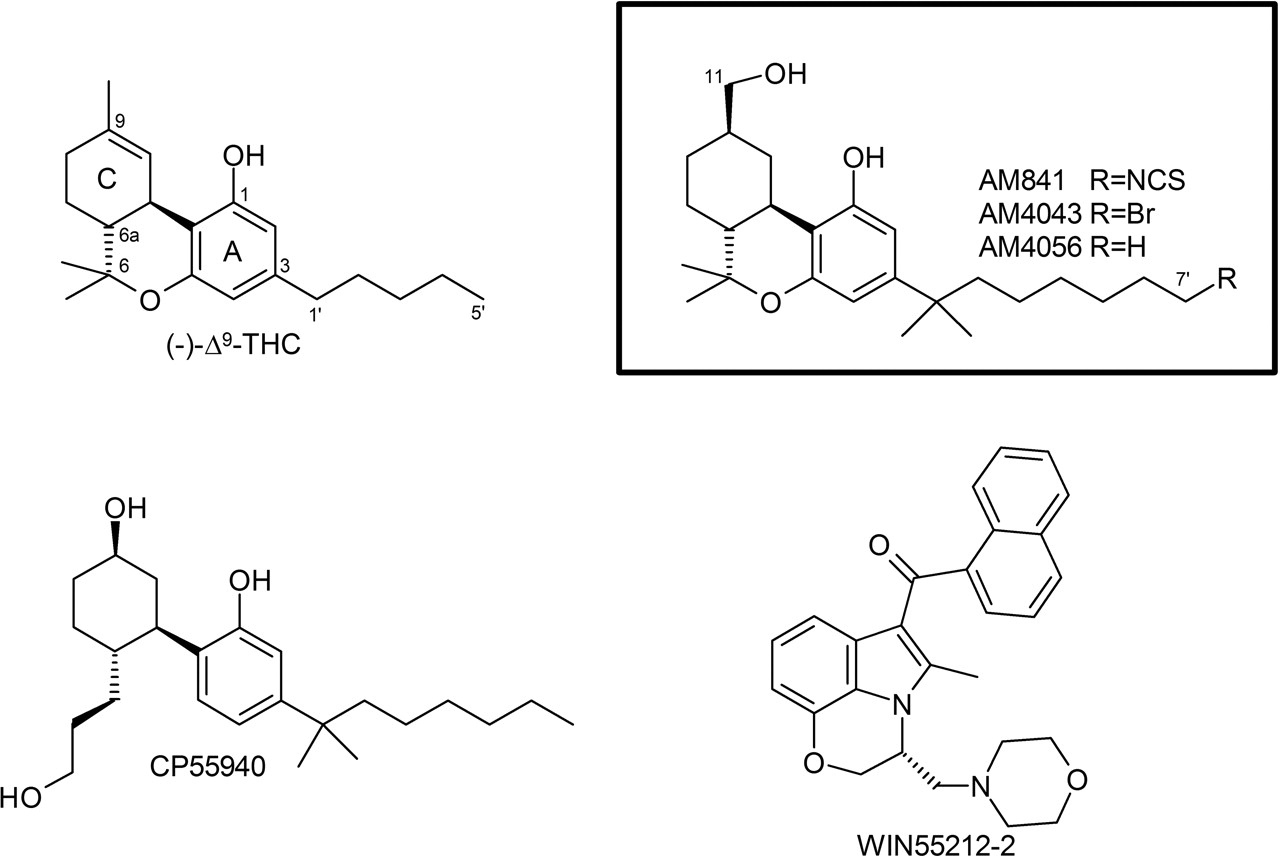
Chemical structures of representative cannabinergic ligands and those used in this study. The numbering scheme for classical cannabinoids is illustrated with Δ9-THC, AM841, AM4043, and AM4056.
CHO-K1 cells were maintained in F12K medium supplemented to contain 10% fetal bovine serum, 50 IU/ml penicillin, and 50 mg/ml streptomycin in a 5%/95% CO2/air incubator at 37°C. CB1 expressing CHO cells (hCB1-CHO-K1) were prepared by transfecting CHO-K1 cells with pRC/CMV-hCB1 (WT and C6.47(355) mutants) using LipofectAMINE 2000 according to the manufacturer's protocol. Stable transfectants were selected for using 1 mg/ml G418 (Geneticin) and isolates were picked via selective trypsinization with sterile glass cloning cylinders. Clonal isolates were propagated and receptor expression evaluated by RT-PCR and ligand binding. To confirm that G418 resistance was associated with CB1 receptor expression, RT-PCR was undertaken. RNA from untransfected and transfected CHO-K1 cells was isolated using the RNeasy Kit according to the manufacturer's protocol. RT-PCR was performed using the Thermo-Script RT-PCR system. The primers employed were: RPP-For-hCB1 5′-GTC ACG GCC TCC TTC ACT GC-3′ and RPP-Rev-hCB1 5′-CCT GTA CCT TCC CAT CCT CA-3′. These primers were designed to amplify a 0.9-kilobase fragment of the CB1 gene. PCR products were resolved on a 2% agarose gel with ethidium bromide and visualized.
Cell Membrane Preparations. Cell membranes from hCB1-CHO-K1 cells were prepared by allowing cells to become approximately 85% confluent, after which point they were washed twice with Hanks' balanced salt solution without calcium and magnesium. After the final wash, cells were scraped in Hanks' balanced salt solution without calcium and magnesium in the presence of 0.02% EDTA also containing 0.5% (v/v) protease inhibitor cocktail [100 mM 4-(2-amino-ethyl)benzensulfonyl fluoride, 4 mM bestatin, 1.4 mM E64, 2.2 mM leupeptin, 0.8 mM aprotinin, and 1.5 mM pepstatin A]. Cellular material was sedimented at 1000g for 10 min at 4°C, the resulting pellet was resuspended in TME (25 mM Tris, pH 7.4, 5 mM MgCl2, and 2 mM EDTA) containing 0.5% (v/v) protease inhibitor cocktail and subjected to nitrogen cavitation with a Parr Cell Disruption Bomb at 800 psi for 10 min on ice. Ruptured cells were sedimented at 500g for 10 min at 4°C and the supernatant containing cellular membranes was washed twice at 100,000g for 15 min. Pellets containing membrane protein were resuspended in TME, divided into aliquots, frozen in liquid nitrogen, and stored at -70°C until use in binding assays as reported previously (Shire et al., 1996; Chin et al., 1998; Abadji et al., 1999; Tao et al., 1999; Hurst et al., 2002; McAllister et al., 2002, 2003, 2004). Total protein content was determined with the Bio-Rad detergent-compatible protein assay.
Saturation Binding. hCB1-CHO-K1 membrane preparations were subjected to saturation binding with [3H]CP55940 and [3H]WIN55212-2. Radiolabeled ligands were diluted into TME containing 0.1% (w/v) essentially fatty acid-free bovine serum albumin (BSA) (TME/0.1% BSA). Initiation of the binding reaction began with the addition of membrane protein at a concentration of approximately 35 μg of membrane protein per well in a total volume of 200 ml followed by a 1-h incubation in a 30°C shaking water bath. Binding was terminated by rapid filtration over Unifilter GF/B-96-well filter plates (PerkinElmer Life and Analytical Sciences) using a Filtermate-196 cell harvester. Filter plates were washed four times with ice-cold wash buffer (50 mM Tris base, 5 mM MgCl2, and 0.5% BSA). Bound radioactivity was quantitated in a PerkinElmer Top-Count scintillation counter. Total binding was assessed by equilibrating radioligand and membrane without competing compounds, whereas nonspecific binding was determined in the presence of 1 μM unlabeled homologous competitor. Nonspecific binding was subtracted from total bound radioactivity to determine the specifically bound radioactivity, which was subsequently fitted to the Scatchard-Rosenthal equation to determine the binding parameters Kd and Bmax (Rosenthal, 1967). Data were collected as points in quadruplicate. All binding parameters are presented as mean ± S.E.M. from at least three independent experiments.
Competitive Binding Assays. Ligands were tested for their ability to bind to CB1 receptors via competition binding using [3H]CP55940 as described previously by Lan et al. (1999), with a few modifications. [3H]CP55940 was employed to achieve a final concentration of 3.0 nM in a final volume of 200 μl. Initiation of the binding reaction began with the addition of hCB1-CHO-K1 cell membranes at a concentration of approximately 35 μg of membrane protein per well. All binding and filtering conditions were as described above. IC50 values were determined using the commercially available program GraphPad Prism (Carlsbad, CA). A Ki value was calculated from the IC50, as described by Cheng and Prusoff (1973). All data were collected as points in triplicate with IC50 and Ki values determined from at least three independent experiments.
Affinity Labeling. Labeling methods were adapted from our previously published procedures with a few modifications (Guo et al., 1994; Morse et al., 1995). Membranes containing CB1 receptor were allowed to equilibrate with AM841 for 1 h in a 30°C shaking water bath, after which suspensions were rapidly sedimented (27,200g) at 30°C. The resulting pellets were then washed three times in TME/1.0% BSA at 30°C. Fifteen minutes was allowed between resuspension of pellets and washing to permit equilibration between buffer and membranes. Two final washes were conducted in TME without BSA. Total protein content was then assayed. Radioligand binding was then undertaken as described above except that only a single concentration of radioligand was employed at final concentrations of 6 and 18 nM for [3H]CP55940 and [3H]WIN55212-2, respectively.
cAMP Accumulation Assay. hCB1 expressing HEK293 cells were prepared and grown according to the method of Tao et al. (1999). For functional assays, cells were grown to ∼75% confluence, harvested by scraping and collecting cells in phosphate-buffered saline containing 1 mM EDTA, and counted with a hemacytometer. Cells were washed and resuspended to a concentration of 1 × 106 cells/ml in Dulbecco's modified Eagle's medium containing 20 mM HEPES, pH 7.3, 0.1 mM RO-20-1724, and 1 mM isobutylmethylxanthine and incubated for 30 min at 37°C in a 95%/5% air/CO2 incubator. Aliquots of cells were added to Regisil-treated polypropylene tubes containing 0.5 μM FSK ± drug with 0.5% essentially fatty acid-free BSA and incubated for 15 min. Incubation was quenched via boiling for 4 min, followed by centrifugation. Samples were assayed for cannabinoid attenuation of FSK-stimulated cAMP production using a competitive protein binding assay as described previously (Tao et al., 1999).
Statistical Analysis. Results were subjected to one-way ANOVA and Bonferroni's post hoc test using the Prism software package (GraphPad Software, San Diego, CA). A p value less than 0.05 was deemed significant. In the case of the covalent binding studies, results between buffer-treated (control) and AM841-treated cells were subjected to Unpaired t test with p values less than 0.05 considered significant.
Molecular Modeling. Aspects of the CB1 computational model have been described previously (Bramblett et al., 1995; Hurst et al., 2002; McAllister et al., 2003, 2004); additional details relevant to the current study are presented below. A model of the R form of CB1 was created using the 2.8-Å crystal structure of Rhodopsin (Palczewski et al., 2000). Because TMH6 figures prominently in the R to R* transition (Jensen et al., 2001), we have studied the conformations accessible to TMH6 in CB1 using CM, a biased Monte Carlo/simulated annealing method (Barnett-Norris et al., 2002). These studies revealed that TMH6 in CB1 has high flexibility because of the small size of residue G6.49(357) immediately preceding P6.50(358). Two families of conformers were identified by CM for TMH6 in CB1. Cluster 1 showed a pronounced proline kink (40 members of 100, 71.2° average kink angle). Cluster 2 contained helices with less pronounced kinks (51 members of 100, 30.1° average kink angle). A conformer from the more kinked CM family of CB1 TMH6s (Cluster 1) was used in our model of the R state of CB1. This conformer was selected (Pro kink angle = 53.1°) so that R3.50(214) and D6.30(338) could form a salt bridge at the intracellular ends of TMHs 3 and 6 in the CB1 TMH bundle. An analogous salt bridge has been shown to be an important stabilizer of the inactive state of the β2-adrenergic receptor (Ballesteros et al., 2001) and to be present in Rhodopsin (Palczewski et al., 2000). Because of the extreme flexibility of TMH6 in CB1, we have proposed that an additional TMH3–6 salt bridge, K3.28(192)-D6.58(366), stabilizes the inactive state on the extracellular side of the TMH bundle (Hurst et al., 2002). Agonists are thought to have higher affinity for the activated form of GPCRs (Leff, 1995); as such, agonist ligands in the work reported here were docked in an R* model of CB1. This model was created by modification of our Rhodopsin-based model of the R form of CB1 and guided by the biophysical literature on the R-to-R* transition, which has indicated that activation is accompanied by rigid domain motion of the intracellular portion of TMH6 relative to TMH3 (Farrens et al., 1996; Ghanouni et al., 2001) and by counter-clockwise rotations of TMH3 (Gether et al., 1997; Hulme et al., 1999) and TMH6, placing C6.47(355) in the binding site crevice (Javitch et al., 1997). A TMH 6 conformer from the second major conformational family (less kinked: 21.8° kink angle) identified by CM (Barnett-Norris et al., 2002) was incorporated into the CB1 bundle to represent the R* conformation of TMH6. Additional details of the generation of the CB1 R* model may be found in McAllister et al. (2003).
Ligand Conformations and Docking Positions. Binding site conformations and anchoring interactions within the receptor used for each ligand were based on earlier published computational and experimental SAR studies. AM841 and Δ9-THC were docked in the global minimum energy conformation of their respective tricyclic ring systems (Reggio et al., 1993) and, based on earlier data, K3.28(192) was used as the primary interaction site for the phenolic hydroxyl of each ligand (Huffman et al., 1996; Song and Bonner, 1996).
The locations and subsequent availability of potential cysteine residues for reaction with AM841 in the binding site crevice of CB1 were made by inspection of the computer-derived receptor model, described here, which is based on the crystal structure of Rhodopsin. Cysteine residues are the most likely candidates for reaction with isothiocyanates (Tahtaoui et al., 2003), which provided the rationale for the interactive docking studies to elucidate binding interactions for AM841. Of the 13 cysteines in human CB1, the two found in the E-2 loop and the two in the N terminus were excluded from consideration. It is well accepted that small ligands for family IA GPCRs, such as CB1, do not interact with the N terminus and that, typically, cysteine residues in the extracellular loops can be involved in disulfide bond formation required for the structural integrity of the receptor (Bockaert and Pin, 1999). In CB1, Cys257 and Cys264 are part of the E-2 loop and are likely candidates for an intraloop disulfide bridge (Shire et al., 1996, 1999; Fay et al., 2005). Cysteine residues in the intracellular loops, including one in I-1 and three in the C terminus, were also eliminated from consideration because ligand binding to family IA GPCRs is widely accepted to be localized to TMHs and extracellular loops (Bockaert and Pin, 1999). Five cysteine residues are located in the transmembrane regions [C1.55(139), C4.47(238), C6.47(355), C7.38(382), and C7.42(386)] and are used here as guides for identifying the interactions of AM841 with CB1. C1.55(139) and C4.47(238) are on the intracellular side of the seven TMH bundle and face out toward the lipid domain as predicted by the positions of analogous residues in the crystal structure of Rhodopsin [Y1.55(60) and A4.47(158)] (Bramblett et al., 1995; Palczewski et al., 2000). The other three cysteine residues are located in the TMHs within a few turns from the extracellular side. In the R* bundle, C6.47(355) and C7.42(386) are oriented toward the binding site crevice, making them potential targets for AM841. In SCAM studies, C6.47(355) has been shown to become accessible from within the binding site cavity in the β2-adrenergic receptor in the activated (R*) state (Javitch et al., 1997). C7.42(386) has recently been shown to be accessible from within the binding site of CB1 (Fay et al., 2005). On the other hand, as illustrated in the Rhodopsin crystal structure, C7.38(382) [P7.38(291) in Rhodopsin] is located at the TMH6–7 interface and is less likely to interact with the ligand. To explore which reactive cysteine within the binding site crevice might be the site for covalent bond formation, the alkyl tail of AM841 was independently covalently attached to C6.47(355) or C7.42(386) (see Discussion). Within the constraints of a phenolic hydroxyl hydrogen bonding interaction with K3.28(192) interactive computer graphics were used to identify a thermodynamically most favored docking orientation which is accompanied with the formation of a maximal number of hydrogen bonds for AM841. Thereafter, Δ9-THC was docked in a similar orientation.
Energy Minimization. The energy of the ligand/CB1 R* TMH bundle complex was minimized using the AMBER* united atom force field in Macromodel 6.5 (Schrödinger Inc., Portland, OR). A distance-dependent dielectric, 8.0-Å extended nonbonded cutoff (updated every 10 steps), 20.0-Å electrostatic cutoff, 4.0-Å hydrogen bond cutoff, and explicit hydrogens on sp2 carbons were used. The first stage of the calculation consisted of 1000 steps of Polak-Ribier conjugate gradient minimization, in which a force constant of 1000 kJ/mol was used on the helix backbone χ, Ψ, and ω torsions to restrain the TMH backbones while permitting the side chains to relax. The second stage of the calculation consisted of 100 steps of conjugate gradient in which the force constant on the helix backbone torsions was reduced to 250 kJ/mol to allow the helix backbones to adjust. Stages 1 and 2 were repeated until a gradient of 0.04 kJ/(mole ×Å2) was reached.
Results
Preparation of CB1-Expressing Cell Lines. After transfection and selection, cells were propagated and total RNA was isolated and probed using gene-specific primers in a sensitive RT-PCR assay to detect CB1 gene expression. Each of the lines prepared was found to express the CB1 gene (data not shown). Cell lines found to give the greatest amount of CB1 expression were chosen for further propagation and pharmacologic characterization. As expected, RT-PCR did not amplify a CB1 gene product using RNA isolated from untransfected CHO-K1 cells (data not shown), consistent with the absence of a CB1 gene.
Ligand Binding. CB1 WT and each C6.47(355) mutant were evaluated for their abilities to bind the two widely used cannabinoid receptor radioligands, [3H]CP55940 and [3H]WIN55212-2, using membrane homogenates, because this method has been found to provide consistently reproducible specific binding (Shire et al., 1996; Chin et al., 1998; Abadji et al., 1999; Tao et al., 1999; Hurst et al., 2002; McAllister et al., 2002, 2003, 2004). In WT and C6.47(355) mutants, the observed radioligand binding was found to be specific and saturable. Binding parameters are given in Table 1, and representative isotherms are shown in Fig. 3. These findings are consistent with data on CB1 expressed in CHO-K1 cells (Chin et al., 1998; Abadji et al., 1999) and in HEK293 cells (Song and Bonner, 1996; McAllister et al., 2002). C6.47(355)S, C6.47(355)A, and C6.47(355)L were well expressed and able to recognize and bind [3H]WIN55212-2 with remarkable similarity to WT CB1, as evidenced by their respective binding parameters, Kd and Bmax. Conversely, the nonclassical cannabinoid [3H]CP55940 had similar affinities for WT CB1 and the C6.47(355)S and C6.47(355)A mutants but exhibited a 3-fold reduced affinity for the C6.47(355)L mutant. There were no differences in the observed Bmax values for either radioligand across the spectrum of mutations compared with WT indicating comparable levels of detectable membrane expression. Specific binding was typically 70 to 80% of the observed total binding. No detectable specific binding was observed for either radioligand in untransfected CHO-K1 cells (data not shown).
Comparison of ligand binding parameters for WT and C6.47(355) mutant human CB1 receptors Binding parameters were determined in cellular membranes prepared from WT and mutant CB1 transfected CHO cells as described under Materials and Methods. The binding affinities shown represent the mean ± S.E.M. of at least three independent determinations each performed in quadruplicate.

Saturation binding isotherms for human CB1 WT and C6.47(355) mutants with [3H]CP55940 and [3H]WIN55212-2. Conditions for membrane preparation and radioligand binding are described under Materials and Methods. Curves are representative of an individual experiment independently replicated a minimum of three times. Data represent the mean ± S.E.M. of radioligand specifically bound from points collected in quadruplicate.
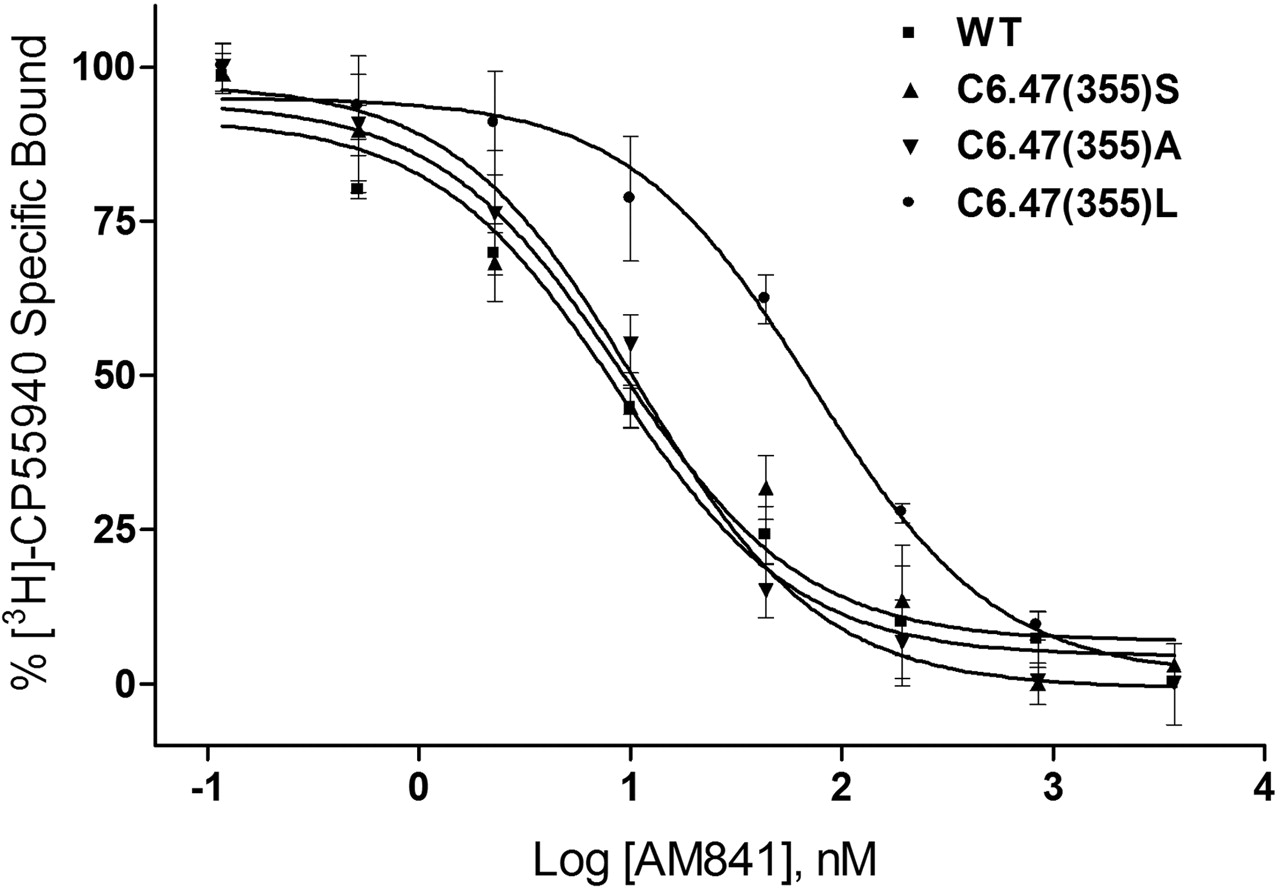
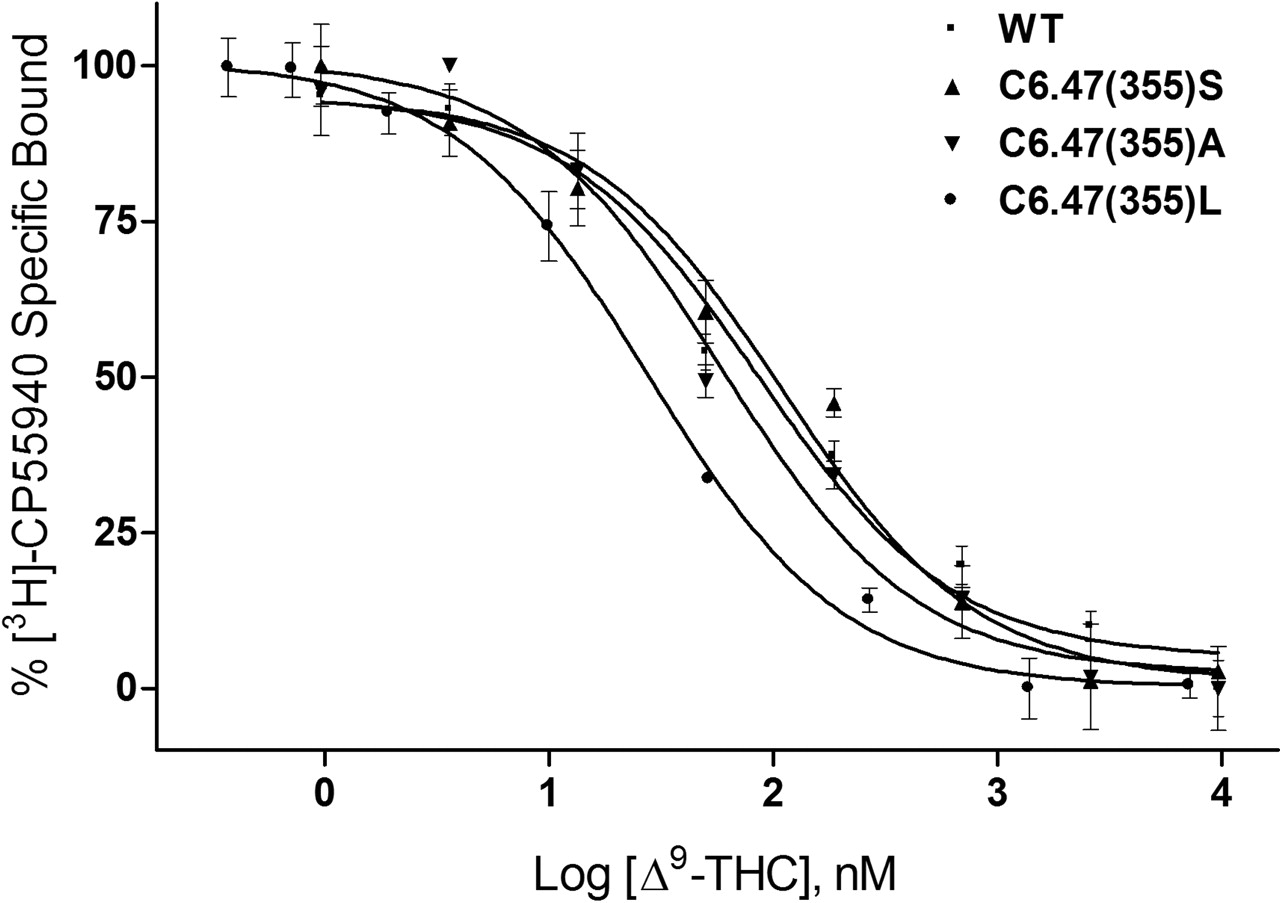
The ability of AM841 to recognize CB1 was also examined in this study as a prelude to covalent binding experiments. Binding with [3H]CP55940 was undertaken to assess the extent (if any) to which mutations would alter recognition and subsequent covalent binding potential of this ligand. Consistent with results obtained using rat brain synaptosomal membranes as a source of CB1 (R. P. Picone, D. J. Fournier, and A. Makriyannis, unpublished observations), AM841 exhibited high affinity for the WT CB1 receptor using [3H]CP55940 as a radioligand (Table 1). It should be noted that because of the irreversible nature of the binding of AM841, the affinity for WT CB1 is presented as an apparent Ki value (Cheng and Prusoff, 1973). The binding affinities of AM841 for C6.47(355)S and C6.47(355)A were indistinguishable from those of WT. However, the ability of AM841 to compete with [3H]CP55940 was attenuated, nearly 6-fold, in the C6.47(355)L mutant with the bulkier hydrophobic leucine side chain compared with the respective cysteine of WT, as shown in Fig. 4. Likewise, the affinities of AM4043 and AM4056 for WT and C6.47(355) mutants were found to follow this trend, but a slight improvement was noted overall, indicating that these AM841 analogs, in which the isothiocyanate group was substituted with -Br and -H groups, possessed higher affinities than the parent compound (Table 1). It is interesting that the affinity of Δ9-THC was determined for CB1 WT and the C6.47(355) mutants where competition binding studies with [3H]CP55940 exhibited a reverse trend compared with that observed in the other cannabinoid ligands. In accordance with our results with [3H]CP55940, AM841, AM4043, and AM4056, the affinity of Δ9-THC was significantly different in the C6.47(355)L mutant and was improved nearly 3-fold, relative to WT, rather than diminished, as was the case with ligands with the various dimethylheptyl C-3 alkyl chains. Moreover, an improvement in affinity of nearly 2-fold was noted for Δ9-THC with C6.47(355)A, in which the WT cysteine is mutated to alanine, resulting in a more hydrophobic methyl group in this position (Fig. 5, Table 1). Unlike the results when using [3H]CP55940 as the radioligand, there were no observed differences in affinity between WT and the mutants when [3H]WIN55212-2 was employed in saturation binding studies (Table 1). This suggests that the structurally dissimilar cannabinergic ligands [3H]CP55940 and [3H]WIN55212-2 exhibit different binding motifs with the CB1 receptor, where the amino acid in position 6.47 can play a prominent role in the recognition of cannabinoid analogs such as [3H]CP55940, AM841, AM4043, AM4056 and Δ9-THC but is not critical for the aminoalkylindole [3H]WIN55212-2.
The Role of C6.47(355) in Covalent Binding of AM841. Exposure of CB1 WT to 90 nM AM841 followed by a 1-h incubation and subsequent washing resulted in a near total elimination of specific binding for both [3H]CP55940 and [3H]WIN55212-2 consistent with irreversible occupation with spontaneously reacting isothiocyanates (Zhu et al., 1996; Picone et al., 2002; Tahtaoui et al., 2003). Specific binding of [3H]CP55940 was reduced from 303 ± 58.4 pmol/g (buffer-treated) to 50.33 ± 27.8 pmol/g (Fig. 6A), whereas binding of [3H]WIN55212-2 was reduced to 70.6 ± 74.8 pmol/g from 408 ± 97.9 pmol/g (buffer-treated) (Fig. 6B). Conversely, there was no statistically significant reduction in the observed binding of either radioligand after exposure to AM841 for C6.47(355)S, C6.47(355)A, or C6.47(355)L compared with the untreated (buffer-treated) controls (Fig. 6, C-H). The data clearly indicate that although AM841 is well recognized by the CB1 mutants, this process is reversible. Unlike the case with WT CB1, where AM841 binds irreversibly, C6.47(355)S, C6.47(355)A, and C6.47(355)L mutants are unable to engage this ligand in a covalent interaction. Incubation of either AM4043 or AM4056 with CB1 WT membranes indicated that these nonelectrophilic analogs of AM841 were incapable of irreversible association as well. Specific binding of [3H]CP55940 and [3H]WIN55212-2 in WT CB1 containing membranes treated with either AM4043 or AM4056 was found to be no different compared with their corresponding buffer-treated controls (Fig. 7). Unlike AM841, the above two ligands, in which the electrophilic isothiocyanate group is substituted for a -Br or -H, respectively, behave as fully reversible, wash-sensitive ligands indicative of their inability to irreversibly interact with CB1 (Guo et al., 1994; Picone et al., 2002; Tahtaoui et al., 2003).
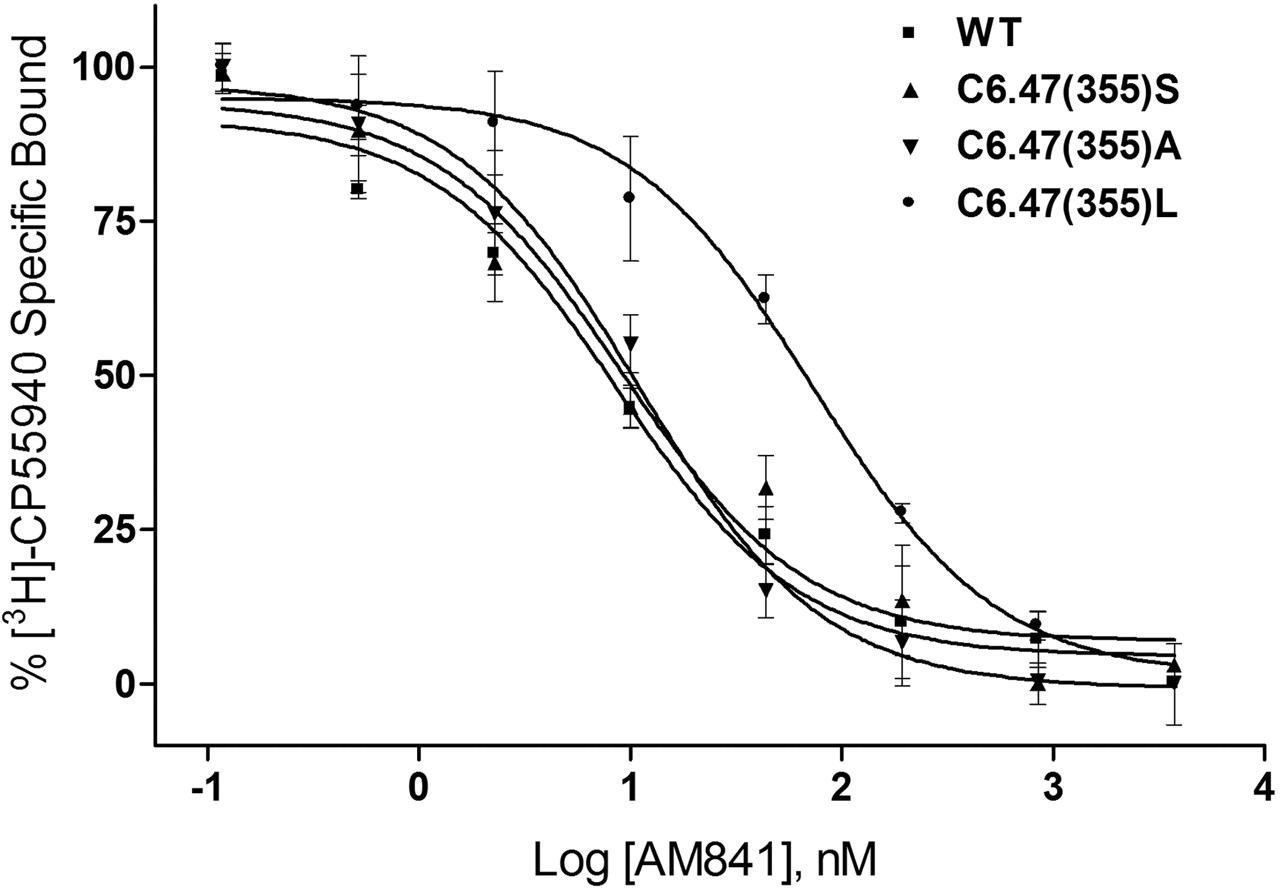
WT human CB1 and C6.47(355) mutants recognize AM841. Membranes containing WT and C6.47(355) mutants were subjected to [3H]CP55940 binding in the presence of AM841 as described under Materials and Methods. Data points represent the mean ± S.E.M. of radioligand specifically bound determined in quadruplicate. The curves shown are a representative example of at least three independent experiments.
cAMP Accumulation. Addition of AM841, AM4043, AM4056, CP55940, or Δ9-THC to cells expressing CB1 led to an approximately equal reduction of accumulated intracellular cAMP after FSK stimulation. AM841, our irreversible agonist, proved to be more potent than any reversible cannabinergic agonist tested in this study. cAMP production determined from FSK-stimulated cells was found to be 3.563 ± 0.132 pmol/106 cells. When incubated with 10 nM CP55940, cAMP production was reduced to 1.808 ± 0.078 pmol/106 cells. Although AM841, at a significantly lower concentration (1.3 nM), produced a nearly equal reduction in FSK-stimulated cAMP to 2.225 ± 0.128 pmol/106 cells relative to CP55940. The cAMP accumulation observed for AM4043 (10 nM), AM4056 (10 nM), and Δ9-THC (100 nM) was reduced to only 2.456 ± 0.333, 2.547 ± 0.219, and 2.181 ± 0.09 pmol/106 cells, respectively, and required a significantly higher concentration compared with AM841 (Fig. 8). Consistent with the above, AM841 produced a concentration-dependent reduction in FSK-stimulated cAMP accumulation with an EC50 of 0.94 nM (0.39–2.27 nM; 95% CI) (Fig. 8, inset). It is interesting to note that although each of the reversible dimethylheptyl-containing ligands possessed higher affinity for CB1, they were all found to be less potent than the irreversible analog AM841 (Table 2). Combined, these results further indicate that AM841 is more potent than AM4043, AM4056, CP55940 (Tao and Abood, 1998), and Δ9-THC.
Comparison of CB1 WT binding affinity and agonist efficacy for classical and nonclassical cannabinoid ligands in this study Affinities reported here represent mean ± S.E.M. described under Materials and Methods, whereas efficacies are reported as EC50 (95% confidence intervals), which were determined as FSK-stimulated reduction in cAMP accumulation as described under Materials and Methods.
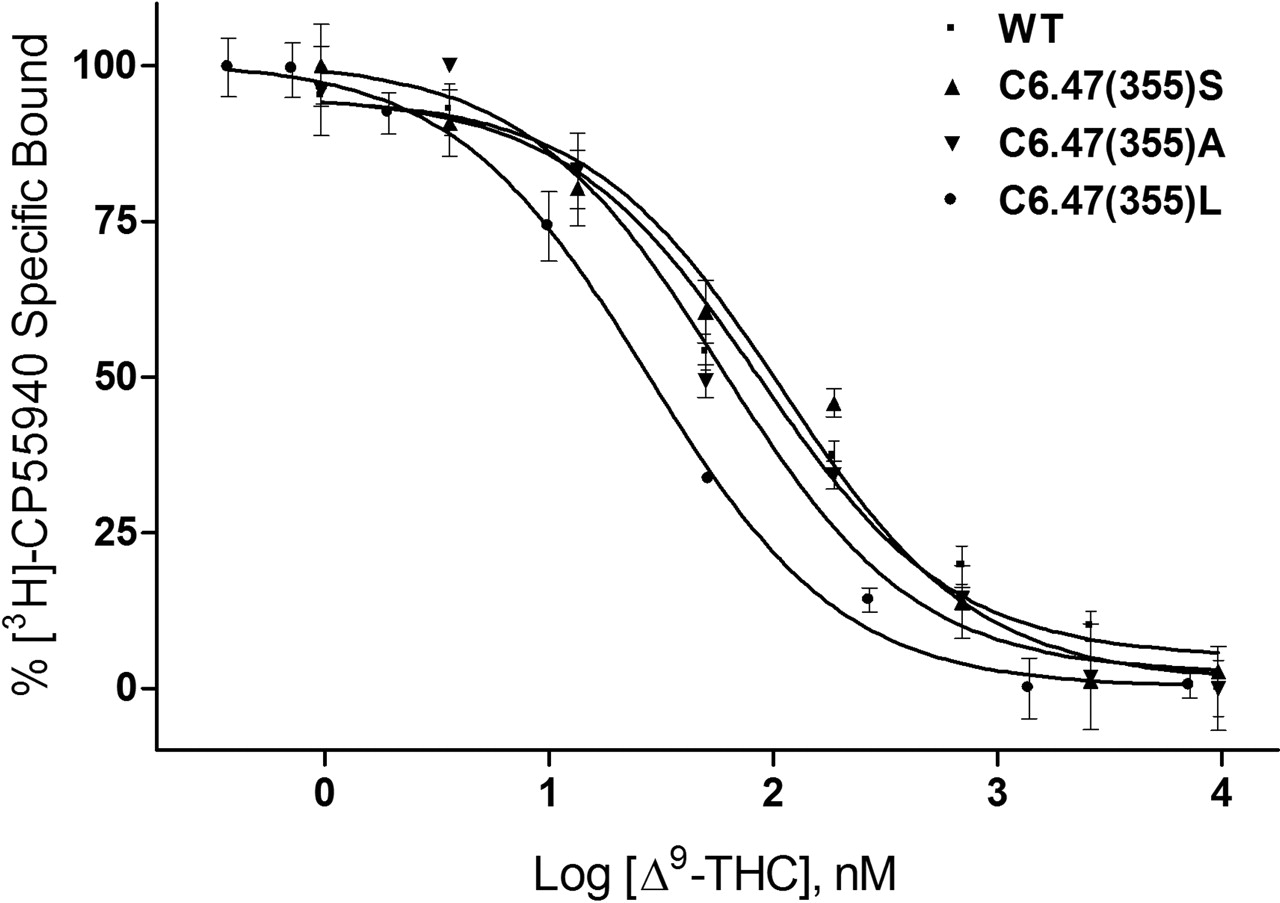
WT human CB1 and C6.47(355) mutants recognize Δ9-THC. Membranes containing WT and C6.47(355) mutants were subjected to competition binding between [3H]CP55940 and Δ9-THC as described under Materials and Methods. Data points represent the mean ± S.E.M. of radioligand specifically bound determined in quadruplicate. The curves shown are a representative example of at least three independent experiments.
Molecular Modeling of CB1 R*-Ligand Complexes. CP55940 has been proposed to bind in the TMH3-6-7 region of CB1, with its dimethylheptyl side chain interacting with a hydrophobic pocket formed collectively by V6.43(351), C6.47(355), L6.51(359), L7.41(385), and L7.44(388) (Tao et al., 1999). Based on this model, it is expected that the C-3 alkyl side chain of classical cannabinoids interacts similarly in this pocket. It can also be argued that enlargement of the residue at position 6.47 from a cysteine to a leucine, as with C6.47(355)L, is expected to produce steric crowding and diminished affinity of [3H]CP55940, a prediction confirmed by our experimental data. Conversely, modeling studies in conjunction with mutation data have suggested that WIN55212-2 binds in the TMH3-4-5-6 aromatic microdomain region of CB1, with direct interactions with W5.43(280), F3.36(201), and W6.48(356) but do not point to any interactions with C6.47(355) (McAllister et al., 2003). For this reason, structural modifications of C6.47(355) in CB1 are not expected to and indeed do not affect the binding of [3H]WIN55212-2. Our combined observations on the binding behavior of [3H]CP55940 and [3H]WIN55212-2 with CB1 WT and the C6.47(355) mutants provide a direct line of evidence that these two structurally dissimilar ligands occupy distinctly different sites within CB1. These results indicate that these two cannabinergic classes (i.e., the classical cannabinoids and the aminoalkylindoles) engage the CB1 receptor through distinct binding motifs (Song and Bonner, 1996; Shire et al., 1996; Chin et al., 1998; Tao et al., 1999; McAllister et al., 2003).

AM841-treatment eliminates binding of [3H]CP55940 and [3H]WIN55212-2 to human CB1 WT but not to C6.47(355) mutants. Exposure of AM841 to membranes prepared from WT and C6.47(355) mutants of human CB1 expressing cells for 60 min was followed by repetitive sedimentation-washing and subjected to radioligand binding using either 6 nM [3H]CP55940 or 18 nM [3H]WIN55212-2, as described under Materials and Methods. Data shown are the mean ± S.E.M. from two independent experiments conducted with six replicates each. *, p < 0.0005, unpaired t test; ‡, p < 0.05, unpaired t test.
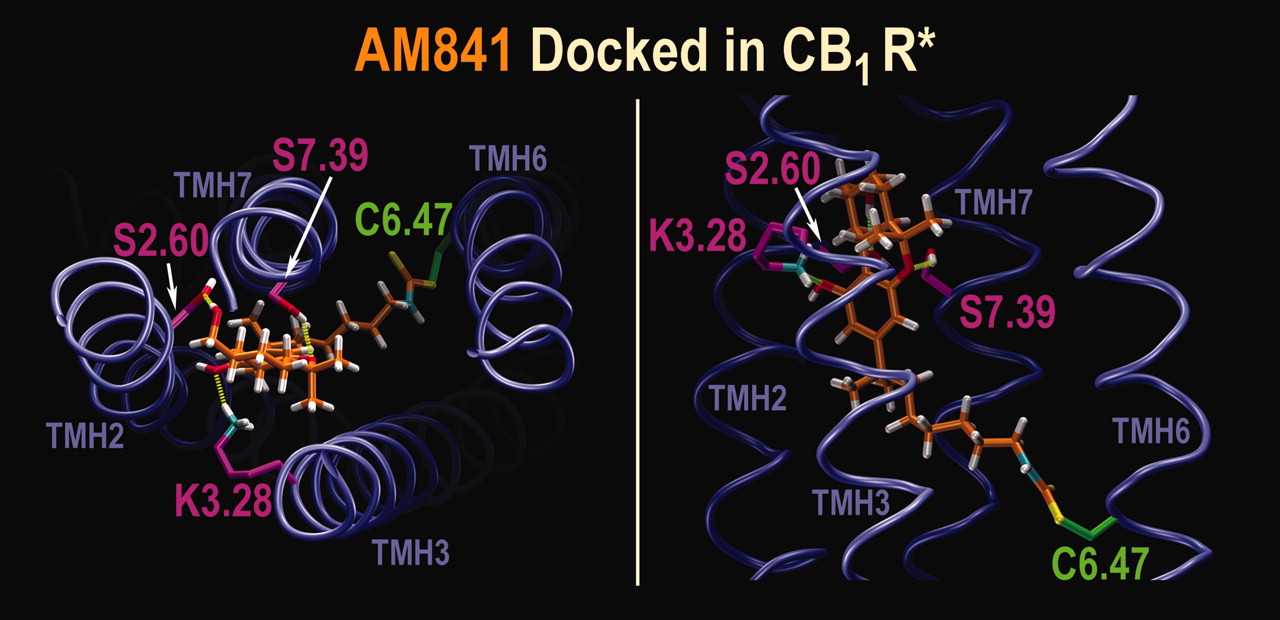
In accord with our modeling, when AM841 is covalently attached at C6.47(355) (Fig. 9) in the R* model of CB1, it is capable of establishing three hydrogen bonding interactions that involve its phenolic hydroxyl group with K3.28(192), its pyran oxygen with S7.39(383), and its 11-hydroxyl with S2.60(173). In this docked position, AM841 sterically occludes the binding of other ligands for which K3.28(192) is a primary interaction site, including CP55940 (Song and Bonner, 1996; Chin et al., 1998; Hurst et al., 2002). It also blocks part of the [3H]WIN55212-2 binding site, although this aminoalkylindole does not directly interact with K3.28(192) (McAllister et al., 2003). When docked in the C6.47(355) mutants, AM841 maintains the same hydrogen bonding interactions as in WT CB1. However, in the C6.47(355)L mutant, the presence of the larger hydrophobic side chain of leucine causes crowding in the alkyl tail region of AM841 and requires a conformational readjustment of the alkyl tail with concomitant reduction in affinity. Such a readjustment is not required with the serine and alanine isosteric mutations.
Docking studies of Δ9-THC in the R* model of CB1 WT indicated that this ligand could adopt an orientation within the binding pocket similar to that of AM841 (Fig. 10), allowing the phenolic hydroxyl and pyran oxygen of Δ9-THC to hydrogen bond with K3.28(192) and S7.39(383), respectively. In such an orientation, the n-pentyl tail of Δ9-THC occupies the same general region as the alkyl tail of AM841. However, because of its shorter length, it encounters a fewer number of hydrophobic interactions in the TMH3-6-7 region, as shown in Figs. 9 and 10. One can thus argue that this, combined with the absence of an 11-hydroxy hydrogen bonding interaction with S2.60(173), can account for the reduced CB1 affinity of Δ9-THC compared with AM841. Congruent with this argument is the enhanced affinity of Δ9-THC in the C6.47(355)L mutant, relative to WT, which can be attributed to a tighter optimized interaction between the shorter alkyl side chain of Δ9-THC with the longer hydrophobic side chain of leucine. Likewise, the two-fold improvement in the affinity of Δ9-THC observed with C6.47(355)A, can be attributed to the hydrophobic nature of the alanine side chain compared with the more polar WT cysteine and C6.47(355)S mutant, which interacts more favorably with the hydrophobic C-3 side chain of Δ9-THC. Altering the binding environment by removing the polar nature of the residue in position 6.47(355) seems to be sufficient to afford a slightly enhanced binding profile for Δ9-THC. The binding data for the two congeners of AM841, which incorporate only small modifications in the distal region of its seven-carbon side chain, are very congruent with the above conclusions. They also argue that both AM4043 and AM4056 engage CB1 in binding motifs similar to those engaged by AM841.
Discussion
To obtain information on the molecular features of the ligand:CB1 receptor complex, we have designed and synthesized high-affinity molecular probes capable of forming a covalent bond at or immediately adjacent to the receptor binding site(s). One of our most successful designs encompasses a classical cannabinoid scaffold functionalized in the terminal position of the C-3 alkyl side chain (Charalambous et al., 1992; Guo et al., 1994; Morse et al., 1995; Picone et al., 2002). Together, these covalent probes have been very effective in irreversibly and selectively binding to CB1 in a highly specific manner. AM841, the cannabinergic covalent probe in this study has been optimized for high affinity for the CB1 receptor. It possesses the tricyclic hexahydrocannabinol ring structure characteristic of the classical cannabinoids, a 1′,1′-dimethylheptyl side chain at the C-3 position of the phenolic A ring and a β-hydroxymethyl group at the 9-position of the C-ring that imparts improved affinity over nonhydroxylated analogs. The electrophilic isothiocyanate group, chosen to permit a selective nucleophilic addition reaction between the ligand and receptor, was introduced at the terminal 7′-position of the side chain. This moiety will selectively react with more reactive nucleophiles, such as the thiol group of cysteine, but not with the hydroxyl of serine side chains or water. Preliminary studies with AM841 (R. P. Picone, D. J. Fournier, and A. Makriyannis, unpublished observations), as well as our earlier generations of C-3 side-chain functionalized isothiocyanates, demonstrated significant efficacy at irreversibly occupying CB1 binding sites in rat brain synaptosomes (Guo et al., 1994; Morse et al., 1995; Picone et al., 2002). This binding was not recoverable with sedimentation washing, which is widely accepted as evidence of irreversible occupation with nonradiolabeled spontaneously reacting isothiocyanate ligands (Guo et al., 1994; Morse et al., 1995; Zhu et al., 1996; Picone et al., 2002; Tahtaoui et al., 2003).
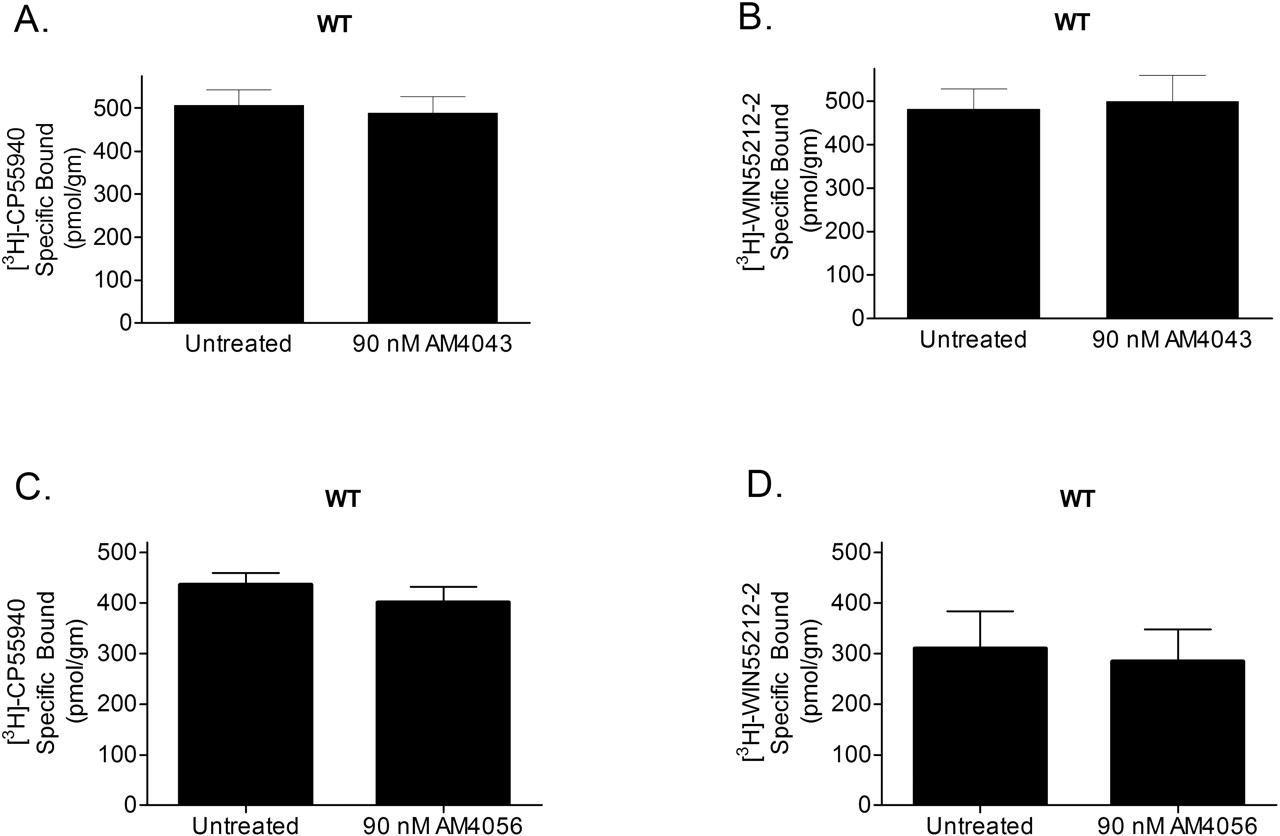
AM4043 and AM4056 treatment has no effect on the binding of [3H]CP55940 and [3H]WIN55212-2 to human CB1 WT. Exposure of AM4043 or AM4056 to membranes prepared from WT human CB1 expressing cells for 60 min was followed by repetitive sedimentation-washing and subjected to radioligand binding using either 6 nM [3H]CP55940 or 18 nM [3H]WIN55212-2 as described under Materials and Methods. Data shown are the mean ± S.E.M. from two independent experiments conducted with six replicates each.
The primary candidate amino acid residues capable of undergoing a nucleophilic addition reaction with AM841's isothiocyanate group are cysteines through their highly nucleophilic thiol or the less nucleophilic amino-containing lysines. Recent studies have proposed that cysteine can be used as a selective chemical sensor for sulfhydryl-reactive affinity labels, such as the isothiocyanate moiety (Tahtaoui et al., 2003). We have considered the possibility that a lysine residue is involved as a nucleophile in this addition reaction. However, the nucleophilic character of this residue is expected to be significantly reduced at physiological conditions, where it is likely to exist in a protonated state. More importantly, in CB1, the only lysine present in the TMH bundle available to interact with cannabinoid ligands is K3.28(192), which has been shown in numerous studies to be involved in a hydrogen-bonding interaction with the phenolic hydroxyl of classical cannabinoids, such as AM841 (Huffman et al., 1996; Song and Bonner, 1996; Chin et al., 1998). Therefore, this important lysine is not expected to be available for interaction with the 7′-isothiocyanate of AM841. The effort to identify the site of covalent bond formation between AM841 and CB1 was accordingly focused on two TMH cysteine residues.
As an agonist, AM841 is expected to bind preferentially to the R* form of CB1, which was used as the model for our ligand/receptor studies. The predictive power of computational models has been very useful in discerning GPCR ligand binding sites and as guides for mutational analyses because the crystal structure of Rhodopsin was solved (Palczewski et al., 2000). AM841 was docked so that its phenolic hydroxyl can accept a hydrogen bond from K3.28(192), which was considered as the primary interaction site for this ligand. This residue has been shown by mutational analysis to be crucial for the binding of classical cannabinoids such as Δ9-THC and AM841 (Song and Bonner, 1996). To identify which reactive cysteine is available within the binding site crevice for reaction with AM841, its isothiocyanate-containing alkyl tail was covalently attached to C6.47(355) and C7.42(386), the most likely nucleophilic candidates, in separate simulations. Docking studies showed that using the phenolic hydroxyl/K3.28(192) hydrogen bond motif as the primary interaction for AM841, C7.42(386) was too high in the binding pocket to be accessible by the isothiocyanate moiety of AM841 without placing the C-3 alkyl side chain in a very tightly folded, high energy conformation. On the other hand, C6.47(355) was optimally positioned to allow for a favorable interaction with the isothiocyanate group, whereas the side chain is maintained in a low-energy conformation (see Fig. 9). Moreover, the existence of a hydrophobic groove composed, in part, of β-branched amino acids that can accommodate the alkyl side chain of cannabinoid agonists localized to TMH6, particularly V6.43(351) and I6.46(354), has been proposed and hypothesized to trigger receptor activation (Barnett-Norris et al., 2002). A covalent reaction between C6.47(355) and AM841 allows the alkyl side chain of the ligand to interact with these β-branched amino acids. This was not possible when covalent bond formation was modeled using C7.42(386) as the target nucleophile. Based on this rationale, a series of mutations was designed to test the hypothesis that C6.47(355) was the site of covalent bond formation between AM841 and CB1. The chosen mutations, serine, alanine, and leucine, were expected to allow AM841 to bind to CB1 only reversibly without the formation of a covalent bond yet varying the physicochemical properties of the side chain in position 6.47(355) to investigate the chemical requirements necessary for covalent bond formation between AM841 and CB1. Although serine retains the polar character of cysteine, it is a relatively weak nucleophile with low reactivity toward the isothiocyanate group, whereas alanine is not polar and has no nucleophilic properties. Leucine, with the longer and bulkier hydrophobic side chain, was intended to probe any steric requirements for ligand interaction in the region of the putative CB1 binding site.

AM841 behaves as an agonist at the human CB1 receptor. Incubation of human CB1 expressing HEK293 cells (described in detail under Materials and Methods) with either 1.3 nM AM841, 10.0 nM AM4043, 10.0 nM AM4056, 10.0 nM CP55940, or 100.0 nM Δ9-THC caused a reduction in FSK-stimulated cAMP production. Data shown are the mean ± S.E.M. from experiments conducted in triplicate and repeated three times independently. *, p < 0.001, Bonferroni's post-hoc analysis. Inset, AM841 produces a concentration-dependent reduction in FSK-stimulated cAMP accumulation. The curve shown is representative of two independent experiments, the data from which are presented as mean ± S.E.M. of five individual replicates.
Introduction of a point mutation within a receptor binding site has the potential of altering ligand binding properties. However, it could also confer undesirable and frequently unpredictable consequences on the global conformation of the receptor that may affect ligand binding in a manner unrelated to binding site structure. Maintenance of the WT affinity of one or more ligands is one indicator that the mutation is not likely to have had an effect on the global conformation of the receptor. In addition, certain mutations have been shown to negatively affect proper membrane localization or insertion of CB1 (Shire et al., 1996; Andersson et al., 2003; McAllister et al., 2003) In the current study, the binding profile obtained for CB1 mutants with [3H]WIN55212-2 was indistinguishable from that of WT. The same held true for [3H]CP55940 binding to C6.47(355) mutants, excluding C6.47(355)L. This strongly suggests that the global conformation of CB1 is not markedly altered by any of the introduced mutations. Moreover, the Bmax values obtained were approximately 1000 pmol/g in all cases in this study, indicating similar levels of membrane expression. These combined results strengthen our interpretation that the differences observed with the C6.47(355)L mutant represent a specific perturbation on ligand binding rather than a nonspecific disturbance of receptor conformation or localization. Our data showing that the C6.47(355)L mutant exhibits different relative binding profiles with respect to [3H]CP55940 and [3H]WIN55212-2 suggests that these two structurally dissimilar ligands interact with CB1 through different binding motifs. This observation corroborates recent studies using mutational and modeling approaches (Shire et al., 1996; Song and Bonner, 1996; Chin et al., 1998; Tao et al., 1999; McAllister et al., 2003).
To engage in a covalent interaction with CB1, AM841 must first recognize and bind to its binding site with the electrophilic isothiocyanate group properly aligned with the reactive cysteine residue. The ensuing nucleophilic addition reaction of the cysteine thiol group is then expected to lead to the formation of a thiocarbamate-derivative. AM841 binds with high affinity to the CB1 WT receptor and the three mutants. Furthermore, treatment of CB1 WT with AM841 followed by equilibration and subsequent washing completely prevented specific binding of either [3H]CP55940 and [3H]WIN55212-2, thus confirming the irreversible occupation of CB1 by this covalent probe. It is important to note that WT, C6.47(355)S, and C6.47(355)A possess nearly identical affinities for AM841, clearly indicating that this ligand occupies the binding sites of all three proteins equally effectively, in a concentration-dependent manner. The data also provide evidence that the ability of AM841 to bind irreversibly can be eliminated by mutating the nucleophilic cysteine from position 6.47(355) to either non-nucleophilic residues, such as alanine or leucine, or to a weak nucleophile such as serine. Additional support for our conclusion regarding the requirements for covalent bond formation is provided by the observation that both AM4043 and AM4056 are readily washed from the binding site of CB1 WT under conditions identical to those used in the AM841 covalent binding experiments. Both AM4043 and AM4056 have identical structural features with AM841 but lack its reactive electrophilic isothiocyanate moiety. The results thus confirm that the formation of a covalent bond between our high-affinity ligand and CB1 requires the presence of a specific nucleophile within the receptor binding site and an electrophilic group situated in a suitable position on the ligand. Our reciprocal experiments demonstrating that elimination of either of these two groups prevents covalent bond formation provide strong support for our hypothesis regarding the role of C6.47(355) in this reaction.

AM841 Docked in CB1 WT R*. Left, an extracellular view of the AM841/CB1 WT R* complex is shown here as predicted by molecular modeling. In addition to the covalent attachment of its alkyl tail to C6.47(355), AM841 forms a hydrogen bond between its phenolic hydroxyl and K3.28(192) [N-O distance = 2.90 Å; O-H-N angle = 163°]; between its pyran oxygen and S7.39(383) [N-O distance = 2.90 Å; O-H-N angle = 143°]; and, between its 11-hydroxyl group and S2.60(173) [N-O distance = 2.80 Å; O-H-N angle = 177°]. Right, side view of the AM841/CB1 WT R* complex as predicted by molecular modeling is shown. This view illustrates the depth of the alkyl tail of AM841 in the binding site identified by modeling studies.

Δ9-THC docked in CB1 WT R*. Left, an extracellular view of the Δ9-THC/CB1 WT R* complex is shown here as predicted by molecular modeling. Δ9-THC adopts an orientation in the CB1 WT R* binding pocket similar to that of AM841. Δ9-THC forms a hydrogen bond between its phenolic hydroxyl and K3.28(192) [N-O distance = 2.78 Å; O-H-N angle = 162°] and between its pyran oxygen and S7.39(383) [N-O distance = 2.89 Å; O-H-N angle = 150°]. Right, side view of the Δ9-THC/CB1 WT R* complex as predicted by molecular modeling is shown. A comparison of this view with that in Fig. 9, right, illustrates the difference in the length of the alkyl tail between Δ9-THC and AM841.
The data reveal a reduction in binding affinity of [3H]CP55940, AM841, AM4043, and AM4056 with the C6.47(355)L mutant, whereas a significant enhancement in affinity was noted for Δ9-THC in this mutant. Moreover, Δ9-THC exhibited an improved affinity for the C6.47(355)A mutant compared with either WT or C6.47(355)S. The observed differences in the manner in which these cannabinergic ligands interact with CB1 WT and the C6.47(355) mutants can be attributed to structural differences in their respective C-3 alkyl chains. Whereas in AM841, AM4043, AM4056, and [3H]CP55940 the C-3 side chains are seven carbons long, with two additional methyl groups in the 1′-position, Δ9-THC has a significantly shorter n-pentyl C-3 substitution. It thus seems that the cannabinoid analogs with longer C-3 alkyl chains optimally interact with WT CB1 and its isosteric mutants C6.47(355)A and C6.47(355)S but are not as well accommodated by the bulkier C6.47(355)L mutant. Conversely, Δ9-THC interacts optimally with the C6.47(355)L mutant but not as well with the mutants in which shorter and more polar side chains are present in position 6.47. Reduction in the length and size of the C-3 alkyl side chain leads to progressively enhanced binding affinity for the C6.47(355)L mutant in the following rank order: AM841 < AM4043 ≈ AM4056 < [3H]CP55940. The apparent relatively higher affinity of [3H]CP55940 can be attributed to a binding contribution by the additional hydroxypropyl pharmacophore within this molecule. On the other hand, the observed 3-fold improvement of binding with Δ9-THC in the C6.47(355)L mutant relative to WT, can be explained by the presence of the longer hydrophobic leucine side chain engaging in a higher affinity interaction with the n-pentyl chain of Δ9-THC. The overall SARs observed for the different ligands and receptor mutants are very congruent with the CB1 receptor model proposed here. It is important to note that whereas we demonstrate that the C6.47(355)L mutation affected CP55940 binding, Fay et al. (2005) very recently showed that a C7.42(386)M mutation had no effect on binding of this analog. These results lend further support to the identification of C6.47(355) and not C7.42(386) as a residue located at the binding site of the classical and nonclassical cannabinoids. Our results also argue that the interaction of the cannabinoid C-3 alkyl side chain with the CB1 receptor is of a preferably hydrophobic nature and is governed by strict size considerations with respect to the ligand-receptor interacting groups. In their totality, the binding data with the cannabinergic ligands used in this study provide validation for the generally accepted cannabinoid SAR according to which major changes in ligand affinity, efficacy, and potency are modulated by manipulations of the key C-3 side chain pharmacophore (Papahatjis et al., 1998; Martin et al., 1999; Luk et al., 2004).
It is noteworthy that C6.47 is one of the amino acid residues of the widely conserved CWXP motif of family 1A GPCRs which has been shown to be critical in ligand binding and receptor dynamics, particularly in mechanisms involving receptor activation (Gether et al., 1997; Jensen et al., 2001; Shi et al., 2002; McAllister et al., 2004). The covalently attached ligand in this study thus identifies a specific interaction site for binding that is part of a motif known to be important in CB1 activation mechanisms (McAllister et al., 2004). In the case of CB1, it has been postulated that receptor activation is accompanied by a counter-clockwise rotation of TMH6, placing C6.47(355) within the binding crevice (Bramblett et al., 1995; Barnett-Norris et al., 2002; McAllister et al., 2004). In our computational work, AM841 is docked in the R* form of CB1 with the alkyl side chain isothiocyanate moiety appropriately oriented to allow formation of the cysteinyl thiocarbamate receptor-AM841 adduct with C6.47(355) while maintaining intact a hydrogen bond network between AM841 with S2.60(173), K3.28(192), and S7.39(383) (Huffman et al., 1996; Song and Bonner, 1996; Chin et al., 1998; Tao et al., 1999). This is in agreement with work reported by Javitch et al. (1997) whereby activation of the β2-adrenergic receptor results in an increase in susceptibility of C6.47 labeling with cysteine-modifying reagents, leading the authors to conclude that C6.47 is present in the binding crevice in the R* form. When combined with our experimentally obtained data, the computer modeling experiments confirm that AM841, while incorporating all the key pharmacophoric features of a CB1 agonist, binds with high affinity to the R* state where it then anchors itself through the covalent interaction of its electrophilic isothiocyanate group with an optimally positioned cysteine residue within the binding crevice. Thus, AM841 successfully fulfills its dual role of first recognizing and interacting with the CB1 receptor binding site through a structural motif, the key features of which are shared by all ligands within the class of cannabinoids, and subsequently attaching itself irreversibly to a single specific amino acid within that site, C6.47(355). It also provides a direct means for probing the identity of residues present in either the R or R* states of the CB1 binding domain as well as their role(s) in receptor function.
To summarize, the work reported here reveals a number of interesting findings related to the structure and function of the CB1 receptor:
-
AM841 attaches itself covalently to CB1 through C6.47(355) and activates CB1. The result reinforces the importance of the CWXP motif in receptor activation, an observation that may prove to be of more general relevance with other GPCRs.
-
Ligands belonging to the class of classical and nonclassical cannabinoids interact with CB1 through a binding motif distinct from that of the aminoalkylindoles. The results reinforce accumulating evidence, indicating that GPCRs can be activated by agonist ligands through multiple binding motifs.
-
This report represents the first direct evidence identifying a binding contact for the C-3 alkyl side chain of cannabinoids, C6.47(355). It also provides a structural basis for interpreting existing SAR data, according to which this important pharmacophore plays a key role in modulating ligand affinity, efficacy, and potency at CB1 and explains why seven and eight carbon side chains are optimal. This information should assist in the design of improved later generation CB1 ligands.
Acknowledgments
We are very grateful to Dr. Tom Bonner for providing the pRC/CMV-hCB1 plasmid used in these studies as well as to Dr. Ebru Caba for advice on the RT-PCR.
Footnotes
-
This work has been supported by National Institute on Drug Abuse grants DA05955 (to R.P.P.) DA00355 (to A.D.K.), DA09158, DA03801, DA07215, DA07312 (to A.M.), DA03934, DA00489 (to P.H.R.), DA05274, and DA09978 (to M.E.A.).
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.105.014407.
-
ABBREVIATIONS: GPCR, G-protein-coupled receptor; Δ9-THC, (-)-trans-Δ9-tetrahydrocannabinol; CP55940, (1R,3R,4R)-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-4-(3-hydroxypropyl)cyclohexan-1-ol; WIN55212-2, [2,3-dihydro-5-methyl-3-[(4-morpholinyl)methyl]pyrrolo-[1,2,3-de]-1,4-benzoxazin-6-yl](naphthyl)methanone; SR141716A, N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide; RO-20-1724, 4-[(3-butoxy-4-methoxyphenyl)-methyl]-2-imidazolidinone; SAR, structure-activity relationship; AM841, (-)-7′-isothiocyanato-11-hydroxy-1′,1′-dimethylheptylhexahydrocannabinol; AM4056, 11-hydroxy-1′,1′-dimethylheptylhexahydrocannabinol; AM4043, (-)-7′-bromo-11-hydroxy-1′,1′-dimethylheptylhexahydrocannabinol; CHO, Chinese hamster ovary; TMH, transmembrane helix; WT, wild type; CM, conformational memories; RT, reverse transcription; PCR, polymerase chain reaction; TME, Tris/MgCl2/EDTA; BSA, bovine serum albumin; HEK, human embryonic kidney; FSK, forskolin; R, inactive state; R*, activated state.
-
↵1 Current affiliation: Center for Drug Discovery, Northeastern University, Boston, Massachusetts.
- Received April 28, 2005.
- Accepted September 12, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}