


Abstract
Many membrane proteins incur a folding problem during biosynthesis; only a fraction thereof is exported from the endoplasmic reticulum (ER), because quality control is stringent. This is also true for G protein-coupled receptors. Here, we identify the deubiquitinating enzyme Usp4 as an interaction partner of the A2a adenosine receptor, a Gs-coupled receptor. Usp4 binds to the carboxyl terminus of the A2A receptor and allows for its accumulation as deubiquinated protein. This relaxes ER quality control and enhances cell surface expression of functionally active receptor. The effect of Usp4 on the A2A receptor was specific because 1) it was not seen in C-terminally truncated versions of the receptor; 2) it was not mimicked by Usp14, another member of the ubiquitin-specific protease family; and 3) it was not seen with the metabotropic glutamate receptor-5, another G protein-coupled receptor with a high propensity for intracellular retention. These observations show that deubiquinating enzymes can regulate quality control in the ER.
Membrane proteins have to be inserted cotranslationally into the endoplasmic reticulum. This occurs via the translocon, a channel formed by the Sec61 subunits. During and after synthesis of membrane proteins in the endoplasmic reticulum, they undergo a strict quality control to ensure correct folding before they are transported to their definitive site of action. Several aspects of this quality control are incompletely understood; nevertheless, it is clear that incorrect folding of a membrane protein is sensed by the machinery of the endoplasmic reticulum (that is by chaperons, presumably). This leads to the activation of ubiquitinating enzymes on the cytoplasmic side. These transfer ubiquitin to the cytoplasmic peptide chain of the incorrectly folded protein, which is retrotranslocated and degraded by the 26S proteasome (Kostova and Wolf, 2003). This scheme relies predominantly on observations that were made in Saccharomyces cerevisiae. Based on several pieces of experimental evidence, it is reasonable, however, to assume that higher eukaryotes use a related machinery to eliminate misfolded proteins (Lilley and Ploegh, 2004; Ye et al., 2004).
It has been increasingly appreciated that many human diseases can be linked to mutations, which result in the retention of the aberrant protein in the endoplasmic reticulum (ER). Cystic fibrosis is most commonly cited as the model disease: more than 1000 mutations have been identified in the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) (Rowntree and Harris, 2003), but the majority of the patients (∼70%) have the ΔF508-mutation of the CFTR. The resulting protein fails to reach the plasma membrane because of a stringent ER quality-control mechanism, but it can function properly if it reaches the plasma membrane (Pasyk and Foskett, 1995). There are many more examples that lead to defective ER export of membrane proteins; these include mutations of the V2 vasopressin receptor (associated with diabetes insipidus; Oksche and Rosenthal, 1998), of the low-density lipoprotein receptor (resulting in hypercholesterolemia; Hobbs et al., 1990), of the human ether-a-go-go-related gene K+ channel (resulting in long QT-syndrome-2; Kupershmidt et al., 2002), and many others. It is unclear why these mutated proteins are retained and eventually degraded, although they are—at least in part—functionally active (see Pasyk and Foskett, 1995). However, the available evidence suggests that the quality-control machinery in the endoplasmic reticulum is stringent, because it prefers to err on the side of rapidly degrading a protein that, when given time, may fold into a functionally active transporter or channel.
G protein-coupled receptors have been documented to incur a folding problem: a large portion of newly synthesized protein (≥50%) is subject to degradation in the endoplasmic reticulum and does not reach the plasma membrane (Petaja-Repo et al., 2000, 2001; Pankevych et al., 2003). This is similar to the situation with many other membrane proteins with multiple transmembrane spans, specifically with CFTR (Rowntree and Harris, 2003). During folding, the intracellular carboxyl terminus of membrane proteins plays a prominent role, because it allows for docking of the ER-associated degradation machinery that eliminates incorrectly folded proteins. In addition, in several instances, segments within the carboxyl terminus of G protein-coupled receptors have been shown to be essential for proper folding of the protein: mutations within the carboxyl terminus of structurally unrelated G protein-coupled receptors result in receptors that fail to adopt a conformation capable of ligand binding (Krause et al., 2000; Pankevych et al., 2003; Duvernay et al., 2004). Here, we identify the deubiquinating enzyme Usp4 as an interaction partner of the A2A-adenosine receptor, a prototypical Gs-coupled receptor: Usp4 binds to the carboxyl terminus of the A2A-adenosine receptor; deubiquitination of the receptor relaxes quality control in the ER and enhances cell surface expression. Hence, ER-associated ubiquitination is reversible, an observation that implies that quality control in the ER may be subject to dynamic regulation.
Materials and Methods
Materials and Reagents. Guanine nucleotides including guanosine-5′-(3-O-thio)triphosphate (GTPγS), adenosine deaminase, insulin-transferrin-sodium selenite supplement, and Complete protease inhibitor tablets were from Roche Diagnostics and Roche Molecular Biochemicals (Mannheim, Germany). CGS21680 and labeled [3H]ZM241385 (specific activity, 27.4 Ci/mmol) were from Tocris Cookson Ltd. (Bristol, UK). Papain was from Worthington Biochemicals (Freehold, NJ), HEPES was from Biomol (Munich, Germany), and xanthine amino congener was from Research Biochemicals (Natick, MA). The materials required for SDS-polyacrylamide gel electrophoresis were from Bio-Rad (Richmond, CA). Fetal calf serum was from PAA Laboratories (Linz, Austria); Dulbecco's modified Eagle medium, nonessential amino acids, β-mercaptoethanol, gentamicin, G418 (Geneticin), LipofectAMINE reagent, and LipofectAMINE Plus reagent were from GIBCO-BRL (Grand Island, NY). Forskolin, chloroquine, kynurenate, progesterone, putrescin, lysozyme, RO201724, cAMP, l-glutamine, streptomycin, Triton X-100, phenylmethylsulfonyl fluoride, anti-FLAG M2 affinity gel, and anti-FLAG monoclonal antibody peroxidase-conjugated, monoclonal anti-HA peroxidase-conjugated antibody were from Sigma Chemical Co. (St. Louis, MO). MG132 was from Calbiochem (Darmstadt, Germany), [3H]adenine was from DuPont NEN (Boston, MA), and cytosine-β-d-arabinofuranoside was from ICN Biomedicals (Irvine, CA). ER-Tracker Blue-White DPX, a fluorescent dye that specifically stains the endoplasmic reticulum, was from Molecular Probes (Leiden, The Netherlands); pMAL vector and amylose resin for expression and purification of maltose binding protein (MBP) were from New England Biolabs Inc. (Beverly, MA). The Micro BCA protein assay reagent kit was from Pierce (Rockford, IL). Buffers and salts were from Merck (Darmstadt, Germany). Centrifuge tubes and tissue culture plates were from Greiner (Vienna, Austria) and Corning Costar (Acton, MA). Plasmid preparation kits were from QIAGEN (Hilden, Germany). Rabbit anti-GFP living colors A.V. peptide antibody and the MATCHMAKER yeast two-hybrid system were from Clontech (Mountain View, CA). Horseradish peroxidase-conjugated anti-mouse and anti-rabbit immunoglobulin antibodies were from Amersham Life Science (Little Chalfont, Buckinghamshire, UK). The immunoreactive bands on nitrocellulose blots were detected by chemoluminescence using SuperSignal chemoluminescence substrate from Pierce.
DNA Constructs. To perform epitope tagging of the A2a adenosine receptor, the sequence of the receptor, encompassing the coding region, was cleaved with HindIII/SalI from pEYFP-N1/human A2aR cDNA (5) and ligated into HindIII/SalI-digested pCMV-tag 2B vector (Stratagene, La Jolla, CA), linking the A2aR in frame to the N terminus of the FLAG epitope. The DNA fragment encoding the C-terminal part of A2aR was amplified by polymerase chain reaction (PCR) from pcDNA3-A2aR vector (5), which bears the full length of A2aR, using the primers 5′-CCAGAATTCCGTATCCGCGAGTTC-3′ and 5′-TTTAACTCGAGT TCAGGACACTCCTGC-3′. After restriction digestion of the PCR fragment with EcoRI and XhoI, A2aR was inserted into pEG202. An identical strategy was used for the generation of mutants in which the carboxyl terminus was truncated by using the following 3′-primers: 5′-CGC GCCTCGAGTTCATTGGTGCCTCAGGACGTGG-3′ [for A2aRct(1–311)] and 5′-TTTTTCTCG AGTTCAGCCATTGGGCCTCCGGTC-3′ [for A2aRct(1–360)]. The integrity of the engineered sequences was confirmed by fluorescent sequencing. The following plasmids were generous gifts: the plasmid for mammalian expression of HA-tagged ubiquitin was from J. Bies (Cancer Research Institute, Bratislava, Slovak Academy of Sciences, Bratislava, Slovakia), GFP- and FLAG-tagged mouse homolog Usp4 was from D. Gray (Ottawa Regional Cancer Center, Ottawa, ON, Canada), and Usp14 was from J. Blahos (UEM AVCR, Praha, Czech Republic).
Yeast Two-Hybrid Interaction Screen. Yeast two-hybrid analysis was performed using the MATCHMAKER yeast two-hybrid system. In this system, yeast strain EGY48 (MATα trp1 his3 ura3 leu2 6 LexAop-LEU2) was used for transformation of bait, prey, and reporter plasmids pEG202, pJG4-5, and pSH18-34, respectively. Yeast strains were grown at 30°C in either glucose minimal medium or induction medium; transformation was performed by using the lithium acetate method in accordance with the manufacturer's instruction. EGY48 containing the lacZ reporter plasmid (pSH18-34) and the A2aRct bait plasmid (pEG202/A2aRct) was transformed with prey plasmids bearing a human brain cDNA library. To verify the two-hybrid interactions, positive transformants grown on selection medium were assayed directly in situ for β-galactosidase activity assay by brief exposure of the plates to chloroform followed by incubation with 100 μM X-gal in the medium as a substrate.
Cell Culture, Cellular Transfection, and Membrane Preparations. HEK293 cells were maintained in Dulbecco's modified Eagle medium at 5% CO2/95% air and 37°C. Culture media were supplemented with 10% fetal calf serum, 2 mM l-glutamine, β-mercaptoethanol, nonessential amino acids, 50 mg/l gentamicin, and, to maintain the selection pressure, with 0.2 mg/ml Geneticin (G418) for the culture of stably transfected cells. PC-12 cells were propagated as described previously (Kudlacek et al., 2001). One day before transfection, cells were replated to obtain subconfluent cultures either on glass coverslips (22 mm in diameter and placed into six-well plates) or 10 cm diameter cell culture dishes. Transient transfections were done using the CaPO4 precipitation method or with LipofectAMINE Plus (Invitrogen). When required in cotransfections with several plasmids, the appropriate empty vectors were added to keep the amount of DNA per dish constant. The medium was changed to remove excess DNA precipitates in 5 h after transfection. Serum starvation and incubation with adenosine deaminase was initiated on day 1 after transfection. The subsequent starvation under serum-free conditions lasted for 12 to 24 h; thereafter, cAMP accumulation was stimulated as outlined below. Where indicated, cells were kept in the medium containing proteosomal inhibitor (50 μM MG132) or lysosomal inhibitor (100 μM chloroquine) at 37°C for 3 h before harvesting. Cells were harvested, and membranes were prepared as described previously (Klinger et al., 2002a). Primary neuronal cultures were obtained from hippocampi of neonatal rats as described earlier (Boehm and Betz, 1997); 4 days after plating, the cultures were treated with 1 μM cytosine-β-d-arabinofuranoside to deplete glial cells. Neurons were seeded on polylysine-coated coverslips and transfected with LipofectAMINE 2000 as outlined previously (Farhan et al., 2004).
Radioligand Binding Assays. PC-12 cells and HEK293 cells transiently transfected with the appropriate plasmids were lysed by three freeze-thaw cycles followed by brief sonication; membranes were collected by centrifugation (30 min at 50,000g), as outlined previously (Klinger et al., 2002a). Hence, the particulate fraction contained intracellular membranes and the plasma membrane. Membranes were prepared from 100 μg/assay and were incubated in a final volume of 0.3 ml containing 50 mM Tris-HCl, pH 8.0, 1 mM EDTA, 5 mM MgCl2, 8 μg/ml adenosine deaminase, and concentrations of [3H]ZM241385 covering the range of 0.2 to 20 nM in the presence of 100 μM GTPγS. After 60 min at room temperature, the reaction was terminated by rapid filtration over glass fiber filters. Nonspecific binding was determined in the presence of 10 μM xanthine amino congener and amounted to 40% at the highest concentration of [3H]ZM241385. The data points were fitted by nonlinear regression to the equation describing a rectangular hyperbola. Assays were performed in duplicate.
Agonist-Mediated Cellular cAMP Accumulation. Cells were grown in six-well plates. The adenine nucleotide pool was metabolically labeled by incubating confluent monolayers for 16 h with [3H]adenine (1 μCi/well) as described previously (Kudlacek et al., 2001). After the preincubation, fresh medium was added that contained 100 μM RO201724 (a nonxanthine phosphodiesterase inhibitor) and adenosine deaminase (2 U/ml) to remove any endogenously produced adenosine. After 1 h, cAMP formation was stimulated by the A2A-selective agonist CGS21680 (1 nM to 1 μM) for 15 min, and the reaction was stopped by adding 2.5% perchloric acid with 100 μM cAMP (1 ml/dish). The supernatant (0.9 ml) was aspirated, neutralized with 100 μl of 0.4 M KOH, and diluted with 1.5 ml of 50 mM Tris-HCl, pH 8.0; [3H]cAMP was isolated by sequential chromatography on Dowex AG 50W-X4 and neutral alumina columns. Assays were performed in triplicate.
Immunoprecipitation of the Epitope-Tagged A2A-Adenosine Receptor. HEK293 cells stably expressing FLAG-tagged A2A-adenosine receptor were washed three times with phosphate-buffered saline; subsequently, the membranes were solubilized in ice-cold lysis buffer [50 mM Tris-HCl, pH 7.5, 1 mM EDTA, 150 mM NaCl containing 1% Nonidet P-40 (v/v), Complete protease inhibitors (Roche Molecular Biochemicals), and, where indicated, 10 mM N-ethylmaleimide] for 1 h on ice. The insoluble material was collected by centrifugation at 16,000g for 10 min at 4°C. The supernatant was processed for immunoprecipitation, each step of which was conducted with constant rotation at 4°C. Then, 40 μl of a 50% (v/v) suspension of anti-FLAG M2 Affinity Gel was added, and the sample was incubated overnight. The beads were collected by centrifugation and washed three times in 1 ml of Tris-buffered saline. Immune complexes were dissociated in SDS-polyacrylamide sample buffer containing 20 mM dithiothreitol by incubation for 1 h at 37°C or for 5 min at 95°C. Proteins were transferred to nitrocellulose membranes by using a semidry transfer system; immunodetection was achieved by using monoclonal peroxidase-conjugated anti-FLAG and anti-HA antibodies to detect the FLAG epitope of the A2aR and the HA-epitope of ubiquitin, respectively. The GFP moiety in Usp4 was detected with an anti-GFP antiserum and a horseradish peroxidase-conjugated anti-rabbit IgG secondary antibody. The immunoreactive bands were visualized with enhanced chemiluminescence.
Purification of Fusion Proteins and In Vitro Binding Assays. The C-terminal part of the A2A receptor was amplified by PCR and subcloned into the EcoRI and XhoI sites in the multicloning site of the pMAL-cri vector. Expression of the resulting construct in Escherichia coli (BL21) produced an MBP-A2aR-C terminus fusion protein, which was purified by affinity chromatography on an amylose resin. MBP-A2aR-C terminus bound to the amylose resin was eluted with buffer A (20 mM Tris, pH 7.4, 200 mM NaCl, 1 mM EDTA, and 10 mM 2-mercaptoethanol) containing 10 mM maltose. Maltose was removed by repeated cycles of concentration and dilution in a Centricon microconcentrator with a nominal cutoff of 30 kDa; the protein was concentrated to 1 to 2 mg/ml in buffer A, frozen in liquid nitrogen, and stored at -80°C. Unfused MBP and a fusion protein comprising MBP and the C terminus of the A1-adenosine receptor were expressed and purified in an analogous way.
HEK293 cells were transiently transfected with a plasmid driving the mammalian expression of Usp4-FLAG, harvested 48 h after transfection, and lysed in Tris-buffered saline (20 mM Tris-HCl, pH 7.4, and 150 mM NaCl), containing 1 mM EDTA, 1% Triton X-100, and Complete protease inhibitors (Roche Molecular Biochemicals). The lysate was subsequently centrifuged at 16,000g for 10 min at 4°C. The supernatant was applied to an anti-FLAG epitope immunoaffinity column with 1 ml of bed volume (anti-FLAG M2 affinity Gel; Sigma). After washing the column with Tris-buffered saline (10 bed volumes), Usp4 was eluted with 6 × 1 ml of 0.1 M glycine/HCl, pH 3.5.
For pull-down assays, purified fusion proteins (3 μg of MBP-A2aR-C terminus, MBP-A1-R-C terminus, or MBP) were incubated with 30 μl of amylose resin (50% slurry); after removal of the unbound proteins, purified Usp4-FLAG (0.3 μg) was then added to the immobilized fusion proteins. Thereafter, the resin was washed three times with 1 ml of MBP buffer (20 mM Tris, pH 7.4, 200 mM NaCl, 1 mM EDTA, and 10 mM 2-mercaptoethanol); 10 mM maltose or the sample buffer for SDS-polyacrylamide gel electrophoresis was used to release the proteins bound to the resin.
Endoglycosidase H Sensitivity. HEK293 cells were collected in buffer containing 20 mM HEPES, pH 7.2, and 0.37 M sorbitol. The cells were lysed by sonication on ice; after centrifugation (10 min at 40,000g), the membrane pellet was resuspended in buffer (50 mM Tris-HCl, pH 7.4, 1 mM EDTA, and 5 mM MgCl2) and frozen in liquid nitrogen. Membrane (20 μg/assay) assays were pelleted and resuspended in 20 μl of buffer (50 mM sodium citrate, pH 5.6, 0.6% SDS, and 0.15 M 2-mercaptoethanol). Samples were incubated for 5 min at 95°C. After cooling at 37°C, phenylmethylsulfonyl fluoride (0.5 mM final concentration) and endo-β-N-acetylglucosaminidase-H (endoglycosidase H, endoH; 10 mU) was added to the reaction; the mixture was incubated at 37°C for 18 h before gel electrophoresis and immunoblotting. Western blot films were scanned by a Bio-Rad scanning densitometer and processed with QuantiScan software (Biosoft, Cambridge, UK).
Fluorescence Microscopy. Transiently transfected HEK293 cells and hippocampal neurons were investigated 1 day after transfection on an inverted epifluorescence microscope (Zeiss Axiovert 200M) using a 63-fold oil immersion objective and filter sets, which discriminate between CFP and YFP fluorescence (Chroma Technology Corporation, Brattleboro, VT). Images were captured with a cooled charge-coupled device camera (CoolSNAP fx; Photometrics, Roper Scientific, Tucson, AZ) and stored in and processed with Meta-Series software (release 4.6, Metafluor and Metamorph; Universal Imaging, Downingtown, PA).
Preparation of RNA, PCR, and RNA Interference. Newborn rat pups were killed, and their striata and hippocampi were dissected and frozen in liquid nitrogen. Cells (HEK293 and PC-12 cells) or tissues were lysed in TriReagent (Sigma), and total RNA was prepared according to the manufacturer's instruction. For HEK293 cells, reverse transcription for HEK cells was performed with 0.5 μg of total RNA; the primer pair for PCR was ATGGCGGAAGGTGGAGGCTG (forward primer) and ACGCTCCGCAGGGATGTTGA (reverse primer). The reverse transcription for hippocampi, striata, and PC-12 cells was performed with 1 μg of total RNA; the subsequent polymerase chain reaction done with the forward primer was AGCAGAAATTTCACTACCTCTTCAAAAC, and the reverse primer was CAGGAAGACCTCCATAATCCG. For RNA interference, 3 × 105 HEK293 cells stably expressing YFP-tagged A2A receptors at low levels were seeded in a six-well plate and transfected with 500 pmol stealth siRNA (Invitrogen) with LipofectAMINE 2000 according to the manufacturer's instructions. Several siRNA sequences were used: CCGAGGCGUAUAAACUACUAAA (referred to as siRNA1) and CCAUUUCAGCAAGGCAGACACCAUU (siRNA2) were investigated in several experiments as a pair of inactive (siRNA1) and active (siRNA2) molecules. Forty-eight hours after transfection, RNA was isolated to quantify the knockdown of mRNA coding for Usp4 by reverse transcription-polymerase chain reaction (RT-PCR). At the same time, the distribution of YFP-tagged A2A receptor was visualized by fluorescence microscopy.
Results
The A2A Receptor Is Subject to Proteasomal Rather than Lysosomal Degradation. The rat pheochromocytoma cell line PC-12 expresses the A2A receptor endogenously. At steady state, substantial amounts of receptor can be visualized within the cell (Arslan and Fredholm, 2000; see also below). These may represent recycling receptors, receptors en route to lysosomal degradation, or receptors subjected to ER-associated degradation. To discriminate between these possibilities, we treated PC-12 cells with the proteasome inhibitor MG132 and with the lysosomal inhibitor chloroquine and determined the number of binding sites with the antagonist radioligand [3H]ZM241385 (Fig. 1A). The addition of the proteasome inhibitor MG132 resulted in an increase in the membrane levels of the A2A receptor (▴, Fig. 1A); on average, binding increased by approximately 50% (Bmax = 2.04 ± 0.29 and 2.9 ± 0.37 pmol/mg in the absence and presence of MG132, respectively). In contrast, the presence of choloroquine did not affect the A2A receptor level (▾, Fig. 1B). The model of quality control in the endoplasmic reticulum predicts a prominent role of the carboxyl terminus of membrane proteins because it serves as the docking site for the machinery that targets the protein for proteasomal degradation (Kostova and Wolf, 2003). Thus, a truncation of the carboxyl terminus of the A2A receptor might render the receptor insensitive to the action of MG132. We tested this prediction by transiently expressing the wild-type (human) A2A receptor and a truncated version thereof, the A2A receptor (1–311), in HEK293 cells. In the A2A receptor (1–311), the last 101 amino acids have been eliminated, but the 20 amino acids adjacent to the seventh transmembrane segment have been retained; the signaling properties of this receptor only differ in a subtle manner from the wild-type receptor (Klinger et al., 2002a). As shown in Fig. 1B, the expression level of the A2A receptor (1–311) was not increased by the addition of MG132. Thus, the carboxyl terminus (i.e., the last 100 amino acids) of the A2A receptor renders the expression levels sensitive to a proteasome inhibitor.
Yeast-Two Hybrid Screen for a Binding Partner. Taken together, the observations summarized in Fig. 1 suggest that proteasomal degradation of the receptor is preceded by one (or several) reversible step(s) in which the receptor committed to degradation can nevertheless be rerouted to increase the number of available receptors. To understand the underlying mechanism, we searched for a candidate protein that bound to the carboxyl terminus of the A2A adenosine receptor by using the last 120 amino acids as bait in yeast two-hybrid interaction hunt. Because the A2A receptor is abundantly expressed in several brain regions, most notably the basal ganglia (Fredholm et al., 2005), we screened a human brain library and identified more than five different interaction partners that reproducibly bound to the fusion protein comprising lexA and the carboxyl terminus of the A2A receptor. We focused on Usp4, because Usp4 is a deubiquinating enzyme, albeit of ill-defined function. It was the only interactor that was related to proteosomal degradation; we surmised that its deubiquinating activity may be relevant for the effects observed with the proteasome inhibitor MG132 summarized in Fig. 1.
Usp4 is the human homolog of murine UNP (>90% amino acid identity), despite the eponymous description (ubiquitous nuclear protein; Gupta et al., 1994), in all cell lines that we tested, Unp/Usp4 was found in the cytosol rather than in the nucleus (see also below). The ratio of nuclear-to-cytosolic distribution is dependent on the cell type (Soboleva et al., 2005). The data shown in Fig. 1B predict that truncation of the bait construct should eliminate the interaction with Usp4 if there were any link between the binding of Usp4 to the A2A receptor and the regulation of receptor levels by inhibition of proteasomal degradation. This was the case: the interaction between the Usp4-containing prey construct and the C terminus of the A2A receptor induced β-galactosidase activity to levels which approached that of the constitutive and inducible controls (compare the first and second streaks with the third and fourth streaks in Fig. 2A). In contrast, truncation of the bait by 101 amino acids eliminated induction of β-galactosidase (seventh and eighth streaks in Fig. 2A); in fact, the truncation of bait by the last 52 amino acids sufficed to abolish the interaction between prey and bait (fifth and sixth streaks in Fig. 2A). It is worth noting that other interactors were still capable of binding to the truncated bait constructs and thus to induce β-galactosidase activity to levels comparable with those seen with the wild-type carboxyl terminus (Gsandtner et al., 2005).

Proteasome inhibition increases expression of the A2A-receptor. A, saturation hyperbolae for specific binding of [3H]ZM241385 to membranes from PC-12 cells (50 μg/assay) endogenously expressing the A2A receptor. Membranes prepared from PC-12 cells, which were kept in the presence or absence of 50 μM MG132 or 100 μM chloroquine for 3 h, were incubated in buffer containing the indicated concentrations of [3H]ZM241385 in the presence of 100 μM GTPγS. B, saturation hyperbolae for specific binding of [3H]ZM241385 to membranes from transiently transfected HEK293 cells. Membranes (50 μg/assay) were prepared from HEK293 cells that expressed the full-length A2A receptor and the truncated version A2A receptor (1–311) [A2AR(1–311)] and that were maintained in the presence or in the absence of MG132 for 3 h. These membranes (50 μg/assay) were incubated in buffer containing the indicated concentrations of [3H]ZM241385 in the presence of 100 μM GTPγS. Data are means from duplicate determinations in a representative experiment which was repeated three times.
Interaction of the A2A-Receptor with Usp4. To obtain independent evidence for an interaction between the A2A receptor and Usp4, we coexpressed the FLAG epitope-tagged A2A receptor and GFP-tagged Usp4 in HEK293 cells and subjected cellular lysates to immunoprecipitation with an anti-FLAG antibody immobilized on Sepharose beads. The eluate contained approximately 5 to 10% of the total Usp4—as detected by an antiserum directed against GFP— provided that Usp4 and the A2A receptor were coexpressed (Fig. 2B, lane 3). In contrast, no immunoreactive material was recovered from cells that did not express the A2A receptor (Fig. 2B, lane 2) or Usp4-GFP (Fig. 2B, lane 1). These findings indicate that the A2A receptor and Usp4 do not only interact when expressed as fragments in yeast but that they are also capable of forming a complex when coexpressed as full-length proteins in mammalian cells.
Neither the yeast two-hybrid interaction assay nor the coimmunoprecipitation from mammalian cell extracts proves that the binding of Usp4 to the carboxyl terminus of the A2A receptor occurs via a direct interaction. We therefore also have detected the physical association between the carboxyl terminus and Usp4 by using a pull-down assay. Because a fusion protein comprising glutathione S-transferase, the carboxyl terminus of the A2A receptor, only yielded insoluble inclusion bodies upon expression in E. coli, we fused the carboxyl terminus to the MBP to obtain MBP-A2AR-CT. As a control, we used the MBP fusion protein containing the carboxyl terminus of the A1-adenosine receptor (MBP-A1-R-CT). The MBP fusion proteins were immobilized on amylose resin and were subsequently incubated with immunopurified FLAG-tagged Usp4. The beads were washed three times, and the last wash was analyzed for the presence of Usp4 (Fig. 2C, top blot, lanes 1 and 2, for suspensions containing MBP-A2AR-CT and MBP-A1R-CT). Usp4 was specifically recovered in the eluate (to approximately 30–50% of the input) of immobilized MBP-A2AR-CT (Fig. 2C, top blot, lane 3) but not of MBP-A1R-CT (Fig. 2C, top blot, lane 4). We verified that comparable amounts of MBP fusion protein had been used by immunoblotting with an antiserum directed against MBP (Fig. 2C, bottom blot).
Usp4 Enhances the Cell Surface Expression of the A2A Receptor. As mentioned above, in living cells, a large portion of A2A-adenosine receptors was visualized within the cell rather than at the plasma membrane (Fig. 3A). This was true irrespective of whether stably or transiently transfected cells were used. If Usp4 was coexpressed, the A2A receptor level in the membrane increased at the expense of the intracellular fraction of the receptor (Fig. 3B). The following observations show that the effect of Usp4 was specific: 1) it was not mimicked by coexpression of another deubiquitinating enzyme, Usp14 (Fig. 3C). Usp14 is the functional homolog of yeast Ubp6p, associates with the 26S proteasome complex (Borodovsky et al., 2001), and is apparently specific for monoubiquitinated substrates (Wilson et al., 2002); 2) we used another G protein-coupled receptor that has a high propensity to accumulate within the cell, namely the type-5 metabotropic glutamate receptor (mGluR5); CFP-tagged mGluR5 was predominantly found within the transfected cells, regardless of whether the receptor was expressed in the absence (Fig. 3D) or presence of Usp4 (Fig. 3E) or Usp14 (Fig. 3F); and 3) based on the yeast two-hybrid screen (Fig. 2A), Usp4 is not expected to affect the subcellular distribution of carboxyl terminally truncated versions of the A2A receptor; this prediction has been verified: Usp4 decreased neither the intracellular levels of the A2A-receptor (1–360) (Fig. 3, G and H) nor those of the A2A receptor (1–311) (Fig. 3, I and J). Finally, we tested the effect of combining expression of Usp4 and inhibition of proteosomal degradation. MG132 should relax quality control and thus allow the receptor to escape from the endoplasmic reticulum. Addition of the proteasome inhibitor MG132 augmented the amount of receptor at the cell surface (compare Fig. 3, K with A); in the presence of both Usp4 and MG132, essentially all of the receptors were found at the cell surface (Fig. 3L).

The A2A receptor interacts with Usp4 in vitro. A, the yeast indicator strain pSH18-34 was cotransformed with various combinations of bait and prey vectors. The left semicircle denotes as a mirror image the individual conditions: streaks 1 and 2, a constitutive and a galactose-inducible control, respectively [i.e., yeast cells transformed with a plasmid encoding the lexA binding domain fused to the transactivation domain and with plasmids encoding the regulatory Bα protein phosphatase 2A fused to lexA and Aα subunit of protein phosphatase fused to the transactivation domain (Yeong et al., 2003), respectively]. Streaks 3 and 4, pEG202-A2aRct and pJG45-Usp4; streaks 5 and 6, pEG202-A2aRct (291–311); streaks 7 and 8, pJG45-Usp4, pEG202-A2aRct (291–360), and pJG45-Usp4. All transformants were grown on selection medium, and β-galactosidase assays were performed with these yeast colonies. Note that the yeast expressing wild-type and truncated A2A receptor C termini were streaked in duplicate. B, coimmunoprecipitation of the A2A receptor and Usp4: FLAG-tagged A2A receptor (A2aR-FLAG) and GFP-tagged Usp4 (Usp4-GFP) were transiently expressed either individually (lanes 1 and 2, respectively) or coexpressed (lane 3) in HEK293 cells. Cellular lysates were subjected to immunoprecipitation with the anti-FLAG M2 antibody (lane 3). Lane 4 shows the cellular lysate prepared from cotransfected HEK293 cells (input to reaction in lane 3). Immunoblotting was done with an anti-GFP antibody. C, MBP pull-down assay. Purified fusion protein (3 μg/reaction) comprising the C terminus of the A2A receptor (MBP-A2aRct, lanes 1 and 3) or of the A1-receptor (MBP-A1Rct, lanes 2 and 4) were incubated in 30 μl of amylose beads (50% slurry); purified FLAG-tagged Usp4 (0.3 μg) was added, and the mixtures were incubated for 3 h at 4°C. The slurry was washed, and the last wash was applied onto the gel (lanes 1 and 2, devoid of MBP in the bottom). SDS sample buffer was used to release the proteins bound to the beads (lanes 3 and 4, containing MBP in the bottom). Immunoreactive bands were detected with the anti-FLAG antibody (top) or with an antiserum directed against MBP (bottom). The experiments were carried out three times with similar results.

Cell surface of the A2A receptor after coexpression of Usp4 or of Usp14 in HEK293 cells. HEK293 cells were transiently transfected with plasmids encoding the following sets of proteins: CFP-tagged A2aR(Aand K); CFP-tagged A2aR and GFP-tagged Usp4 (B and L); CFP-tagged A2aR and GFP-tagged Usp14 (C); CFP-tagged mGluR5 (D); CFP-tagged mGluR5 and Usp4 (E); CFP-tagged mGluR5 and Usp14 (F); CFP-tagged A2aRct (1–360) (G); CFP-tagged A2aRct (1–360) and GFP-tagged Usp4 (H); CFP-tagged A2aRct (1–311) (I); and CFP-tagged A2aRct (1–311) and GFP-tagged Usp4 (J). Cells were incubated in the presence of MG132 (50 μM) for 3 h (K and L). Images were captured 24 h later with the appropriate filter settings. The bar diagram summarizes the data from three independent experiments; cells were photographed as in A through L. Prints were assessed by a blinded observer who scored the number of cells indicated on the y-axis for predominant expression of the receptor at the plasma membrane (□, M) or within the cell (□, C); data are from three separate transfections each.
To test whether endogenous levels of Usp4 sufficed to regulate cell surface expression of A2A receptors, we used RNA interference. We were not successful in reducing mRNA levels for Usp4 by RNA interference in PC-12 cells (three primers tested). When expressed at levels comparable with those seen in PC-12 cells or in striatal membranes, the A2A receptor accumulates in large amounts within HEK293 cells (Fig. 3A). Under these conditions, RNA interference is unlikely to yield any detectable increment in intracellular accumulation. Therefore, we selected a HEK293 cell line that expressed A2A receptors at low levels. In these cells, the A2A receptor is predominantly found at the plasma membrane (Fig. 4B). Of several siRNAs, siRNA2 caused a significant reduction in the mRNA encoding Usp4 (Fig. 4A). In cells in which Usp4 mRNA had been lowered by RNA interference (Fig. 4A), the A2A receptor appeared within the cell (Fig. 4C).
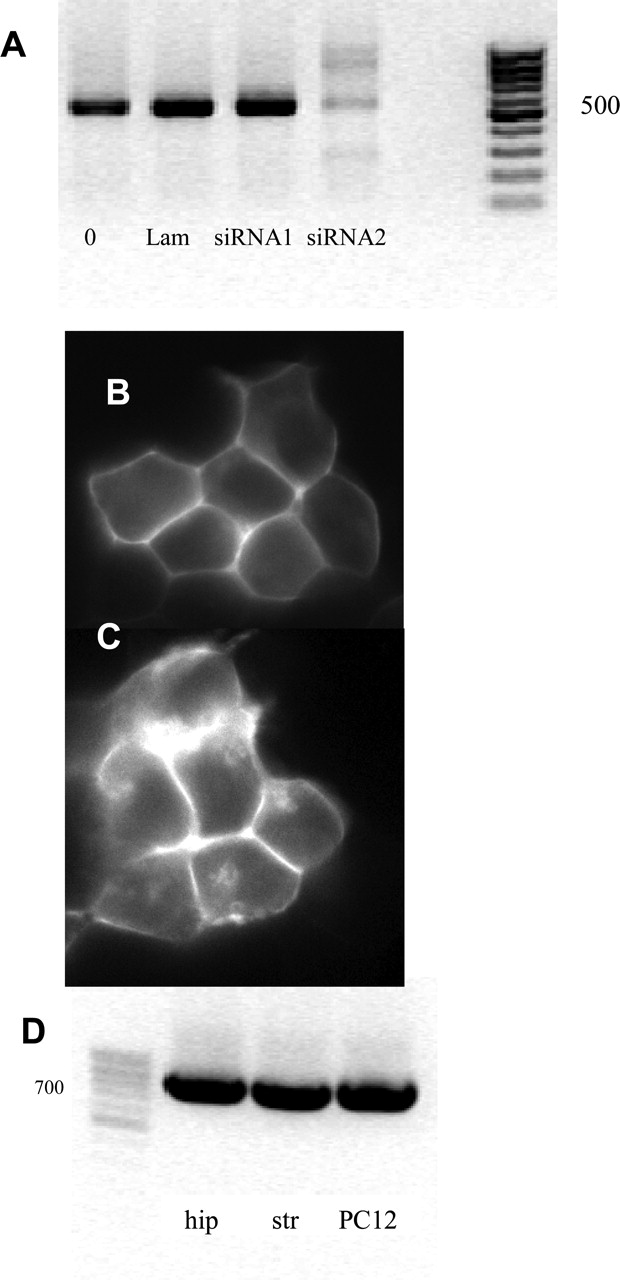
siRNA-mediated knockdown of Usp4 mRNA in HEK293 cells (A–C) and detection of Usp4 mRNA in hippocampus, striatum, and PC-12 cells (D). A, HEK293 cells (3 × 105 cells) stably expressing low levels of YFP-tagged A2A receptors were transiently transfected with LipofectAMINE 2000 (lane 0) or the combination of LipofectAMINE 2000 and 500 pmol each siRNA directed against lamin (irrelevant siRNA, lane Lam), siRNA1 (directed against Usp4, but inactive), and siRNA2. After 48 h, cells were lysed, and the level of mRNA coding for Usp4 examined by RT-PCR (A). The right lane is the marker; the approximate size of the amplicon was 500 base pairs. In parallel, sham-transfected cells (B) and cells treated with siRNA2 were examined by fluorescence microscopy (C). D, RT-PCR was conducted to document the expression of Usp4 in hippocampus (hip), striatum (str), and PC-12 cells. Data are representative of at least three separate transfections.
Usp4 Increases the Cell Surface Level of A2A Receptors in Hippocampal Neurons. The intracellular accumulation of the A2A receptor in HEK293 cells may arise from the fact that these cells do not endogenously express the receptor and are more likely to retain the receptor. Usp4 is abundantly expressed in hippocampus, striatum, and PC-12 cells (Fig. 4D). We therefore expressed the CFP-tagged A2A receptor in hippocampal neurons, in which the receptor is endogenously expressed (Rosin et al., 1998), and visualized the distribution of receptors in living cells in the absence and presence of coexpressed GFP-tagged Usp4. In the absence of Usp4, the A2A receptor was mainly found within the soma (Fig. 5A). Coexpression of Usp4 (shown in Fig. 5C) increased the level of the fluorescence at the cell surface (Fig. 5B). In contrast, the presence of Usp4 (shown in Fig. 5F) did not enhance the cell surface level of the truncated version A2A receptor (1–311), which was predominantly detected within the somata of the neurons (compare Fig. 5, D and E). Finally, we also visualized CFP-tagged mGluR5, which was found within the neuronal soma (Fig. 5G); the coexpression of Usp4 (shown in Fig. 5I) did not lead to the appearance of mGluR5 at the cell surface (Fig. 5H).
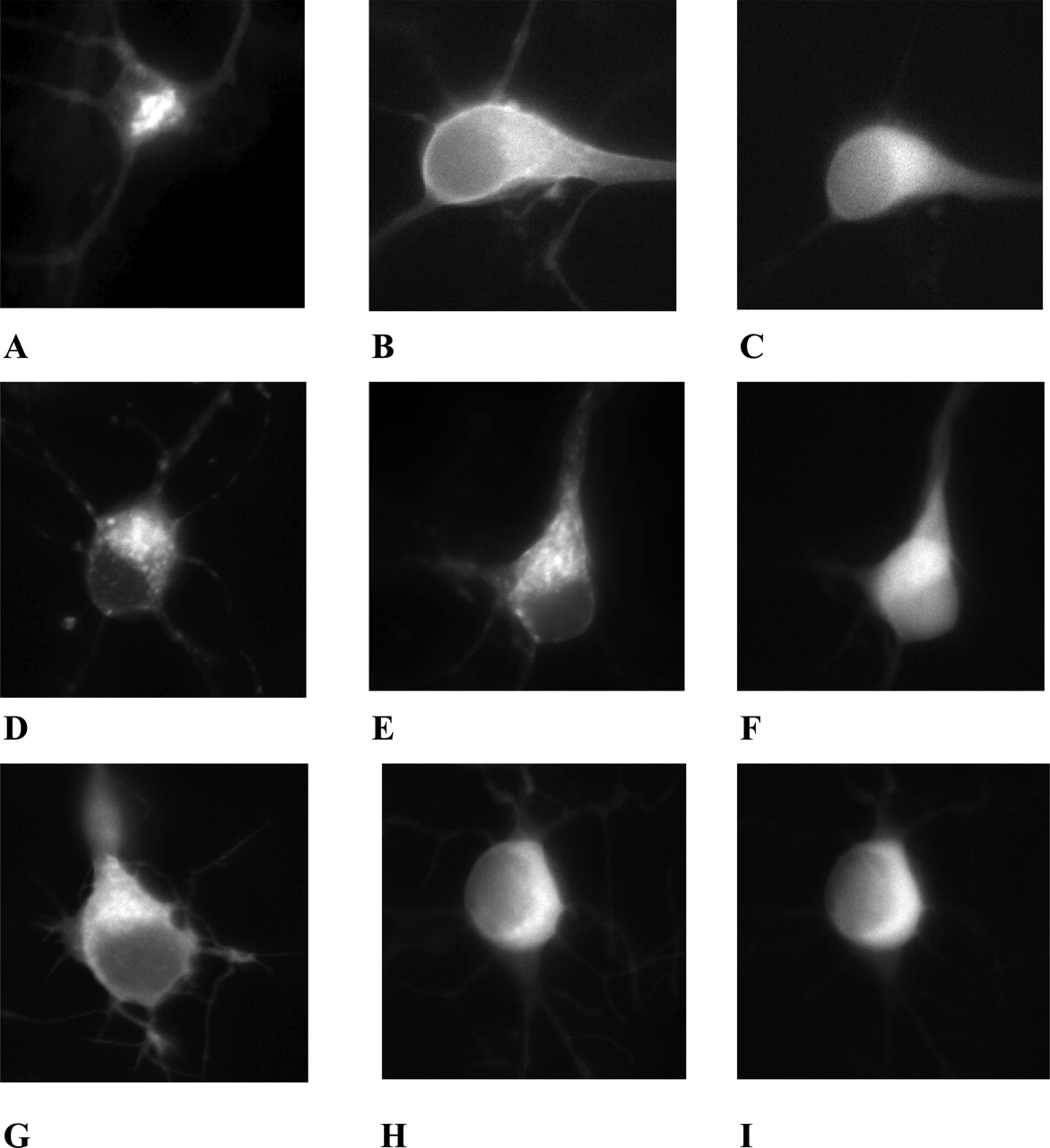
Expression of the A2A receptor or of mGluR5 in the absence and presence of Usp4 in cultured hippocampal neurons. Neurons were prepared from rat neonatal hippocampus and cultured as outlined under Materials and Methods, transfected using LipofectAMINE Plus with CFP-tagged A2A-receptor (A2aR-CFP; A), A2aR-CFP and GFP-tagged Usp4 (Usp4-GFP) (B and C); A2aR(1–311)-CFP (D), A2aR(1–311)-CFP and Usp4-GFP (E and F); mGluR5-CFP (G); and mGluR5-CFP and Usp4 (H and I). All images were captured 24 h later with the appropriate filter settings, that is CFP filter settings in A, B, D, E, G, and H (excitation at 430 nm, emission at 476 nm), and YFP-filter settings in C, F, and I were taken using (excitation at 510 nm, emission at 530 nm) to document the expression of Usp4-GFP in the neurons shown in B, E, and H.
Coexpression of Usp4 Results in the Accumulation of Deubiquitinated A2A Receptor. To show that Usp4 used the A2A receptor as substrate, we transiently cotransfected HEK293 cells with plasmids encoding the FLAG-tagged A2A receptor, HA-tagged ubiquitin, and GFP-tagged Usp4. The A2A receptor was immunoprecipitated with anti-FLAG antibodies from detergent lysates of cells that either coexpressed only HA-tagged ubiquitin (Fig. 6A, lanes 1 and 2) or the combination of HA-tagged ubiquitin and Usp4 (Fig. 6B, lanes 4 and 5). Receptor bands were detected with anti-FLAG antibody (blots shown on top); in the absence of Usp4, the FLAG-reactive immunostaining was seen in the range of ∼48 to 50 kDa (Fig. 6A top, lanes 1 and 2). In the presence of Usp4, the FLAG-tagged receptor migrated at ∼40 to 42 kDa (Fig. 6B top, lanes 4 and 5). Lanes 3 and 6 represent the negative controls, showing immunoprecipitation that was carried out with cellular lysates that lacked the A2A adenosine receptor but contained HA-tagged ubiquitin and, in lane 6, Usp4. Regardless of the conditions, immunoreactivity was recovered neither in the ∼40- to 42-kDa nor in the ∼48- to 50-kDa range. Thus, the immunostaining was specific. The nitrocellulose membranes were stripped and stained with anti-HA antibodies (Fig. 6, A and B, bottom blots). In cells cotransfected with the plasmids encoding the FLAG-tagged A2A receptor and HA-tagged ubiquitin, the HA-antibody stained a ∼48- to 50-kDa band. This corresponded to the ubiquitinated form of the A2A receptor, because this band was also stained with the anti-HA antibody (compare Fig. 6A, top and bottom blots). In contrast, when coexpressed with Usp4, the A2A receptor, which migrated as a band of 40 to 42 kDa (Fig. 5B, top, lanes 4 and 5), was not detected with the anti-HA antibody (Fig. 6B, bottom, lanes 4 and 5). This band therefore represents the deubiquitinated species of the receptor.

The A2A receptor is deubiquitinated by Usp4 in vitro and is insensitive to cleavage by endo-β-N-glucosaminidase H when coexpressed with Usp4. A and B, immunoprecipitation of A2aR from HEK293 cells transiently transfected with the following sets of plasmids: FLAG-tagged A2aR, HA-tagged ubiquitin (lines 1 and 2); FLAG-tagged A2aR, HA-tagged ubiquitin, and GFP-tagged Usp4 (lines 4 and 5); and GFP-tagged Usp4 and/or HA-tagged ubiquitin (lines 6 and 3). Cells were collected 48 h after transfection, and membrane preparation and immunoprecipitation were made as described under Materials and Methods. After the transfer, membranes with proteins were stained with anti-FLAG antibody (1:500 dilution) to reveal A2a receptor (top) and then stripped for 30 min at 50°C and incubated with anti-HA antibody to stain ubiquitin (bottom). Data are from a representative experiment that was reproduced three times. C, immunoblot of membranes from HEK293 cells transiently expressing A2aR-FLAG, Usp4-GFP, and treated with endoH. Cells were collected 48 h after transfection, membranes were prepared, and extracts thereof were subjected to endoH treatment. After the transfer, immunoreactive bands were revealed with the anti-FLAG antibody. Data are from a representative experiment that was reproduced three times. Images were scanned and processed with QuantiScan software. The average densities in optical units (o.u.) are shown in D for these four independent experiments. Error bars indicate S.E.M.
Coexpression of Usp4 Increases the Surface Expression of A2A Receptors. As documented in Fig. 3, Usp4 caused a redistribution of the CFP-tagged A2A receptor to the cell surface. Two scenarios can be envisaged: Usp4 enhances ER export of the de novo synthesized A2A receptor. On the other hand, the A2A receptor may undergo rapid constitutive endocytosis such that, even in the absence of agonist, a high proportion of the receptor is found within the cell at steady state. In contrast with newly synthesized receptors which are either en route to the Golgi or to ER-associated degradation, recycling receptors have already passed through the Golgi; thus, they should be fully glycosylated. As can be seen from Fig. 6, C and D, the bulk of the A2A receptor that was recovered from cellular lysates was cleaved by endoglycosidase H (Fig. 6, C and D, compare first and second left-hand lanes and set of bars, respectively). However, in cells that coexpressed Usp4, the A2A receptor became resistant to deglycosylation by endoglycosidase H (Fig. 6, C and D, compare the third and fourth lanes and set of bars, respectively). Thus, the presence of Usp4 allows the A2A receptor to escape the endoplasmic reticulum and rapidly traffic through the Golgi to the plasma membrane.

Usp4 enhances the expression of A2A receptor binding sites. Saturation hyperbolae for specific binding of [3H]ZM241385 to membranes from transiently transfected HEK293 cells. Membranes prepared from HEK293 cells expressing the full-length A2A receptor (A) and truncated versions A2aR (1–360) and A2aR (1–311) with or without Usp4 (B) in the presence or absence of MG132 were incubated in buffer containing the indicated concentrations of [3H]ZM241385 in the presence of 100 μM GTPγS. The Bmax values from these binding experiments are summarized in C. Results are means ± S.D. from four independent experiments that were carried out in parallel and with duplicate determinations. *, significant difference from the full-length A2aR at p = 0.001 (unpaired t test). Inset, HEK293 cells expressing YFP-tagged A2A receptor (same cell line as used in Fig. 4) were transfected with a plasmid driving the expression of HA-ubiquitin. On the next day, the cells were incubated for 3 h in the presence of 50 μM MG132, and the receptor was immunoprecipitated by using an anti-GFP antiserum. The amount of ubiquitinated receptor recovered in the immunoprecipitate was visualized by blotting with an antibody directed against the HA epitope.
It is conceivable that relaxing quality control by coexpressing Usp4 allowed unfolded receptors to escape from the endoplasmic reticulum. To rule out this possibility, binding assays were performed with [3H]ZM241385, a specific and selective A2A receptor antagonist (Palmer et al., 1995). Figure 7A shows a set of representative saturation curves for specific binding of [3H]ZM241385 to membranes from HEK293 cells that were either solely transfected with a plasmid driving the expression of either the CFP or the FLAG-tagged A2A receptor or of the receptor and Usp4. The coexpression of Usp4 (Fig. 7A, ▴) increased Bmax but did not affect the affinity of the radioligand. KD values were, for the wild-type A2A receptor, 1.97 ± 1.2 and 2.12 ± 0.96 nM (n = 7); for the A2A receptor (1–360), 2.17 ± 1.16 and 1.84 ± 0.57 nM (n = 5); and for the A2A receptor (1–311), 2.70 ± 1.57 and 3.26 ± 1.51 nM (n = 4), in the absence and presence of Usp4, respectively. The effect of Usp4 depended on the carboxyl terminus of the A2A receptor, for it was not seen with the truncated forms A2A receptor (1–311) or A2A receptor (1–360). Representative saturation curves are shown in Fig. 7B; Bmax averaged from several saturation experiments are shown in the bar diagram in Fig. 7C.
The model of quality control in the endoplasmic reticulum predicts that all steps are reversible provided that the carboxyl terminus of the membrane protein has not yet been engulfed by the proteasome (Kostova and Wolf, 2003). Accordingly, the action of Usp4 and of proteasome inhibition is expected to be additive. This was the case. As can be seen from the average Bmax values summarized in Fig. 7C, the sole addition of MG132 caused a pronounced increase in the amount of A2A receptor binding sites. This was associated with a substantial increase in ubiquitinated receptor (Fig. 7C, inset). The combined presence of both Usp4 and MG132 resulted in an even more dramatic increase in the number of receptors.
Coexpression of Usp4 Enhances the A2A Receptor-Mediated cAMP Accumulation. The A2A receptor is a prototypical Gs-coupled receptor (Klinger et al., 2002b); thus, activation of the receptor leads to the stimulation of adenylyl cyclase. The binding data showed that coexpression of Usp4 increased the number of cell surface binding sites. We verified that this translated into an increase in functional receptors by measuring agonist-induced cellular cAMP accumulation. In cells that expressed Usp4 (Fig. 8A, triangles), the agonist CGS21680 elicited a larger maximum effect than in cells that only expressed the A2A adenosine receptor (Fig. 8A, ○). We stress that this is not a nonspecific effect that can, for instance, be accounted for by an increased responsiveness of the catalytic moiety of adenylyl cyclase in the presence of Usp4. Control experiments revealed that cells expressing solely the A2A receptor or the A2A receptor and Usp4 did not differ in their responsiveness to forskolin (Fig. 8B). When expressed in HEK293 cells, the A2A receptor displays some appreciable constitutive activity, which is magnified in the presence of forskolin (Klinger et al., 2002a). Therefore, the antagonist ZM241385 (1 μM) was included to eliminate any possible confounding effect, which may result from the fact that A2A receptor levels differ substantially in the absence and presence of coexpressed Usp4.
Discussion
The family of deubiquitinating enzymes is categorized into three subfamilies: the smaller ubiquitin C-terminal hydrolase (UCH) subfamily, the larger ubiquitin-specific processing protease (USP/UBP) subfamily, and the recently described subfamily of OTU-domain containing proteins. UCH enzymes are generally small, containing only a proteolytic core domain that functions to cleave small molecular weight adducts from the ubiquitin C terminus. In contrast, USPs/UBPs, such as Usp4, are physically much larger and contain substantial and divergent sequences beyond their catalytic core. The analysis of genomic sequences predicts the presence of more than 90 deubiquinating enzymes in a given mammalian genome (Chung and Baek, 1999), the vast majority of which belong to the USP/UBP family. In fact, cDNAs have recently been cloned that encode 22 additional members of the human USP family (Quesada et al., 2004). Whereas the catalytic activity has been tested using artificial substrates, very little is known about their physiological substrates and thus their physiological functions. In fact, the number of identified substrates is remarkably small: the largest list exists for fat facets/FAM/USP9X, which deubiquitinates the ras effector AF-6 (Taya et al., 1998), β-catenin (Taya et al., 1999), liquid facets (a Drosophila melanogaster homolog of the mammalian endocytotic protein epsin; Chen et al., 2002), and Vasa (an RNA-helicase; Liu et al., 2003). The substrates of the mammalian enzymes USP7/HAUSP (herpes virus-associated USP) and USP2a are p53 (Li et al., 2002) and fatty acid synthase (Graner et al., 2004), respectively. Membrane-embedded receptors, in general, and G protein-coupled receptors, in particular, are subject to ubiquitination (Hicke, 1999; Wojcikiewicz, 2004). However, to the best of our knowledge, specific deubiquitination reactions have not yet been reported for transmembrane receptors: the A2A receptor is the first example of a G protein-coupled receptor that is shown to be a specific substrate for a deubiquinating enzyme of the USP/UBP family.

Usp4 enhances A2A receptor-mediated cAMP accumulation. Stimulation of cAMP accumulation in transiently transfected HEK293 cells by the A2A agonist CGS21680 (A) or by 10 μM forskolin in the presence of the antagonist ZM241385 (1 μM). Cells expressing solely the full-length A2A receptor (•) together with Usp4 (triangles) were seeded in six-well dishes, and the cellular adenine nucleotide pool was metabolically prelabeled for 16 h with [3H]adenine. After a preincubation of 30 min in fresh medium containing adenosine deaminase (2 U/ml), cAMP production was stimulated by the indicated concentrations of the A2A-selective agonist CGS21680 (A) or 10 μM forskolin in the presence of 1 μM ZM241385. Data are means ± S.D. from four (A) and three (B) independent experiments that were done in triplicate and carried out in parallel.
In many instances, deubiquinating enzymes accelerate proteasomal degradation by removing ubiquitin from proteasome-bound substrate or proteolytic intermediates thereof. Deubiquinating enzymes may thereby allow the proteasome to engulf the substrate more efficiently and to operate at the maximal rate. However, there is also precedent for an antagonism between proteasomal degradation and deubiquinating enzymes: the developmental defect, which is caused by fat facet mutants in the D. melanogaster compound eye, can be suppressed by a reduction in proteosomal activity (Huang et al., 1995). Likewise, USP7/HAUSP prevents the degradation of p53 (Li et al., 2002). The effect of Usp4 on the A2A receptor is also consistent with a preproteasomal action of Usp4: Usp4 rescues the receptor from degradation and thereby enhances its cell surface levels. This conclusion is based on the following sets of observations: 1) Usp4 binds to the carboxyl terminus of the A2A-receptor, and the interaction requires the most distal portion of the receptor (i.e., the last 50 amino acids); 2) when heterologously expressed, a substantial portion of the A2A receptor is detected as ubiquitinated protein. The coexpression of Usp4 and the A2A receptor allows for the accumulation of deubiquinated receptor, and this is associated with enhanced levels of the A2A receptor at the cell surface, increased amounts of functional receptor that can bind ligand, and an augmented biological response; 3) as an additional control, we used mGluR5, a group I mGluR, because, like the A2A receptor, mGluR5 also has a propensity to accumulate within the cell. In addition, group I-metabotropic glutamate receptors are subject to modification by the seven-in-absentia homolog 1A, a ring domain containing E3 ligase. This modification accelerates their rate of degradation, whereas blockage of the proteasome prevents their degradation (Moriyoshi et al., 2004). Usp4 does not act on mGluR5; we used another USP/UBP-family member, namely Usp14. The rationale for choosing Usp14 was that murine Usp14 is encoded by the ataxia (ax) gene, the mutated form of which is associated with ataxia in mice (Wilson et al., 2002); the absence of the mGluR1, a Gq-coupled receptor, also causes ataxia in mice (Aiba et al., 1994). We thus surmised that Usp14 may act on group I mGluRs, which it did not. More importantly, Usp14 did not affect the subcellular distribution of the A2A receptor. Thus, raising deubiquinating activity in the cell did not suffice to enhance the accumulation of the A2A receptor at the cell surface.
Ubiquitination plays a prominent role in supporting endocytosis of membrane receptors (Hicke, 1999). A more recent survey of several G protein-coupled receptors indicates that ubiquitination can either target the receptors during biosynthesis to proteasomal degradation or redirect them on the endocytotic pathway to the lysosome (Wojcikiewicz, 2004). Our experiments were not designed to address a possible role of Usp4 in regulating the rate of endocytosis of the A2A receptor, and it is conceivable that Usp4 also affects the rate of receptor recycling. It is unlikely, however, that the effects observed in the present study are accounted for by a Usp4-induced inhibition of endocytosis. First, endocytosed receptors are still capable of binding antagonist radioligands. In contrast, a large fraction of newly synthesized receptors in the endoplasmic reticulum fails to bind antagonist with appreciable affinity (Pankevych et al., 2003). Thus, the finding that Usp4 (and the proteasome inhibitor) increased the level of antagonist binding is consistent with an action during synthesis and subsequent quality control in the ER. Second, coexpression of Usp4 affected the proportion of receptors that were resistant to the action of endoglycosidase H. In the absence of Usp4, the bulk of the receptors were sensitive to deglycosylation by endoglycosidase H and thus had not yet been delivered to the Golgi apparatus. In contrast, in the presence of Usp4, a substantial portion of the receptor was resistant to endoglycosidase H and had thus left the ER compartment and trafficked through the Golgi. Finally, our experiments were done in the absence of agonist (i.e., under conditions in which the endocytosis of G protein-coupled receptors is not prominent). Similar to other misfolded membrane proteins, incorrectly folded G protein-coupled receptors are retrotranslocated through the sec61 translocon and degraded by the proteasome (Petaja-Repo et al., 2001). The latter step is contingent on ubiquitination. From a teleological perspective, it seems most plausible to target the carboxyl terminus of a G protein-coupled receptor for ubiquitination. This allows for cotranslational quality control because the accumulation of misfolded intermediates can be sensed and trigger the ubiquitination of the carboxyl terminus that is still tethered to the Sec61 channel. Point mutations are known to render G protein-coupled receptors more prone to misfold. Mutations in the V2-vasopressin receptor, which lead to diabetes insipidus, are among the most prominent examples (Oksche and Rosenthal, 1998). Attempts to rescue these mutant receptors were inspired originally by nonspecific manipulations that were successful in CFTR, including dimethyl sulfoxide, glycerol and other polyols, trimethylamine-N-oxide, and phenylbutyrate (Gelman and Kopito, 2002). G protein-coupled receptors, however, have the advantage that folding may be aided by receptor ligands provided that they are membrane-permeable (Bernier et al., 2004). The current observations suggest another mechanism that may represent a viable alternative, namely, to target the deubiquinating enzyme that can recognize the receptor as a substrate. As shown here for the A2A receptor, this approach is, in principle, also capable of rescuing a receptor from ER-associated degradation and of thereby increasing its cell surface level. We assume that the current observations are of relevance for G protein-coupled receptors in general and for other membrane proteins that are subject to stringent quality control in the endoplasmic reticulum.
Acknowledgments
We thank J. Bies, D. Gray, and J. Blahos for generous gifts of plasmids.
Footnotes
-
This work was supported by Austrian Science Foundation (Fonds zur Förderung der Wissenschaftlichen Forschung) grant P15034 and by a grant from the fifth framework program of the European Union (Epileptosome) (to M.F.) and by BioDevelops GmbH.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.105.015818.
-
ABBREVIATIONS: ER, endoplasmic reticulum; A2aR, A2a adenosine receptor; CFP, cyan fluorescent protein; UBP, ubiquitin-specific processing protease; USP, ubiquitin-specific protease; YFP, yellow fluorescent protein; MBP, maltose binding protein; GFP, green fluorescent protein; PCR, polymerase chain reaction; RT-PCR, reverse transcription-polymerase chain reaction; HEK, human embryonic kidney; endoH, endo-β-N-acetylglucosaminidase-H; HA, hemagglutinin; siRNA, small interfering RNA; GTPγS, guanosine-5′-(3-O-thio)triphosphate; CFTR, cystic fibrosis transmembrane conductance regulator; mGluR5, type-5 metabotropic glutamate receptor; [3H]ZM241385, 4-(2-[7-amino-2-[2-furyl]-[1,2,4]triazolo[2,3-a][1,3,5] triazin-5-yl-amino]ethyl)phenol; RO201724, 4-(3-butoxy-4-methoxybenzyl)imidazoline-2-one; MG132, N-benzoyloxycarbonyl (Z)-Leu-Leu-leucinal; CGS21680, 2-[p-(2-carboxyethyl)phenethylamino]-5′-N-ethylcarboxamidoadenosine.
- Received June 16, 2005.
- Accepted December 8, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}