


Abstract
SK channels are small conductance Ca2+-activated K+ channels important for the control of neuronal excitability, the fine tuning of firing patterns, and the regulation of synaptic mechanisms. The classic SK channel pharmacology has largely focused on the peptide apamin, which acts extracellularly by a pore-blocking mechanism. 1-Ethyl-2-benzimidazolinone (1-EBIO) and 6,7-dichloro-1H-indole-2,3-dione 3-oxime (NS309) have been identified as positive gating modulators that increase the apparent Ca2+ sensitivity of SK channels. In the present study, we describe inhibitory gating modulation as a novel principle for selective inhibition of SK channels. In wholecell patch-clamp experiments, the compound (R)-N-(benzimidazol-2-yl)-1,2,3,4-tetrahydro-1-naphtylamine (NS8593) reversibly inhibited recombinant SK3-mediated currents (human SK3 and rat SK3) with potencies around 100 nM. However, in contrast to known pore blockers, NS8593 did not inhibit 125I-apamin binding. Using excised patches, it was demonstrated that NS8593 decreased the Ca2+ sensitivity by shifting the activation curve for Ca2+ to the right, only slightly affecting the maximal Ca2+-activated SK current. NS8593 inhibited all the SK1-3 subtypes Ca2+-dependently (Kd = 0.42, 0.60, and 0.73 μM, respectively, at 0.5 μM Ca2+), whereas the compound did not affect the Ca2+-activated K+ channels of intermediate and large conductance (hIK and hBK channels, respectively). The site of action was accessible from both sides of the membrane, and the NS8593-mediated inhibition was prevented in the presence of a high concentration of the positive modulator NS309. NS8593 was further tested on mouse CA1 neurons in hippocampal slices and shown to inhibit the apaminand tubocurarine-sensitive SK-mediated afterhyperpolarizing current, at a concentration of 3 μM.
The excitability and the patterns of action potential firing of neurons as well as the function and plasticity of synapses are balanced by the activity of many ion channels, including small conductance Ca2+-activated K+ (SK) channels (Faber et al., 2005; Ngo-Anh et al., 2005). SK channels mediate a Ca2+-activated afterhyperpolarizing current, IAHP, in neocortical, hippocampal, and amygdala pyramidal cells, whereas large conductance Ca2+-activated K+ (BK) channels are responsible for an earlier component of the afterhyperpolarization (the fast Ca2+- and voltage-dependent K+ current, FAHP). The channels mediating the long-lasting, Ca2+-dependent afterhyperpolarization (the slow afterhyperpolarizing K+ current, sAHP) in these neurons have not yet been identified (for recent reviews on SK channels, see Stocker, 2004; Bond et al., 2005).
Three SK channel subunits have been cloned (SK1-3), which exhibit distinct, but partly overlapping distributions in the CNS (Kohler et al., 1996; Stocker and Pedarzani, 2000; Sailer et al., 2004). The three subunits are highly homologous and have indistinguishable biophysical properties in terms of single channel conductance, rectification, selectivity and gating, which is governed by Ca2+ binding to calmodulin constitutively attached to the C terminus of each subunit (Xia et al., 1998). Despite the structural and functional similarities of homomultimeric SK channels, various peptide toxins block the SK1, SK2, and SK3 subtypes with different potencies. The classic blocker of SK channels is apamin, an alkaline 18-amino acid peptide from the venom of the honey bee, Apis mellifera. Apamin is a pore blocker carrying positively charged amino acids (Arg13 and Arg14) as well as a neutral amino acid (Gln17), which are thought to interact with negative (Asp341) and neutral (Asn368) amino acids, positioned in the outer pore mouth of SK channels (Ishii et al., 1997). The interaction is highly potent (affinities in the picomolar to nanomolar range) and selective with the blocker selectivity sequence: SK2 > SK3 > SK1 (for review, see Stocker et al., 2004). The intermediate conductance Ca2+-activated K+ channel (IK), which is closely related to SK channels, is not blocked by apamin, and neither is any other known mammalian K+ channel. This makes apamin an SK-specific blocker, and it has therefore been extensively used as a reliable and valuable tool for the identification of SK-mediated physiological responses in isolated cells, tissues, and, to a lesser extent, in whole animals (for review, see Blank et al., 2004). The potent scorpion toxins scyllatoxin (from Leiurus quinquestriatus; Chicchi et al., 1988) and the recently described tamapin (from Mesobuthus tamulus; Pedarzani et al., 2002), both displace apamin from its binding site and exhibit blocker selectivity similar to apamin. Based on scyllatoxin, an artificial peptide called Lei-Dab7, with considerably improved selectivity for SK2 channels, has been synthesized (Shakkottai et al., 2001). Furthermore, a number of lower potency (micromolar) compounds, such as the anticholinergic d-tubocurarine and dequalinium (Castle et al., 1993), and quaternary methyl derivatives of bicuculline (Seutin and Johnson, 1999), block SK channels and displace apamin from its binding site (Finlayson et al., 2001). Finally, SK channels have been the focus for rational drug design targeting the apamin site. This has resulted in very potent, positively charged, small-molecule blockers with potencies around 1 nM, such as the bisquinolinium cyclophane UCL 1684 (for review, see Liegeois et al., 2003; Campos Rosa et al., 2000).
The SK and IK channels can be positively modulated by a number of small heterocyclic organic molecules of which the prototype compound is the benzimidazolinone 1-EBIO (Syme et al., 2000; Pedarzani et al., 2001), and the most potent compound described thus far is the indole-oxime NS309 (Strøbæk et al., 2004; Pedarzani et al., 2005). The positive modulation is due to a concentration-dependent increase in the Ca2+ sensitivity of the SK channels. Mutagenesis studies suggest a binding site for 1-EBIO involving the C-terminal region of the SK channels (Pedarzani et al., 2001). In contrast to the peptide blockers, the positive modulators are slightly more potent on IK channels compared with the SK channel subtypes.
In the present article, we describe a new potent and selective inhibitor of recombinant SK channels and of the SK-mediated, apamin-sensitive IAHP in hippocampal CA1 neurons. Thus, as a result of our medicinal chemistry effort, we identified the compound (R)-N-(benzimidazol-2-yl)-1,2,3,4-tetrahydro-1-naphtylamine (NS8593), which is chemically distant from all previously known small-molecule blockers of SK channels and does not carry permanent positive charges (Sørensen et al., 2006). NS8593 acts through a novel mechanism by decreasing the Ca2+ sensitivity of SK channels rather than blocking their pore. NS8593 is therefore the first inhibitory gating modifier of SK channels described so far. The mechanism of action of NS8593 on SK channels is discussed together with possible implications for drug development.
Materials and Methods
Chemical Synthesis of NS8593. NS8593 (1) was synthesized in a one-step reaction (Scheme 1) from commercially available 2-chlorobenzimidazole (2) and (R)-1,2,3,4-tetrahydro-1-naphtylamine (3). Thus, 2 (5.0 g; 32.8 mmol) and 3 (5.8 g; 39.3 mmol) were suspended in acetonitrile (5 ml) in a closed vial and heated to 170°C for 40 min by microwave irradiation (Emrys Optimizer EXP, Biotage, Sweden). After cooling the reaction mixture to room temperature, the precipitated solid was filtered off and washed with acetonitrile to give 1 as a hydrochloride salt in 65% yield.
To evaluate the enantiomeric purity of 1, its optical rotation was measured and compared with that of the corresponding (S)-enantiomer prepared from 2 and commercially available (S)-1,2,3,4-tetrahydro-1-naphtylamine (reacting for 6 days in refluxing toluene using conventional heating). The optical rotation of the (S)-enantiomer and that of 1 were determined at -55.8 and 58.7° (MeOH; 25°C), respectively. In the compound characterization of 1 the following additional data were obtained (for the HCl salt). Melting point 263-265°C; MS(ES+) m/z 264 ([M + 1]+, 100%); 1H NMR [dimethyl sulfoxide (DMSO)-d6] δ 1.75-1.85 (m, 1H), 1.88-1.97 (m, 2H), 2.06-2.13 (m, 1H), 2.72-2.88 (m, 2H), 5.01-5.07 (m, 1H), 7.16-7.28 (m, 5H), 7.34-7.43 (m, 3H), 9.48 (m, 1H), 12.8 (br s, 2H); 13C NMR (DMSO-d6) δ 19.7, 28.8, 30.0, 51.9, 111.7, 123.2, 126.4, 127.9, 128.8, 129.4, 130.4, 135.5, 137.6, 150.0. Elemental analysis (Department of Chemistry, University of Copenhagen): Anal. Calcd. for C17H17N3·HCl: C, 68.11; H, 6.05; N, 14.02; Found: C, 67.87; H, 5.97; N, 13.98.
Cell Cultures. Apamin binding and patch-clamp studies using recombinant channels were performed on HEK293 cells stably expressing the various channels. Establishment of hSK1, rSK2, hSK2, rSK3, hSK3, and hIK channel cell lines was described in Strøbæk et al. (2004). Selectivity studies were performed on BK channels (for cloning and expression, see Strøbæk et al., 1996), Kv7.2 + 7.3 channels (Schrøder et al., 2001), human ether-a-go-go-related gene channels (T. Ljungstrøm, PhD thesis, 2002; University of Copenhagen), rNav1.2 channels (M.P.G. Korsgaard, PhD thesis, University of Copenhagen, 2001) as well as native voltage-dependent Na+, Ca2+, and K+ channels in rat embryonic dorsal root ganglion (eDRG) neurons.
Cell lines were cultured in Dulbecco's modified Eagle's medium (Cambrex Bio Science Walkersville, Inc., Walkersville, MD) supplemented with 10% fetal calf serum (Sigma-Aldrich, Vallensbæk Strand, Denmark). At 60 to 80% confluence, cells for patch-clamp experiments were harvested by trypsin treatment and seeded on coverslips (diameter, 3.5 mm; custom made at VWR International APS, Albertslund, Denmark) placed in a Petri dish. Cells for apamin binding assays were cultured to 80 to 90% confluence, the medium was removed, and cells were rinsed with 10 ml of phosphate-buffered saline (PBS) and harvested by scraping. The cell suspension was centrifuged for 10 min (27,000g) at 4°C, and the pellet was resuspended by homogenization in 50 mM Tris-HCl, pH 7.4, and centrifuged. The final hSK3 pellet was suspended in buffer and stored at -80°C.
125I-Apamin Binding. Male Wistar rats (150-200 g; Taconic M&B, Ry, Denmark) were used for rat brain membrane preparation. The animals were sacrificed by cervical dislocation, their brains were removed and placed on ice, and the corpi striati were dissected. The tissue was homogenized for 5 to 10 s in 100 volumes of ice-cold 0.32 M sucrose using a motor-driven glass/Teflon homogenizer. The homogenate was centrifuged at 1000g for 10 min (4°C), and the resulting supernatant was centrifuged at 27,000g for 50 min. The membrane pellet was resuspended in 2000 volumes of 50 mM Tris-HCl buffer, pH 8.5, containing 5 mM KCl and 0.1% bovine serum albumin (BSA) and used for binding experiments.
The hSK3 membrane suspension was thawed on the day of experiment, centrifuged for 10 min (27,000g) at 4°C, and the final pellet was resuspended in ice-cold assay buffer (25-50 μg of protein/assay).
125I-Apamin binding conditions were as described previously (Finlayson et al., 2001). In brief, binding assays were performed in a total volume of 2.2 ml consisting of 2.0 ml of tissue suspension, 100 μl of test drug solution or buffer, and 100 μl of 125I-apamin (2.7-8.3 pM final concentrations). Samples were incubated for 90 min at 2°C, and binding was terminated by rapid filtration onto glass fiber filters (Whatman GF/C; Whatman, Maidstone, UK) presoaked 90 min with 0.25% polyethylenimine, followed by three washes (5 ml) with icecold 50 mM Tris-HCl buffer, pH 8.5, containing 5 mM KCl. Nonspecific binding was determined using unlabeled apamin (100 pM final concentration). The radioactivity on the filters was determined using a Packard Cobra auto-gamma counter (PerkinElmer Life and Analytical Sciences, Boston, MA). Compounds were tested at eight to 12 concentrations ranging from 0.03 pM to 10 μM. All apamin solutions (in incubation buffer) contained 0.1% BSA to avoid adsorption to glass and plastic. All test drug stock solutions were prepared and diluted in 48% ethanol. Final ethanol concentration in the assay was 2%.
Electrophysiology on Recombinant Channels. Experiments were performed in the whole-cell, inside-out and outside-out configurations of the patch-clamp technique. Cells seeded on coverslips were transferred to a 15-μl recording chamber grounded by an integrated AgCl pellet and superfused at 1 ml/min by gravity from an inlet tube connected to a 10-position solution exchanger (VICI Valco Instruments, Houston, TX). Patch pipettes (1.8-2.3 MΩ) were pulled from borosilicate tubes (Modulohm, Copenhagen, Denmark) on a horizontal electrode puller (Zeitz Instruments, Augsburg, Germany) and positioned on the cells by an electronically controlled micromanipulator (Eppendorf; Radiometer, Copenhagen, Denmark). Experiments were controlled by an EPC-9 amplifier (HEKA, Lambrecht/Pfalz, Germany) connected to a Macintosh computer by an ITC-16 interface. Data were filtered at 3 kHz. In whole-cell experiments, the series resistance was below 8 MΩ, and 80% compensation was performed before application of each voltage ramp or step.
The extracellular solution for whole-cell experiments on SK1-SK3 and IK channels contained 144 mM NaCl, 4 mM KCl, 0.1 mM CaCl2, 3 mM MgCl2, and 10 mM HEPES. pH was adjusted to 7.4 with NaOH. For experiments with inside-out or outside-out patches a high K+ solution (154 mM KCl, 2 mM CaCl2, 1 mM MgCl2, and 10 mM HEPES with pH adjusted to 7.4 with KOH) was used on the extracellular side of the patch.
The intracellular solutions all contained 154 mM KCl and 10 mM HEPES as well as 10 mM EGTA or a combination of EGTA and nitrilotriacetic acid (10 mM in total). Concentrations of CaCl2 and MgCl2 needed to yield the desired free concentrations of Ca2+ (Mg2+ always 1 mM) were calculated by EqCal Software (Cambridge, UK) and added. The free Ca2+ concentrations were controlled by a Ca2+-sensitive electrode (WPI, Sarasota, FL). For whole-cell experiments, a constant free concentration of 0.4 μM was used in the pipette. This concentration was chosen to yield an intermediate activation of the SK channels. Excised patches were exposed to free [Ca2+]i in the range from 0.01 to 10 μM to cover the dynamic range of SK channel activation. All intracellular solutions were adjusted to pH 7.2 with concentrated HCl or KOH.
Slice Preparations and Electrophysiology. Transverse dorsal hippocampal slices (300 μm in thickness) were prepared from adult C57BL/6J mice (20-60 days old) using a VT1000S vibroslicer (Leica, Wetzlar, Germany), as described previously (Stocker et al., 1999). Throughout this procedure, the brain hemispheres were maintained in oxygenated (95% O2, 5%CO2) artificial cerebrospinal fluid (ACSF) at ∼4°C. The ACSF contained 125 mM NaCl, 1.25 mM KCl, 2.5 mM CaCl2, 1.5 mM MgCl2, 1.25 mM KH2PO4, 24 mM NaHCO3, and 16 mM glucose. The slices were subsequently incubated in a humidified interface chamber at room temperature (∼24°C) for ≥1h.
Giga-seal whole-cell recordings were obtained from the somata of CA1 pyramidal cells using the “blind” patch-clamp technique (Blanton et al., 1989). Patch pipettes (4.0-6.5 MΩ) were pulled from borosilicate glass capillary tubes in a two-stage vertical puller (PP-830; Narishige, Tokyo, Japan) and were filled with a pipette solution containing 135 mM potassium methylsulfate, 10 mM KCl, 10 mM HEPES, 1 mM MgCl2, 2mMNa2-ATP, and 0.4 mM Na3-GTP; pH 7.2-7.3 with KOH; osmolarity 280-295 mOsM. The pipette solution contained 50 μM 8-(4-chlorophenylthio)-cAMP to inhibit sIAHP (Stocker et al., 1999). Recordings were performed in a submersion chamber with a constant superfusion with oxygenated 2 ml/min ACSF at room temperature. Once the whole-cell configuration had been established, the access resistance was regularly monitored and maintained at ≤25 MΩ for the duration of the experiment. No series resistance compensation and no corrections for liquid junction potentials were made.
Pyramidal cells were clamped at a membrane holding potential of -50 mV and depolarized to +10 mV for 100 ms every 30 s to activate voltage-gated Ca2+ channels. The membrane potential was then stepped back to -50 mV where the IAHP was visualized as a Ca2+-activated tail current. These experiments were conducted in the presence of 0.5 μM tetrodotoxin (TTX) and 1 mM tetraethylammonium (TEA) to block voltage-gated Na+ channels and some voltagegated K+ channels. Data were generated and acquired using an EPC9 amplifier (HEKA) and the software Pulse (HEKA). Data were filtered at 1 kHz and sampled at 4 kHz.

.Synthesis of NS8593.
Chemicals.125I-Apamin (2200 Ci/mmol) was purchased from PerkinElmer Life and Analytical Sciences. Apamin and TTX for slice electrophysiology were obtained from Latoxan (Rosans, France), d-tubocurarine (dTC) was from Fluka (Buchs, Switzerland), and XE991 was from Tocris Cookson Inc. (Bristol, UK). Apamin and dTC for binding assays and experiments on cell lines, charybdotoxin (ChTX), TEA, clotrimazole (Clot), dequalinium chloride, Na2ATP, Na3GTP, DMSO, and 8-(4-chlorophenylthio)-cAMP were purchased from Sigma-Aldrich. Potassium methylsulfate was purchased from ICN (Aurora, OH), and HEPES was from BIOMOL Research Laboratories (Hamburg, Germany). UCL 1684 (Campos Rosa et al., 2000) and NS309 (Jensen et al., 2000) were synthesized at NeuroSearch (Ballerup, Denmark). All other chemicals were purchased from regular commercial sources and were of the purest grade available.
For the electrophysiological experiments, apamin, TTX, dTC, XE991, ChTX, and TEA was dissolved in the salt solutions used. Clot, dequalinium chloride, UCL 1684, NS309, and NS8593 were dissolved in DMSO and diluted at least 1000 times in the experimental solutions. The presence of 0.1% DMSO had no effect on the currents recorded.
Calculations, Fitting Routines, and Statistics. IC50 values were estimated from a nonlinear regression fit to a Hill-type equation: 
where B is the fraction of relative current in patch-clamp experiments or specific binding in apamin binding experiments, C is the test concentration, and n is the Hill coefficient.
Ki values for binding of test ligands were calculated from IC50 values using the Cheng and Prusoff (1973) equation: 
The dissociation constant (Kd) values determined for 125I-apamin binding to striatal tissue and hSK3 membranes were 4.1 and 4.0 pM, respectively.
Kd values for functional inhibition of SK channels were obtained by fitting SK current versus time data to eq. 3 (Jenkinson, 1996): 
where the Kd is defined as koff/kon [the dissociation (s-1) and association (M-1 s-1) rate constants, respectively]. For the equilibrium situation (t → ∞) and for n = 1, eq. 3 reduces to eq. 1 (and thereby IC50 = Kd).
The potency of inhibition is given and discussed as the IC50 value herein when it is obtained by patch-clamp equilibrium concentration-response experiments, as the Kd value when obtained by patch-clamp kinetics experiments, and finally as the Ki value when obtained from displacement of radioactive apamin in equilibrium binding experiments.
Fitting routines were performed with the IGOR software (Wave-metrics, Lake Oswego, OR) using customized macros, with Excel (Microsoft, Redmond, WA), SigmaPlot (SPSS Inc., Chicago, IL), and InStat and Prism (GraphPad Software Inc., San Diego, CA). Averaged data are given as means ± S.E.M.; statistical differences were determined by the Student's t test with p = 0.05 taken as level of significance.
Results
The novel compound NS8593 (Fig. 1A) was synthesized as part of a medicinal chemistry program (Sørensen et al., 2006). It is a chiral 2-aminobenzimidazole derivative without structural and physiochemical resemblance to any known SK channel blocker. In particular, NS8593 is a small (mol. wt. 263 g/mol), lipophilic compound (calculated log D = 3.9; pH 7.4) with basic properties, although without permanent positive charges.
Inhibition of Recombinant SK3 Channels by NS8593. The effect of NS8593 on typical whole-cell SK3 currents is illustrated in Fig. 1B. A prominent K+ current (control; Vrev of ca. -85 mV) was elicited by voltage ramps (-120 mV to +30 mV) in a cell exposed to physiological salt gradients and perfused with a pipette solution buffered at 0.4 μM free Ca2+. Addition of 100 nM NS8593 reduced the current and shifted the Vrev toward more positive potentials, consistent with decreasing a K+ conductance. Superfusion with high concentrations of bicuculline methobromide (BMB; 100 μM) and 30 nM apamin to the same cell completely blocked the current elicited at potentials more negative than -30 mV, demonstrating that it was mediated by SK channels. At more positive potentials, a residual K+ current was observed in the presence of apamin and BMB because of activation of endogenous voltage-dependent K+ currents in HEK293 cells. The endogenous HEK293 K+ current was not significantly affected by NS8593 in the applied concentration range. The NS8593-induced inhibition of SK3 current was analyzed at -30 mV (Fig. 1B, arrow) and the entire time course of the experiment is depicted in Fig. 1C. Because BMB is an extremely fast and fully reversible blocker of SK channels, it was applied at the beginning of the experiment to identify the SK-mediated current. After washing out BMB, a reversible inhibition was induced by a 3-min application of 100 nM NS8593. Finally, application of 30 nM apamin fully blocked the SK3 current.
Figure 1D shows a concentration-response curve for the NS8593-induced reduction of the SK3-mediated current. An IC50 value of 91 nM was calculated by fitting to eq. 1. The Hill coefficient was 1.15, implying that the effect of NS8593 does not show cooperativity. Figure 1D furthermore shows that full inhibition can be obtained.
The kinetics of the NS8593-induced inhibition was investigated from the same set of experiments as used for Fig. 1D. Kd values were calculated by fitting the time courses of the NS8593-induced decrease in current to eq. 3 and are plotted as a function of the compound concentration in Fig. 1E. Very similar Kd values are obtained when NS8593 is applied in the concentration range from 30 nM to 3 μM. The data in Fig. 1, D and E, furthermore pinpoint that the mean of the Kd values (90 ± 8.0 nM; n = 33), determined in this concentration range, is identical to the IC50 value obtained from the equilibrium concentration-response calculation (91 nM). Therefore, the kinetic approach was used in all subsequent experiments. The mean association and dissociation rate constants for NS8593 were 1.4 × 105 M-1 s-1 and 0.012 s-1, respectively. For comparison, the kon and koff values obtained for apamin (applied at 0.3-1 nM) with the same method were 1.4 × 107 M-1 s-1 and 0.0046 s-1, respectively. The (S)-enantiomer of NS8593 also inhibited SK3 channels, although less potently than NS8593 (Kd = 448 ± 89 nM; n = 5; data not shown).
All data in Fig. 1 were obtained with the rSK3 isoform, but identical results were obtained with NS8593 and apamin on hSK3 channels (Table 2). The affinity of NS8593 for rSK3 was also tested in symmetrical 154 mM K+ solutions as used in the excised patch experiments (see below). Under these conditions, the Kd value was slightly higher (169 ± 53 nM; n = 4).
Inhibition of hSK3 currents and of 125I-apamin binding to hSK3 and striatal membranes
Kd values were calculated from whole-cell hSK3 measurements as shown in Fig. 1, and Ki values were from binding experiments using rat striatal membranes and membranes from HEK293 cells stably expressing hSK3. All values shown are mean ± S.E.M. from three to seven experiments. In binding experiments, the calculated Hill coefficients were in the range 0.88 to 1.24.
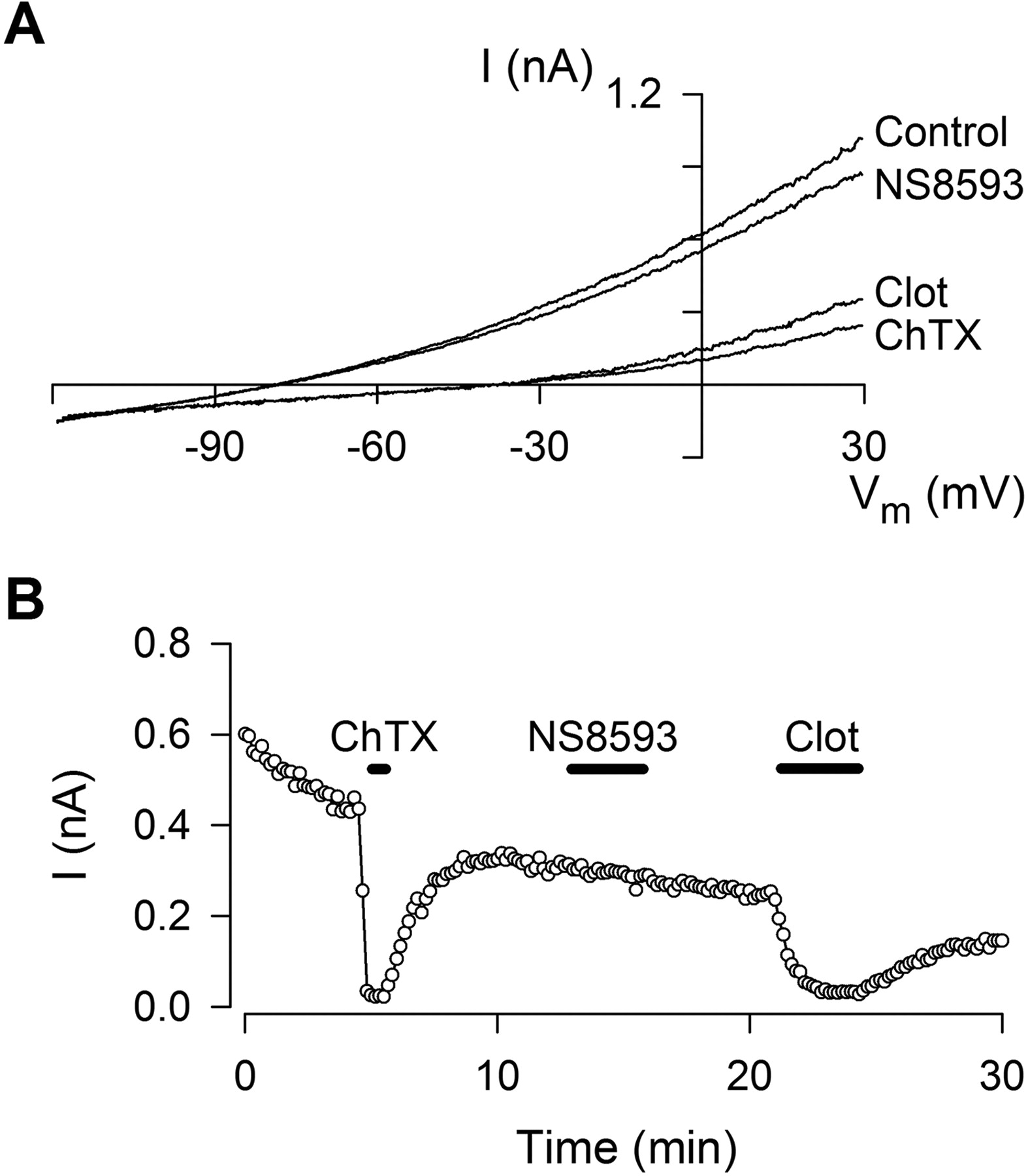
Selectivity of NS8593 toward Other Ion Channels. The selectivity of NS8593 toward the other members of the SK/IK family was tested. Figure 2 shows a representative whole-cell experiment performed on hIK channels expressed in HEK293 cells. The experimental strategy is similar to the strategy used in Fig. 1. After stabilization of the whole-cell hIK current upon Ca2+ equilibration, the channel was blocked by addition of 100 nM ChTX, a reversible IK channel peptide blocker. After washout, 10 μM NS8593 was applied for 4 min, during which there was no associated decrease in current or change in the reversal potential. The experiment was terminated by addition of 1 μM Clot. Thus, NS8593 discriminates between IK and SK types of voltage-independent, Ca2+-activated K+ channels. Table 1 further shows that NS8593 is inactive also on the voltage-dependent BK channel and lists the effects of NS8593 on a number of other K+ channels as well as on voltage-dependent Na+ and Ca2+ channels measured in the whole-cell patch-clamp mode. NS8593 exhibits considerable selectivity for SK channels, but it is not a specific inhibitor at concentrations exceeding 10 μM.
Effect of NS8593 on selected ion channels
NS8593 was tested in whole-cell experiments, and Kd values were obtained from a fit to eq. 3. Voltage-ramp protocols were used for SK3, IK, and BK channels, whereas standard voltage-step protocols were used for the other channels listed. Recombinant channels were expressed in HEK293 cells, and the native channels were measured in rat eDRG neurons.
SK Subtype Selectivity. Apamin is the reference SK channel pore blocker, and it shows a characteristic SK channel subtype selectivity: SK2 = 0.074 ± 0.01 nM (n = 10); SK3 = 0.33 ± 0.05 nM (n = 18); SK1 = 2.9 ± 0.3 nM (n = 14) based on Kd values from pooled whole-cell data (rat/human, 4 mM/150 mM extracellular K+). For comparison, the SK subtype selectivity of NS8593 was explored in a series of experiments using excised multichannel patches, where the channel activity is independent of the intracellular milieu. Because the recombinant rSK1 channels do not readily express, the studies were performed on the human SK isoforms to avoid potential species-related differences. Symmetrical [K+] solutions were used to increase current amplitude, and the holding potential was 0 mV, which largely inactivated the endogenous KV channels. Figure 3 shows the current-voltage (I-V) relationship for the three SK channel subtypes obtained from inside-out patches in the presence of 0.5 μM free Ca2+ before and after application of 3 μM NS8593 (left). There is no significant difference between the degree and kinetics of inhibition by NS8593 for all SK channel subtypes. The Kd values (at -75 mV, nanomolar) were 415 ± 101 (hSK1; n = 4), 598 ± 51 (hSK2; n = 4), and 726 ± 115 (hSK3; n = 7). These values are not significantly different. Furthermore, no voltage-dependence of NS8593-induced inhibition was detected for any of the subtypes (comparison of Kd values at -75 and at 75 mV; data not shown).

NS8593 inhibits recombinant SK3 channel activity. A, chemical structure of NS8593. B, whole-cell I-V relationships, measured in HEK293 cells stably expressing rSK3, upon application of 200-ms-long voltage ramps (-120 to +30 mV), elicited every 5 s from a holding potential of -90 mV. The experiment was conducted with 4 mM K+ in the extracellular solution and a pipette solution with a free Ca2+ concentration of 0.4 μM. I-V relationships were measured in the absence of compound (control) and in the presence of 100 μM BMB, 30 nM apamin, or 100 nM NS8593. The arrow indicates the point of analysis. C, SK3 current at -30 mV obtained from voltage ramps (B) as a function of time. The cell was exposed to 100 μM BMB, 100 nM NS8593, and 30 nM apamin as indicated by bars. D, concentration-response relationship for NS8593 obtained from experiments conducted as in C. Residual current in the presence of NS8593 is depicted as a function of NS8593 concentration. The solid line is the fit of the data to the Hill equation, yielding an IC50 value of 91 nM and a Hill coefficient of 1.15. Each data point is the mean ± S.E.M. of 5 to 12 experiments. E, Kd, as a function of the NS8593 concentration. Kd values, estimated from the kinetic fit described under Materials and Methods, were obtained from the same experiments used for the IC50 estimate summarized in D. The dotted line symbolizes a mean Kd of 90 nM.
The Kd values obtained from inside-out patches on SK3 are higher than the corresponding values obtained in the wholecell experiments at high potassium (726 versus 169 nM). In the two configurations NS8593 is applied to the intracellular and extracellular side of the membrane, respectively. Furthermore, in whole-cell experiments the Ca2+ concentration cannot be accurately controlled, whereas the inside-out configuration isolates channel activity from cellular regulation, including changes in Ca2+ concentration and channel phosphorylation. We therefore performed a series of experiments on inside-out and outside-out patches to test for a possible sidedness or a possible Ca2+ dependence of the inhibitory action of NS8593 in the absence of ATP.

NS8593 does not affect recombinant IK channel activity. A, whole-cell I-V relationship, measured in HEK293 cells stably expressing hIK. Voltage protocol and solutions were as described in Fig. 1A. I-V relationships were measured under control conditions and after applications of 100 nM ChTX, 10 μM NS8593 (n = 12), or 1 μM Clot. B, whole-cell IK current at -30 mV plotted as a function of time. The 100 nM ChTX, 10 μM NS8593, and 1 μM Clot were applied as indicated by bars.
Ca2+ Dependence of the NS8593-Mediated Inhibition. In Fig. 4A, it is shown that 3 μM NS8593 induced a prominent reduction of the SK3-mediated current when applied to outside-out patches in the presence of 0.5 μM free Ca2+ in the pipette as observed in inside-out patches. Apamin (100 nM) and 200 μM BMB were added as control of the patch configuration and to pharmacologically isolate the SK-mediated current. NS8593 was tested in the presence of different Ca2+ concentrations, and Fig. 4B shows the average time courses of inhibition plotted for 0.5 and 10 μM intracellular Ca2+ from inside-out (open circles) and outside-out (closed circles) experiments, respectively. The curves represent the best fit to eq. 3. NS8593 (3 μM) caused an almost full inhibition of the SK3-mediated current at 0.5 μM Ca2+, whereas the inhibition was substantially smaller in the presence of 10 μM Ca2+. This is further quantified in Fig. 4C that shows a plot of the calculated Kd values as a function of the intracellular [Ca2+]. The inhibition by NS8593 decreased with increasing intracellular [Ca2+] (0.3-10 μM), but it was not dependent on the side of compound application. Using inside-out patches, the Kd value increased from 0.47 ± 0.11 μM (n = 5) at 0.3 μM Ca2+ to 14 ± 3.4 μM (n = 6) at 10 μM Ca2+. The corresponding values for outside-out patches were 0.70 ± 0.20 μM (n = 7) and 6.6 ± 1.5 μM (n = 6). This shows that the affinity of NS8593 for SK channels is Ca2+-dependent, and the most likely explanation for the higher affinity observed in the whole-cell experiments is that the actual intracellular free Ca2+ concentration near the Ca2+ binding site on calmodulin is substantially lower than the nominal buffered 0.4 μM in the whole-cell pipette. Extrapolating the Kd versus [Ca2+]i curves from the excised patches suggests that the local concentration in the whole-cell configuration is more likely to be 0.1 to 0.2 μM.
The mechanism underlying the Ca2+ dependence of inhibition by NS8593 was analyzed further in inside-out patches. In these experiments, the intracellular [Ca2+] was varied systematically in small steps in the range of 0.01 μM (closed channels) to 100 μM (maximally Ca2+-activated channels) in the absence or in the presence of NS8593. Examples of ramp I-V curves obtained at 0.3, 0.5, and 10 μM Ca2+ in the absence of NS8593 are shown in Fig. 5A. In Fig. 5B, the same Ca2+ concentrations were used but in the presence of 3 μM NS8593. The currents obtained in 0.3 and 0.5 μM Ca2+ were clearly reduced by NS8593, whereas the inhibition was more modest in 10 μM Ca2+ (the dashed control curve represents the current in the same patch obtained in 10 μM Ca2+ in the absence of NS8593).
Figure 5C shows a summary of the effect of NS8593 on the full activation curve for Ca2+. In the control situation, hSK3 channels were highly sensitive to Ca2+ with an EC50 value of 0.43 μM and a Hill coefficient of 5.6 (n = 18). In the presence of 3 μM NS8593, the channels had a reduced Ca2+ sensitivity with an EC50 of 1.6 μM and a Hill coefficient of 2.0 (n = 6). The observed shift in the activation curve for Ca2+ implicates that the effect of NS8593 is pronounced at the low Ca2+ concentrations, whereas the inhibition is essentially abolished at 30 μM Ca2+. It should be noted that very similar Ca2+ activation curves were obtained of hSK1- and hSK2-mediated currents and that the affinity for NS8593 was Ca2+-dependent on these subtypes as well (data not shown).
The pronounced Ca2+ dependence of the NS8593-induced inhibition may reflect that NS8593 binds to and stabilizes the SK channel in a closed state. To elucidate this possibility, we conducted experiments as outlined in Fig. 5D. After establishment of the leak current in 0.01 μM Ca2+, the fast and monophasic response to 3 μM Ca2+ was demonstrated. The channels were then closed by a 4-min superfusion with 0.01 μM Ca2+ in the presence of 3 μM NS8593. The subsequent hSK3 activation by 3 μM Ca2+ was now drastically changed to a biphasic response, consisting of an initial fast component followed by a slow sigmoid phase approaching the current level of the unmodified channel within 5 min. This illustrates that a significant inhibition developed during the closed period and the slow phase is interpreted as Ca2+ displacement of NS8593 gradually reducing the inhibition to 15%. After washout of NS8593, the deactivation/activation procedure was repeated, showing that the monophasic, fast response to 3 μM Ca2+ was completely restored and that NS8593 applied after Ca2+ activation inhibited hSK3 by 17% in good agreement with the results from Figs. 4C and 5C.
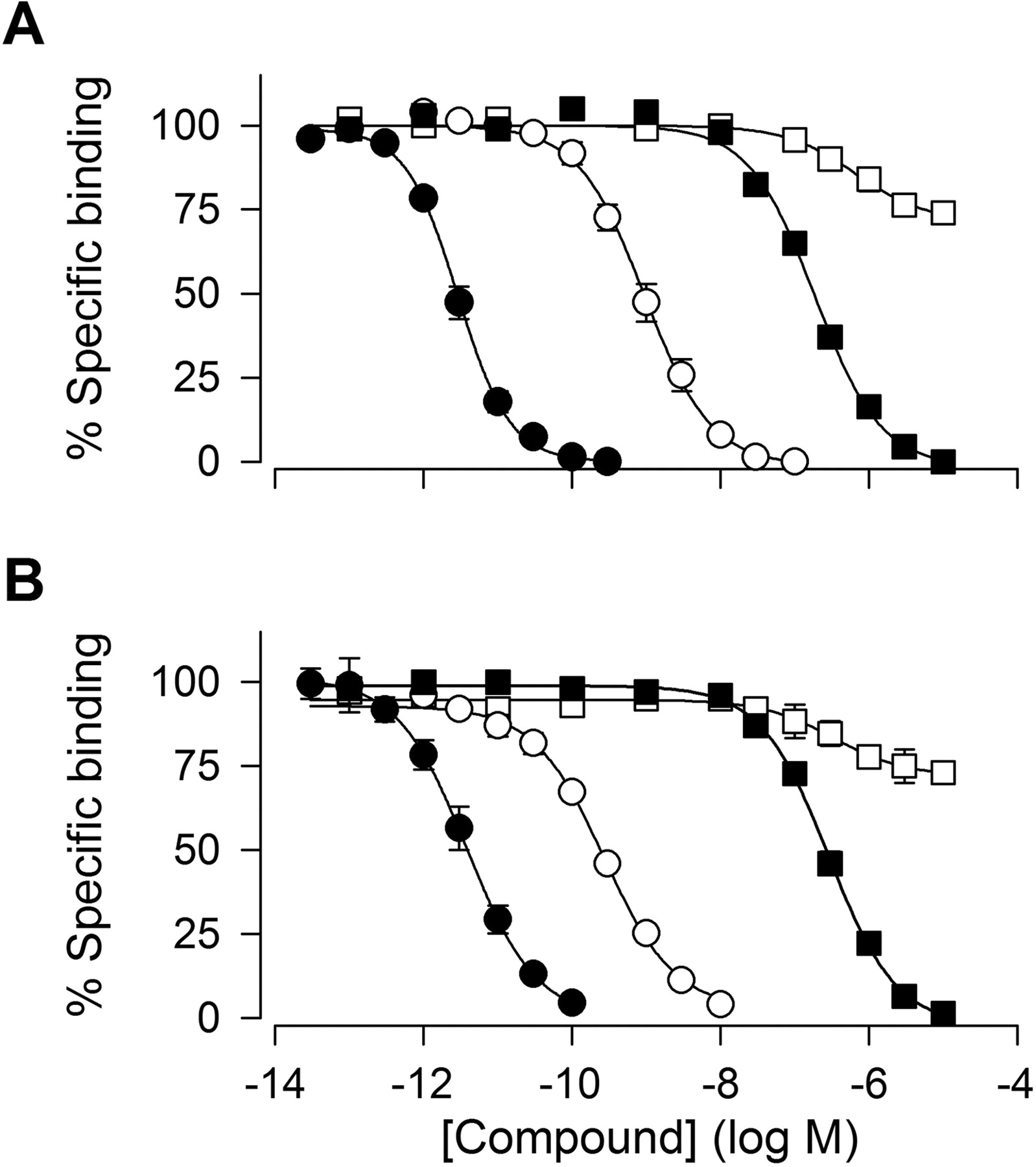
Apamin Binding. To investigate whether NS8593 interacts with the apamin binding site, 125I-apamin binding studies were performed. Figure 6 shows displacement curves for apamin, UCL 1684, dequalinium, and NS8593 on HEK293 cells expressing hSK3 (A) and on rat striatal membranes (B), which predominantly express the SK3 isoform (Sailer et al., 2004). Apamin, UCL 1684, and dequalinium all fully inhibited the specific 125I-apamin binding with Hill coefficients close to 1, whereas NS8593 only caused a 30% reduction at the highest concentrations tested. The rank order of potency in both preparations was apamin, UCL 1684, dequalinium, and NS8593 (>10 μM). A summary of binding Ki values and patch-clamp Kd values is given in Table 2.
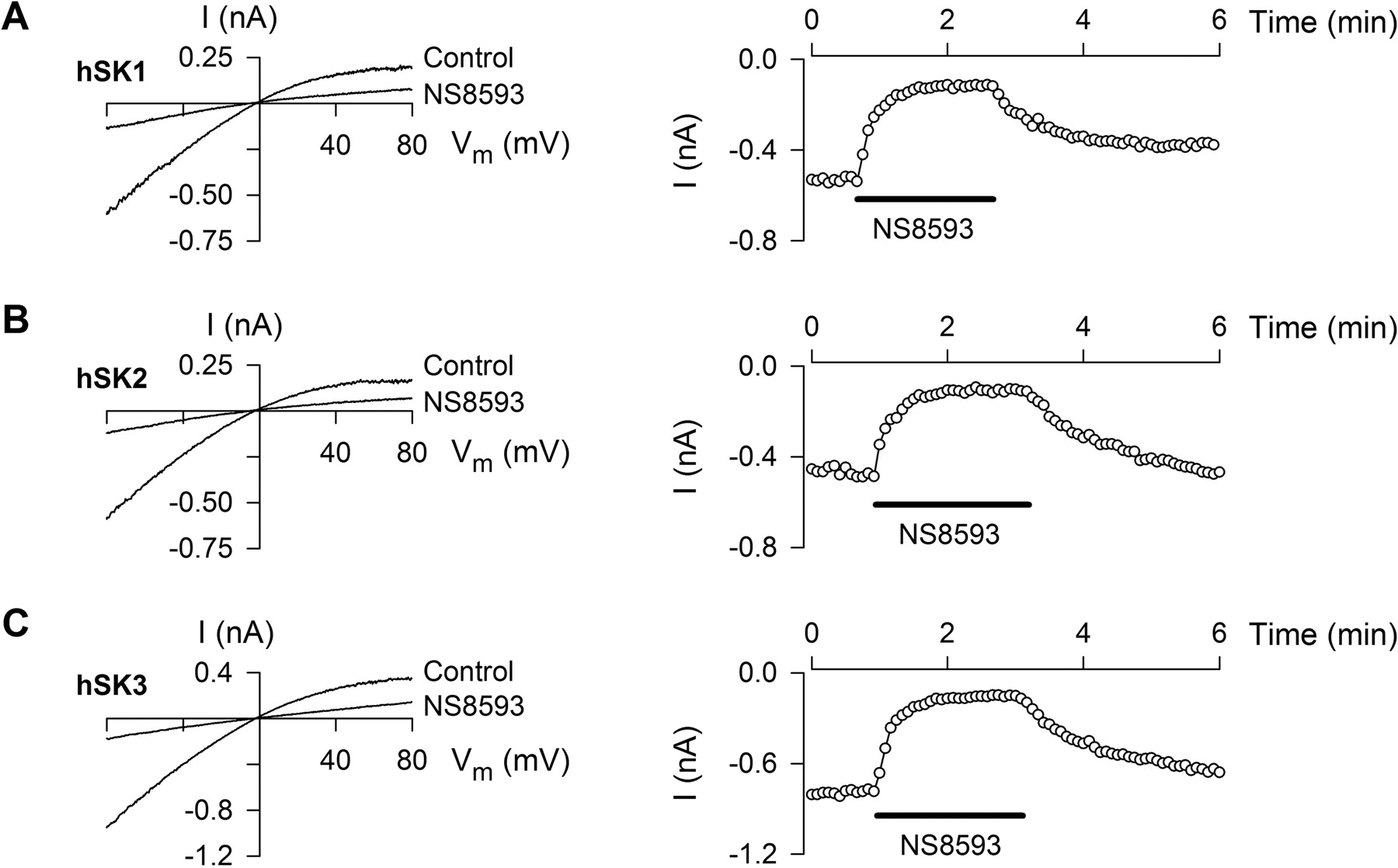
NS8593 inhibits SK1, SK2, and SK3 channels equipotently. Currents measured from inside-out patches obtained from HEK293 cells stably expressing hSK1 (A), hSK2 (B), or hSK3 (C) upon application of 200-ms-long voltage ramps (-80 to +80 mV) elicited every 5 s from a holding potential of 0 mV. Each I-V plot (left) shows a control trace and a trace obtained in the presence of 3 μM NS8593 (applied to the bath/intracellular solution). The free [Ca2+]in the bath/intracellular solution was 0.5 μM in all experiments. Right, the current at -75 mV is depicted as a function of time. NS8593 (3 μM) was applied as indicated by bars. The Kd values for NS8593-induced inhibition are expressed as mean ± S.E.M.: 415 ± 101 nM (SK1; n = 4), 598 ± 51 nM (SK2; n = 4), and 726 ± 115 nM (SK3; n = 7).

NS8593 inhibits SK3 channel activity equally well when applied to the intra- or the extracellular side of the membrane. A, current at -75 mV measured from an outside-out patch obtained from a HEK293 cell stably expressing hSK3. The currents were obtained from 200-ms-long voltage ramps (-80 to +80 mV) elicited every 5 s from a holding potential of 0 mV. [Ca2+]i was 0.5 μM, and 3 μM NS8593, 100 nM apamin, and 200 μM BMB were applied to the extracellular/bath solution as indicated by bars. B, time courses of the NS8593-induced inhibition of SK3 current at a [Ca2+]i of 0.5 μM (left) and 10 μM (right). The two top panels are derived from inside-out (IO) experiments (○), whereas the two bottom panels are from experiments performed in the outside-out (○○) configuration (•). Currents were normalized with respect to the steady-state current before 3 μM NS8593 application. Data points are mean ± S.E.M. of four to nine experiments. C, Kd as a function of [Ca2+]i measured from inside-out (○) and outside-out (•) patches.
Interactions of NS8593 and Apamin with the Positive Modulator NS309. The apparent mirror-like action of the positive and negative modulators on the activation curve for Ca2+ (left and right shift, respectively) may suggest that NS8593 inhibits the SK channel gating by an interaction that shares the mechanism of action or even the physical binding site with the positive modulators such as 1-EBIO and NS309. It was tested whether NS8593 was able to inhibit SK3 channels, which were fully activated by a high concentration of the positive modulator NS309, in the presence of a low concentration of intracellular Ca2+ (Fig. 7A). First, the insideout patch was exposed to 0.3 μM Ca2+, partially activating the SK3 channels, and subsequent superfusion with 3 μM NS8593 at this [Ca2+] gave the expected pronounced inhibition (87 ± 1%; n = 5). After washout of NS8593 and a short exposure to 0.01 μM Ca2+, the channel was activated by a combination of 0.3 μM Ca2+ (as before) and 10 μM NS309. Under these conditions, the inhibition by NS8593 was profoundly reduced (16 ± 3%; n = 3). After sequential washout of both compounds and a brief exposure to 0.01 μM Ca2+, the channels were fully activated with 10 μM Ca2+ (in the absence of NS309). In the presence of such high [Ca2+], NS8593 exerted only a weak inhibitory effect (26 ± 6%; n = 6). The experiment was concluded with a repetition of the initial NS8593 application in the presence of 0.3 μM Ca2+. Similar experiments were performed using outside-out patches. In the experiment shown in Fig. 7B, profound inhibitions of the hSK3 current activated by 0.3 μM Ca2+ in the pipette solution were first obtained upon superfusion with 100 μM BMB and subsequently with 3 μM NS8593. The channels were then further activated by application of 10 μM NS309, and, as observed with the inside-out experiments, this diminished the inhibition obtained by 3 μM NS8593 from 77 ± 4.3% (n = 7) to 17 ± 2.0% (n = 7). In contrast to this, the addition of 0.3 nM apamin in the continued presence of NS309 resulted in the same Kd value (0.26 ± 0.10 nM; n = 5) as observed in whole-cell experiments without NS309 (Table 2), indicating that apamin-mediated inhibition of SK3 channels, in contrast to NS8593, is independent of full occupancy by NS309 at the positive modulator site.

NS8593 inhibits SK3 channel activity in a Ca2+-dependent manner. A, I-V relationship measured from inside-out patches obtained from HEK293 cells stably expressing hSK3. The currents were obtained from 200-ms-long voltage ramps (-80 to +80 mV) elicited every 5 s from a holding potential of 0 mV. Left, inside-out patch exposed to a [Ca2+]i of 0.3, 0.5, or 10 μM as indicated. B, 3 μM NS8593 was present in the solutions containing 0.3, 0.5, or 10 μM free Ca2+, except for the dashed trace, which was obtained in 10 μM Ca2+ after NS8593 washout. C, Ca2+ concentration-response curves measured under control conditions or in the presence of 3 μM NS8593. The current at -75 mV was measured from inside-out patches exposed to increasing [Ca2+]i either in the absence (○) or in the presence of 3 μM NS8593 (•). Currents from individual patches were normalized with respect to the effect of 10 μM Ca2+, which induces maximal SK3 activity. Data points are mean ± S.E.M. of 18 experiments under control conditions and 6 experiments in the presence of 3 μM NS8593. The solid lines are the fit of data to the Hill equation, yielding an EC50 value for Ca2+ of 0.42 μM and a Hill coefficient of 5.6 under control conditions and of 1.6 μM and 2.0 in the presence of 3 μM NS8593. D, inhibition of the channels in the deactivated state. The current at -75 mV was measured from an inside-out patch exposed 0.01 and 3 μM [Ca2+]i as indicated at the abscissa axis. NS8593 (3 μM) was present in the bath solutions as indicated by the bars.
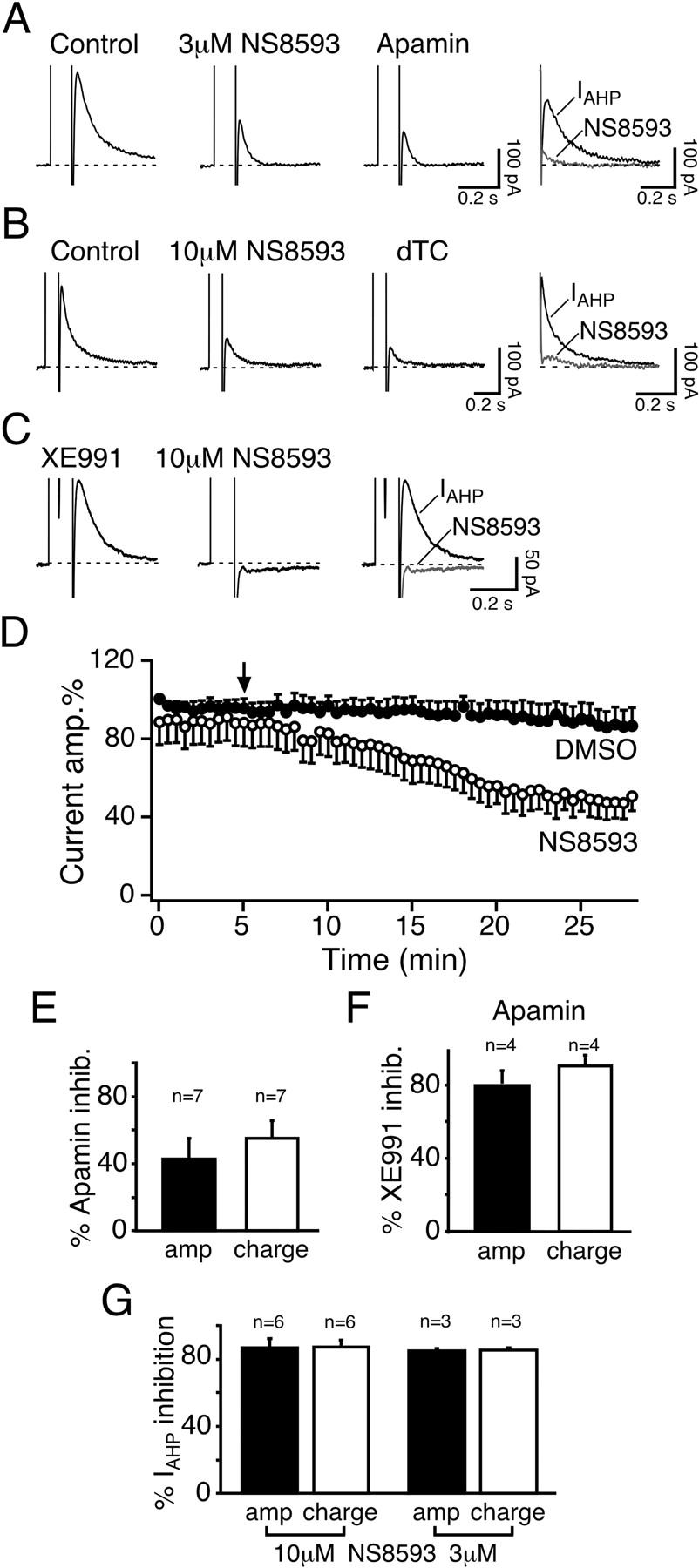
Effect of NS8593 on the SK-Mediated IAHP in Hippocampal Slices. Apamin-sensitive SK channels mediate IAHP, a voltage-independent, Ca2+-activated K+ current, in hippocampal pyramidal neurons (Stocker et al., 1999, 2004). In whole-cell voltage-clamp recordings from mouse CA1 pyramidal neurons the tail current after a depolarizing step to 10 mV was only partly suppressed by apamin (50 nM; 42.9 ± 12.1% peak amplitude inhibition; n = 7; Fig. 8, A and E). The current remaining after SK channel blockade (Fig. 8A, apamin, and B, dTC) was not Ca2+-dependent, as tested by applying a nominally Ca2+-free ACSF containing 5 mM Mg2+ (n = 4; data not shown). It was largely suppressed by higher concentrations of 5 mM TEA [inhibition of residual current amplitude: 74.0 ± 9.7%; n = 7; p < 0.01; inhibition of residual current integral (charge transfer): 96.6 ± 3.4%; n = 7; p < 0.0001; data not shown], and by the Kv7/KCNQ/M channel blocker XE991 [5 μM; inhibition of residual current amplitude: 80.5 ± 7.6%; n = 4; p < 0.01; inhibition of residual current integral (charge transfer): 90.4 ± 5.9%; n = 4; p < 0.001; Fig. 8F]. Given its pharmacological profile, this residual current is most likely IM, a noninactivating, voltage-dependent K+ current that is mediated by KCNQ channels (Gu et al., 2005). We did not investigate this current component in further detail and focused instead on IAHP that we isolated by subtracting the apamin- or dTC-resistant current from current traces obtained before and after NS8593 application (Fig. 8, A and B, right) or by applying XE991 to block the IM component (Fig. 8C).

Apamin, UCL 1684, dequalinium, but not NS8593, inhibit 125I-apamin binding. Inhibition of 125I-apamin binding to membranes from HEK293 cells stably expressing hSK3 (A) and rat striatal membranes by apamin (•), UCL 1684 (○), dequalinium (•), and NS8593 (□) (B). hSK3 or striatal membranes were incubated with 125I-apamin (2.7-8.3 pM) in 50 mM Tris-HCl buffer, pH 8.5, containing 5 mM KCl, 0.1% BSA, and competing compounds. Binding was terminated after 90 min by filtration. Each data point represents the mean ± S.E.M. for three to four experiments. Curve fitting was performed as described under Calculations, Fitting Routines, and Statistics. IC50 values for hSK3 were as follows: apamin, 2.9 pM; UCL 1684, 950 pM; dequalinium, 190 nM; and NS8593 > 10 μM; and for rat striatal membranes: apamin, 3.8 pM; UCL 1684, 270 pM; dequalinium, 290 nM; and NS8593 > 10 μM.
Application of 10 μM NS8593 significantly reduced the total tail current by inhibiting IAHP (IAHP amplitude inhibition: 86.7 ± 5.1%; n = 6; p < 0.0001; IAHP charge transfer inhibition: 87.2 ± 4.0%; n = 6; p < 0.0001; Fig. 8, B, D, and G). Subsequent application of the SK channel blocker dTC (50 μM) had no further effect on the remaining current amplitude or charge transfer (n = 6; p > 0.1; Fig. 8B), indicating that 10 μM NS8593 had completely suppressed the SK-mediated IAHP. This was further confirmed by experiments performed in the presence of 5 μM XE991 to suppress IM, whereby the residual IAHP was fully inhibited by 10 μM NS8593 (IAHP amplitude inhibition: 98.4 ± 1.6%; n = 3; p < 0.001; IAHP charge transfer inhibition: 100 ± 0%; n = 3; p < 0.0001; Fig. 8C). Although the concentration used in this set of experiments is lower than the Kd values reported for Kv7 and Cav channels (Table 1), we were concerned that NS8593 might still produce slight effects on these conductances at 10 μM. We therefore performed a second set of experiments using a lower concentration of NS8593 (3 μM). As shown in Fig. 8A, application of 3 μM NS8593 resulted in a significant reduction of the total tail current amplitude by 49.9 ± 6.0% and the corresponding charge transfer by 67.8 ± 9.2% (n = 3; p < 0.02 for both measures). Subsequent application of apamin (50 nM) resulted in a slight further reduction of the tail current amplitude (p < 0.03; n = 3), but with no significant effect on the residual charge transfer (p > 0.1; n = 3). NS8593 (3 μM) suppressed the amplitude of the SK-mediated IAHP, obtained by subtraction, by 84.7 ± 1.4% (n = 3; p < 0.0003; charge transfer inhibition: 85.2 ± 1.3%, n = 3; p < 0.0001), as shown in Fig. 8A, right, and Fig. 8G. No significant difference was observed between the effect of 3 and 10 μM NS8593 (Fig. 8G), suggesting that 3 μMisa nearly saturating concentration. The compound was not tested at lower concentrations, however, because the time course of inhibition in the slices was rather slow, as illustrated in Fig. 8D, where inhibition by 10 μM NS8593 develops over 15 to 20 min. Control currents were therefore recorded in the presence of DMSO at the same final concentration used for the application of 10 μM NS8593 (0.1%; Fig. 8D). DMSO had no significant effect on the amplitude and only a small effect on the charge transfer of the recorded currents (amplitude: 86.1 ± 8.9%; n = 4; p > 0.2; charge transfer: 89.0 ± 2.5%; n = 4; p < 0.03). Altogether, these data show that NS8593 targets and strongly suppresses the SK-mediated IAHP in mouse hippocampal pyramidal neurons.
Discussion
In this study, we have identified and characterized a novel compound, NS8593, which is a potent and selective inhibitor of SK channels. In contrast to well established pore blockers, such as apamin and d-tubocurarine, NS8593 acts as an inhibitory gating modifier, interacting at a site distinct from the apamin binding site in SK channels. The evidence for this conclusion can be summarized as follows: 1) The potency of inhibition by NS8593 is strongly dependent on the intracellular [Ca2+]. 2) NS8593 causes a shift to the right of the Ca2+ activation curve, in analogy with the positive modulators, which cause a shift to the left. 3) Closed channel inhibition can be reversed by high Ca2+. 4) NS8593 does not displace 125I-apamin in the concentration range used to inhibit the SK currents in electrophysiological experiments. 5) In contrast to apamin, NS8593 exerts no selectivity toward the SK channel subtypes. 6) Also in contrast to apamin, NS8593 is a weak inhibitor of NS309-modulated SK channels. 7) NS8593 is structurally distant from the known SK blockers, and it does not fit into a recently described pharmacophore model for the external blocker site (Dilly et al., 2005). 8) NS8593 is the first inhibitory compound described that can reach its high-affinity binding site from both the inside and the outside of the cell membrane. Furthermore, this study shows that the inhibition by NS8593 follows the Hill-Langmuir formalism closely, because the nonequilibrium kinetics were monoexponential for both inhibition and washout and the potency determined from the kinetic measurements (Kd = koff/kon) equaled the IC50 determined from the concentration-response experiments.
The tail current measured from mouse CA1 pyramidal neurons was shown to be the sum of two components, IM and IAHP, being sensitive to XE991 and apamin/tubocurarine, respectively. The negative modulation by NS8593 was effective in inhibiting the SK-mediated IAHP. The Ca2+ dependence of the NS8593 potency suggests that neuronal SK channels mediating the IAHP are not exposed to saturating concentrations of intracellular Ca2+ after the depolarizing pulses used in this study. This is in good agreement with previous experiments, in which the positive modulators 1-EBIO (Pedarzani et al., 2001), 5,6-dichloro-1-ethyl-1,3-di-hydro-2H-benzimidazol-2-one, and NS309 (Pedarzani et al., 2005) increased the apamin-sensitive IAHP elicited by a similar depolarization protocol.

NS8593-mediated inhibition of SK3 is diminished by the positive modulator NS309. A, current at -75 mV measured from an inside-out patch obtained from a HEK293 cell stably expressing hSK3. The inside-out patch was exposed to a [Ca2+]i of 0.01, 0.3, or 10 μM as indicated. At 0.3 μM Ca2+, 0.3 μM Ca2+ + 10 μM NS309, or 10 μM free Ca2+, 3 μM NS8593 was applied as indicated by bars. The Kd values for NS8593-induced inhibition were 0.30 μM at 0.3 μM Ca2+, 27 μM at 0.3 μM Ca2+ with 10 μM NS309, and 24 μM at 10 μM Ca2+. B, SK3 current from an outside-out patch exposed to 100 μM BMB and 3 μM NS8593 in the absence of NS309 and then 3 μM NS8593 and 0.3 nM apamin in the presence of 10 μM NS309 as indicated by the bars. The Kd values for NS8593-induced inhibition were 0.43 μM in the absence and 8.9 μM in the presence of NS309. The Kd value for apamin was 0.16 nM. The pipette solution contained 0.3 μM free Ca2+. In both A and B, the currents were obtained from 200-ms-long voltage ramps (-80 to +80 mV) elicited every 5 s from a holding potential of 0 mV. Outside-out and inside-out experiments are labeled OO and IO, respectively.
From an enzyme kinetics point of view, the effect of NS8593 on SK channels can be described as a reduction in the Ca2+ affinity occurring concomitantly with a reduced cooperativity of Ca2+ binding. This is very much in analogy with the activators (1-EBIO), which accordingly cause an increase in Ca2+ affinity and slightly increase cooperativity (Pedarzani et al., 2001). However, the nature of the molecular mechanisms mediating the observed inhibitory gating modulation of SK channels by NS8593 is unknown as is also the case for the positive modulators, although the C terminal has been shown to be involved for 1-EBIO (Pedarzani et al., 2001). The present study, which furthermore demonstrates that high concentrations of NS309 (at low Ca2+) can also prevent NS8593 inhibition, shows that a functional interaction exists between the action of positive and negative modulators, an interaction that does not exist between the positive modulators and apamin (Fig. 7B). The simplest possibility is that positive and negative modulators share their binding site and that the functional interaction represents a direct competition between the two compounds. Such an interaction is well described for other ion channels such as the benzodiazepine site on GABA receptors, where agonists, inverse agonists as well as antagonists bind at the same site (for review, see Sieghart, 1994). Another example is the dihydropyridine site on L-type Ca2+ channels, which binds both negative and positive modulators (Greenberg et al., 1984). If the positive and negative modulators of SK channels share the same site it is noteworthy that NS8593 shows a high degree of selectivity for SK channels toward the IK channel, whereas the known positive modulators 1-EBIO and NS309 are more potent on IK than on SK channels (Strøbæk et al., 2004). However, even though the general gating mechanism depending on constitutively bound calmodulin is shared between SK and IK, it may be important to pay attention to the observed smaller steepness of the IK Ca2+ activation curve relative to SK (Ishii et al., 1997). Most likely this significant physiological difference reflects a structural variation among the SK and IK subtypes, a difference that may be related to the distinct gating-based pharmacology observed here. Future studies on the molecular mechanism may involve mutagenesis studies using SK/IK chimeras and/or mutated calmodulin analogs.
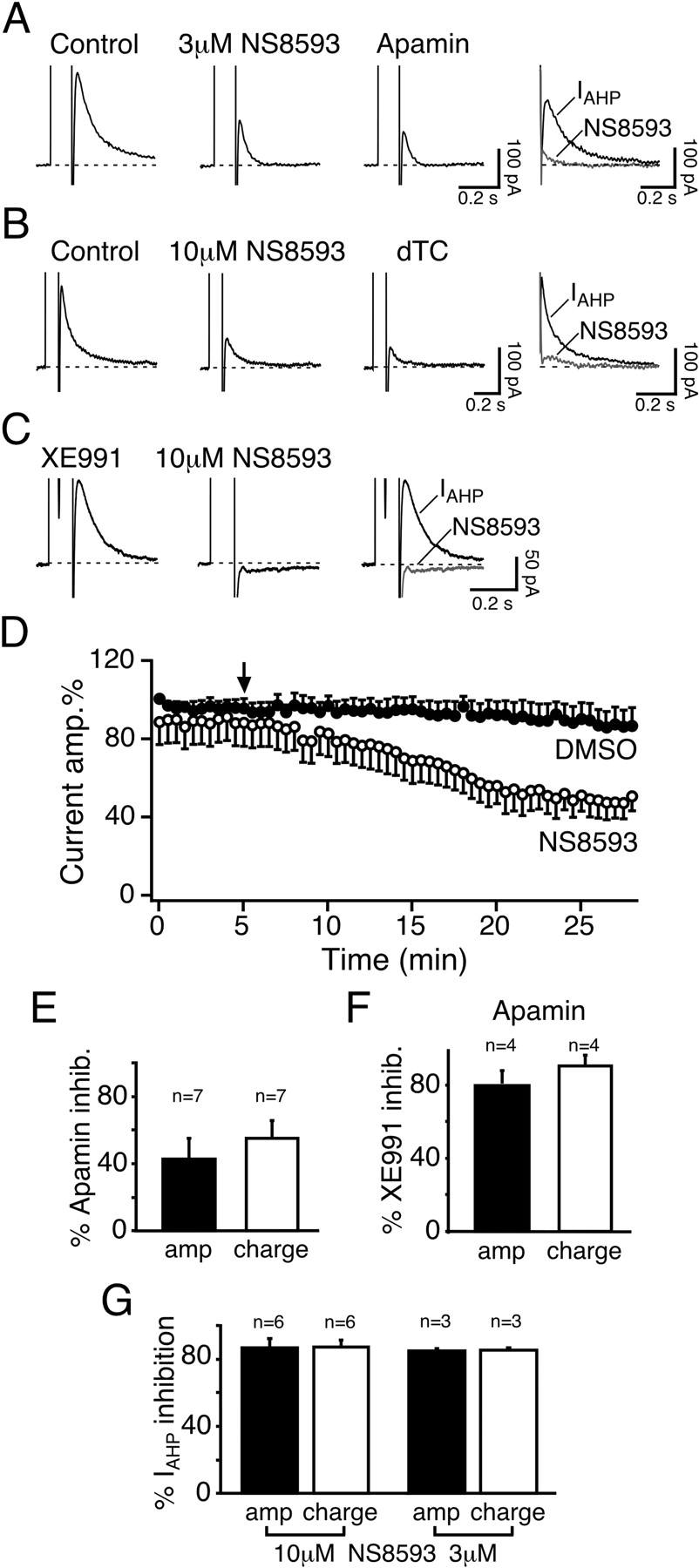
NS8593 inhibits the SK-mediated neuronal IAHP. A, tail currents measured in a mouse CA1 pyramidal neurons activated by 100-ms depolarizing pulses to 10 mV elicited every 30 s from a holding potential of -50 mV. The tail currents before and after application of 3 μM NS8593 is shown in the two left panels and the combined inhibition by NS8593 and 50 nM apamin. Right, SK-mediated component of the tail current, IAHP, was obtained by subtracting the current trace obtained in the presence of apamin from the control and NS8593 traces. B, tail currents obtained as in A, but with 10 μM NS8593 and using 50 μM dTC as a reference SK blocker. Right, IAHP was obtained by subtracting the current trace obtained in the presence of dTC from the control and NS8593 traces (also see A). C, tail currents obtained as in A, but in the presence of the Kv7/KCNQ/M channel blocker XE991 (5 μM; left). Further addition of 10 μM NS8593 led to a full suppression of IAHP (middle). Right, superimposed traces obtained in the presence of XE991 before (IAHP) and after (NS8593) the application of 10 μM NS8593. Similar results were obtained in three cells. D, time course showing the effect of 10 μM NS8593 on the normalized amplitude of the total tail current (comprising the SK-mediated I and a Ca2+AHP-independent, TEA-sensitive component) (○; n = 6). Control data were recorded in the presence of 0.1% DMSO (•; n = 4). The depolarizing step was applied every 30 s. NS8593 (10 μM) or 0.1% DMSO was applied at the time point indicated by the arrow. E, bar diagram summarizing the effect of 50 nM apamin on the total tail current. In seven cells, apamin suppressed 42.9 ± 12.1% of the current peak amplitude and 55.1 ± 9.9% of the charge transfer, measured as the area underneath the outward current. F, bar diagram summarizing the effect of 5 μM XE991 on the tail current elicited as described in A and remaining after the application of 50 nM apamin. In four cells, in the presence of apamin, XE991 suppressed 80.5 ± 7.6% of the residual current peak amplitude and 90.4 ± 5.9% of the charge transfer. G, bar diagram summarizing the maximal effect of different concentrations of NS8593 (3 μM, n = 3 and 10 μM, n = 6) on the amplitude (black bars) and charge transfer (white bars) of the SK-mediated IAHP obtained by subtraction (see A and B). Error bars are S.E.M.
Apamin was known as a very potent neurotoxin even before recognition of its molecular target. Intracerebral ventricular injections cause severe convulsions and seizures, and the peptide also exerts CNS toxicity after systemic administration. However, at low nonconvulsive doses, apamin has distinct behavioral effects, indicating a potential therapeutic value of blocking SK channels. Apamin increases the cognitive performance of mice in the Morris water maze and in the object recognition test (Stackman et al., 2002) as well as in the Y-maze (Deschaux and Bizot, 2005). SK channels have been shown to exert a postsynaptic feedback regulation of the N-methyl-d-aspartate receptor-mediated depolarization and Ca2+ influx in dendritic spines (Ngo-Anh et al., 2005). The augmented cognitive performance induced by apamin might therefore be linked to a facilitation of the induction of longterm potentiation. Apamin also reduced immobility time in the forced swimming test (Galeotti et al., 1999), an acute model for behavioral despair and a routine assay for the evaluation of the antidepressant potential of new drugs. This effect may be linked to an increased or changed pattern of electrical activity of monoaminergic neurons, which all express the SK3 subtype (Stocker and Pedarzani, 2000; Tacconi et al., 2001).
Despite these promising findings, however, apamin remains a very toxic compound, and all behavioral testing occurs at doses close to or even overlapping with the adverse effects (van der Staay et al., 1999). The reason for the limited therapeutic window may be the high potency and specific SK subtype selectivity of apamin, favoring SK2 over SK3 and SK1 channels. However, it cannot be excluded that the mode of action (pore blocking rather than modulatory inhibition) may in itself contribute to the development of uncontrolled neuronal firing leading to seizures. The molecular mechanism of action is thought to be an important safety and efficacy parameter for several CNS acting drug classes, such as use-dependent NaV blockers (for review, see Taylor and Narasimhan 1997) and glutamate receptor antagonists (for review, see Parsons 2001). An ion channel block without any feedback from the physiological activation process is generally less tolerable than competitive antagonism or allosteric modulation. Thus, Ca2+-dependent, inhibitory gating modulation, as described here for NS8593, may be anticipated to be most effective at neurons with relatively low firing frequencies, but ineffective at neurons with sustained, highfrequency firing. NS8593 may therefore be effective in subtly changing firing patterns (e.g., switch a dopaminergic neuron from pace-making to burst mode; Waroux et al., 2005; Ji and Shepard, 2006), without eliciting widespread neuronal hyperexcitability. Because NS8593 is a small molecule that does not carry any permanent positive charges and permeates cell membranes, it is potentially a compound that readily crosses the blood-brain barrier in vivo. A gating-modifying, reversible SK channel inhibitor of this type, and in particular a compound that exhibits SK subtype selectivity and further improved selectivity toward other ion channels, may help the elucidation of SK channels as possible therapeutic targets for the treatment of psychiatric and neurological diseases.
Acknowledgments
We thank Dr. Martin Stocker for useful discussions and Dr. Gayle M. Passmore for advice on the use of XE991. We thank Helle D. Rasmussen and Tine Sparre for technical assistance with the synthesis of NS8593. Jette Sonne, Anne Stryhn Meincke, and Vibeke Meyland-Smith are acknowledged for assistance with patch-clamp experiments, and we thank Susanne Kalf Hansen and Anne B. Fischer for running the apamin binding experiments.
Footnotes
-
This work was supported by the Medical Research Council (career establishment grant to P.P.; Ph.D. fellowship to R.T.)
-
ABBREVIATIONS: SK, small conductance Ca2+-activated K+;IAHP, afterhyperpolarizing current; BK, large conductance Ca2+-activated K+; AHP, afterhyperpolarization; CNS, central nervous system; IK, intermediate conductance Ca2+-activated K+; UCL 1684, 6,10-diaza-3(1,3),8(1,4)-dibenzena-1,5(1,4)-diquinolinacyclodecaphane; 1-EBIO, 1-ethyl-2-benzimidazolinone; NS309, 6,7-dichloro-1H-indole-2,3-dione 3-oxime; NS8593, (R)-N-(benzimidazol-2-yl)-1,2,3,4-tetrahydro-1-naphtylamine; DMSO, dimethyl sulfoxide; HEK, human embryonic kidney; h, human; r, rat; eDRG, embryonic dorsal root ganglion; PBS, phosphate-buffered saline; BSA, bovine serum albumin; TTX, tetrodotoxin; ACSF, artificial cerebrospinal fluid; TEA, tetraethylammonium; dTC, d-tubocurarine; XE991, 10,10-bis(4-pyridinylmethyl)-9(10H)-anthracenone dihydrochloride; ChTX, charybdotoxin; Clot, clotrimazole; BMB, bicuculline methobromide; I-V, current-voltage; IO, inside-out.
- Received May 24, 2006.
- Accepted August 22, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}