


Abstract
Long-term exposure to nicotinic ligands results in up-regulation of neuronal nicotinic α4β2 receptors in the brain and in heterologous expression systems. Up-regulation can be produced by a variety of drugs, including nicotinic agonists and competitive antagonists, but previous studies have indicated that the signal for up-regulation does not reflect a traditional nicotinic pharmacology, and the initial steps in signal transduction are not clear. We examined the signal for up-regulation and the possible involvement of the nicotine binding site in signal reception using mutated subunits transiently expressed in human embryonic kidney 293 cells. The data indicate that receptor activation and desensitization are not part of the signaling pathway. However, mutations to residues in the binding site can affect up-regulation. These results provide evidence that the binding site is directly involved in receiving the signal for up-regulation.
Long-term exposure to nicotine or other nicotinic drugs increases the numbers of high-affinity nicotine binding sites in the brains of experimental animals (Gentry and Lukas, 2002), and the density is also increased in postmortem brains of humans who smoke. The majority of these sites contain the α4 and β2 subunits, and up-regulation is accompanied by an increase in the amount of subunit antibody immunoreactive material but not mRNA. Up-regulation occurs for some other types of neuronal nicotinic receptors as well, although usually to a lesser extent (Gentry and Lukas, 2002).
In addition to occurring in the animal, up-regulation of receptors containing the α4 and β2 subunits (termed α4β2 receptors) also occurs in cultured neurons and in heterologous expression systems after transient or stable expression of cDNA for the subunits in somatic cells or even injection of mRNA into Xenopus laevis oocytes (Gentry and Lukas, 2002). These observations suggest that the signal for up-regulation involves recognition of the ligand by the α4β2 receptor itself and that the mechanism for up-regulation involves processes common to many types of cells. We are interested in defining the nature of the signal for up-regulation—the structural features of the ligand, the nature of the ligand binding site, and the initial response—to gain insight into pharmacological approaches to controlling up-regulation in vivo or in vitro. The mechanism of up-regulation [i.e., the subsequent processes that eventually result in the appearance of an increased number or responsiveness (or both) of the α4β2 receptor] is under investigation in a number of laboratories (see below).
Previous studies (Gopalakrishnan et al., 1997; Whiteaker et al., 1998; Kuryatov et al., 2005) have provided evidence that a number of agonists (e.g., nicotine, cytisine) and antagonists (e.g., dihydro-β-erythroidine, dHbE) can produce upregulation. This has suggested that either the binding site for ligand is not the same as the nicotine-binding site involved in activation or that the immediate response to signal is not receptor activation. Furthermore, the dependence of up-regulation on ligand concentration does not match the concentration dependence for other known receptor processes, such as activation, competitive inhibition, or desensitization (however, see Fenster et al., 1999, in the case of desensitization).
We have used transient expression of subunits with mutations to determine some structural features of the receptor which are involved in up-regulation. Overall, the results indicate that residues in the known nicotine binding site can influence up-regulation, indicating that binding to the nicotine binding site is a critical step in initiating up-regulation.
Materials and Methods
Unless otherwise noted, all chemicals were obtained from Sigma Chemical Co. (St. Louis, MO).
Cells, Constructs, and Transient Transfections. HEK293 cells (CRL-1573; American Type Culture Collection, Manassas, VA) were maintained in a mixture of Dulbecco's modified Eagle's medium and Ham's F-12 (1:1, also containing 5 mM HEPES), with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA), penicillin (100 units/ml), and streptomycin (100 μg/ml) in a humidified atmosphere containing 5% CO2 at 37°C. Cells for transient transfections were carried at less than 75% confluence and were used for 20 or fewer passages.
The human α4 and β2 constructs were kindly provided by Dr. J. Lindstrom (University of Pennsylvania) (α4 accession number NM000744, β2 accession number NM000748). The out-of-frame initiation site at the 5′ end of the β2 subunit was mutated by Dr. Lindstrom. Subunit cDNAs were transferred to pcDNA3 (Invitrogen, Carlsbad, CA) for expression. Mutations were made using the QuikChange kit (Stratagene, La Jolla, CA), and all mutated subunits were sequenced over the full-length of the subunit to confirm that only the desired mutation had been produced.
We report results on a total of 11 mutations. In the Results section, we present the numbering of the residues in terms of the full length, including the signal sequence. To obtain the position in the mature form, subtract 27 from the number for α4 and 26 for β2. For studies of the role of desensitization, we produced α4 L283T (position in the predicted mature subunit is α4 L256T; this residue is also denoted as position 9 of the second membrane spanning helix, or M2 L9′T), α4 S280F (α4 S253F or M2 S6′F), α4 S284L (α4 S257L or M2 S10′F), β2 L274C (β2 L248C or M2 L9′C), and β2 V287L (β2 V261L in the M2-M3 linker). The mutation expected to affect calcium permeability was α4 E273A (α4 E246A or M2 E-1′A). Mutations in the agonist binding site were α4 Y126F (α4 Y99F), α4 W182F (α4 W155F), α4 Y223F (α4 Y196F), α4 Y230F (α4 Y203F), and β2 W82F (β2 W56F).
Transient transfections were performed in 48-well plates [Biocoat poly(d-lysine); BD Biosciences, Palo Alto, CA]. Wild-type HEK cells were plated at a density of 10,000 cells/well 1 day before transfection. A total of 0.1 μg of plasmid DNA was mixed with 2.5 μl of Effectene reagent and 0.8 μl of Enhancer of Effectene (QIAGEN, Valencia, CA) per well. Twenty-four hours later, cells were washed with culture medium. Drugs were applied 48 h after the wash. In each experiment, a control transfection was performed using empty pcDNA3.
Drug Treatment and Cell Surface ELISA. Drugs were applied to transiently transfected cultures 72 h after transfection (48 h after washing the cultures) and incubated for 24 h. The ELISA assay was performed as for stably transfected cells. Preliminary experiments indicated that expression had reached a steady level at 72 h.
Cell surface ELISA was performed basically as described by Bonnert et al. (1999). Cells were fixed with 3% paraformaldehyde in phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, and 1.4 mM KH2PO4, pH 7.3) for 15 min, washed with PBS once, blocked with 5% skim milk in PBS for 1 h, incubated with mAb 290 anti-β2 subunit antibody (Sigma, N8533; 1:2000 dilution in milk/PBS) for 2 h, washed with PBS, incubated with anti-rat IgG peroxidase-conjugated goat antibodies (Sigma, A9037; 1:100 dilution in milk/PBS) for 1 h, washed with PBS four times, and then assayed using the 1-Step Ultra TMB-ELISA kit (Pierce, Rockford, IL). Color development was stopped by adding2MH2SO4 stop solution. The ELISA signal was converted into an equivalent volume of secondary antibody using a standard curve generated in parallel. For each experiment, the ELISA signal was obtained from triplicate wells and the total cell protein (bicinchoninic acid method; Pierce) from triplicate wells. The ELISA signal was normalized to protein.
Data Analysis. Cells transiently expressing some of the mutated subunits (see Results) gave lower ELISA signals than for wild-type subunits. Because in some cases, the signal for cells which had not been treated with drugs was comparable with that for cells transfected with pcDNA3, it was not always possible to subtract the nonspecific signal. To present the data for all transient transfection experiments in a consistent fashion, nonspecific binding was not subtracted, and data for all constructs are presented as the ratio of the signal after drug treatment to the signal without drug.
The possible cell toxicity of drug treatments or constructs was examined by determining whether the cell protein per well was significantly reduced from control (either untreated cells or cells transfected with empty pcDNA3 vector). None of the constructs showed a significant reduction. However, higher concentrations of lobeline (greater than 100 μM) did reduce total protein. Lobeline appeared to be directly cytotoxic, because the total protein was reduced in untransfected HEK293 cells by 1 mM lobeline (B. W. Ma and J. H. Steinbach, unpublished observations).
Data were analyzed in Excel (Microsoft, Redmond, WA), the Hill equation was fit using SigmaPlot (SSPS, Chicago, IL), and multiple comparisons and the Dunnett post hoc correction were made using Systat (SSPS). In studies of receptors containing mutated receptors, two statistical tests were applied. To determine whether expression for mutated receptors was higher than control (pcDNA3 transfection), a paired two-tailed t test was used. To determine whether up-regulation by a given drug differed from wild-type receptors, data from cells treated with 10 μM nicotine or lobeline or 100 μM carbamylcholine or dHbE were compared. Significance was assessed using a two-tailed test with a Dunnett post hoc correction for the comparison of all the mutated receptors to control for a given drug (6 or 11 comparisons). Data are plotted as mean ± 1 S.E. Fit parameter values are the best fitting value with the standard error estimated from the fit.
Results
Pharmacological Studies of Up-Regulation of Transiently Expressed Wild-Type α4β2 Receptors. Our assay for up-regulation was the binding of mAb 290 to the surface of intact cells. This monoclonal antibody binds to a conformationally sensitive epitope on the β2 subunit of the α4β2 receptor (Sallette et al., 2005) and recognizes a mature conformation of the receptor. As discussed below, the up-regulation we study might reflect an increase in the number of physical receptors on the surface, an increase in the proportion of receptors on the surface which are in a given conformational state, or both.
HEK cells transiently transfected with wild-type α4β2 subunits bind mAb 290 at levels significantly greater than for cultures transfected with empty vector (pcDNA3) alone (Table 1). Binding is increased by nicotine, carbamylcholine, lobeline, and dHbE (Fig. 1).
Expression and up-regulation α4β2 receptors with mutations expected to affect desensitization or calcium permeability
Results are shown for cultures transiently transfected with either WT receptors or receptors containing the indicated mutated subunit (with wild-type complementary subunit). The left set of columns shows the ratio of mAb 290 binding to intact cells, normalized to the binding to cultures transfected with empty vector in the same experiment. The column headed P vs 1 gives the probability that the binding was identical for cells transfected with pcDNA3 or with subunits. The column headed P vs WT gives the probability that the ratio differs to that for cultures transfected with wild-type receptors computed using all data and corrected using a post hoc Dunnett correction. The right columns give the mean ratio of the binding to cultures treated with 10 μM nicotine, normalized to the binding to cultures transfected with the same subunits in the same experiment but not treated with nicotine. Values are presented as mean ± S.D. (n).
The EC50 value for up-regulation by nicotine (approximately 1 μM) is much higher than the affinity for steadystate binding to intact cells (1 nM; Zhang and Steinbach, 2003), as found by others (Gopalakrishnan et al., 1996). Nicotine is a full agonist at α4β2 receptors and is highly efficacious and potent at producing desensitization (Paradiso and Steinbach, 2003). The EC50 value for up-regulation by nicotine is more similar to the EC50 value for activation (Buisson et al., 1996; Gopalakrishnan et al., 1996). Carbamylcholine has been less studied, but the EC50 value for up-regulation seems to be greater than 10 μM, whereas the affinity for steady-state binding is approximately 70 nM (Gopalakrishnan et al., 1996).
dHbE is a competitive antagonist at the agonist binding site, with an apparent affinity to inhibit nicotine binding or to block the activation of less than 100 nM (Buisson et al., 1996; Gopalakrishnan et al., 1996; Sabey et al., 1999). dHbE produced up-regulation, albeit at higher concentrations than nicotine (EC50 value greater than 10 μM; Fig. 1), and much higher than the affinity for block.
Lobeline was chosen for examination because of reports that it is a high-affinity inhibitor of agonist binding to rat brain α4β2 receptors (Kd value of approximately 5 nM) (Damaj et al., 1997), which shows essentially no agonist activity (Damaj et al., 1997). It has not been studied in vitro, although it is reported to be ineffective at producing upregulation in vivo (Auta et al., 1999). Lobeline was a potent up-regulator (EC50 value of approximately 100 nM; Fig. 1), although, again, the EC50 value is much higher than the affinity for inhibiting agonist binding. (We note that lobeline seemed to be cytotoxic at concentrations greater than 100 μM, as cell protein was reduced.)

Up-regulation of transiently expressed wild-type human α4β2 receptors. Data are shown for two agonists and two blockers. The data shown are the mean (±1 S.E.M.) of the ratio of the ELISA signal after drug treatment to that of untreated cells in the same experiment (see Materials and Methods).
Overall, these pharmacological studies confirm previous work to indicate that the ability of a drug to activate the α4β2 receptor is not critical to up-regulation. This is most clearly shown by the abilities of dHbE and lobeline to up-regulate without detectable activation. The concentration dependence for up-regulation does not match the steady-state affinities for any of the drugs, suggesting that the known binding site in a desensitized conformation is not involved. We are left then with the questions of which site and which receptor response might be involved in the signal for up-regulation.
Pharmacological Studies of Mutations that Affect Desensitization. We undertook two sets of studies. The first was to produce receptors with mutations previously shown to affect desensitization or calcium ion permeability. The second was to mutate residues in the classic ligand binding site.
We produced five subunits with mutations that had been reported to alter desensitization (and, in some cases, other properties of the receptor). (Note that numbering is from the initiating methionine of the unprocessed subunit; to obtain the position in the mature form, subtract 27 from the number for α4 and 26 for β2.) We produced α4 L283T, α4 S280F, and α4 S284L, which, respectively, produce slower desensitization than wild type (Labarca et al., 2001; Moroni et al., 2006), faster desensitization (Kuryatov et al., 1997), and faster desensitization (Matsushima et al., 2002). We also produced β2 L274C (slower desensitization; De Fusco et al., 2000) and β2 V287L (slower desensitization; Khiroug et al., 2002). Most of these mutations are in the second membrane-spanning region (M2). In terms of the location in M2 (numbered from the predicted amino terminus), they correspond to the following: α4 L283T (position 9, or M2 L9′T), α4 S280F (M2 S6′F), α4 S284L (M2 S10′F), β2 L274C (M2 L9′C), and β2 V287L (in the M2-M3 linker). We note that a recent study of the α4 S280F subunit indicated that, in HEK cells, it acts as a loss of function mutation (Kuryatov et al., 2005). The other mutations have not been examined in HEK cells.
Each mutated subunit was cotransfected with wild-type complementary subunit (e.g., mutated α4 plus wild-type β2). The binding of mAb 290 was greater than that for cultures transfected with vector alone and comparable with that for wild-type subunits (Table 1). First, we obtained concentration-response curves for up-regulation by nicotine (10 nM to 100 μM) and carbamylcholine (100 nM to 100 μM; Fig. 2). These curves did not show any difference from those for wild type. We then tested additional cultures with 10 μM nicotine to compare with the up-regulation of wild-type receptors. The up-regulation produced did not differ from that for wild-type receptors for any of the mutants studied (Table 1). These results confirm the previous report that HEK cells stably transfected with the α4 S280F receptor show normal upregulation (Kuryatov et al., 2005). Therefore, it seems unlikely that desensitization is critical for the signal involved in up-regulation. The mutations α4 L283T and β2 L274C also are expected to markedly shift the concentration-dependence for activation to lower agonist concentrations (Labarca et al., 2001; Moroni et al., 2006).
A possible signal is calcium-ion entry from the external medium, resulting in activation of a variety of intracellular signaling pathways. A mutation that has been reported to reduce calcium permeability of the α7 receptor channel is homologous to α4 E273A (M2 E-1A; Bertrand et al., 1993). However, transfection of cultures with this mutated α4 subunit did not significantly reduce the ability of nicotine to up-regulate (Table 1). Therefore, it is not likely that calcium entry is critical for the signal.
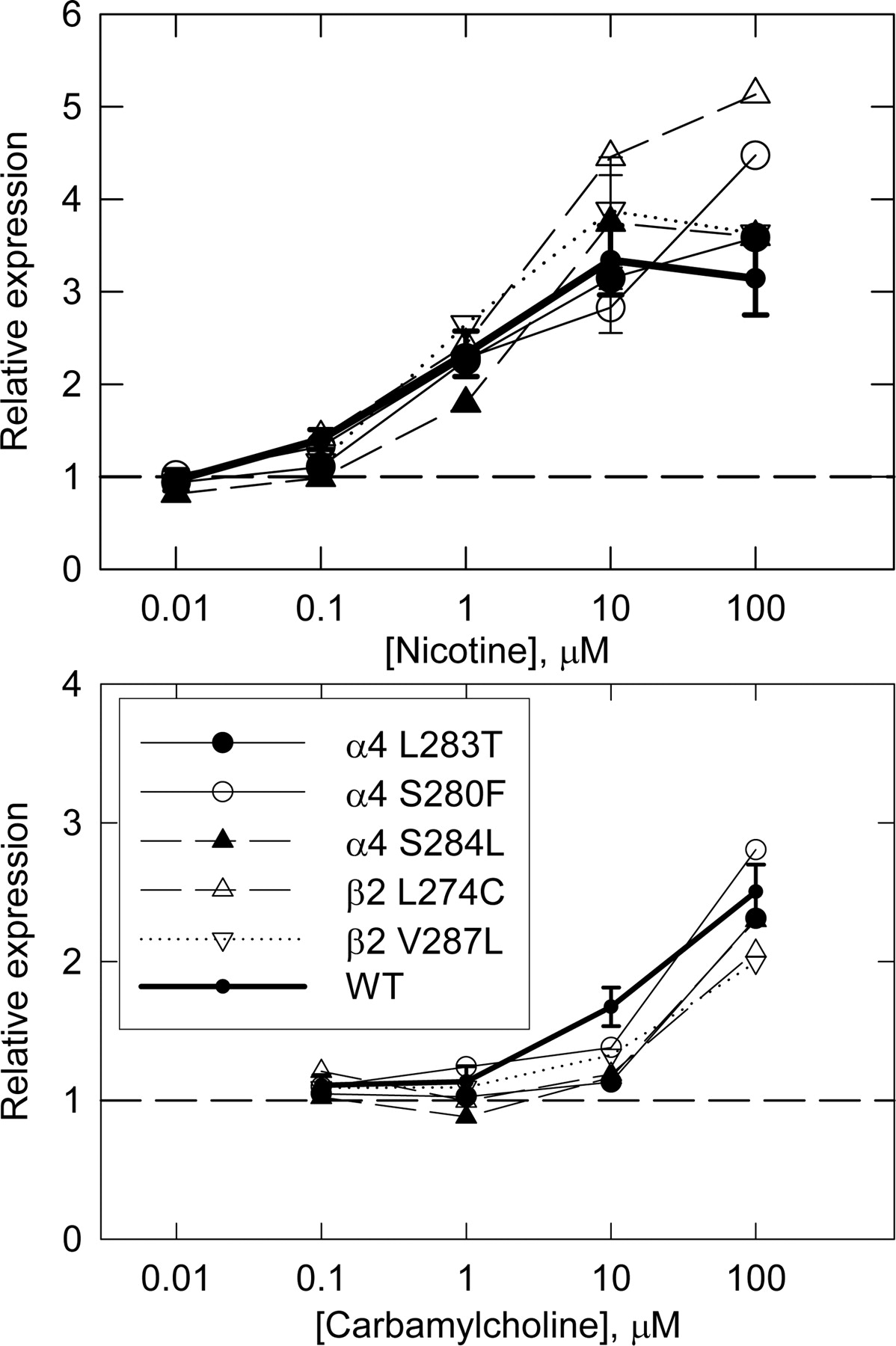
Up-regulation of transiently expressed receptors with mutations which affect desensitization. The data are shown as in Fig. 1 for the receptors containing mutated receptors as indicated and for wild-type receptors (heavy line with error bars, data from Fig. 1). Results for receptors with mutated subunits are from single experiments, except for values for 10 μM nicotine (see Table 1).
Pharmacological Studies of Mutations in the Agonist Binding Site. We then undertook mutations of residues believed to be located in the acetylcholine and nicotine binding site (Le Novere et al., 2002; Sine, 2002; Lester et al., 2004). These sites are located at the interface between the α and non-α (e.g., β2) subunits, and the binding residues seem to be conserved among members of the nicotinic receptor family. Therefore, we based our decisions as to appropriate residues from work on muscle type receptors and other neuronal nicotinic receptors, because relatively little work has been performed directly with α4β2 receptors. There are three domains in the α subunit (A, B, and C) and four domains in the non-α subunit (D, E, F, and G), which are brought into close proximity in the folded and assembled receptor. We made five residue changes, each of which should result in a reduction of affinity for ligands. In the α4 subunit, we produced α4 Y126F (A domain, the homologous residue in the mature muscle subunit is α1 Tyr93; Galzi et al., 1991), α4 W182F (B domain, α1 Trp149; Galzi et al., 1991), α4 Y223F (C domain, α1 Tyr190; Sine et al., 1994), and α4 Y230F (C domain, α1 Tyr198; Galzi et al., 1991). In the β2 subunit, we produced β2 W82F (E domain, γ Trp55; Corringer et al., 1995).
In the absence of added drugs, three of these mutated receptors expressed at levels not significantly different from wild type (α4 Y126F, α4 Y230F, and β2 W82F; Table 2). In contrast, α4 W182F and α4 Y223F showed reduced levels of surface mAb 290 binding (Table 2), which were not significantly different from that of cells transfected with pcDNA3.
Expression of α4β2 receptors containing mutations in the ligand binding site
Data are expressed as in Table 1.
We tested for up-regulation by nicotine, lobeline, carbamylcholine, and dHbE to use a range of ligands for the receptors. Receptors containing each of the mutated subunits could be up-regulated, but the ability of the different ligands and the extent of up-regulation differed among the constructs (Fig. 3). To quantitatively compare the amount of up-regulation, we compared the enhancement produced by 10 μM nicotine, 10 μM lobeline, 100 μM carbamylcholine, and 100 μM dHbE (Fig. 4 and Table 3). Receptors containing the α4 Y230F mutated subunit behaved indistinguishably from wild type. In contrast, α4 Y126F, α4 Y223F, and β2 W82F showed increased up-regulation by lobeline and reduced up-regulation by carbamylcholine, whereas α4 W182F was, in general, less responsive to all treatments. Overall, lobeline was the most effective drug at up-regulating the mutated receptors, carbamylcholine was most severely affected by mutations, whereas nicotine and dHbE produced relatively normal amounts of up-regulation. The observation that lobeline was effective at up-regulating all of the mutated receptors indicates that the mutations had not fatally affected receptor assembly but had selectively altered the efficacy of carbamylcholine.
Up-regulation of α4β2 receptors containing mutations in the ligand binding site
Data are expressed as in Table 1.
With the data available, it is not clear whether potency (EC50 value) or efficacy (maximal up-regulation) is affected. The problem is that the maximal effect often is not clear for mutations that impair up-regulation. Up-regulation by lobeline generally shows no shift in the concentration-effect curves, although efficacy is affected, except possibly a decrease in potency for α4 W182F. dHbE also shows no change, except again a possible decrease in potency for α4 W182F. The concentration-effect curves for nicotine suggest a decrease in efficacy (although not reaching statistical significance; see Table 3). Finally, for carbamylcholine, none of the relationships shows a maximal up-regulation, and those for four mutations are so flat that no inference can be drawn.
Discussion
The new information these studies provide is evidence that mutations to residues in the acetylcholine binding site on the human α4β2 receptor can affect the ability of carbamylcholine to stimulate up-regulation of the receptor. The present work and previous studies have found that both agonists and competitive antagonists can stimulate up-regulation, indicating that receptor activation was not a required element of the initial signal for up-regulation. Furthermore, the concentration-dependence for up-regulation did not match well with the concentration-dependencies for binding of drugs to resting or desensitized receptors. We have tested the effects of five mutations that should affect the rate or extent of desensitization and found that none of the mutations affected up-regulation. We also note that several of these mutations are expected to shift the concentration of agonist producing half-maximal activation to lower concentrations (e.g., α4 L283T and β2 L274C; Labarca et al., 2001; Moroni et al., 2006), whereas the mutations in the ligand binding site shift activation to higher concentrations. Therefore, it seems unlikely that steady-state activation is a critical part of the signal. These observations, although consistent with previous results, raise the question of whether the drugs producing up-regulation interact with the classic agonist binding site on the receptor or with some other site.

Up-regulation of transiently expressed receptors with mutations in the ligand binding domain. Data are presented in four panels to show the ability of the drugs to up-regulate. Data are shown as in Fig. 2, and the results for wild-type receptors are shown for comparison (heavy line with error bars, data from Fig. 1).
We found that mutations of residues in the agonist binding site can affect the ability of drugs to produce up-regulation. The most detrimental mutation was α4 W182F. Homologous mutations in the α7or α1 subunit produce large deficits in activation (Galzi et al., 1991; Akk, 2001), including reductions in both affinity and in gating efficacy, and this residue provides a critical part of the ligand binding site for acetylcholine or carbamylcholine (Le Novere et al., 2002; Lester et al., 2004). The least detrimental mutation was α4 Y230F, and homologous mutations also have lesser effects on activation or ligand binding (Galzi et al., 1991; Sine et al., 1994). Of the remaining three mutations, mutations homologous to α4 Y223F have relatively large effects on binding or activation (Galzi et al., 1991; Sine et al., 1994; Chen et al., 1995), whereas the other two positions show somewhat smaller effects (Sine et al., 1994; Corringer et al., 1995; Akk, 2001). There is a reasonable correlation between the effects on upregulation and the severity of the functional defect produced. In addition, the two mutations that produced the largest reductions in up-regulation by carbamylcholine (α4 W182F and α4 Y223F) also produced the largest reductions in control expression. This observation is reminiscent of studies of kainate receptors in which mutations in the ligand binding domain reduced normal assembly (Valluru et al., 2005). It is somewhat disappointing that mutations did not show more ability to distinguish among ligands, but this is perhaps expected because they were chosen based on relatively strong effects.

Relative expression and up-regulation for transiently expressed receptors with mutations in the ligand binding domain. The ratios for expression in the absence of drug (Table 2) or up-regulation (Table 3) are shown normalized to the mean value for wild-type receptors to facilitate a qualitative comparison of results.
Based on the pharmacology of up-regulation, the drugrecognition properties of the receptor involved in up-regulation do not accord with known properties of the mature ligand binding site, for which ligand binding results in activation or desensitization. However, it is not necessary that the signal for up-regulation involves any of the other responses of the receptor. Indeed, the present study has examined the roles for activation and desensitization by mutating the receptor structure (to complement pharmacological studies) and found no evidence that they are involved. An implication is that the structural features required of the ligand for up-regulation may not be identical with those for activation or desensitization. Therefore, the conformation of the residues in the receptor that provide the docking site for ligands involved in signaling for up-regulation may not reflect either a “mature” or an “immature” conformation for the residues involved in activation or desensitization.
Mechanism of Up-Regulation. Our studies do not address the mechanism by which up-regulation occurs, although the mechanism may provide some insights into the location of the relevant receptor for the signal. This question is presently under active investigation in several laboratories, and there are significant issues to be considered. A major one is whether there is a increase in the number of physical α4β2 receptors. This question is raised by the methods used to assay for receptors. In most studies, the methods are sensitive to the conformation of the receptor, either in terms of possession of a high affinity for radio-labeled probes (cytisine, nicotine, or epibatidine) or possession of a conformation-sensitive epitope (mAbs 270, 290, 299). One study that did not have this concern used subunits labeled by inclusion of modified fluorescent proteins in the major cytoplasmic loop (Nashmi et al., 2003). After treatment with nicotine, there was an increase in fluorescence, indicating increased numbers of subunits, and an increase in fluorescence resonance energy transfer between α4 and β2 subunits, indicating increased assembly. These observations could, however, be produced by a stabilization of the receptor after initial assembly, resulting in reduced intracellular degradation. There is also a brief mention that surface expression of an epitope labeled β2 subunit increases after nicotine treatment (Harkness and Millar, 2002). In contrast, a thorough examination of expression of epitope-labeled subunits or biotinylated surface receptors found that there was no significant increase in the numbers of physical surface receptors during up-regulation of the numbers of high-affinity ligand binding sites (Vallejo et al., 2005). More recent studies report results consistent with an increase in the physical numbers of receptors (Kuryatov et al., 2005). However, strong data support the idea that there is a change in the properties of receptors after long-term treatment with nicotine, most remarkably a shift in the activation of the receptors to a population with greater sensitivity to agonists (Buisson and Bertrand, 2001). The functional change has been ascribed to a conformational change in the receptor (Buisson and Bertrand, 2001; Vallejo et al., 2005) or to a change in subunit stoichiometry for assembly (Nelson et al., 2003; Moroni et al., 2006). If the mechanism involves increased assembly, then the signal is likely to be received by an incomplete receptor. In this case, the present observation that residues in either the α4or β2 subunit can affect up-regulation suggests that the signal may be mediated by partially assembled receptors (e.g., dimers). If the mechanism involves an induced conformational change in already assembled receptors, then the agonist binding site is present, although the present observations do not directly address the conformation of the site.
The debate over the nature of up-regulation—an increase in number or a change in properties—also has some significance for the localization of action for the signal. An initial study (Peng et al., 1994) found that quaternary amine drugs could produce up-regulation and concluded that drugs that act solely on surface receptors are effective. This observation was confirmed in subsequent examinations (Gopalakrishnan et al., 1997; Whiteaker et al., 1998). Whiteaker et al. (1998) also reported that several effective drugs did not enter cells during the period required for up-regulation. However more recent articles have found that quaternary drugs do enter cells over this time (Kuryatov et al., 2005; Sallette et al., 2005; Vallejo et al., 2005). Therefore, up-regulation could involve signaling to either surface (fully assembled) or internal (possibly unassembled or conformationally immature) receptors, or both.
Irrespective of the mechanism for up-regulation or the localization of the signal, most published reports have concluded that binding of a drug to the α4β2 receptor provides a signal that results in a conformational change in the receptor which promotes up-regulation, either directly or by enhancing assembly.
These studies do not provide additional information with which to interpret the present results regarding the structural requirements for the receipt of and initial transduction of the signal for up-regulation. However, the present data are consistent with the conclusion, reached by others, that the signal for up-regulation is received by a receptor, which has an agonist binding site whose conformation differs from the mature conformation. This consistency, however, must be tempered by the caveat that transduction of the signal for up-regulation does not necessarily correspond to characterized functions of the receptor, and so the conformation of the site necessary for up-regulation may directly reflect the conformation required for other functional consequences (discussed above).
The Signal for Up-Regulation. Overall, the present data indicate that residues in the classic acetylcholine binding site on the α4β2 receptor are involved in the initial signal for up-regulation. This provides direct evidence for the involvement of this site in the signal for up-regulation. However, the concentration dependence suggests that the binding properties of the site are not identical with those of the mature receptor in any functionally identified state. The pharmacological properties, and the lack of effect of several mutations which would be expected to alter desensitization or activation, suggest that neither activation nor desensitization is required for the initial step of signal transduction. The data support the suggestion made previously that nicotinic ligands bind to the agonist binding sites on receptors and induce a conformational change that facilitates maturation and/or assembly (Harkness and Millar, 2002; Kuryatov et al., 2005; Sallette et al., 2005; Vallejo et al., 2005).
Acknowledgments
We thank Beiwen Ma for assistance with cell culture and Gustav Akk and John Bracamontes for discussions and comments on the manuscript.
Footnotes
-
This research was supported by National Institutes of Health NS 22356. J.H.S. is the Russell and Mary Shelden Professor of Anesthesiology.
-
ABBREVIATIONS: dHbE, dihydro-β-erythroidine; HEK, human embryonic kidney; ELISA, enzyme-linked immunosorbent assay; PBS, phosphatebuffered saline; mAb, monoclonal antibody; WT, wild type; N.S., not significant.
- Received July 25, 2006.
- Accepted September 11, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}