


Abstract
Approaches to identify G protein-coupled receptor oligomers rather than dimers have been lacking. Using concatamers of fluorescent proteins, we established conditions to monitor sequential three-color fluorescence resonance energy transfer (3-FRET) and used these to detect oligomeric complexes of the α1b-adrenoceptor in single living cells. Mutation of putative key hydrophobic residues in transmembrane domains I and IV resulted in substantial reduction of sequential 3-FRET and was associated with lack of protein maturation, prevention of plasma membrane delivery, and elimination of signaling function. Although these mutations prevented cell surface delivery, bimolecular fluorescence complementation studies indicated that they did not ablate protein-protein interactions and confirmed endoplasmic reticulum/Golgi retention of the transmembrane domain I plus transmembrane domain IV mutated receptor. The transmembrane domain I plus transmembrane domain IV mutated receptor was a “dominant-negative” in blocking cell surface delivery of the wild-type receptor. Mutations only in transmembrane domain I did not result in a reduction in 3-FRET, whereas restricting mutation to transmembrane domain IV did result in reduced 3-FRET. Mutations in either transmembrane domain I or transmembrane domain IV, however, were sufficient to eliminate cell surface delivery. Terminal N-glycosylation is insufficient to determine cell surface delivery because both transmembrane domain I and transmembrane domain IV mutants matured as effectively as the wild-type receptor. These data indicate that the α1b-adrenoceptor is able to form oligomeric rather than only simple dimeric complexes and that disruption of effective oligomerization by introducing mutations into transmembrane domain IV has profound consequences for cell surface delivery and function.
In recent times, it has become widely accepted that many (Milligan, 2004; Park et al., 2004) but perhaps not all (Meyer et al., 2006) members of the rhodopsin-like family A G protein-coupled receptor (GPCR) group possess quaternary structure. It has been suggested that these receptors exist, and probably function, as dimers (Baneres and Parello, 2003; Fotiadis et al., 2004). The application of atomic force microscopy to membranes of murine rod outer segments demonstrated complex in situ quaternary organization of rhodopsin in that paracrystalline rows or oligomers of dimers were observed (Liang et al., 2003). This allowed interaction interfaces to be predicted and modeled (Fotiadis et al., 2004). The rod outer segment, however, is a highly specialized structure in which rhodopsin comprises nearly 50% of the total protein. In contrast, many GPCRs in other tissues are expressed in relatively low amounts. It is therefore unclear whether the in situ oligomeric organization of rhodopsin is relevant to other GPCRs or to other environments. However, molecules of rhodopsin reconstituted into asolectin liposomes were shown to self-associate into dimers or multimers (Mansoor et al., 2006), and this has been suggested to provide evidence that rhodopsin can spontaneously self-associate in membranes other than the rod outer segment (Mansoor et al., 2006). Although approaches used to examine interactions between GPCR monomers have developed from initial coimmunoprecipitation studies to the application of a range of resonance energy transfer-based techniques (Milligan and Bouvier, 2005), these are generally unable to determine whether quaternary structure is restricted to dimers or whether higher-order oligomers may exist (Milligan and Bouvier, 2005). Recent studies on the α1b-adrenoceptor, based on the ability of fragments of this GPCR to self-associate, have indicated that both transmembrane domain (TMD) I and TMD IV can provide symmetrical interaction interfaces and these observations generated a “daisy-chain” model in which repeating TMD I–I and then TMD IV–IV interactions could potentially produce oligomers rather than simple dimers (Carrillo et al., 2004).
One key role that has been suggested for GPCR dimerization/oligomerization is to promote the correct folding and maturation of the receptor (Bulenger et al., 2005) and, subsequently, to allow cell surface delivery. Alterations in GPCR structure can result in retention of the protein in the endoplasmic reticulum (Conn et al., 2006), and hence, modification of interfaces involved in GPCR dimerization/oligomerization might be anticipated to limit receptor maturation and cell surface delivery.
Although, 2 chromophore fluorescence resonance energy transfer (FRET) imaging (Herman et al., 2004) has become a popular approach to observe GPCR protein-protein interactions in single cells (Milligan and Bouvier, 2005), it is only with the very recent development of three-chromophore FRET (Galperin et al., 2004) that it has become conceptually possible to image complexes containing at least three polypeptides. Herein we develop and optimize a sequential three-color FRET imaging approach and use this to demonstrate the presence of oligomeric complexes of the α1b-adrenoceptor in single cells. Introduction of mutations into both TMD I and TMD IV of the α1b-adrenoceptor results in a substantial reduction in the normalized sequential three-protein FRET signal, indicating alterations in the oligomeric organization. This was accompanied by an inability of the modified receptor to mature correctly, to be delivered to the cell surface, and, hence, to function. However, use of bimolecular fluorescence complementation indicated that although these mutations block transport from the endoplasmic reticulum/Golgi, they do not inherently eliminate receptor quaternary interactions. Deconstruction of the TMD I + TMD IV receptor mutant indicated the key mutations that alter α1b-adrenoceptor organization are those in TMD IV and that terminal N-glycosylation of the α1b-adrenoceptor (Bjorklof et al., 2002) is not a definitive indication of a fully mature receptor that will be successfully delivered to the plasma membrane.
Materials and Methods
Materials. All materials for tissue culture were from Invitrogen (Paisley, UK). [3H]Prazosin was from PerkinElmer Life and Analytical Sciences (Boston, MA).
Fluorescent Protein Concatamers. Different concatamers containing eCFP-dsRed2, eYFP-dsRed2, or eCFP-eYFP-dsRed2 as single open-reading frames were constructed in a similar way as the eCFP-eYFP concatamer described previously (Carrillo et al., 2004). For the construction of the concatamer containing the three fluorescent proteins, the original eCFP-eYFP concatamer subcloned into pcDNA3 (Invitrogen) was digested with KpnI and NotI, allowing the liberation of the eYFP-encoding nucleotide region. Subsequently, eYFP was amplified by PCR from its original vector (Clontech, Mountain View, CA) using as a forward primer CGGGGTACCATGGTGAGCAAGGGCGAGGAG that includes a KpnI restriction site (underlined) and as a reverse primer CCGGAATTCCTTGTACAGCTCGTCCATGCC, where the stop codon had been removed and an EcoRI restriction site had been added. Likewise, dsRed2, a variant of the originally described dsRed1, (Clontech) was amplified using as a forward primer CCGGAATTCATGGCCTCTTTGCTGAAGAAC containing an EcoRI restriction site and as a reverse primer TTTTTCCTTTTGCGGCCGCTCAGTTGTGGCCCAGCTTGGA that contains a NotI site. All of these PCR products were purified and subsequently digested with the corresponding endonuclease enzymes. Afterward, the digested PCR products corresponding to eYFP and dsRed2 were ligated to the pcDNA3-eCFP vector obtained after the digestion of eCFP-eYFP concatamer with KpnI and NotI (see above). This ligation resulted in the three fluorescent proteins in tandem within a unique open-reading frame. To generate the eCFP-dsRed2 concatamer, eYFP was removed from the original eCFP-eYFP construct by digestion with KpnI and NotI and substituted by dsRed2 containing a KpnI site at the 5′-end and a NotI site at the 3′-end. Likewise, the eYFP-dsRed2 concatamer was constructed after removing eCFP from the eCFP-dsRed2 construct by digestion with HindIII and KpnI and subsequent insertion of eYFP without a stop codon containing a HindIII site at the 5′-end and a KpnI site at the 3′-end.
α1b-Adrenoceptor Fusions with Fluorescent Proteins. Forms of the α1b-adrenoceptor fused to eCFP or eYFP were described in Carrillo et al. (2004). To generate α1b-adrenoceptor C-terminally tagged with dsRed2, we removed eCFP from the α1b-adrenoceptor-eCFP construct by digesting with KpnI and NotI endonucleases and inserted a dsRed2 nucleotide sequence containing KpnI and NotI restriction sites at the 5′- and 3′-ends, respectively. In the same way, for bimolecular fluorescence complementation studies, α1b-adrenoceptor constructs were generated by subcloning the sequence encoding for the N-terminal 172 amino acid fragment or the C-terminal 67 amino acid fragment of eYFP. These two eYFP fragments were selected according to Hu et al. (2002).
Site-Directed Mutagenesis. To produce amino acid substitutions in the primary structure of the different proteins, site-directed mutagenesis of the encoding nucleotide sequence was performed using the QuikChange II site-directed mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer's instructions. The following primers were designed for the mutagenesis of the α1b-adrenoceptor: GCCATTGTGGGCAACATCGCAGCCATCCTGTCAGTGGCCTGC (forward) and GCAGGCCACTGACAGGATGGCTGCGATGTTGCCCACAATGGC (reverse) that substitute leucine and valine at positions 65 and 66, respectively, in transmembrane domain I with two consecutive alanines (underlined bases); and AGCAAGGCCATCTTGGCAGCAGCAAGTGTGTGGGTTTTGTCC (forward) and GGACAAAACCCACACACTTGCTGCTGCCAAGATGGCCTTCCT (reverse) that exchange two consecutive leucine residues at positions 166 and 167 in transmembrane domain IV with two consecutive alanine residues.
The same strategy was used to mutate eYFP into a nonfluorescent form by substitution of tyrosine at position 67 with cysteine. In this case, the PCR primers used were CCACCTTCGGCTGCGGCCTGCAGTG (forward) and CACTGCAGGCCGCAGCCGAAGGTGG (reverse).
Transient Transfection of HEK293 Cells. HEK293 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 0.292 g/l l-glutamine and 10% (v/v) newborn calf serum at 37°C ina5%CO2 humidified atmosphere. Cells were grown to 60 to 80% confluence before transient transfection. Transfection was performed using Effectene transfection reagent (QIAGEN GmbH, Hilden, Germany) according to the manufacturer's instructions.
Epifluorescence Imaging: Two-Step FRET Microscopy in Living Cells. HEK293 cells were grown on poly(d-lysine)-treated coverslips (number 0) and transiently transfected with appropriate eCFP/eYFP/dsRed2 constructs. Coverslips were placed into a microscope chamber containing physiological HEPES-buffered saline solution (130 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 20 mM HEPES, and 10 mM d-glucose, pH 7.4). Cells were then imaged using an inverted Nikon TE2000-E microscope (Nikon Instruments, Melville, NY) equipped with a 40× (numerical aperture = 1.3) oilimmersion Fluor lens and a cooled digital photometrics Cool Snap-HQ charge-coupled device camera (Roper Scientific, Trenton, NJ).
Epifluorescence excitation light was generated by an ultra-high-point intensity 75 W xenon arc Optosource lamp (Cairn Research, Faversham, Kent, UK) coupled to a computer-controlled Optoscan monochromator (Cairn Research). Monochromator was set to 425/10 nm, 495/9 nm, and 580/10 nm for the sequential excitation of eCFP, eYFP, and dsRed2, respectively. eCFP, eYFP, and dsRed2 excitation light was transmitted through the objective lens using a custom-designed DCXR 86006 polychroic (Chroma Inc., Rockingham, VT). eCFP, eYFP, and dsRed2 fluorescence emission was controlled via a high-speed filterwheel device (Prior Scientific Instruments, Cambridge, UK) containing the following bandpass emitters: HQ470/30 nm for eCFP; HQ535/30 nm for eYFP; and HQ630/60 nm for dsRed2. This excitation/emission setup made it possible to leave the polychroic in place in the microscope. This enhanced image acquisition and therefore lessened the potential threat of motion. Images were collected using a Cool Snap-HQ digital camera operated in 12-bit mode. Computer control of all electronic hardware and camera acquisition was achieved using Metamorph software (version 6.3.3; Molecular Devices, Sunnyvale, CA). The illumination time was 230 ms, and binning was set to 2 × 2.
To detect sequential FRET in living cells, six filter channel combinations were established (i.e., dsRed2: Ex = 580/10 nm, Em = 630/60 nm; eYFP: Ex = 495/9 nm, Em = 535/30 nm; eCFP: Ex = 425/10 nm, Em = 470/30 nm; eCFP/eYFP/FRET: Ex = 425/10 nm, Em = 535/30 nm; eCFP/eYFP/DsRed2 FRET: Ex = 425/10 nm, Em = 630/60 nm; eYFP/dsRed2/FRET: 495/9 nm, Em = 630/60 nm), which allowed acquisition of images from all three protein fluorophores and three raw FRET images. These images were used to calculate three corrected FRET images. Metamorph imaging software was used to quantify the FRET images and to apply the necessary algorithms, pixel-by-pixel-based, to obtain a final image corresponding to the corrected and normalized FRET (see below and Supplementary information).
Determination of Bleed-Through Coefficients. Bleed-through coefficients were defined as the ratio between the amount of fluorescence detected in the corresponding FRET filter set (Fx) and the fluorescence detected for each single fluorescent protein at its own filter set (eCFP, eYFP, and dsRed2). To obtain these coefficients, cells were transfected with each of the fluorescent proteins individually, and the fluorescence measurements resulted in the following parameters: FCFP-YFP/CFP = 0.82, FCFP-YFP/YFP = 0.12, FYFP-dsRed2/YFP = 0.08, FYFP-dsRed2/dsRed2 = 0.05, FCFP-dsRed2/YFP = 0.01, FCFP-dsRed2/DsRed2 = 0.04.
FRET Signal Correction and Normalization. Because the fluorescence detected in the FRET channel (raw FRET) comprises not only the actual FRET but also the bleed-through from the fluorescent proteins expressed, this signal was corrected using the bleed-through coefficients determined previously. In addition, this corrected FRET was also normalized to generate a final value independent of protein expression levels. For this purpose, the equation RFRET = raw FRET/Σ(FP*Bx) was used, where FP is the intensity of each fluorescent protein involved in the final FRET, and Bx is its corresponding bleed-through coefficient (i.e., fluorescence intensity in the background-corrected FRET channel image divided by the total spillover from the relevant FRET partners). Thus, in the absence of energy transfer, FRETN has a predicted value of 1, whereas values greater than 1 would reflect the occurrence of FRET.
Hoechst 33342 and RQAPB Imaging. Glass coverslips bearing HEK293 cells expressing wild-type or mutant versions of the α1b-adrenoceptor were rinsed several times in PBS. Cell nuclei were stained by incubating the cells for 20 min at room temperature (RT) with fresh PBS containing the nuclear DNA-binding dye Hoechst 33342 (10 μg/ml). After 20 min, cells were washed 3 to 4 times with PBS and reincubated (at RT) for 70 to 90 min with fresh PBS supplemented with 10 nM concentration of the red fluorescent α1-AR antagonist ligand RQAPB (Pediani et al., 2005). Using the epifluorescence microscope system, Hoechst 33342 and RQAPB were sequentially detected using the appropriate excitation wavelength of light, beam splitter (BS), and emitter (Hoechst: Ex = 355 nm, BS = 400 DCLP, Em = 470/30 nm; RQAPB: Ex = 550 nm, BS = Q570LP, Em = 610/75 nm). Sequential images (no binning) were collected, and exposure to excitation light was 150 to 200 ms/image. Metamorph imaging software was used for image data processing.
[Ca2+]i Ratio Imaging. HEK293 cells were transfected with FLAG-Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala, α1b-adrenoceptor-eCFP, or FLAG-α1b-adrenoceptor-eYFP. After 24 h, cells were harvested, mixed, and plated onto coverslips where they grew for an additional 24 h before experimentation. Transfected cells were loaded with the Ca2+-sensitive dye Fura-2 acetoxymethyl ester (1.5 μM) by incubation (30 min, 37°C) under reduced light in Dulbecco's modified Eagle's growth medium. Using the epifluorescence microscope system, loaded cells were essentially illuminated, excited, and imaged as described previously above. Optoscan monochromator was used to rapidly alternate the excitation wavelength between 340 and 380 nm and to control the excitation bandpass (340 nm, band pass = 10 nm; 380 nm, band pass = 8 nm). Fura-2 fluorescence emission at 510 nm was monitored using a Cool Snap-HQ digital charge-coupled device camera. MetaFluor imaging software was used for control of all electronic hardware and image-data processing. Sequential images (3 × 3 binning) were collected every 2 s, exposure to excitation light was 100 ms/image, and all experiments were undertaken in HEPES-buffered saline solution (composition detailed above).
[Ca2+]i Image Analysis. Ratio images were presented in MetaFluor intensity-modulated display mode, which associates the color hue with the excitation ratio value and the intensity of each hue with the source image brightness. Background fluorescence was subtracted from each raw image as follows: a region of interest was manually drawn in a region of no fluorescence adjacent to the fluorescent cells in the 380 nm channel image and then selected and transferred to the matched image acquired at 340 nm. The background amount was then subtracted from each pixel in each channel. Background-subtracted images were then used for calculating the 340/380 nm ratio of each pixel, which was used to indicate changes in [Ca2+]i. After determination of the upper and lower thresholds, the ratio value of each pixel was associated with 1 of the 24 hues from blue (low [Ca2+]i) to red (high [Ca2+]i). Pooled average intensity-modulated display ratio intensity values measured from single cells were expressed as the mean of at least 10 cells with the vertical lines representing S.E.M.
Immunocytochemistry Fluorescence. Cells grown on coverslips and expressing the appropriate receptor fusion protein were fixed in 4% paraformaldehyde in PBS containing 5% sucrose for 10 min at RT. When indicated, cells were permeabilized for 10 min in 0.15% Triton X-100 and 3% nonfat milk in PBS; otherwise, the blocking step was performed avoiding the detergent (3% nonfat milk in PBS). Cells were incubated with a rabbit anti-FLAG polyclonal antibody (1:100; Sigma-Aldrich, Poole, Dorset, UK) for 1 h at room temperature and then washed twice with PBS. Cells were then incubated for a further 1 h with an Alexa594-conjugated anti-rabbit antiserum (1:400) (Molecular Probes, Eugene, OR). After washing with PBS, coverslips were mounted onto glass slides and viewed using the epifluorescence microscope. For detection of c-myc-α1b-adrenoceptor-Tyr67Cys-eYFP, the same procedure was followed but substituting a rabbit anti-c-myc polyclonal antiserum (1:100; Cell Signaling Technology Inc., Danvers, MA).
Western Blotting. After heating samples at 65°C for 15 min, both cell membrane samples and cell lysates were subjected to SDS-PAGE analysis using 4 to 12% Bis-Tris gels (NuPAGE; Invitrogen) and MOPS buffer. After electrophoresis, proteins were transferred onto nitrocellulose membranes that were incubated in 5% nonfat milk and 0.1% Tween 20/PBS solution at room temperature on a rotating shaker for 2 h to block nonspecific binding sites. The membrane was incubated overnight with a polyclonal anti-GFP antiserum and detected using a horseradish peroxidase-linked anti-goat IgG secondary antiserum (GE Healthcare, Little Chalfont, Buckinghamshire, UK). Immunoblots were developed by the application of enhanced chemiluminescence solution (Pierce Chemical, Rockford, IL).
Endoglycosidase Treatment. Endoglycosidase treatment was carried out on membrane preparations using PNGase F (Roche Diagnostics, Mannheim, Germany) at a final concentration of 1 U/μlat room temperature for 14 to 16 h.
[3H]Prazosin Binding Studies. Binding assays were initiated by the addition of 15 to 20 μg of cell membranes to an assay buffer (50 mM Tris-HCl, 100 mM NaCl, and 3 mM MgCl2, pH 7.4) containing [3H]prazosin (0.02–1 nM in saturation assays and 0.4 nM for competition assays) in the absence or presence of increasing concentrations of phenylephrine. Nonspecific binding was determined in the presence of 100 μM phentolamine. Reactions were incubated for 60 min at 30°C, and bound ligand was separated from free ligand by vacuum filtration through GF/B filters (Semat, St. Albans, Hertsfordshire, UK). The filters were washed twice with assay buffer, and bound ligand was estimated by liquid scintillation spectrometry.
Results
Development of Sequential 3 Color FRET Imaging Using Chromophore Concatamers. Previous studies based on the ability of coexpressed fragments of the hamster α1b-adrenoceptor to interact (Carrillo et al., 2004) allowed us to propose a model for the organization of this GPCR that consists of a chain of polypeptide monomers linked by alternating symmetrical TMD I–TMD I and TMD IV–TMD IV interactions (Fig. 1). To establish appropriate conditions and algorithms to monitor the transfer of energy within oligomeric complexes involving at least three polypeptides in living cells, we generated a concatamer (Fig. 2A) in which the dsRed2 fluorescent protein (Erickson et al., 2003) (a variant of the original dsRed1 from the reef coral Discosoma species that has improved solubility and a reduced tendency to form aggregates) was linked in-frame to the C terminus of a single open-reading frame containing the sequences of both the enhanced cyan (eCFP) and enhanced yellow (eYFP) variants of the Aequoria victoria green fluorescent protein. We have used previously the eCFP-eYFP single open-reading frame (Fig. 2A) as a positive control for imaging conventional, CFP to YFP, two-protein FRET (Carrillo et al., 2004). Furthermore, concatamers of all possible combinations of two of the three individual fluorescent proteins used in these studies (Fig. 2A) were also generated and analyzed (see Materials and Methods) to account for possible spillover and bleed-through. When expressed in HEK293 cells, the three-protein eCFP-eYFP-dsRed2 concatamer was distributed evenly throughout the cytoplasm but was excluded from the nucleus (Fig. 2B, A1–A6). Excitation of the eCFP element of this concatamer with 425 nm light (monochromator set to 425/10 nm) (Fig. 2C) followed by cell imaging and appropriate correction for bleed-through and spillover between channels (see Materials and Methods and Supplementary Data for details) allowed detection of eCFP to eYFP FRET (Fig. 2B, A4) and eCFP to dsRed2 FRET (Fig. 2B, A6). Excitation of the eYFP component of the concatamer with 495 nm (monochromator set to 495/9 nm) light allowed detection of eYFP to dsRed2 FRET (Fig. 2B, A5).
eCFP to dsRed2 FRET may represent a combination of direct eCFP-dsRed2 FRET and sequential eCFP-eYFP-dsRed2 FRET (Fig. 2C), but only sequential three-protein FRET can provide information on oligomeric rather than dimeric interactions. To establish the component of eCFP to dsRed2 FRET signal reflecting direct two-protein eCFP to dsRed2 versus sequential eCFP to eYFP to dsRed2, three-protein FRET (Fig. 2C), a Tyr67Cys mutation known to ablate fluorescence, was introduced into the eYFP segment of the concatamer (Fig. 2A). When this concatamer, in which the distance between and relative orientation of eCFP and dsRed2 is unchanged from the wild-type concatamer, was expressed in HEK293 cells, both the eCFP and dsRed2 elements retained fluorescence (Fig. 2B, B1 and B3), but, as anticipated, minimal signals were obtained in each of the eCFP to eYFP, eCFP to dsRed2, and eYFP to dsRed2 FRET channels (Fig. 2B, B4–B6). Because Tyr67Cys YFP is not fluorescent (Fig. 2B, B2), the minimal signal obtained at 630 nm (Em = 630/60 nm) after excitation of cells expressing the eCFP-Tyr67CysYFP-dsRed2 concatamer at 425 nm must represent direct eCFP to dsRed2 FRET, and therefore, the difference in signal obtained at 630 nm when using eCFP-eYFP-dsRed2 and eCFP-Tyr67CysYFP-dsRed2 concatamers represents the true sequential three-protein FRET signal (Fig. 2, C and D3). In addition, the possible contribution of an eYFP component being transferred to the dsRed2 as a result of excitation of eYFP by 425 nm light (i.e., as a concomitant of excitation of eCFP) was assessed using the eYFP-dsRed2 two-protein concatamer (Fig. 2A), but no FRET was detected (data not shown). As a further control, we cotransfected cells with isolated forms of each of eCFP, eYFP, and dsRed2. All of these three proteins were coexpressed in individual cells (Fig. 2B, C1–C3), and in this case, in addition to even distribution throughout the cytoplasm, some nuclear localization was observed. However, as for the eCFP-Tyr67Cys YFP-dsRed2 concatamer, negligible FRET signals were obtained in each channel (Fig. 2B, C4–C6), consistent with the concept that the three fluorescent proteins do not have significant inherent affinity for each other and because individual polypeptides were not constrained to be within FRET-competent distances. Quantitation of the extent of normalized eCFP to eYFP and eYFP to dsRed2 two-protein FRET and eCFP to eYFP to dsRed2, three-protein FRET (Fig. 2D) was achieved by imaging cells expressing various of the two-protein open-reading frame concatamers (Fig. 2A) and the three-protein concatamers described above and by coexpression of different pairs of the three individual fluorescent proteins. Of central importance to the current studies, with the parameters and corrections used, a substantially higher value was obtained for eCFP-dsRed2 FRET in cells expressing eCFP-eYFP-dsRed2 than in those expressing the eCFP-Tyr67Cys eYFP-dsRed2 concatamer (Fig. 2D3). Because dsRed2 retains some capacity to self-associate, we also coexpressed the eCFP-dsRed2 and eYFP-dsRed2 two-protein concatamers. No significant sequential three-protein or single eCFP to eYFP FRET was recorded (data not shown), eliminating the formal possibility that dsRed2 self-association brought all three fluorescent polypeptides into a complex within FRET-competent distances. These results demonstrate the ability to image and quantitate sequential eCFP to eYFP to dsRed2 three-protein FRET in individual living cells and hence to generate a system that can be applied to detect the presence of at least three appropriately tagged polypeptides in a quaternary structure complex, to define the appropriate background for subtraction, and to eliminate any contribution of direct eCFP-dsRed2 FRET to three-protein FRET data sets.

Proposed oligomeric organization of the α1b-adrenoceptor. Based on interactions between fragments of the α1b-adrenoceptor, in which TMD I and TMD IV (yellow) were shown to contribute symmetrical protein-protein interaction interfaces, Carrillo et al. (2004), proposed a “daisy-chain” structure that may link α1b-adrenoceptor monomers into higher-order oligomers. Cartoons of such oligomers are displayed: Top, viewed from the extracellular space; bottom, viewed as a section through the plasma membrane. Such organizational structure is reminiscent of the arrays of rhodopsin in murine rod outer segments observed by atomic force microscopy (Fotiadis et al., 2004).

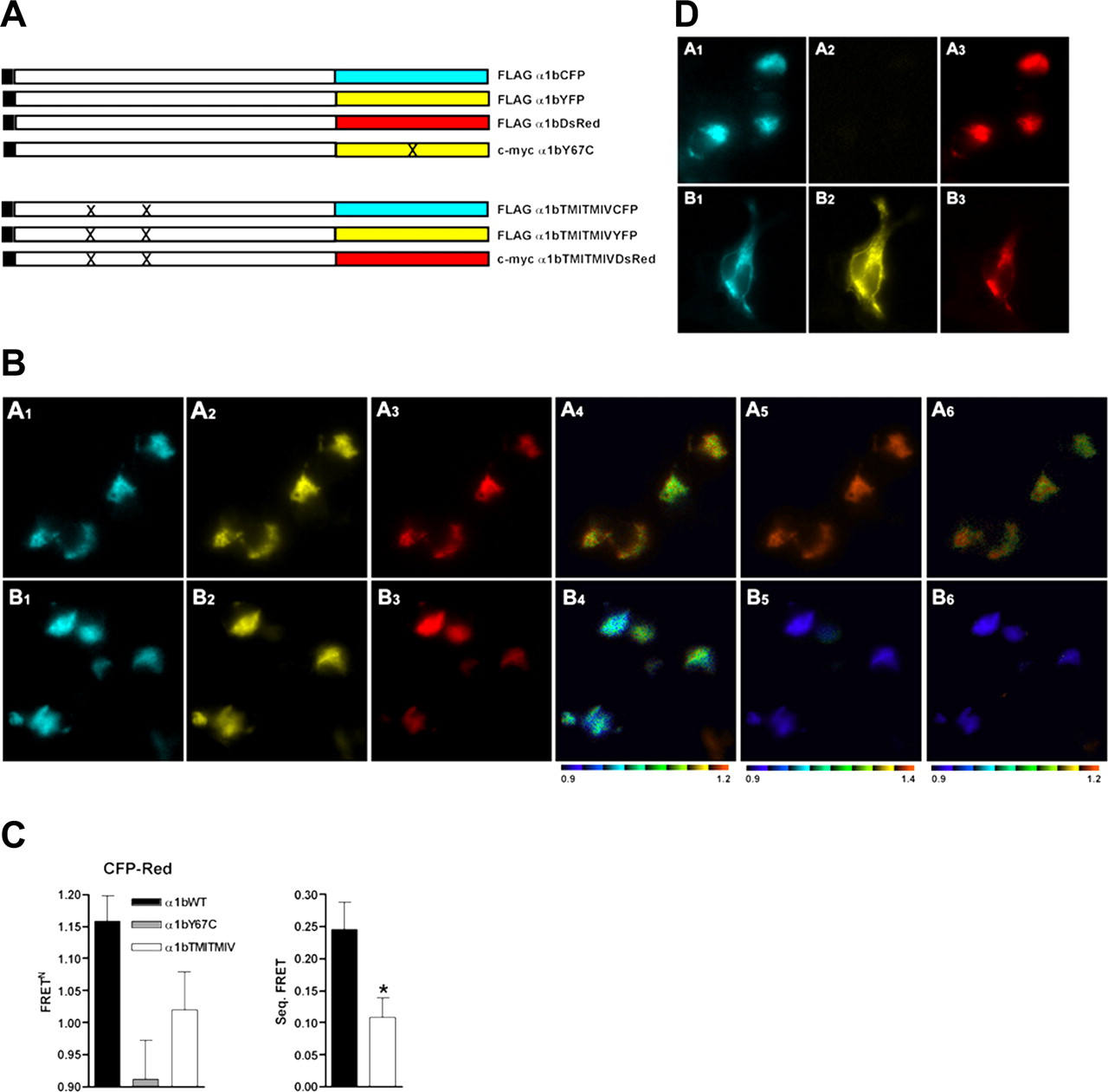
Establishment of single-cell three-color FRET imaging using concatamers of fluorescent proteins. A, concatamers of eCFP, eYFP, and dsRed2 and eCFP, Tyr67Cys eYFP with dsRed2, and a number of two-protein open-reading frames were generated. B, A1–A6, the eCFP-eYFP-dsRed2 concatamer was expressed in HEK293 cells and imaged. Each element of the concatamer (A1, eCFP; A2, eYFP; A3, dsRed2) could be monitored. Excitation at 425 nm allowed imaging of eCFP-eYFP (A4) and eCFP-dsRed2 (A6) FRET. Excitation at 495 nm allowed imaging of eYFP-dsRed2 (A5) FRET. B1–B6, the eCFP-Tyr67Cys eYFP-dsRed2 concatamer was expressed in HEK293 cells and imaged. Although the eCFP (B1) and dsRed2 (B3) elements could be visualized, Tyr67Cys eYFP is not fluorescent (B2). B4–B6, FRET signals akin to A4–A6 are shown. C1–C6, individual forms of eCFP (C1), eYFP (C2), and dsRed2 (C3) were coexpressed (C4–C6). FRET signals were monitored. Pseudocolor images of FRET signals are coded according to the normalized FRET calculations (see Materials and Methods and Supplementary Data). C, the basis of direct eCFP-dsRed2 and sequential eCFP-eYFP-dsRed2 FRET is illustrated. D, various two-protein open-reading frames and the three-protein concatamers were expressed, or the individual fluorescent proteins were coexpressed. FRET signals were quantitated and normalized (see Materials and Methods and Supplementary Data). These were used to establish appropriate conditions to measure both direct two-protein eCFP-dsRed2 and sequential three-protein eCFP-eYFP-dsRed2 FRET; 1.0 is equivalent to no FRET signal.
The α1b-Adrenoceptor Is Able to Form Oligomers and the Organizational Structure Is Altered by Combined Mutations in TMDs I and IV. We next used three-protein FRET to garner support for the presence of oligomeric rather than dimeric α1b-adrenoceptor organization in single transfected cells and to explore the molecular basis for this. Coexpression of each of C-terminally tagged eCFP, eYFP, and dsRed2 forms of the α1b-adrenoceptor (Fig. 3A) in HEK293 cells followed by excitation at 425 nm resulted in each of eCFP to eYFP and eCFP to dsRed2 FRET signals (Fig. 3B, A1–A6). Excitation with 495 nm light resulted in eYFP to dsRed2 FRET (Fig. 3B, A1–A6). With coexpression of α1b-adrenoceptor-eCFP, α1b-adrenoceptor-Tyr67Cys eYFP, and α1b-adrenoceptor-dsRed2, eCFP to dsRed2 FRET was virtually eliminated (Fig. 3, B and C). Because any eCFP to dsRed2 FRET signal obtained in the presence of α1b-adrenoceptor-Tyr67Cys eYFP must represent direct eCFP to dsRed2 FRET, then subtraction of this value from that obtained after coexpression of the eCFP-, eYFP-, and dsRed2-tagged forms of the receptor provided the true sequential three-protein FRET GPCR oligomer signal (Fig. 3C). The lack of eCFP-dsRed2 FRET signal in the presence of α1b-adrenoceptor-Tyr67Cys eYFP did not reflect lack of expression of this construct. Although not fluorescent, incorporation of an N-terminal c-myc epitope tag into the construct allowed immunological detection of expression of α1b-adrenoceptor-Tyr67Cys eYFP with the same pattern of distribution as α1b-adrenoceptor-eCFP and α1b-adrenoceptor-dsRed2 (Fig. 3D). Furthermore, in the quantified data, FRET signals were normalized (see Materials and Methods and Supplemental Data) to take into account any variation in construct expression levels. Because the difference in eCFP to dsRed2 FRET in these cases must correspond to sequential eCFP to eYFP to dsRed2 three-protein FRET, these results demonstrate that at least a proportion of these receptors were within an oligomeric complex comprising at least three receptor monomers within FRET-competent distances.
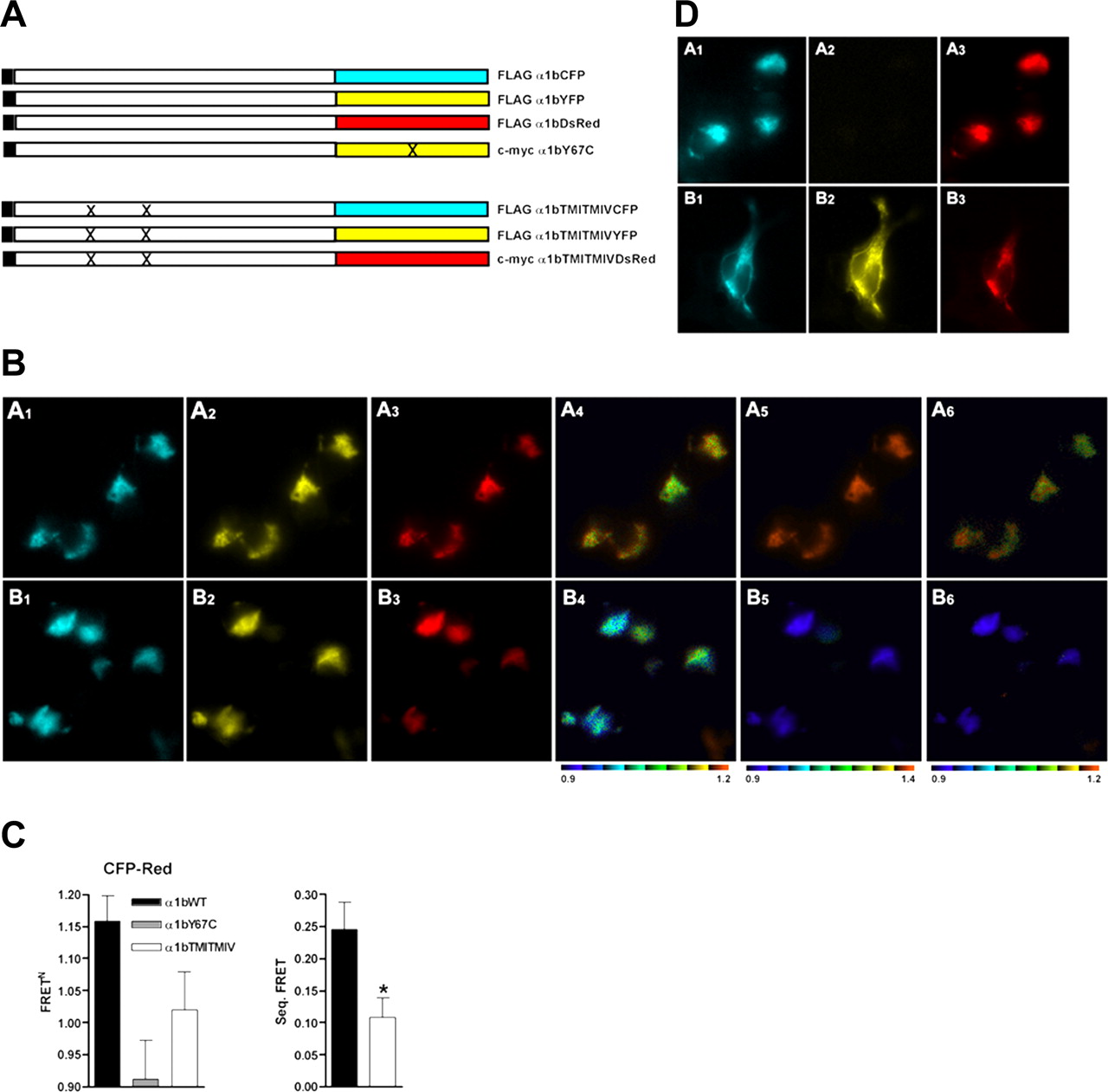
Single-cell imaging of oligomers of the α1b-adrenoceptor containing at least three monomers. Combined mutations in TMD I and TMD IV modify oligomeric organization. A, forms of the α1b-adrenoceptor N-terminally tagged (black) with the FLAG epitope were C-terminally tagged with eCFP, eYFP, or dsRed2, whereas an N-terminally c-myc-tagged form of the α1b-adrenoceptor was C-terminally tagged with Tyr67Cys eYFP. FLAG-tagged forms of Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor were C-terminally tagged with either eCFP or eYFP and a c-myc-tagged form of Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor with dsRed2. X indicates sites of mutation. B, A1–A6, FLAG-tagged forms of α1b-adrenoceptor-eCFP (A1), α1b-adrenoceptor-eYFP (A2), and α1b-adrenoceptor-dsRed2 (A3) were coexpressed in HEK293 cells. B1-B6, equivalent forms of Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor were expressed and imaged: B1, eCFP; B2, eYFP; B3, dsRed2. FRET signals after excitation as in Fig. 2 were imaged (A4–A6, B4–B6). C, eCFP-dsRed2 FRET signals were normalized for the wild-type α1b-adrenoceptor (black), α1b-adrenoceptor with Tyr67CysYFP as one of the constructs (gray), and Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor (white). Sequential three-protein FRET was calculated by subtracting the signal in cells expressing α1b-adrenoceptor-Tyr67CysYFP. *, significantly lower, p < 0.05. D, α1b-adrenoceptor-Tyr67Cys eYFP is nonfluorescent but is expressed. α1b-Adrenoceptor-eCFP (A1, B1) and α1b-adrenoceptor-dsRed2 (A3, B3) were coexpressed with c-myc-α1b-adrenoceptor-Tyr67Cys eYFP (A2, B2). A1–A3, fluorescence imaging; B1–B3, cells were permeabilized and immunoblotted with anti-c-myc and secondary antibody, and either direct fluorescence (B1 and B3) or immunofluorescence (B2) images were obtained.
Based on our prediction of key TMD helices for oligomerization of the α1b-adrenoceptor (Carrillo et al., 2004) and the bioinformatic identification of the role of specific amino acids in both TMDs I and IV in the quaternary structure of the CCR5 receptor (Hernanz-Falcon et al., 2004; de Juan et al., 2005), we generated a Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor. This contains point mutations in hydrophobic residues in both TMD I (Leu65Ala, Val66Ala) and TMD IV (Leu166Ala, Leu167Ala) (Fig. 3A) that are similar to those modified by Hernanz-Falcon et al. (2004) and are reported to eliminate two-protein FRET signals corresponding to protein-protein interactions for the CCR5 receptor. Coexpression of C-terminally eCFP-, eYFP-, and dsRed2-tagged forms of the Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor resulted in a substantial decrease of normalized eCFP-dsRed2 FRET (Fig. 3B, B6, and 3C) compared with wild-type α1b-adrenoceptor, although there was no reduction in expression of the mutants based on the fluorescence corresponding to the eCFP, eYFP, or dsRed2 tags (Fig. 3B, A1–A3 versus B1–B3). Applying the direct eCFP-dsRed2 FRET correction provided by the Tyr67Cys eYFP mutant, sequential three-protein eCFP to eYFP to dsRed2 FRET was reduced by 56% (p < 0.05) in cells expressing the tagged forms of Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor (Fig. 3C). These results indicate that the introduced mutations reduced the proximity and/or altered the orientation of the fluorescent proteins linked to the mutant α1b-adrenoceptor and hence are consistent with mutations causing disruption of the organizational structure of the α1b-adrenoceptor oligomeric complex.

The wild-type α1b-adrenoceptor but not Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor is able to reach the cell surface. A, HEK293 cells were transfected to express FLAG-tagged forms of wild-type α1b-adrenoceptor-eYFP (A and B) or Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eYFP (C and D). Nonpermeabilized (A and C) and permeabilized (B and D) cells were stained with anti-FLAG and secondary antibody and imaged to detect eYFP (A1, B1, C1, and D1) or anti-FLAG (A2, B2, C2, and D2). eYFP and anti-FLAG images were also merged (A3, B3, C3, and D3). B, cells expressing wild-type α1b-adrenoceptor-eYFP (A1–A4) or Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eYFP (B1–B4) were treated with Hoechst 33342 nuclear stain and RQAPB for 30 min and imaged; A1 and B1, YFP; A2 and B2, Hoechst 33342; A3 and B3, RQAPB; A4 and B4, merged images.
The Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-Adrenoceptor Fails to Mature, Is Not Delivered to the Cell Surface, and Does Not Signal. A key question in understanding the importance of GPCR quaternary structure is whether modulation of protein-protein interactions can modify receptor maturation and/or cell surface delivery and therefore function (Bulenger et al., 2005). At least in some cell lines and native tissues, α1-adrenoceptor subtypes are known to recycle to and from the plasma membrane via endosomal pools both rapidly and constitutively, and therefore, at steady state, a large proportion of these GPCRs is inside the cell rather than at the plasma membrane (Morris et al., 2004; Pediani et al., 2005). Comparing the distribution pattern of N-terminally epitope-tagged forms of wild-type α1b-adrenoceptor-eYFP with that of Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eYFP in nonpermeabilized and permeabilized transfected HEK293 cells, cell surface antiepitope-tag staining in nonpermeabilized cells could be shown for the wild-type α1b-adrenoceptor (Fig. 4A, A2) but not for Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor (Fig. 4A, C2). Furthermore, α1-adrenoceptors are able to bind and internalize the ligand BODIPY-FL prazosin (QAPB) only if they previously had reached the cell surface (Pediani et al., 2005). QAPB displays significant fluorescence only when bound to receptor. A form of QAPB (RQAPB) that displays red fluorescence when bound to α1-adrenoceptor subtypes (Pediani et al., 2005) was provided to cells expressing wild-type α1b-adrenoceptor-eYFP. Living cells subsequently displayed intracellular red fluorescence (Fig. 4B, A3). This did not occur in cells expressing Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eYFP (Fig. 4B, B3). This was not a reflection that Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor is simply denatured and is inherently unable to bind antagonist ligands. However, although direct measures of expression levels based on fluorescence corresponding to α1b-adrenoceptor-eYFP and Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eYFP (Fig. 5A) indicated that the mutations did not hinder protein expression, the capacity of Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eYFP to bind [3H]prazosin was substantially reduced (wild type = 2.02 ± 0.37; Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor = 0.36 ± 0.03 pmol/mg of membrane protein, means ± S.E.M., n = 5) (Fig. 5B). However, the affinity of [3H]prazosin for the Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eYFP binding sites it was able to label was not reduced (Kd value for wild type = 85 ± 13 pM; Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eYFP = 33 ± 3 pM). Likewise, competition studies using a single concentration of [3H]prazosin and varying concentrations of RQAPB demonstrated that the affinity of RQAPB was actually slightly higher for the Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor (pKi = 8.85 ± 0.18) than for the wild-type α1b-adrenoceptor (pKi = 8.33 ± 0.02).
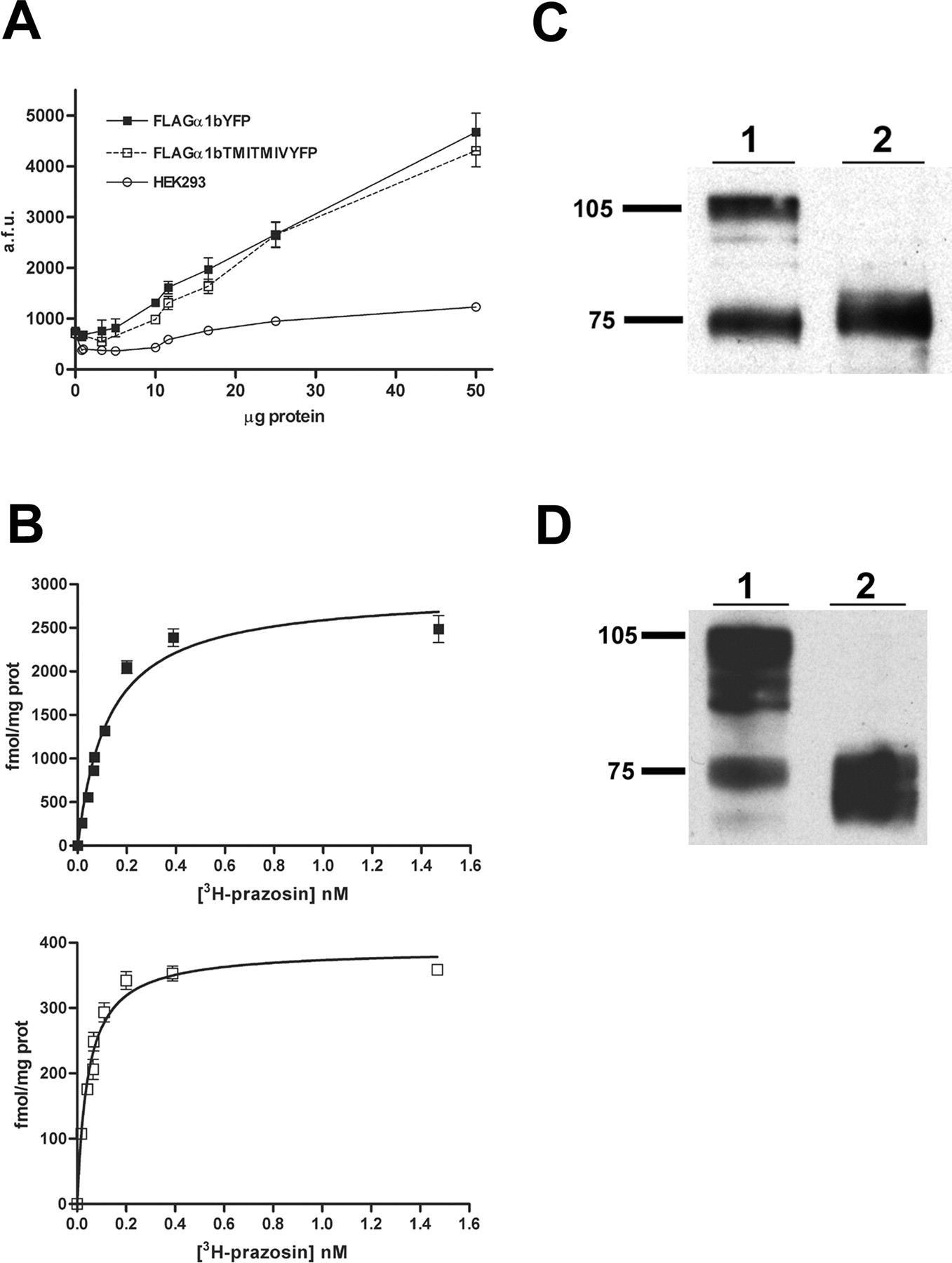
The Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor is not terminally glycosylated. A, membranes from mock-transfected HEK293 cells (○) and those transfected to express FLAG-α1b-adrenoceptor-eYFP (▪) or FLAG Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eYFP (□) were used to monitor fluorescence corresponding to eYFP. B, the specific binding of varying concentrations of [3H]prazosin to FLAG-α1b-adrenoceptor-eYFP (top) and FLAG-Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eYFP (bottom) was assessed. C, membranes expressing α1b-adrenoceptor-eYFP (1) or Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eYFP (2) were resolved by SDS-PAGE and immunoblotted with anti-GFP. D, for α1b-adrenoceptor-eYFP, the band of apparent molecular mass of 75 kDa is the immature form of the receptor and the band of apparent molecular mass of 105 kDa is the mature, terminally N-glycosylated form. Membranes expressing wild-type α1b-adrenoceptor-eYFP were treated with vehicle (1) or N-glycosidase F (2) and then resolved and immunoblotted as in C.
It is interesting that when samples expressing wild-type α1b-adrenoceptor-eYFP and Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eYFP were resolved by SDS-PAGE and immunoblotted with an anti-GFP antiserum, the patterns were distinct. Whereas Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eYFP migrated predominantly as a single band of some 75 kDa, wild-type α1b-adrenoceptor-eYFP was represented by a mixture of 75 and 105 kDa polypeptides (Fig. 5C). Previous studies have indicted these to represent the core-glycosylated (lower molecular mass) precursor form and mature (higher molecular mass) form of the receptor (Bjorklof et al., 2002). In accord with this, treatment of membranes expressing wild-type α1b-adrenoceptor-eYFP with N-glycosidase F eliminated the 105-kDa band, with all the immunoreactivity now migrating with apparent molecular mass between 70 and 75 kDa (Fig. 5D).
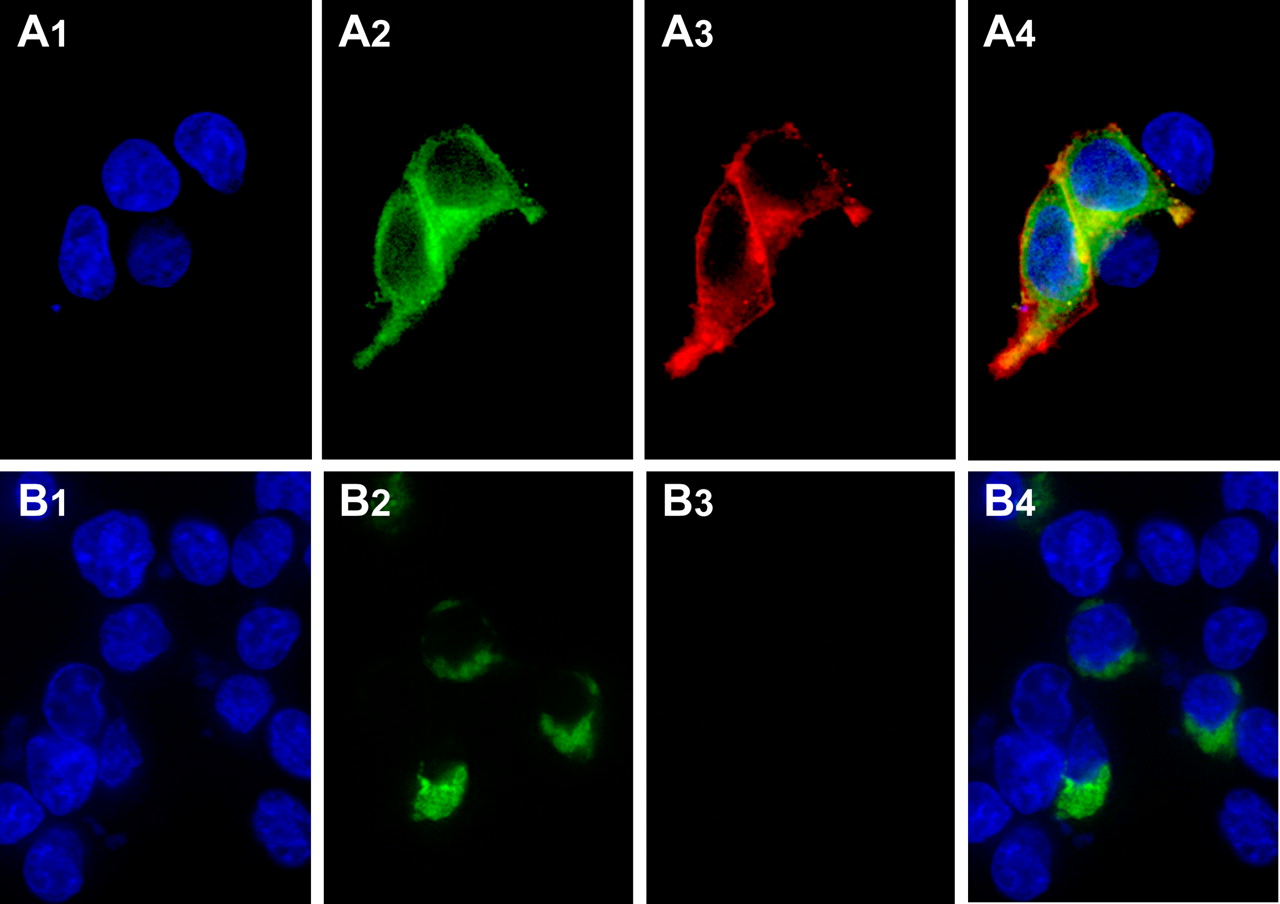
To further explore whether the mutations introduced into TMDs I and IV eliminated α1b-adrenoceptor protein-protein interactions and quaternary structure, we used bimolecular fluorescence complementation (Kerppola, 2006). When eYFP is split into N-terminal 172 amino acid and C-terminal 67 amino acid fragments, neither displays inherent autofluorescence. However, if they are linked to polypeptides that form a protein complex, this can be sufficient to bring the two fragments of eYFP into proximity and allow partial reformation of the fluorescent protein chromophore (Kerppola, 2006). When N-terminally FLAG-tagged forms of the α1b-adrenoceptor with either N-terminal 172-eYFP or C-terminal 67-eYFP fragments linked to the receptor C terminus were expressed individually in HEK293 cells, no yellow autofluorescence was observed (Fig. 6, A1 and B1), although successful expression of the constructs could be shown via anti-FLAG immunocytochemistry (Fig. 6, A2 and B2). However, after coexpression of these two constructs, eYFP fluorescence was generated (Fig. 6, C1). This dimeric/oligomeric α1b-adrenoceptor complex was able to reach the surface of transfected HEK293 cells because the addition of RQAPB to these cells resulted in the appearance of red fluorescence at the cell surface and inside the cells (Fig. 7). Merging of the fluorescent signals corresponding to the complemented eYFP, and hence the dimeric/oligomeric α1b-adrenoceptor, and RQAPB resulted in a strong overlap correlation coefficient (r2 = 0.84) (Fig. 7, A4) consistent with RQAPB being bound to the receptor when it was at the plasma membrane followed by internalization and cycling of the α1b-adrenoceptor dimer/oligomer with the ligand still bound. In contrast, when c-myc-Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-N-terminal 172-eYFP and FLAG-Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-C-terminal 67-eYFP were coexpressed, although fluorescence complementation was achieved, indicating the two forms of the receptor were in proximity, the complemented eYFP signal was restricted entirely to a perinuclear endoplasmic reticulum/Golgi location, and the addition of RQAPB failed to generate red fluorescence (Fig. 7, B3), consistent with the premise that the mutant form of the receptor had never reached the cell surface.

Monitoring α1b-adrenoceptor quaternary structure via bimolecular fluorescence complementation. N-terminally FLAG-tagged forms of the α1b-adrenoceptor were C-terminally tagged with either the N-terminal 172 amino acids (A and C) or C-terminal 67 amino acids (B and C) of eYFP. Cells were either imaged to detect bimolecular fluorescence complementation (top row) or permeabilized cells were used to detect the FLAG epitope (bottom row).
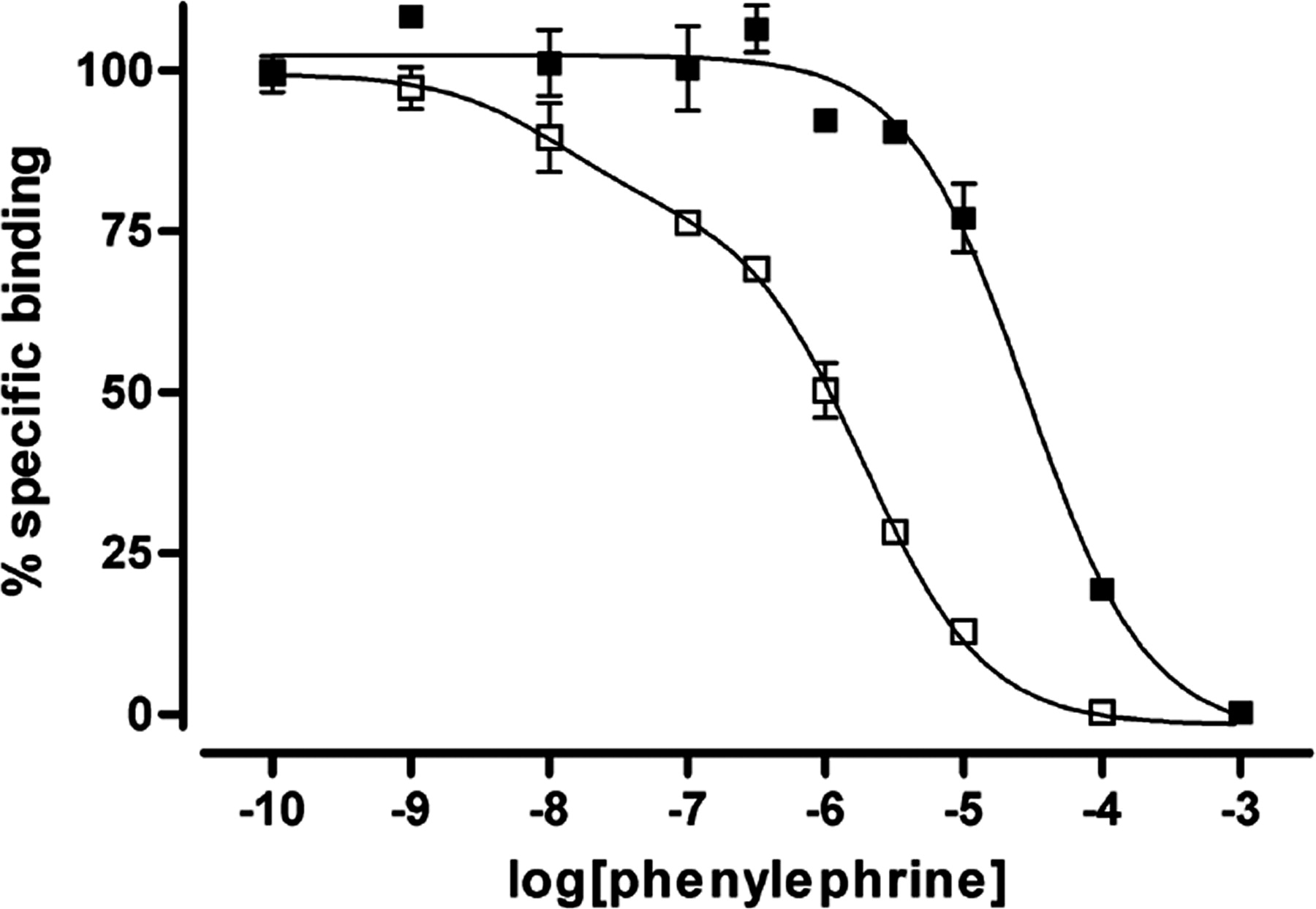
If only the oligomeric wild-type α1b-adrenoceptor matures and is able to reach the cell surface, then only the wild type would be anticipated to generate signals in response to agonist ligands. Cells expressing either wild-type α1b-adrenoceptor-eYFP or Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eCFP were plated onto different sections of a single coverslip, imaged (Fig. 8A), and loaded with the Ca2+ indicator dye FURA-2. In cells expressing wild-type α1b-adrenoceptor-eYFP, addition of the α-adrenoceptor agonist phenylephrine resulted in elevation of intracellular [Ca2+] (Fig. 8, B and C). In contrast, no such signal was produced in equivalent cells expressing Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eCFP (Fig. 8, B and C). This did not reflect that expression of Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eCFP somehow ablated the potential for receptor-mediated [Ca2+] elevation. In cells expressing either wild-type or Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor, after challenge with phenylephrine, the addition of ATP to activate the endogenously expressed P2Y purinoceptor population resulted in equivalent elevations of intracellular [Ca2+] (Fig. 8C). Again, the lack of capacity of Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor to respond to phenylephrine did not reflect an inherent inability to bind the agonist ligand. Phenylephrine was actually substantially more potent in competing for the binding of [3H]prazosin in membrane preparations of cells expressing Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor than the wild-type receptor (Fig. 9).
Although we were unable to detect Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor at the cell surface, we wished to ascertain whether small amounts of the mutant might reach the cell surface and the limits of detectability. First, we transfected HEK293 cells with varying amounts of cDNA corresponding to the wild-type α1b-adrenoceptor-eYFP construct, generated membranes, and performed [3H]prazosin binding studies. A 4-fold reduction in cDNA amount translated to an approximately 10-fold reduction in [3H]prazosin binding levels (Fig. 10A). Parallel studies directly measured eYFP fluorescence and provided similar conclusions (Fig. 10B). Scaling the amounts of cDNA used to transfect cells for [Ca2+] mobilization imaging/assays showed that phenylephrine-mediated elevation of [Ca2+] was easily detected, even with amounts of cDNA that resulted in an inability to resolve total from nonspecific [3H]prazosin binding and hence is at the limit of receptor detection (Fig. 10C). However, [Ca2+] elevation did require some level of wild-type α1b-adrenoceptor-eYFP expression because no response was observed to phenylephrine in mock-transfected cells (Fig. 10C). As such, the inability of phenylephrine to cause elevation of intracellular [Ca2+] in cells expressing the Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor implies that if any of this mutant reaches the cell surface, it must be in vanishingly small amounts.
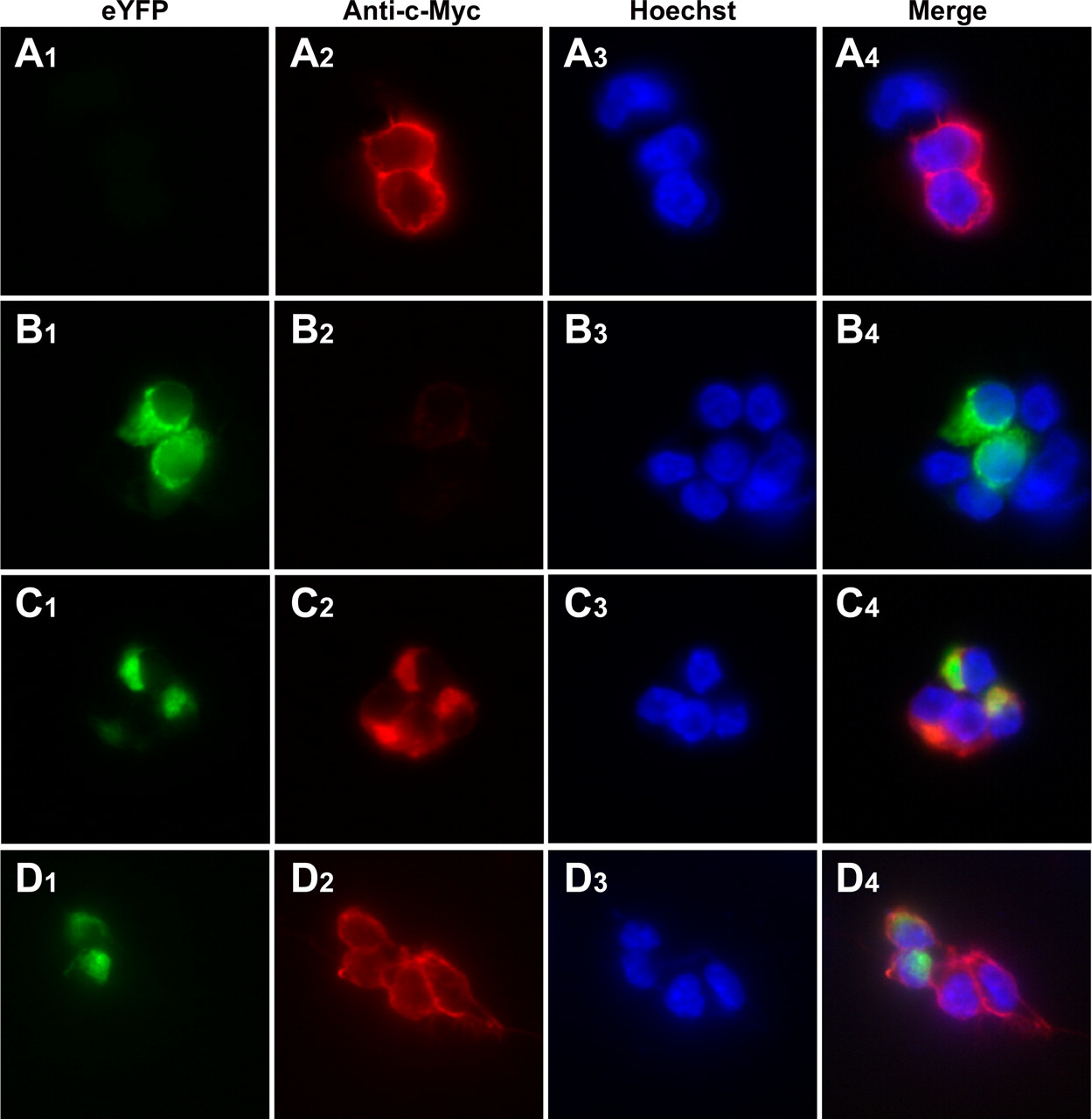
In a number of cases, receptor mutants that cannot escape the endoplasmic reticulum/Golgi apparatus after synthesis can act as “dominant-negatives” and limit cell surface delivery of a coexpressed, wild-type version of the same receptor. This is both relevant to a number of human diseases and has provided evidence for a requirement for protein-protein interactions between GPCR monomers before cell surface delivery (Bulenger et al., 2005). Because the bimolecular fluorescence complementation studies had shown that the Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor was still able to self-dimerize/oligomerize to some extent, we asked whether this mutant would prevent cell surface delivery of wild-type α1b-adrenoceptor. HEK293 cells were transfected to express either c-myc-α1b-adrenoceptor-Y67C-eYFP alone (Fig. 11A) or together with FLAG-Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eYFP in a 1:3 ratio (Fig. 11, B and C). Immunocytochemistry using an anti-c-myc antibody and a secondary antibody conjugated to Alexa594 in nonpermeabilized cells detected the wild-type receptor at the cell surface in the absence of the mutant (Fig. 11A), whereas cell surface wild-type α1b-adrenoceptor was undetectable in cells coexpressing FLAG-Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eYFP (Fig. 11B), the expression of which was detected by monitoring eYFP fluorescence (Fig. 11B). Anti-c-myc immunocytochemistry in permeabilized cells coexpressing the wild-type and mutant forms of the receptor confirmed the expression and intracellular location of c-myc-α1b-adrenoceptor-Y67C-eYFP (Fig. 11C), whereas merging of the anti-c-myc and eYFP signals confirmed the codistribution of the two forms (Fig. 11C). As a control for these studies, we transiently transfected cells with FLAG-Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eYFP and c-myc-CXCR1 because we have demonstrated previously negligible interactions between CXCR1 and α1-adrenoceptors (Wilson et al., 2005). The presence of FLAG-Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eYFP failed to interfere with cell surface delivery of anti-c-myc immunoreactivity corresponding to the CXCR1 receptor (Fig. 11D).
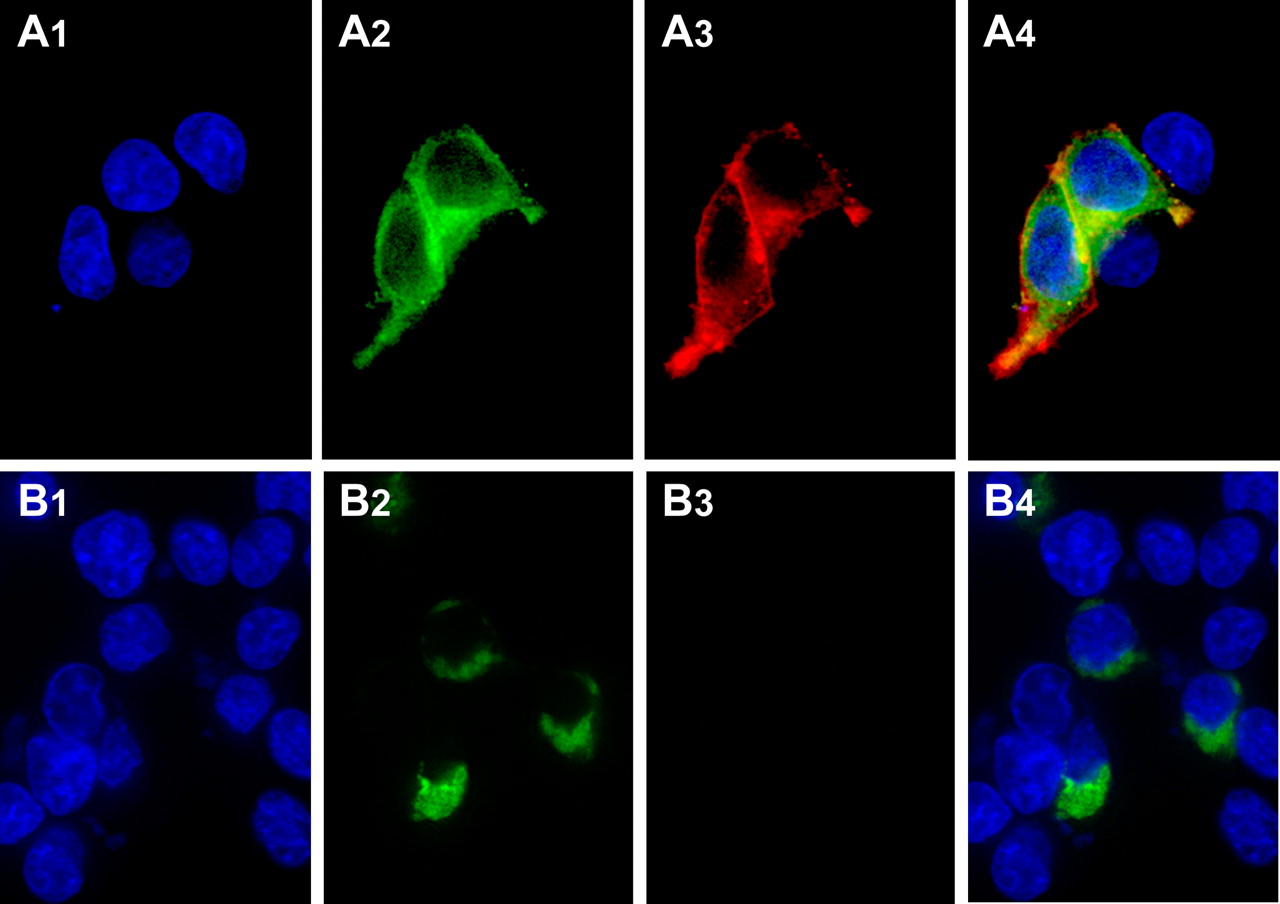
Bimolecular fluorescence complementation and RQAPB labeling demonstrates the α1b-adrenoceptor is recycling in cells as a dimer/oligomer and Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor retains quaternary structure but is retained in the endoplasmic reticulum. N-terminally FLAG-tagged forms of the α1b-adrenoceptor were C-terminally tagged with the N-terminal 172 or C-terminal 67 amino acids of eYFP and expressed in HEK293 cells (A). RQAPB was subsequently added. Cell nuclei (A1) were stained with Hoechst 33342 nuclear dye, bimolecular fluorescence complementation (A2) was assessed via eYFP fluorescence, whereas previous cell surface delivery of the α1b-adrenoceptor was shown by the appearance (A3) of red fluorescence corresponding to ligand binding. Merging of the images (A4) produced a strong overlap correlation coefficient for the receptor dimer/oligomer and bound RQAPB (r2 = 0.84). B, c-myc-Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor was C-terminally tagged with the N-terminal 172 amino acids of eYFP, whereas FLAG-Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor was C-terminally tagged with the C-terminal 67 amino acids of eYFP. After transfection into HEK293 cells, RQAPB was added. Cell nuclei (B1) were stained with Hoechst 33342 nuclear dye, bimolecular fluorescence complementation (B2) was assessed via eYFP fluorescence, whereas lack of cell surface delivery of Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor was shown by the lack of development (B3) of red fluorescence corresponding to ligand binding. The merged image (B4) confirms the perinuclear location of the Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor dimer/oligomer.
The Capacity to Be TerminallyN-Glycosylated Does Not Determine Cell Surface Delivery and Function of the α1b-Adrenoceptor. To attempt to ascertain individual contributions of the TMD I and IV mutations to the pheno-type of the combined Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor, both Leu65Ala, Val66Ala α1b-adrenoceptor and Leu166Ala, Leu167Ala α1b-adrenoceptor mutants were constructed. After the addition of each of eCFP, eYFP, or dsRed2 to the C-terminal tail of each mutant, these potentially FRET-competent forms were coexpressed in HEK293 cells and 3-FRET studies performed. The sequential eCFP to eYFP to dsRed2 3-FRET signal for the Leu65Ala, Val66Ala α1b-adrenoceptor was indistinguishable from the wild-type α1b-adrenoceptor (Fig. 12), whereas the Leu166Ala, Leu167Ala α1b-adrenoceptor mutant generated a reduced 3-FRET signal that was not statistically different from the reduction in 3-FRET produced by the Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor (Fig. 12). It is interesting, however, that although both the Leu65Ala, Val66Ala α1b-adrenoceptor and the Leu166Ala, Leu167Ala α1b-adrenoceptor failed to be delivered to the plasma membrane (Fig. 13, A and B), immunoblotting studies demonstrated that both these forms were able to mature into the 105-kDa form of the receptor, and the ratios of 105- to 75-kDa species were not different from the wild-type α1b-adrenoceptor (Fig. 13C).

Cells expressing the wild-type but not Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor respond to phenylephrine. Cells transfected to express wild-type α1b-adrenoceptor-eYFP (A and C, green,) or Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eCFP (A and C, red) were loaded with Fura-2 and stimulated sequentially with 10 μM phenylephrine (B and C) and ATP (C). In A, the group of cells on the left of the image (red) express Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eCFP, and the group on the right (green) express wild-type α1b-adrenoceptor-eYFP. B, peak Ca2+ signals after addition of phenylephrine. Only cells expressing α1b-adrenoceptor-eYFP respond to phenylephrine (arrow). Picture taken 30 s after the addition of phenylephrine.
Discussion
The lack of ability to discriminate between dimeric and oligomeric GPCR organization has generally resulted in these terms being used interchangeably (Milligan, 2006). Although combined immune capture and coimmunoprecipitation of three differently epitope-tagged and coexpressed forms of the M2 muscarinic acetylcholine receptor (Park and Wells, 2004) was certainly consistent with oligomeric interactions, this approach has not been used frequently and is limited by the same issues that have dogged analysis of coimmunoprecipitation of pairs of GPCRs (Milligan and Bouvier, 2005). Many recent efforts to explore GPCR quaternary structure have, therefore, centered on the use of either bioluminescence or fluorescence resonance energy transfer with eYFP acting as energy acceptor for light produced either by excitation of eCFP or oxidation of a substrate by a luciferase (Milligan and Bouvier, 2005). Bioluminescence resonance energy transfer techniques are not currently compatible with cell imaging, and although FRET techniques are, only a limited number of studies have attempted to expand two-protein FRET imaging to three-protein FRET imaging (Galperin et al., 2004). In addition to the inherently small signals anticipated in these studies, this reflects, in part, the necessity for custom filter sets to define and optimally limit spill-over of emitted light into the individual FRET channels, and requirements to define that eCFP to red fluorescent protein FRET signals actually represent sequential energy transfer between appropriate resonance energy transfer pairings. As described previously by Galperin et al. (2004), we developed sequential 3-FRET and optimized filter sets by using concatamers of three fluorescent proteins that are FRET-compatible. We also used the Tyr67Cys eYFP mutation to estimate direct eCFP-dsRed2 FRET versus sequential eCFP-eYFP-dsRed2 FRET. Finally, to generate data with sufficiently low coefficient of variation to allow statistically significant differences in sequential 3-FRET signals to be recorded for oligomers of the wild-type and mutant α1b-adrenoceptor constructs required development of a new modified ratio method for FRET quantification. This greatly improved the coefficient of variation in both our own data and published data from others compared with previous FRET quantification algorithms (see Supplemental Data).
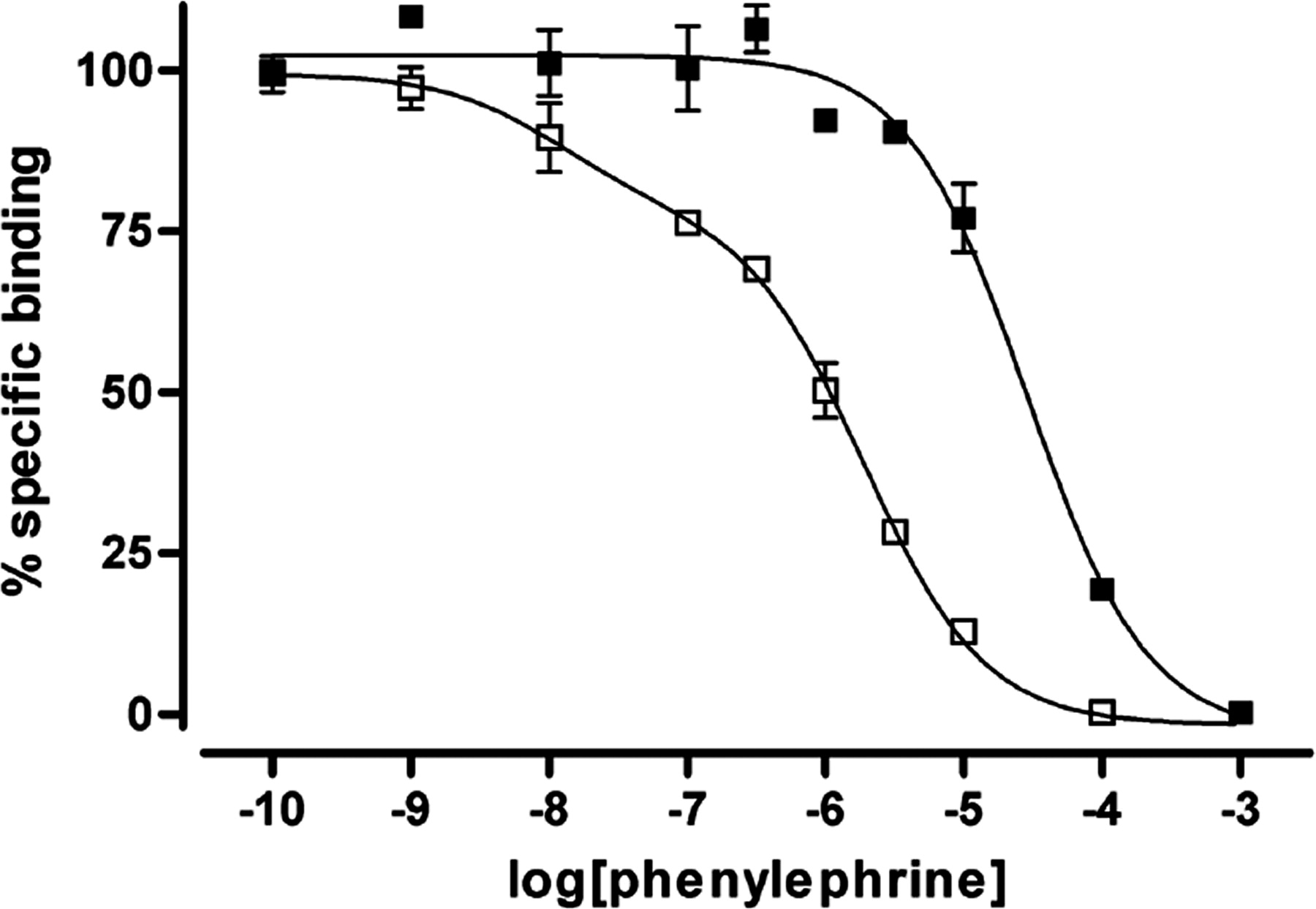
Ligand binding characteristics of the wild-type and Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor. The capacity of phenylephrine to compete for binding with [3H]prazosin to the wild-type (filled symbols) and Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala (open symbols) α1b-adrenoceptor-eYFP was assessed. The graph corresponds to a representative result from three independent experiments. Wild-type α1b-adrenoceptor-eYFP (pKi = 4.82 ± 0.25), Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eYFP (pKi high = 8.41 ± 0.16; pKi low = 6.35 ± 0.14; % high = 26.10 ± 4.40).

Functional cell surface α1b-adrenoceptors can be measured at vanishingly low levels. HEK293 cells were transiently transfected with varying amounts of cDNA encoding FLAG-α1b-adrenoceptor-eYFP. A, membranes of these cells were used to measure the specific binding of a single concentration (0.4 nM) of [3H]prazosin anticipated to bind more than 90% of the available sites (see Fig. 5). B, varying amounts of these membranes were used to monitor fluorescence corresponding to eYFP. C, cells mock-transfected (purple) or transfected with varying amounts of cDNA encoding FLAG-α1b-adrenoceptor-eYFP were loaded with Fura-2 and monitored for intracellular Ca2+ signals after stimulation with 10 μM phenylephrine and subsequently with 10 μM ATP (only in mock-transfected cells).
With such technical information in place, we were able to demonstrate sequential three-protein FRET between appropriately tagged forms of the α1b-adrenoceptor, confirming our previous prediction that oligomers of this GPCR, rather than only dimers, would exist (Carrillo et al., 2004). Because we had also predicted that this would reflect, at least in part, both TMD I–TMD I and TMD IV–TMD IV interactions, we used mutagenesis to test this hypothesis. It is interesting that in studies on the chemokine CCR5 receptor, Hernanz-Falcon et al. (2004) reported using bioinformatics to predict amino acids in both TMD I and TMD IV important for receptor “dimerization” (de Juan et al., 2005) and demonstrated a loss of two-protein FRET signals, potentially consistent with a lack of interaction after mutation. The studies of Hernanz-Falcon et al. (2004) have attracted considerable interest. Although the mutants we introduced into the α1b-adrenoceptor were based, in part, on alignments with the CCR5 receptor, the results we have produced are quite different. For example, Hernanz-Falcon et al. (2004) reported a complete loss of two-protein FRET signal. We observe only a partial loss of sequential three-protein FRET. This may be for a number of reasons. First, these single-point mutations in TMDs seem to be insufficient to fully disassemble a dimer/oligomer. Second, in our previous analysis of the oligomeric potential of the α1b-adrenoceptor we also noted further, nonsymmetrical, interactions between TMD I and/or II with TMD V and/or VI (Carrillo et al., 2004) and concluded these might allow generation of a structure akin to the “arrays of dimers” noted for murine rhodopsin (Fotiadis et al., 2004). Interactions within an α1b-adrenoceptor oligomer may, therefore, be only partially disrupted by point mutations in TMDs I and IV. It is also important to note that the biophysical basis of any resonance energy transfer study is dependent on both distance between the energy donor and acceptor species and their relative orientation (Pfleger and Edine, 2006).
It is interesting that Hernanz-Falcon et al. (2004) also reported the mutated CCR5 receptor that was unable to “dimerize” was delivered effectively to the cell surface. Again our observations are different. The Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor was completely unable to reach the cell surface. This may reflect growing evidence that GPCR dimers/oligomers form during protein synthesis (Salahpour et al., 2004; Wilson et al., 2005) and that this is required for cell surface delivery.

Coexpression with Leu65Ala, Val66Ala, Leu166AlaLeu167Ala α1b-adrenoceptor limits cell surface delivery of the wild-type α1b-adrenoceptor but not the CXCR1 receptor. HEK293 cells were transfected to express either c-myc-α1b-adrenoceptor Y67C eYFP alone (A) or together with FLAG-Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eYFP in a 1:3 ratio (B and C). Immunocytochemistry was performed in nonpermeabilized (A and B) or permeabilized (C) cells using an anti-c-myc antibody and a secondary antibody conjugated to Alexa594 (red), whereas monitoring eYFP (green) provided a measure of FLAG-Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor expression and distribution. Nuclei were identified with Hoechst 33342 nuclear dye (blue). Merging of the images showed clear intracellular colocalization of the two forms of the receptor after their coexpression (C). Cells were cotransfected with FLAG-Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eYFP and c-myc-CXCR1 (D). Cell surface anti-c-myc staining (red), corresponding to the CXCR1 receptor, was observed in nonpermeabilized cells expressing both high and low levels of eYFP fluorescence (green).
In certain cases, an inability to reach the cell surface reflects an inability of receptors to become fully N-glycosylated and hence pass cellular quality control (Petaja-Repo et al., 2002; Pietila et al., 2005). We show herein that Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor is indeed unable to mature and become fully N-glycosylated. In addition to relying on the capacity in intact cells to detect an epitope tag added to the N terminus of the receptor that is expected to be on the extracellular face of the cell if the receptor was successfully delivered to the plasma membrane, we also used a range of other techniques to determine whether the various forms of the α1b-adrenoceptor were able to reach the cell surface. Bimolecular fluorescence complementation demonstrated that although the Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor still maintained quaternary structure, it remained in the endoplasmic reticulum/Golgi. α1-Adrenoceptors recycle rapidly in cells without the presence of an agonist ligand (Pediani et al., 2005) and hence at steady state a substantial fraction of the wild-type receptor is inside the cells. However, treatment of cells expressing bimolecular fluorescence complementation-competent forms of the wild-type α1b-adrenoceptor with the fluorescent α1-adrenoceptor ligand RQAPB confirmed that the receptor had traveled to the cell surface to bind and internalize the ligand, whereas no interaction of QAPB could be detected for Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor, indicating that this mutant had never been at the cell surface.
The Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor also displayed unexpected pharmacological characteristics. Measures of protein expression and amount based on the fluorescence of attached tags clearly demonstrated the Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor to be expressed as well as the wild-type α1b-adrenoceptor. However, this would not have been the conclusion if analysis had been restricted to 3H-antagonist binding studies, in which the capacity, but not affinity, of Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor to bind [3H]prazosin was markedly reduced. Others have reported substantial differences in binding capacity of a single GPCR for different [3H]antagonist ligands, and such data have been used to argue for cooperativity between binding sites and hence GPCR quaternary structure (Park et al., 2004; Strange, 2005; Franco et al., 2006). Disruption of quaternary structure might therefore modulate ligand binding capacity. Despite this, Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor retained the ability to bind the agonist ligand phenylephrine and, indeed, displayed a higher affinity to bind this agonist than the wild-type receptor.

Mutations in TMD IV but not TMD I reduce 3-FRET signals. 3-FRET studies were performed as in Figs. 2 and 3 using HEK293 cells transfected to express each of eCFP, eYFP, and dsRed2 tagged forms of wild-type (WT), Leu65Ala, Val66Ala (TM I), Leu166Ala, Leu167Ala (TM IV), and Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala (TMI TMIV) α1b-adrenoceptor. Data are means ± S.E.M. n = 3, *. significantly lower (p < 0.05) than wild type.

Although both TMD I and TMD IV mutants of the α1b-adrenoceptor mature to be terminally N-glycosylated neither is delivered to the plasma membrane. A, HEK293 cells were transfected to express FLAG-tagged forms of wild-type α1b-adrenoceptor-eYFP (A), Leu65Ala, Val66Ala α1b-adrenoceptor-eYFP (B), or Leu166Ala, Leu167Ala α1b-adrenoceptor-eYFP (C). Nonpermeabilized cells were stained with anti-FLAG and secondary antibody and imaged to detect eYFP (A1, B1, and C1) or anti-FLAG (A2, B2, and C2). B, cells expressing Leu65Ala, Val66Ala α1b-adrenoceptor-eYFP (A1–A4) or Leu166Ala, Leu167Ala α1b-adrenoceptor-eYFP (B1–B4) were treated with Hoechst 33342 nuclear stain and RQAPB for 30 min and imaged; A1 and B1, Hoechst 33342; A2 and B2, YFP; A3 and B3, RQAPB; A4 and B4, merged images. C, membranes expressing α1b-adrenoceptor-eYFP (1), Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor-eYFP (2), Leu65Ala, Val66Ala α1b-adrenoceptor-eYFP (3), or Leu166Ala, Leu167Ala α1b-adrenoceptor-eYFP (4) were resolved by SDS-PAGE and immunoblotted with anti-FLAG.
A significant number of genetic diseases reflect mutations in genes that result in endoplasmic reticulum/Golgi apparatus retention of the expressed protein. In a number of examples, the mutated proteins are GPCRs (Bernier et al., 2004; Ulloa-Aguirre et al., 2004). It is interesting that mutant GPCRs that are expressed but are unable to transit through the endoplasmic reticulum/Golgi apparatus frequently act as “dominant-negatives” for cell surface delivery of a coexpressed wild-type GPCR (Duvernay et al., 2005), and this may also be directly relevant to physiology and disease because certain GPCR splice variants display this characteristic (Ding et al., 2002). This indicates that effective and correct interactions to form GPCR dimers/oligomers are central to effective cell surface delivery of GPCRs. The Leu65Ala, Val66Ala, Leu166Ala, Leu167Ala α1b-adrenoceptor studied herein was also shown to have such a dominant-negative effect on the cell surface delivery of the wild-type α1b-adrenoceptor.
It is interesting that when we studied the effect of mutations in either TMD I or TMD IV in isolation, although both the Leu65Ala, Val66Ala and Leu166Ala, Leu167Ala forms of the α1b-adrenoceptor were poorly, if at all, delivered to the cell surface, both of these mutants seemed to be terminally N-glycosylated as effectively as the wild-type α1b-adrenoceptor. As such, simple analysis of the terminal glycosylation status (Bjorklof et al., 2002) cannot be used as a surrogate marker for complete receptor maturation. The mutations introduced into TMD IV were clearly responsible for the reduction in 3-FRET signal observed in the TMD1 + TMD IV mutant. It is noteworthy that TMD IV has been implicated as a key “dimer” interface in the D2 dopamine receptor (Guo et al., 2003; Lee et al., 2003) and, indeed, agonist and inverse agonist ligands alter the conformational details and structural organization of TMD IV of this receptor (Guo et al., 2005). The current studies demonstrate that the α1b-adrenoceptor is able to organize into oligomers rather than simple dimers and that modification of the effectiveness of this process has major consequences for function. At this stage, such studies have required levels of expression higher than generally reported for the α1b-adrenoceptor in native tissues. The application of 3-FRET imaging with emerging generations of novel fluorescent proteins may allow assay sensitivity to be increased.
Footnotes
-
These studies were supported by the Medical Research Council and the Wellcome Trust.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.106.033035.
-
ABBREVIATIONS: GPCR, G protein-coupled receptor; FRET, fluorescence resonance energy transfer; TMD, transmembrane domain; QAPB, BODIPY-FL prazosin; RQAPB, red BODIPY-FL prazosin; eCFP, enhanced cyan fluorescent protein; eYFP, enhanced yellow fluorescent protein; GFP, green fluorescent protein; PCR, polymerase chain reaction; HEK, human embryonic kidney; Ex, excitation; Em, emission; PBS, phosphate-buffered saline; RT, room temperature; PAGE, polyacrylamide gel electrophoresis; MOPS, 3-(N-morpholino)propanesulfonic acid; BS, beam splitter.
-
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material. - Received November 29, 2006.
- Accepted January 12, 2007.

- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}