


Abstract
In the central nervous system, glutamate transporters terminate the actions of this neurotransmitter by concentrating it into cells surrounding the synapse by a process involving sodium and proton cotransport followed by countertransport of potassium. These transporters contain two oppositely oriented helical hairpins 1 and 2. Hairpin 1 originates from the cytoplasm, but its tip is close to that of hairpin 2, which enters the transporter's lumen from the extracellular side. Here we address the question of whether hairpin 1 and/or domains surrounding it undergo conformational changes during the transport cycle. Therefore, we probed the reactivity of cysteines introduced into hairpin 1 and the cytoplasmic ends of transmembrane domains 6, 7, and 8 of the GLT-1 transporter to membrane-permeant N-ethylmaleimide. In each domain, except for transmembrane domain 6, cysteine mutants were found in which the inhibition of d-[3H]aspartate transport by the sulfhydryl reagent was increased when external sodium was replaced by potassium, a condition expected to increase the proportion of cytoplasmic-facing transporters. Conversely, the nontransportable blocker kainate protected against the inhibition in several of these mutants, presumably by locking the transporter in an outward-facing conformation. Moreover, external potassium decreased the oxidative cross-linking of two cysteines, each introduced at the tip of each hairpin. Our results are consistent with a model based on the crystal structure of an archeal homolog. According to this model, the inward movement of hairpin 1 results in the opening of a pathway between the binding pocket and the cytoplasm, lined by parts of transmembrane domains 7 and 8.
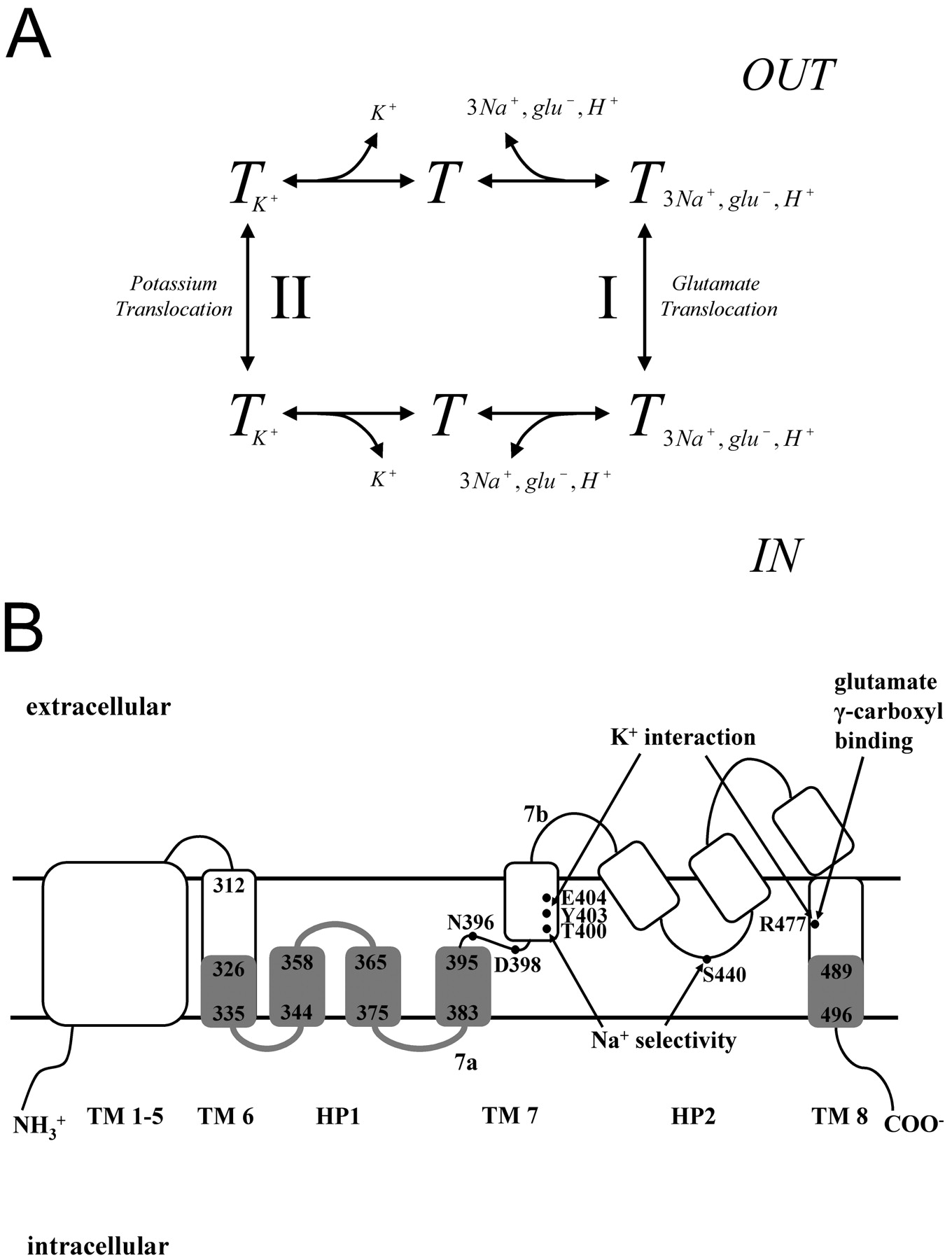
Glutamate is the major excitatory neurotransmitter in the central nervous system, and the sodium-dependent glutamate transporters are essential for terminating the synaptic action of this neurotransmitter, preventing hyperexcitability and neurotoxicity. Glutamate transport is an electrogenic process (Kanner and Sharon, 1978; Brew and Attwell, 1987; Wadiche et al., 1995) in which the transmitter is cotransported with three sodium ions and one proton (Zerangue and Kavanaugh, 1996; Levy et al., 1998) followed by the counter-transport of one potassium ion (Kanner and Bendahan, 1982; Pines and Kanner, 1990; Kavanaugh et al., 1997). The mechanism, involving two half-cycles (Fig. 1A), is supported by the fact that mutants impaired in potassium interaction are locked in an obligatory exchange mode (Kavanaugh et al., 1997; Zhang et al., 1998). Under physiological conditions, the inwardly directed sodium gradient and outwardly directed potassium gradient and the interior negative membrane potential promote the accumulation of the transmitter into the cell against its concentration gradient (Kanner and Sharon, 1978; Zerangue and Kavanaugh, 1996; Levy et al., 1998). According to the transport cycle, when external sodium is replaced by potassium, an increase of the proportion of transporters in the inward-facing conformation is expected (Fig. 1A), and the transporters operate in reverse, mediating glutamate efflux (Kanner and Bendahan, 1982; Szatkowski et al., 1990).
A high-resolution crystal structure of the glutamate transporter homolog GltPh from the archaeon Pyrococcus horikoshii has been published (Yernool et al., 2004). It forms a trimer with a permeation pathway through each of the monomers. The membrane topology of the monomer is quite unusual (Yernool et al., 2004), but it is in excellent agreement with the topology inferred from biochemical studies (Grunewald et al., 1998; Slotboom et al., 1999; Grunewald and Kanner, 2000). It contains eight transmembrane domains (TM) and two oppositely oriented reentrant loops, also termed helical hairpins (Yernool et al., 2004). HP1 is located between TMs 6 and 7, and HP2 is between TMs 7 and 8 (Fig. 1B). In at least one of the conformations of the transporter, the tips of HP1 and HP2 come into close proximity of each other (Brocke et al., 2002; Yernool et al., 2004). Transport activity of many single cysteine mutants of the outward-facing HP2 is sensitive to the impermeant sulfhydryl reagent MTSET (Grunewald et al., 2002; Leighton et al., 2002). On the other hand, transport of most cysteine mutants in the more intracellular oriented HP1 is not sensitive to MTSET (Grunewald and Kanner, 2000). Cysteine residues exposed to the cytoplasmic side can be alkylated by the permeant alkylating reagent NEM, which reacts with the deprotonated form of a sulfhydryl group. In contrast to NEM, thiol groups modified by permeant (2-aminoethyl)methane thiosulfonate can be regenerated by intracellular-reducing species. Alkylation reflects the reactivity and/or accessibility of a cysteine replacement at a given position, which depends on the environment of its thiol group. Any change in reactivity of the thiol group upon binding of substrate or coions is indicative of a change in the local environment.

Transport cycle and membrane topology of glutamate transporters. Model of the glutamate translocation cycle (Kanner and Bendahan, 1982; Kavanaugh et al., 1997). The order of binding of the three sodium ions, glutamate, and the proton is not indicated, although it seems that at least one of the sodium ions binds before glutamate, and this is followed by the binding of one or two additional sodium ions (Watzke et al., 2001). After glutamate and the coions bind to the transporter (T) from the external medium (going clockwise), they are translocated and released into the inside of the cell. These steps represent half-cycle I. Then, potassium binds from the intracellular side and, after translocation to the outside, is released there. After completion of half-cycle II, a new translocation cycle can commence. The steps in this scheme are reversible, and therefore, elevated levels of extracellular potassium can cause the transporter to become inward-facing (going counterclockwise). If the interaction with potassium is abolished by mutation, half-cycle II is not operative, but the transporters can still exchange labeled glutamate (or aspartate), added to the outside with internal glutamate by reversible translocation via half-cycle I (A). The membrane topology shown is based on the high-resolution structure of GltPh (Yernool et al., 2004). TMs are indicated by Arabic numerals, and the two reentrant loops by HP1 and HP2. The gray regions, including the linkers between the structural elements (thick gray lines) are those analyzed in the accessibility studies reported here. HP1a and HP1b are the parts of HP1 closest to TMs 6 and 7, respectively. The residue numbers mark the boundaries of the regions analyzed. Some of the amino acid residues, critical for transport, are indicated together with the functional aspect affected upon mutation. Residues Asn396 and Asp398 play multiple roles in transport (Rosental et al., 2006; Tao et al., 2006). Glu404 is also believed to be important for the interaction with the cotransported proton (Grewer et al., 2003) (B).
The binding pocket of GltPh is predominantly formed by TMs 7 and 8 and the two reentrant loops, which in the crystal structure enclose a nonprotein density presumably corresponding to glutamate (Yernool et al., 2004). It is noteworthy that many of the amino acid residues of the transporter inferred to be important for the interaction with sodium (Zhang and Kanner, 1999; Borre and Kanner, 2001), potassium (Kavanaugh et al., 1997; Zhang et al., 1998), and glutamate (Bendahan et al., 2000) (Fig. 1B) are facing toward the binding pocket and are close to the nonprotein density (Yernool et al., 2004). It has been suggested that re-entrant loop HP1 may form the internal gate of the transporter, which moves to a more intracellular position when the transporter becomes inward-facing (Yernool et al., 2004). In this study, we provide support for this idea by comparing the reactivity of cysteine residues engineered into HP1 and the intracellular-facing parts of TMs 6, 7, and 8 (Fig. 1B) to the membrane-permeant sulfhydryl reagent NEM in the presence of sodium and potassium.
Materials and Methods
Generation and Subcloning of Mutants. Mutations were made by site-directed mutagenesis of the cysteine-less GLT-1 (CL-GLT-1) (Grunewald et al., 1998) in the vector pBluescript SK(–) (Stratagene, La Jolla, CA) using single-stranded uracil-containing DNA as described previously (Kunkel et al., 1987; Kleinberger-Doron and Kanner, 1994). In brief, the parent DNA was used to transform Escherichia coli CJ236 (dut–, ung–). From one of the transformants, single-stranded uracil-containing DNA was isolated upon growth in a uridine-containing medium, according to the standard protocol from Stratagene, using helper phage R408. This yields the sense strand, and consequently, mutagenic primers were designed to be antisense. Mutants were subcloned into the CL-GLT-1 construct in pBluescript SK(–), using unique restriction enzymes and the coding and noncoding strands were sequenced between those unique sites. The double mutants used have been described previously (Brocke et al., 2002).
d-[3H]aspartate Transport in HeLa Cells. HeLa cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 200 U/ml penicillin, 200 μg/ml streptomycin, and 2 mM glutamine. HeLa cells plated on 24-well plates were infected with the recombinant vaccinia/T7 virus vTF7-3 (Fuerst et al., 1986) and transfected with cDNA [pBluescript SK(–) with CL-GLT-1 or single-cysteine mutant transporter inserted downstream to the T7 promotor] using the transfection reagent N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium methylsulfate as described previously (Pines et al., 1995). Uptake of d-[3H]aspartate into the cells was assayed 18 to 20 h after transfection. The cells were washed with a solution containing 150 mM choline chloride, 5 mM potassium phosphate, pH 7.4, 0.5 mM MgSO4, and 0.3 mM CaCl2 (1 ml/well), and subsequently, transport was initiated by the addition of 200 μl of an NaCl-based transport solution (150 mM NaCl, with KPi, MgSO4, and CaCl2 as above), containing 0.4 μCi of d-[3H]aspartate (23.9 Ci/mmol), to each well. Transport was carried out for 10 min at room temperature (22–24°C), and the assay was terminated by washing the cells twice with 1 ml of ice-cold NaCl-based transport solution. Cells were lysed with 1% SDS, and radioactivity was measured by liquid scintillation counting. Transport activity of each of the single cysteine mutants was directly compared with that of CL-GLT-1, indicated in the figures as the percentage of activity of CL-GLT-1 (± S.E.M.), in at least three different experiments, each done in triplicate. It should be noted that the concentration of d-aspartate used (83.7 nM) is more than 2 orders of magnitude lower than its Km value. Therefore, altered transport in the mutants, whether due to reduced Vmax or increased Km values, will be detected under these conditions.
Inhibition by Sulfhydryl Reagents and CuPh. Before the transport measurements, the cells adhering to 24-well plates were washed once with 1 ml of the transport medium containing 150 mM choline chloride instead of NaCl. Each well was then incubated at room temperature with 1 ml of the preincubation medium (NaCl-based transport solution or the same solution with 150 mM KCl instead of NaCl, as indicated in the figures) supplemented with the concentrations of sulfhydryl reagent or CuPh and other additions, as indicated in the figure legends. After 5 min, the medium was aspirated, and the cells were washed twice with 1 ml of the choline-based solution. Afterward, they were assayed for d-[3H]aspartate transport as described above. The CuPh stock solution was prepared by mixing 0.4 ml of 1.25 M 1,10-phenanthroline in water/ethanol (1:1) with 0.6 ml of 250 mM CuSO4. In the case of NEM, a fresh stock solution of 0.1 M was prepared in 50% ethanol for each experiment. In the inhibition studies with NEM, the sulfhydryl reagent was initially used at a concentration of 0.5 mM with all of the mutants. With many of the mutants, the inhibition at this concentration was almost complete. To determine whether there is a difference between the inhibition in the presence of potassium and sodium, lower concentrations of NEM were used in subsequent experiments, and these concentrations are indicated in the figure legends. In other mutants, little inhibition was obtained by 0.5 mM NEM. In this case, the concentration was increased in the subsequent experiments (see figure legends). The statistical evaluation of the difference in inhibition by NEM under different conditions used a one-way analysis of variance with a post hoc Dunnett multiple comparison test (*, p < 0.01; **, p < 0.05). Results were plotted using data for each mutant normalized to its activity after preincubation with the same solution but without NEM or CuPh.
Results
Cysteine Scanning of Re-Entrant Loop HP1. The stretch of HP1 that begins at the cytoplasmic side of the membrane and reaches the interior of the transporter is termed HP1a (Fig. 1B, based on the GltPh structure; Yernool et al., 2004). Using the cysteine-less GLT-1 (CL-GLT-1) (Grunewald et al., 1998) as a parent construct, we have replaced all the residues of HP1a, one at a time, by cysteine. After transient expression of the CL-GLT-1 construct and the 15 cysteine replacement mutants in HeLa cells, transport of d-[3H]aspartate was monitored. The cysteine replacement was tolerated well at most positions, except for the F346C and A357C mutants. Because our aim was to determine the impact of sulfhydryl reagents on transport activity, we have not attempted to characterize whether the defects of the inactive mutants encountered in this study are in expression at the plasma membrane or in intrinsic function. In the other HP1a mutants, significant transport ranging from 30 to 95% of CL-GLT-1 activity was observed (Fig. 2A). The stretch of re-entrant loop HP1, which loops back from the interior of the transporter to the cytoplasmic side (Fig. 1B), is termed HP1b (Yernool et al., 2004). All the residues of HP1b of CL-GLT-1 and those forming the connection between HP1a and HP1b were replaced by cysteine one at a time. Compared with HP1a, the effect of cysteine replacement, at the positions in this stretch, on transport was much more severe (Fig. 2, A and B). In accordance with our previous observations (Grunewald and Kanner, 2000), the four consecutive replacement mutants T360C, A361C, S362C, and S363C were basically inactive. L367C, P368C, T370C, and E375C also had very low transport activity (Fig. 2B). The transport activity of the other nine single cysteine mutants ranged from 40 to 110% of that of CL-GLT-1 (Fig. 2B).
Transport activity of CL-GLT-1 was only slightly inhibited by 0.5 mM NEM (Fig. 3A). The same was true for 13 of the 22 functional replacement mutants in HP1, which exhibited more than 30% of CL-GLT-1 activity (data not shown, but these 13 mutants are listed in the legend to Fig. 3). This slight inhibition was apparently due to a nonspecific effect on the HeLa cells. To determine whether the reactivity of the other nine functional HP1 single cysteine mutants to the sulfhydryl reagent is dependent on the nature of the external cation, it was necessary to decrease the concentration of NEM for those mutants, which were almost totally inhibited by 0.5 mM sulfhydryl reagent. For several of the mutants, we verified that the degree of inhibition increased with the concentration of NEM used (data not shown, but see Fig. 3B). Concentrations of NEM, lower than 0.5 mM and optimized for each mutant, were used in the case of A353C, A364C, G365C, T366C, and L374C (see the legend to Fig. 3). In the case of I350C, the inhibition by 0.5 mM NEM was modest, and therefore a higher concentration (1 mM) was used. When the external sodium is replaced by potassium, the proportion of transporters in the inward-facing conformation is expected to increase (Fig. 1A). When the treatment by NEM, normally performed in the presence of sodium, was compared with that in the presence of potassium, inhibition was similar in the case of G349C, G359C, and A364C. However, in the case of the mutants I350C, A353C, G365C, T366C, S373C [the native cysteine reintroduced at CL-GLT-1; at this position, a serine was introduced during the construction of CL-GLT-1 (Grunewald et al., 1998)], and L374C, there was a marked potentiation of this inhibition in the presence of potassium (Fig. 3A). The potentiation of the inhibition by potassium was dependent on the final concentration of the sulfhydryl reagent, as exemplified for S373C, and the increased sensitivity to NEM in the presence of potassium was observed at all concentrations used (Fig. 3B). We also analyzed the inhibition of transport activity of several of the mutants by (2-aminoethyl)methanethiosulfonate, which also can permeate the membrane. Inhibition by the methanethiosulfonate reagent was not observed in all of the NEM-sensitive mutants. However, significant inhibition by the methanethiosulfonate reagent was seen in A353C and A393C, and this inhibition was potentiated in the presence of potassium (data not shown).
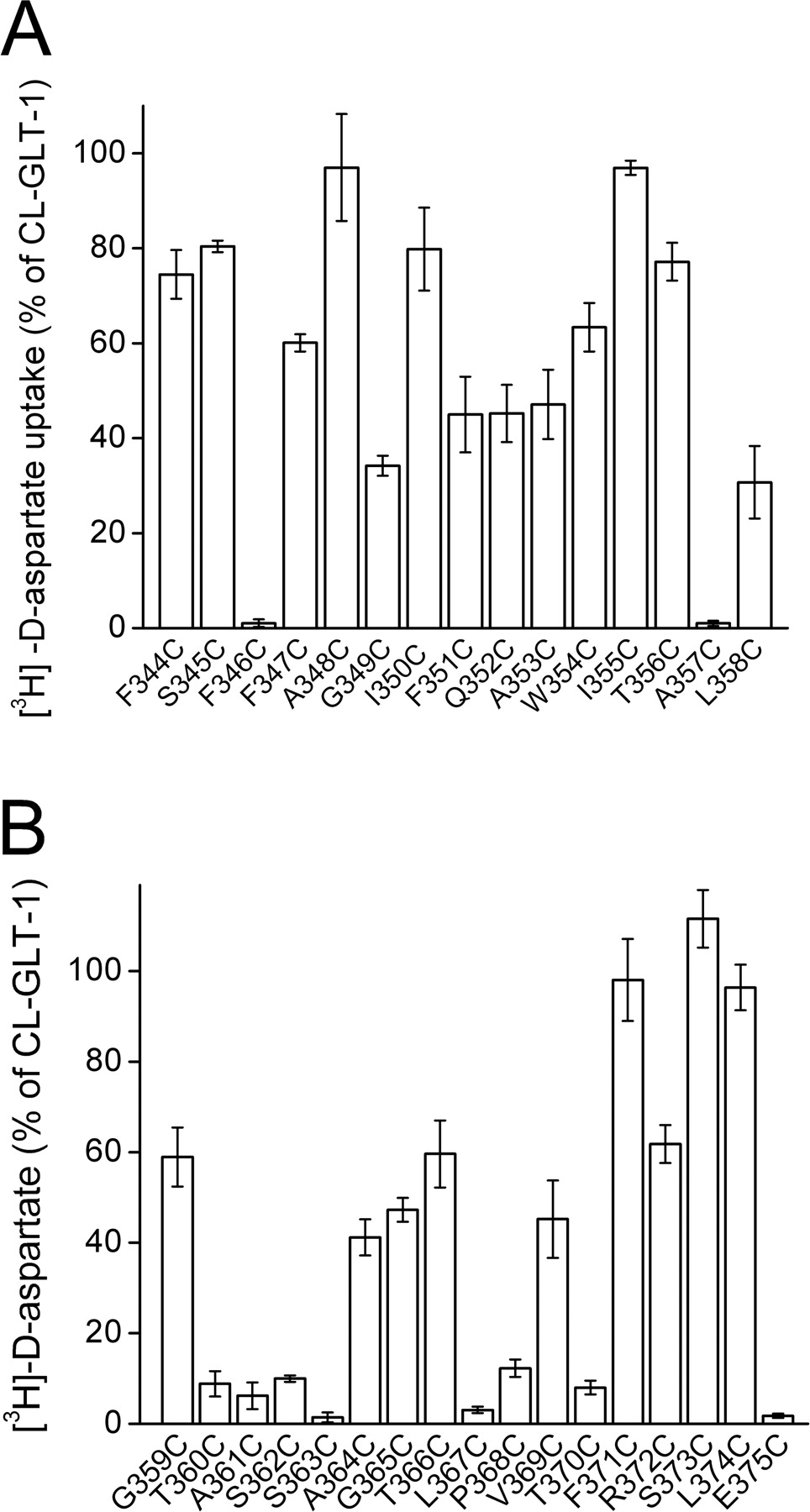
d-[3H]aspartate transport activity of single cysteine mutants in HP1. Transport of d-[3H]aspartate by the single cysteine mutants in HP1a (A) and HP1b, including the linker between HP1a and b (B), was measured as described under Materials and Methods, and the activity of the mutants is expressed as a percentage of activity of CL-GLT-1.
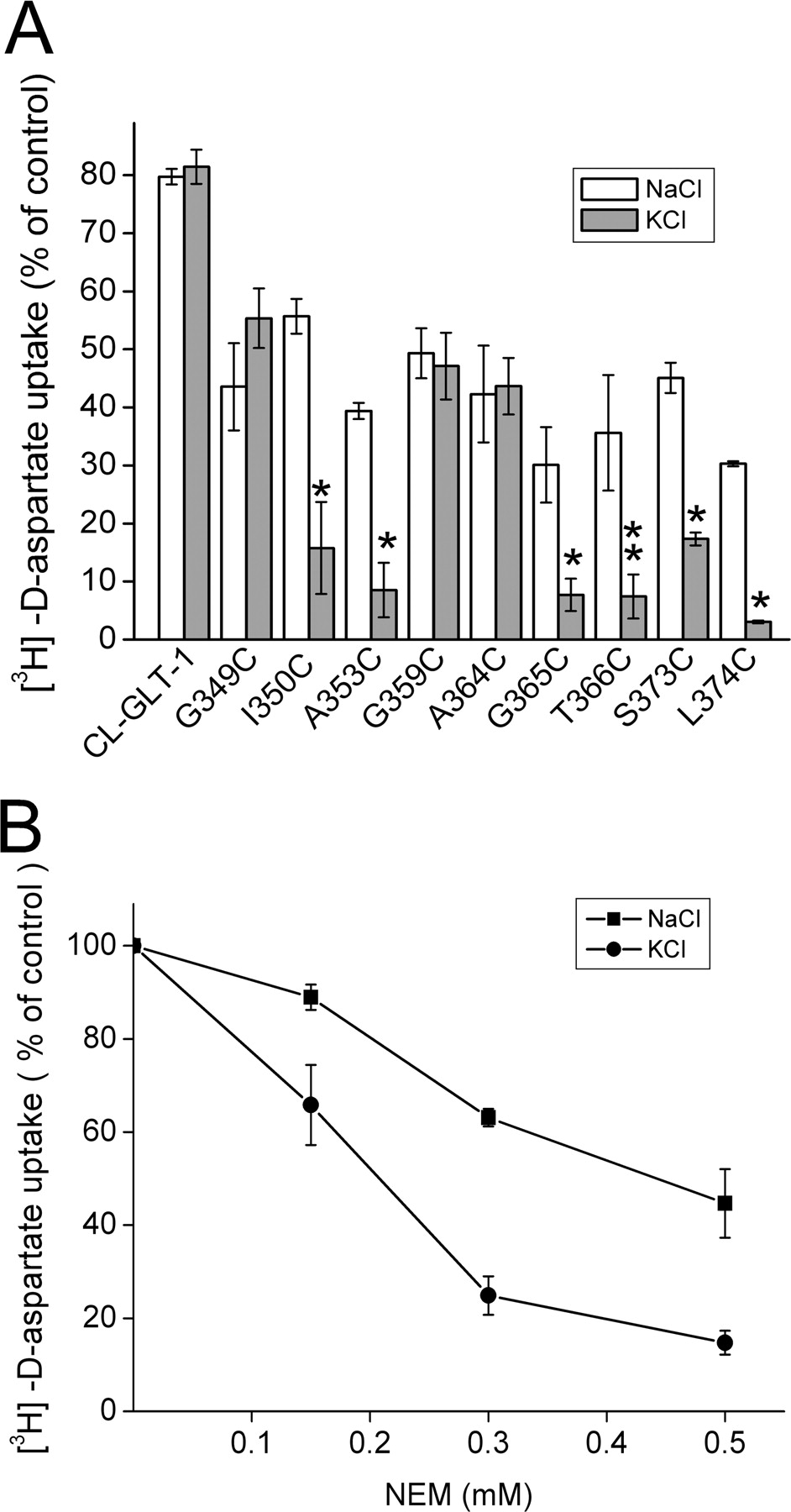
Inhibition of d-[3H]aspartate transport in single cysteine mutants in HP1 by NEM. Inhibition of the transport activity of the single cysteine mutants after treatment by NEM in the presence of either sodium (□) or potassium ( ) was performed as described under Materials and Methods and is given as a percentage of remaining activity of the same mutant, treated under the same conditions but without NEM. The concentration of NEM used was 0.5 mM, except for I350C (1 mM), A353C and T366C (0.15 mM), A364C (0.04 mM), G365C (0.2 mM), and L374C (0.25 mM). No significant inhibition by NEM in the presence of either sodium or potassium was observed with the following active mutants: F344C, S345C, F347C, A348C, F351C, Q352C, W354C, I355C, T356C, L358C, V369C, F371C, and R372C (A). In the case of S373C, the preincubation was done with the concentrations of NEM indicated on the abscissa (B).
) was performed as described under Materials and Methods and is given as a percentage of remaining activity of the same mutant, treated under the same conditions but without NEM. The concentration of NEM used was 0.5 mM, except for I350C (1 mM), A353C and T366C (0.15 mM), A364C (0.04 mM), G365C (0.2 mM), and L374C (0.25 mM). No significant inhibition by NEM in the presence of either sodium or potassium was observed with the following active mutants: F344C, S345C, F347C, A348C, F351C, Q352C, W354C, I355C, T356C, L358C, V369C, F371C, and R372C (A). In the case of S373C, the preincubation was done with the concentrations of NEM indicated on the abscissa (B).
Cysteine Scanning of the Cytoplasmic Ends of TMs 7 and 8. In the GltPh structure, TMs 7 and 8 line a possible permeation pathway connecting the binding pocket of the transporter with the cytoplasm (Yernool et al., 2004). One at a time, cysteine residues were introduced at 13 positions of the “cytoplasmic” part of TM 7 (7a) of CL-GLT-1 and at the 7 positions of the “linker” connecting the intracellular ends of HP1b with TM 7 (Fig. 1B). In 7 of these 20 single cysteine mutants, the transport activity was too low to conduct inhibition studies (Fig. 4A, left), but the transport activity of the remaining 13 mutants was at least 40% of that of CL-GLT-1 (Fig. 4A). In the case of eight of these mutants, a potent inhibition by NEM was observed, and in six of these eight, the inhibition by NEM, using optimized concentrations for each mutant, was increased in the presence of potassium (Fig. 4B). In the case of the “cytoplasmic” part of TM 8, eight mutants were made (at positions 489–496), and six of those exhibited at least 35% of the transport activity of CL-GLT-1 (Fig. 4A, right). In five of these mutants, NEM was inhibitory, and in four of them, potassium significantly increased the inhibition of transport by NEM (Fig. 4B).
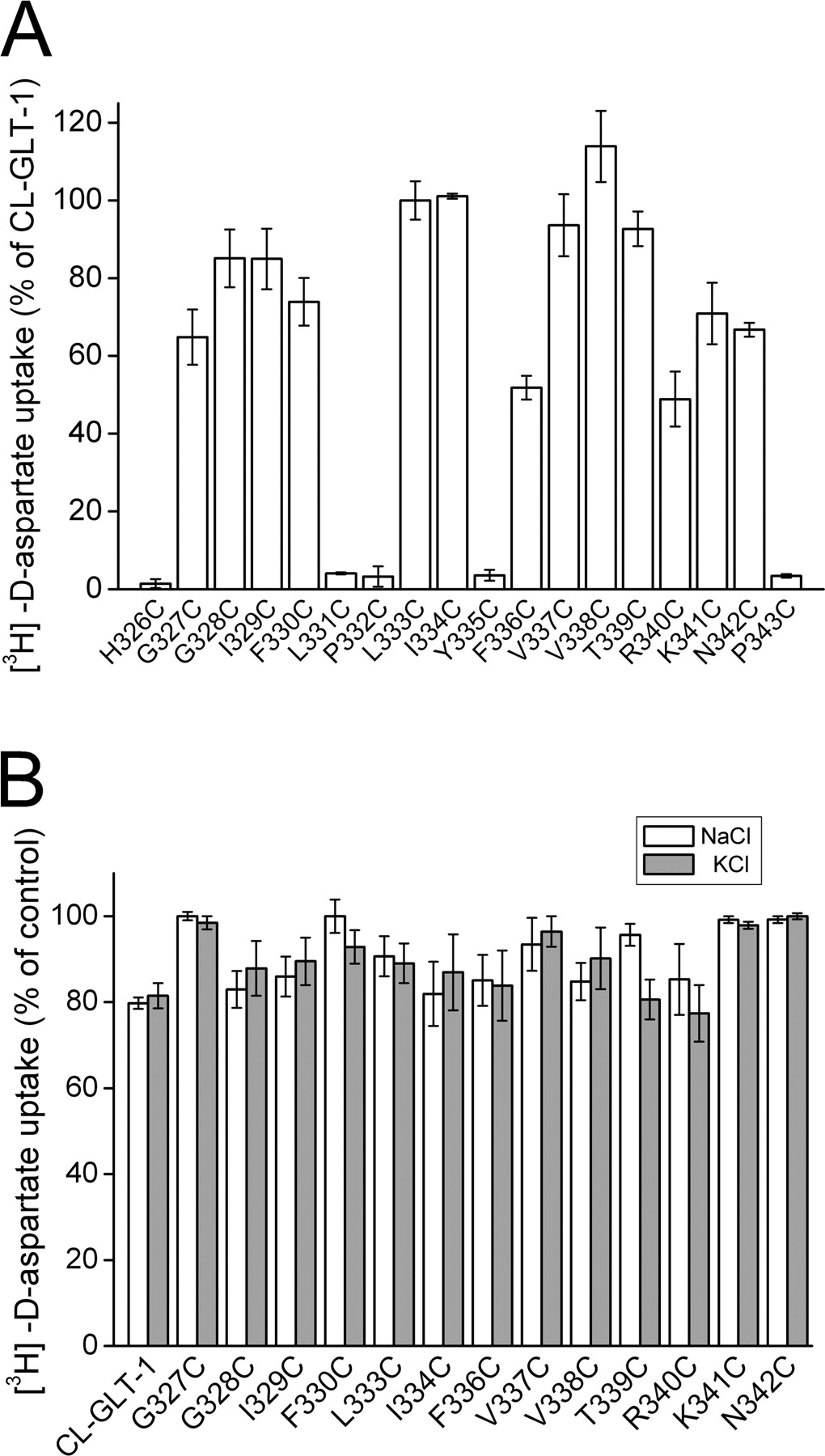
Cysteine Scanning of the Cytoplasmic End of TM 6. TM 6 is more peripheral than TMs 7 and 8 (Yernool et al., 2004), and therefore it is possible that this domain does not undergo conformational changes during transport. Cysteine residues were introduced, one at a time, in 10 positions of the “cytoplasmic” part of TM 6 of CL-GLT-1 and in the stretch of 8 residues connecting the intracellular ends of TM 6 and HP1a (Fig. 1B). In five of those, the activity of transport was severely impaired (Fig. 5A). One of the replacements was at His326, shown previously to be critical for transport activity in the context of the wild-type transporter (Zhang et al., 1994). The transport activity of the other 13 replacement mutants was at least 45% of that of CL-GLT-1 (Fig. 5A). In none of those 13 mutants was the inhibition by 0.5 mM NEM larger than in CL-GLT-1, regardless of the presence of sodium or potassium during the preincubation with the sulfhydryl reagent (Fig. 5B).
Effect of Kainate on Inhibition by NEM. As shown in Figs. 3 and 4, the inhibition of transport in many of the single cysteine mutants studied by NEM was potentiated when external sodium was replaced by potassium, a condition expected to increase the proportion of cytoplasmic facing transporters. One would predict that, if the proportion of outward-facing transporters is increased, less inhibition of transport by NEM should be observed in at least several of the cysteine mutants. For this purpose, we used the substrate analog kainate. This blocker can bind to the transporter instead of glutamate but is not transported. As a consequence, the proportion of outward-facing transporters is expected to increase. Indeed with 9 of the 15 mutants tested, in which potassium potentiated the effect of NEM, a significant protection against the inhibition by NEM was afforded by the blocker (Fig. 6). Moreover, in T385C, in which no significant increase in the sensitivity to NEM was observed in the presence of potassium (Fig. 4B), a significant protection by kainate was seen (Fig. 6). In the mutants A393C, S373C, and L374C, we also tested the effect of 1 mM glutamate during the preincubation with sodium and NEM. However, this transportable substrate had no effect on the inhibition of transport by NEM in these mutants (data not shown).
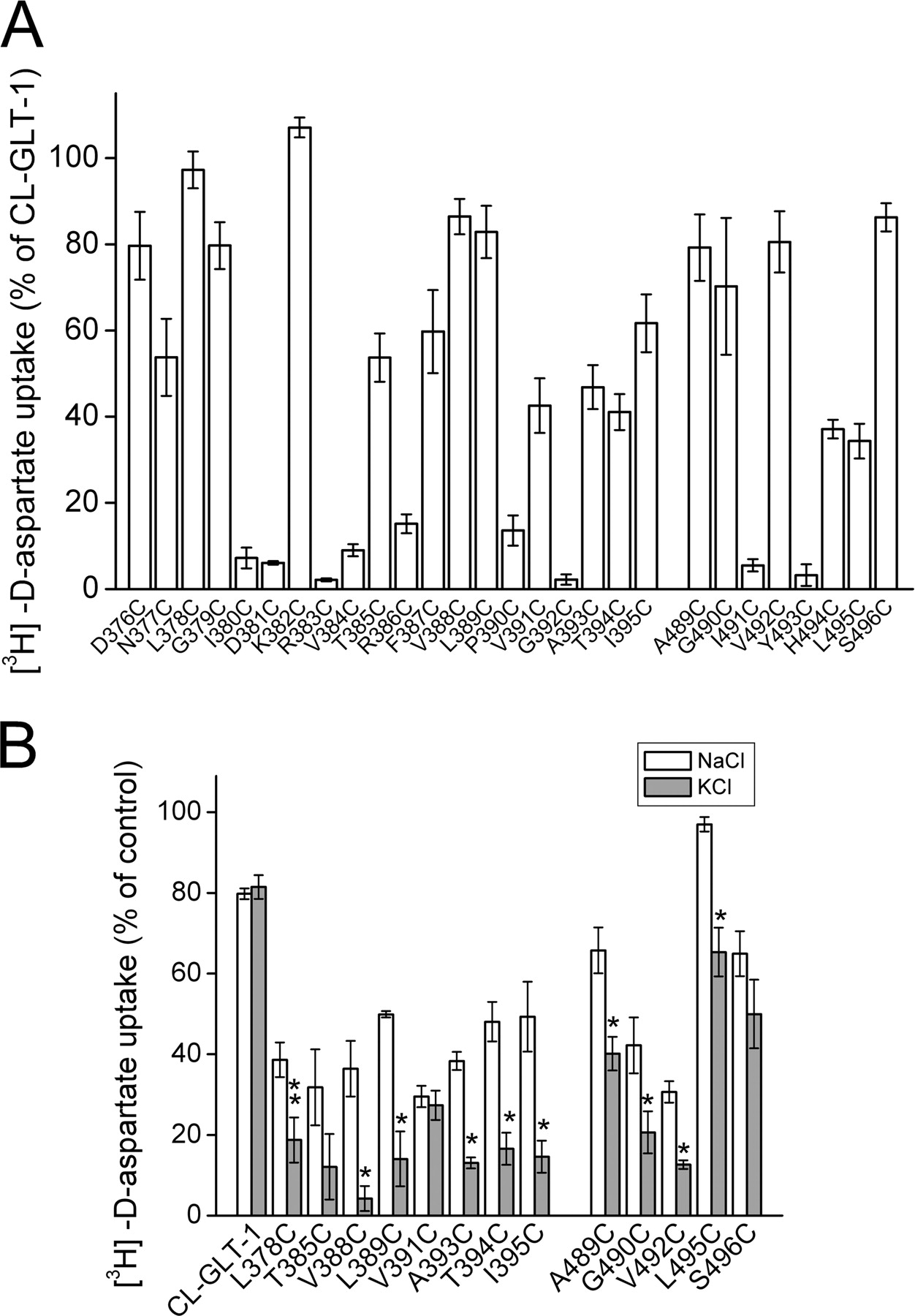
Transport activity and inhibition by NEM in single cysteine mutants in TMs 7 and 8. The mutants were from the “cytoplasmic” parts of TMs 7 (TM 7a), including the linker between HP1b and TM 7a (positions 376–395) and 8 (positions 498–496), as indicated in Fig. 1B. Transport of d-[3H]aspartate by the single cysteine mutants was measured as described under Materials and Methods, and the activity of the mutants is expressed as a percentage of activity of CL-GLT-1 as in Fig. 2 (A). Inhibition of transport by NEM was done as described in the legend to Fig. 3. The concentration of NEM used was 0.5 mM except for L378C, V388C, A393C, T394C, I395C, and V492C (all at 0.3 mM), T385C (0.075 mM), L389C (0.2 mM), V391C (0.4 mM), A489C (1 mM), and G490C (0.15 mM) (B). No significant inhibition by NEM, in the presence of either sodium or potassium, was observed with the following active mutants: D376C, N377C, G379C, K382C, F387C, and H494C.
Reactivity and Oxidative Cross-Linking of A364C and S440C. Even though the inhibition of transport of many single cysteine mutants of HP1 by NEM was increased in the presence of potassium, no change was observed in the case of A364C (Fig. 3A). In contrast with most other HP1 positions, this residue, whose GltPh counterpart is located closest to the external medium (Yernool et al., 2004), was shown to be accessible to membrane impermeant MTSET (Grunewald and Kanner, 2000). Whereas potassium did not increase the inhibition of transport by NEM, this cation decreased the sensitivity of transport of A364C to MTSET (Fig. 7A), consistent with a movement of the tip of HP1 away from the external aqueous cavity. On the other hand, potassium potentiated the inhibition of transport of the single cysteine mutant S440C by MTSET, and the same effect was observed with NEM (Fig. 7B). Position 440 is located at the tip of HP2 and is close to position 364 (Brocke et al., 2002; Yernool et al., 2004). The inhibition of transport of the A364C/S440C double mutant by CuPh, documented to be the result of oxidative cross-linking of the two engineered cysteines within one transporter monomer (Brocke et al., 2002), was reduced in the presence of external potassium (Fig. 8A). In the case of the A412C/V427C double mutant, it has also been shown that the two engineered cysteines, both located on HP2, can be cross-linked intramolecularly by CuPh (Brocke et al., 2002). In contrast to A364C/S440C, attenuation of the CuPh inhibition of transport by potassium was not observed in A412C/V427C (Fig. 8B).

Transport activity and inhibition by NEM in single cysteine mutants in TM 6. The mutants were from the “cytoplasmic” parts of TM 6, as indicated in Fig. 1B. The conditions were the same as described in the legends to Figs. 2 and 3. The concentration of NEM used was 0.5 mM for all single cysteine mutants.

Effect of kainate on the inhibition of transport of single cysteine mutants by NEM. Transport by the indicated mutants was measured after pretreatment of the cells with the concentrations of NEM specified in the legends to Figs. 3 and 4 in sodium-containing medium in the presence and absence of 0.2 mM kainate.

Effect of NEM and MTSET on transport by A364C and S440C. HeLa cells expressing A364C (A) and S440C (B) were pretreated in the presence of sodium or potassium with NEM and MTSET at the concentrations indicated. Then, transport of d-[3H]aspartate was measured.
Discussion
Transport of many mutants of GLT-1, with single cysteines in the cytoplasmic ends of TMs 7 and 8 or in HP1, is inhibited by the membrane-permeant sulfhydryl reagent NEM (Figs. 3 and 4). The functionality of the mutants is underscored by their significant activity and their responsiveness to potassium, indicating that the results are not due to distorted conformations caused by the mutations. The single cysteines were engineered at sites that, both according to the topology determined previously (Grunewald et al., 1998; Slotboom et al., 1999; Grunewald and Kanner, 2000) and the recently determined crystal structure of an archeal homolog (Yernool et al., 2004), are located close to the cytoplasmic side of the membrane. Indeed, transport by many of these single cysteine mutants was shown previously to be insensitive to the membrane-impermeant MTSET, and labeling of several of these cysteines by a biotinylated maleimide could be prevented by NEM but not by MTSET. These and other experiments on HP1 accessibility in GLT-1 (Grunewald and Kanner, 2000) and in the bacterial glutamate transporter GltT (Slotboom et al., 1999) indicate that inhibition of transport by NEM throughout most of HP1 is a result of reactivity/accessibility of the sulfhydryl reagent from the cytoplasmic side. Alkylation by NEM, requires the cysteine to be deprotonated, and therefore the changes in the reactivity observed here seem to be due to changes in their exposure to the cytoplasm. An excellent example of alkylation of cysteines of lactose permease from the intracellular side by NEM added from the extracellular side was published very recently (Kaback et al., 2007).

Inhibition of A364C/S440C and A412C/V427C by CuPh. HeLa cells expressing the double mutants A364C/S440C (A) and A412C/V427C (B) were pretreated with the indicated concentrations of CuPh in the presence of sodium or potassium as described under Materials and Methods. Then, transport of d-[3H]aspartate was measured.
In most of the cysteine mutants of HP1, the extent of inhibition of transport by NEM was dependent on the nature of the external cation. In the presence of external sodium, most transporters are expected to be outward-facing, because in the absence of external glutamate, the translocation complex cannot be formed (Fig. 1A; the intermediate sodium-bound, glutamate-free form of the transporter is not indicated in the figure). External potassium promotes inward translocation via half-cycle II (Fig. 1A). Even though the potassium relocation step is believed to be the rate-limiting step for the transport cycle (Bergles et al., 2002), the proportion of inward-facing transporters is expected to increase when external potassium replaces sodium. Under these conditions, the sensitivity of transport to NEM was increased in 16 of the 22 NEM-sensitive mutants documented in Figs. 3 and 4. It is significant that potassium did not decrease the inhibition any of the NEM-sensitive mutants. It is possible that in some of the positions, the introduction of a cysteine could have resulted in a prolonged dwell time of an accessible state so that this cysteine becomes more accessible. Nevertheless, an increased sensitivity to NEM in the presence of potassium means that also in such mutants, potassium can increase the proportion of inward-facing transporters. Kainate, expected to exert the opposite effect of potassium and to trap the transporter in an outward-facing conformation, has the opposite effect of potassium (Fig. 6). This provides additional support for the idea that NEM acts on most positions from the cytoplasmic side.
The addition of glutamate in the presence of sodium would promote transport and thereby increase the proportion of inward-facing transporters. However, in several mutants in which the NEM-sensitivity of transport was increased by potassium and decreased by kainate, no effect by sodium plus glutamate was observed. A possible explanation could be that in the latter case, many of the inward-facing transporters are likely to be in the glutamate-bound conformation (Fig. 1A, bottom right state) rather than in the potassium bound conformation (Fig. 1A, bottom left state). It is likely that glutamate can physically restrict the access of NEM to the engineered cysteines from the cytoplasm much better than the smaller potassium ion.
Based on the GltPh structure, we predicted that when the transporter becomes inward-facing, HP1 moves away from HP2 to a location closer to the cytoplasm (Yernool et al., 2004). In this study, we have provided experimental evidence consistent with two of the major predictions: 1) an increased cytoplasmic reactivity/accessibility of many positions of HP1 (Figs. 3 and 4); and 2) an increased distance between the tips of HP1 and HP2, respectively. Support for the second prediction comes from the effect of potassium on inhibition of transport activity of the A364C/S440C double mutant by oxidative cross-linking (Fig. 8A). The two cysteines of this double mutant are located on HP1 and HP2, respectively. The most straightforward explanation for the diminished inhibition of transport by CuPh in the presence of potassium is that when the transporter becomes inward-facing, HP1 moves away from HP2 and this is further supported by the opposite effect of potassium on the inhibition of transport of A364C and S440C by MTSET (Fig. 7). On the other hand, external potassium had no effect on the inhibition of transport of the A412C/V427C double mutant by CuPh (Fig. 8B). This case, in which the two cysteine residues are inserted in HP2, illustrates the specificity of the effect of potassium. Therefore, the effect of potassium on the CuPh inhibition of transport by A364C/S440C is probably due to the relative movement of HP1 and HP2 rather than to the movement of more peripheral parts of the transporter around the binding pocket, sealing it off alternatively from the outside and the inside. Alternatively, potassium could bind close to position 364, thereby physically blocking its reactivity/accessibility. However, in the GltPh structure, which reflects the substrate-occluded form of the transporter, the two positions are within 4Å of each other. Therefore, a blockade of accessibility of position 364 by external potassium effectively means that the distance between positions 364 and 440 is increased.
The inhibition of transport of A364C by membrane-impermeant MTSET is diminished in the presence of potassium (Fig. 7A), but inhibition by NEM is unaffected under these conditions (Fig. 7A). Presumably this is because NEM may be able to reach the cysteine at position 364 equally well from the outside or from the inside. In the case of transport activity of S440C, potassium potentiates the inhibition by NEM and that by MTSET (Fig. 7B). It has been proposed that when the intracellular gate is open, HP2 moves toward the center of the trimer, occupying the space vacated by the tip of HP1, thereby preventing the formation of an open transmembrane pore (Yernool et al., 2004). It is possible that in this new position, the cysteine at position 440 is more accessible to either of the two sulfhydryl reagents.
Recent fluorescence energy transfer experiments suggest that glutamate transporters do not undergo large conformational changes during transport (Koch and Larsson, 2005). In these experiments, donor and acceptor fluorophores were attached mostly at the extracellular ends of the transmembrane domains. However, sulfhydryl modification at the tips of HP1 and HP2 by MTSET leads to inactivation of transport (Zhang and Kanner, 1999; Grunewald and Kanner, 2000), and a fluorescently labeled mutant via a cysteine introduced at the tip of HP1 is inactive (Koch and Larsson, 2005). Thus, significant movements of HP1 and HP2 during transport are not ruled out by the fluorescence resonance energy transfer experiments.
It is perhaps surprising that so many HP1 positions retain a low reactivity toward NEM, even in the presence of potassium (legend to Fig. 3). However, these observations are consistent with the proposal that in the inward-facing form of the transporter, this loop may become sequestered within the crevice formed between TMs 1 and 6 on the lipid-exposed face of each subunit (Yernool et al., 2004). Alternatively, the movement by HP1 could be more limited than the prediction.
Many of the positions of the more intracellular parts of TMs 7 and 8, but not of the more peripheral TM 6, become more reactive/accessible to NEM in the presence of potassium (Figs. 4 and 5). It is possible that TMs 7 and 8, which are close to each other in the GltPh structure, could move away from each other when the transporter becomes inward-facing. However, increased reactivity of an engineered cysteine does not necessarily mean that such a residue moves, because it could rather be the result of movement of other parts of the protein around it. Therefore, HP1 could act as a “plug,” obstructing the path connecting the binding pocket with the cytoplasm, which is lined by TMs 7 and 8. Removal of this “plug” would expose the access channel from the cytoplasm and make several positions of TMs 7 and 8 more accessible to permeant sulfhydryl reagents.
The GltPh structure (Yernool et al., 2004) represents one conformation of the transporter. Experiments on transporter dynamics, such as the ones described here, are an important tool toward a better understanding of how glutamate transporters move their substrates from the extracellular medium to the cytoplasm and vice versa.
Footnotes
-
This work was supported by National Institute of Neurological Disorders and Stroke/National Institutes of Health grant NS 16708 and the European Union Consortium EUGINDAT.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.106.032607.
-
ABBREVIATIONS: TM, transmembrane domain; HP, helical hairpin; MTSET, [2-(trimethylammonium)ethyl]methanethiosulfonate; NEM, N-ethylmaleimide; CuPh, Cu(II)(1,10-phenantroline)3; CL-GLT-1, cysteine-less GLT-1.
- Received November 13, 2006.
- Accepted February 1, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}