


Abstract
Target identification and validation remain difficult steps in the drug discovery process, and uncovering the core genes and pathways that are fundamental for cancer cell survival may facilitate this process. Glioblastoma represents a challenging form of cancer for chemotherapy. Therefore, we assayed 16,560 short interfering RNA (siRNA) aimed at identifying which of the 5520 unique therapeutically targetable gene products were important for the survival of human glioblastoma. We analyzed the viability of T98G glioma cells 96 h after siRNA transfection with two orthogonal statistical methods and identified 55 survival genes that encoded proteases, kinases, and transferases. It is noteworthy that 22% (12/55) of the survival genes were constituents of the 20S and 26S proteasome subunits. An expression survey of a panel of glioma cell lines demonstrated expression of the proteasome component PSMB4, and the validity of the proteasome complex as a target for survival inhibition was confirmed in a series of glioma and nonglioma cell lines by pharmacological inhibition and RNA interference. Biological networks were built with the other survival genes using a protein-protein interaction network, which identified clusters of cellular processes, including protein ubiquitination, purine and pyrimidine metabolism, nucleotide excision repair, and NF-κB signaling. The results of this study should broaden our understanding of the core genes and pathways that regulate cell survival; through either small molecule inhibition or RNA interference, we highlight the potential significance of proteasome inhibition.
High-throughput analysis of gene function has deepened our appreciation of the molecular underpinnings associated with particular biological processes in cancer and holds promise for the identification of novel cancer drug targets (Ramadan et al., 2007). Target identification and validation remain difficult steps in the drug discovery process (Rich and Bigner, 2004; Ramadan et al., 2007). Therefore, uncovering the core genes and pathways that are fundamental for cell survival may facilitate this process.
An unbiased approach to explore these essential targets exploits the use of short interfering RNA (siRNA). In cells, siRNA can silence essentially any gene product in the genome through sequence-specific mRNA transcript degradation (Sachse and Echeverri, 2004), and large-scale siRNA screening is made possible by siRNA libraries and automated liquid handlers (Berns et al., 2004; Iorns et al., 2007). This genomic tool offers simultaneous and systematic genome-wide interrogation of the loss-of-function phenotypes associated with protein suppression without requiring a priori knowledge of gene functions or cellular pathways.
Glioblastoma multiforme (GBM) is an excellent cell-based model for identifying these essential genes. GBM is a high-grade brain malignancy arising from astrocytes (Iorns et al., 2007), and despite aggressive surgical approaches, optimized radiation therapy regimens, and the application of cytotoxic chemotherapies, the median survival of patients with GBM from time of diagnosis is approximately 12 months, which has not changed in decades (Mischel and Cloughesy, 2003). Annotation of these essential genes in this glioma cell-based system should facilitate the drug discovery process by rapidly identifying novel targets (Stein, 1979; Weller et al., 1998; Short et al., 1999).
We implemented a siRNA screen using a “druggable” genome library of 16,560 siRNAs targeting 5520 unique human genes to identify the genes and pathways essential for GBM cell survival in the T98G glioma cell line. We employed a therapeutic target genome siRNA library, which comprises siRNAs targeting gene products that are potential drug targets or are disease-modifying, such as ion channels, transporters, receptors, and protein kinases, to facilitate the target identification process (Hopkins and Groom, 2002; Russ and Lampel, 2005; Overington et al., 2006). We also developed two rigorous, orthogonal statistical analysis algorithms that combined reproducibility with magnitude of effect to finalize the hit list. To our knowledge, this is the first study to use a systematic, unbiased siRNA-based screen on glioma cells, and the results of this study should broaden our understanding of the core genes and pathways that are essential for glioma cell survival and possibly other cell types. Indeed, we identified the proteasome as a highly represented essential complex in our glioma cell model, and we highlight the potential significance of proteasome inhibition, both by small molecule inhibition and therapeutic RNA interference, in glioma and other cell types.
Materials and Methods
Reagents.
DharmaFECT 2 transfection reagent and the siGENOME nontargeting siRNA 1 were purchased from Dharmacon (Lafayette, CO). CellTiter-Blue cell viability assay was from Promega (Madison, WI). The Silencer Druggable Genome siRNA Library (Version 1.1) and 5× siRNA resuspension buffer were from Ambion (Austin, TX). Tissue culture-treated 384-well microtiter plates were from Greiner Bio-One GmbH (Frickenhausen, Germany). OptiMEM, McCoy's 5A Medium (modified), Dulbecco's modified Eagle's medium, Eagle's minimal essential medium, PBS, and Hoechst 33342 were from Invitrogen (Carlsbad, CA). ECL Western blotting substrate was from Pierce Biotechnology (Rockford, IL). The well characterized proteasome subunit β-type 4 (PSMB4) mouse monoclonal antibody was from Abcam (Cambridge, UK) (Catlow et al., 2007). Proteasome subunit β1 mouse monoclonal antibody, proteasome subunit β2 mouse monoclonal antibody, and proteasome subunit β5 goat polyclonal antibody were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-β-tubulin antibody was from Cedarlane Laboratories (Burlington, ON, Canada) and anti-PARP was from Cell Signaling Technology (Danvers, MA). All other reagents were from Sigma-Aldrich (St. Louis, MO) unless otherwise noted.
Cell Culture and Treatments.
The cell lines U87, T98G, U373, A172, and A549 were obtained from the American Type Culture Collection (Manassas, VA). LN-Z308 and LN-Z428 were generously provided by Dr. Nicolas de Tribolet (Lausanne, Switzerland). Human astrocytes (HAs) and human umbilical vein endothelial cells (HUVECs) were obtained from ScienCell Research Laboratories (San Diego, CA). T98G, U87, and U373 cells were maintained in Eagle's minimal essential medium supplemented with Earle's basic salt solution, nonessential amino acids, sodium pyruvate, 1% l-glutamine, 100 U/ml penicillin/streptomycin (Invitrogen), and 10% fetal bovine serum (MediaTech, Manassas, VA). A172, LN-Z428, and LN-Z308 were maintained in α-minimal essential medium supplemented with l-glutamine. HAs were grown in astrocyte growth medium and HUVECs in endothelial cell medium (ScienCell Research Laboratories, Carlsbad, CA).
MG-132 (Calbiochem, La Jolla, CA) was dissolved into dimethyl sulfoxide (DMSO) and medium (final DMSO concentration, 0.5%) for cell treatment and added 24 h after cell plating. Cells were incubated in a humidified incubator at 37°C with 5% CO2.
A Zoom MV automated microplate dispenser (Titertek, Huntsville, AL) was used to dispense the cells, transfection reagent, and OptiMEM. V-Prep high-speed automated precision microplate pipetting station (Velocity 11, Menlo Park, CA) was used to make the siRNA-OptiMEM-DharmaFECT2 complexes, replace medium and add CellTiter-Blue. For 384-well experiments, fluorescence readout for cell viability from the CellTiter-Blue viability assay was measured with a SpectraMax M5 multidetection microplate reader and absorbance spectrophotometer (Molecular Devices, Sunnyvale, CA), equipped with a Molecular Devices StakMax robotic plate handler, was used to read cell viability. ABgene SEAL-IT 100 automated microplate sealer was used to reseal siRNA library plates. Cellomics ArrayScan II HCS system (Thermo Fisher Scientific, Waltham, MA) was used for cell counting and screening validation studies.
Lysate Preparation and Western Blotting.
Western blotting was conducted as described previously (Tomko and Lazo, 2008). In brief, six-well plates containing T98G, U87, U373, LN-Z308, LN-Z428, and A172 were placed on ice, washed with ice-cold PBS, collected by scraping into a modified radioimmunoprecipitation buffer (Bansal and Lazo, 2007), and incubated on ice for 30 min with frequent vortexing. Lysates were cleared by centrifugation at 13,000g for 18 to 20 min. Relative protein concentrations of each sample were determined using the a protein assay kit (Bio-Rad Laboratories, Hercules, CA). Equivalent protein amounts (30 μg) from cell lysates were resolved on 12 and 16% SDS-polyacrylamide gels and transferred to nitrocellulose membranes. Membranes were probed with anti-β-tubulin, anti-PSMB4, anti-PSMB1, anti-PSMB2, anti-PSMB5 or poly (ADP-ribose) polymerase (PARP) antibodies. β-Tubulin was used as a loading control. Positive antibody reactions were visualized using peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) and chemiluminescence by ECL Western blotting substrate (Pierce) according to manufacturer's instructions, and membranes were then sbjected to filmless autoradiographic analysis (LAS-3000; Fuji, Tokyo, Japan).
siRNA Sequences.
The 5520 therapeutic targets of the Silencer Druggable Genome siRNA Library comprised three unique siRNA duplexes targeting each gene. The sequences for siRNA duplexes targeting PSMB4 (Ambion) were: duplex A sense, GCUAUAGUCCUAGAGCUAUtt; antisense, AUAGCUCUAGGACUAUAGCtg; duplex B sense, GCUAUUCAUUCAUGGCUGAtt; antisense, UCAGCCAUGAAUGAAUAGCtc; and duplex C sense, GAUGGACACAGCUAUAGUCtt; antisense, GACUAUAGCUGUGUCCAUCtc. The sequence for the siGENOME nontargeting siRNA 1 (negative control) was UAGCGACUAAACACAUCAA. Sequences for the siRNAs targeting PSMA3, DDX39, and RAN are given in Supplementary Table S9.
siRNA Transient Transfection.
T98G cells were wet reverse-transfected with the siRNA library at a final concentration of 20 nM/target in a one-gene, one-well format. This concentration of siRNA was selected to generate the maximum transfection efficiency based on optimization studies using fluorescent siRNA and loss of sentinel protein, namely lamin A/C. For each siRNA target, 1.56 μl of 833.3 nM siRNA was combined with 0.06 μl of DharmaFECT2 transfection reagent and 12.38 μl of OptiMEM. Fifty-one microliters per 450 cells of T98G cell suspension was then added directly onto the siRNA complexes. siRNA complexes were prepared with DharmaFECT2 transfection reagent in pools of three unique siRNA duplexes per well, one gene per well across 16 384-well siRNA library plates. siRNA complexes were prepared at 50 nM per well, and the addition of cell suspension (20–25 min after complex preparation) to the complexes brought the final siRNA concentration to 20 nM per well. Five hours later, medium containing siRNA complexes was removed and replaced with fresh complete medium. Cells were incubated for 96 h to allow for gene silencing in a humidified incubator at 37°C with 5% CO2 with a medium change after 48 h before measuring cell viability with the CellTiter-Blue viability assay and the ArrayScan II.
Reverse Transcription Polymerase Chain Reaction.
T98G cells transfected with scramble siRNA or PSMB4 siRNA were washed with PBS once and the total RNA isolated and purified using RNeasy Mini Kit following the manufacturer's instructions (Qiagen, Valencia, CA). The SuperScript III One-Step reverse transcription PCR System with Platinum Taq DNA Polymerase was used for cDNA synthesis and PCR amplification. Total RNA (100 ng) was reverse-transcribed to cDNA, and the synthesized cDNA was amplified using Mycycler thermal cycler (Bio-Rad Laboratories) in 25 μl of reaction mixture containing SuperScript III reverse transcription/Platinum Taq mix, 2× reaction mix (a buffer containing 0.4 mM concentrations of each dNTP and 3.2 mM MgSO4), and 0.2 μM sense and antisense primers. The sequences of the primers were as follows: PSMB4: forward, 5′-CCTCAGTCCTCGGCGTTAAG-3′; reverse,5′-GCATGGTACTGTTGTTGACTCG-3′; PSMB5: forward, 5′-GTGAAGGGAACCGGATTTCAG-3′; reverse, 5′-CTCGACGGGCCAGATCATAG-3′; PSMB2: forward, 5′-TACCTCATCGGTATCCAAG-3′;reverse, 5′-ATATCCATAGTCACC-3′; and actin: forward, 5′-AAGAGAGGCATCCTCACCCT-3′; reverse, 5′-TACATGGCTGGGGTGTTGAA-3′. Cycling conditions were as follows: cDNA synthesis at 50°C for 30 min (one cycle), denaturation at 95°C for 5 min (one cycle), PCR amplification at 95°C for 1 min, then at 60°C (for PSMB4 and PSMB5) or 52°C (for PSMB2 and actin) for 1 min, and then at 72°C for 1 min (25 cycles), and final extension at 72°C for 7 min (one cycle). The PCR products were confirmed using 2% agarose gel stained with ethidium bromide.
Data Analysis.
The siRNA screen was performed eight times over eight separate weeks. Relative fluorescence units from each targeted siRNA well were normalized to in-plate controls that had received a scrambled siRNA sequence (negative control), which then permitted plate-to-plate comparisons of cell viability. To analyze the screening data, we derived an objective statistical analysis method using two orthogonal statistical methods.
In the first method, cell viabilities for each screen were sorted in ascending order, and the top 2.5% or 138 genes (i.e., genes that when inhibited caused greatest survival inhibition) for each of the eight screens were binned. Genes present in at least five of eight bins were considered for further analysis.
In the second method, outlier values were detected and removed before further analysis. An outlier was defined as an observation that was distant from other data and may be generated by unexpected system errors (Hawkins, 1980). We employed this rigorous method because outliers shift the mean and variance calculated from the observations so that the widely used Z-scores and other mean-variance-based outlier detection methods are not suitable to detect the “true” outliers (Iglewicz and Hoaglin, 1993). The median of the absolute deviation (MAD) method, which is resistant to outliers in the samples, has been implemented to detect the outliers of the gene viability replicates. The method is described below:
Estimate the MAD of each gene viability replicates, defined as:
 Xij was the viability on the ith gene and jth screen, and X̃i was the median of the eight viability replicates of the ith gene from the eight survival gene screens.
Xij was the viability on the ith gene and jth screen, and X̃i was the median of the eight viability replicates of the ith gene from the eight survival gene screens.Calculate the MAD score of each gene viability sample on the ith gene and jth screen, defined as:
The gene viability sample was marked as outlier when the absolute value of its MAD-score was greater than 3.5. Genes selected by both the binning method and MAD method were chosen as screening hits. Two-tailed t tests were used to compare suppression of cell viability from unpooled and pooled siRNA sequences to scrambled siRNA sequences (negative control).


Pathway and Network Analysis.
Pathway and functional network analyses were performed with Ingenuity Pathways Analysis (IPA; Ingenuity Systems, Redwood City, CA). The gene list consisting of RefSeq accession numbers was uploaded to this web-based application and used for generating biological networks. Fisher's exact test was used with α = 0.05 to calculate the statistical probability for the correct functional assignment of the survival genes.
Results
Genome-Wide Therapeutic Target siRNA Screen.
We performed a genome-wide therapeutic target siRNA screen to identify the genes essential for glioma cell survival. This screen was performed in eight replicates over eight separate weeks, and cell viability values were normalized to in-plate controls. We designed the assay to ensure maximal siRNA transfection, which resulted in some transfection-associated cytotoxicity and a median cell viability of 79% for all 5520 genes. Fifty-eight genes (Supplementary Table S1) and 138 genes (Supplementary Table S2) were identified as reproducibly essential genes for glioma cell viability using the binning and MAD methods, respectively (Fig. 1A). Of the total 44,160 targeting siRNA reactions, the MAD method classified 1189 (2.7%) values as outliers. A composite set of 55 genes, defined as “survival genes,” were present in both the binned and MAD gene lists (Table 1, Fig. 1A), TNFRSF10B, DPYSL4, and AGA being selected by only the binning method (Supplementary Table S3). Histogram analysis for this set of survival genes revealed a cell viability distribution between 6 and 35%, which was defined as significant toxicity (Fig. 1B). For a more detailed listing of our survival genes, MAD method results, and binning method results, refer to Supplementary Tables S4, S5, and S6, respectively.

Two-condition analysis and frequency distribution and classification of survival gene siRNAs. A, fifty-eight genes were selected using the binning method, and 138 genes (2.5%) were selected using the MAD method. A composite set of 55 survival genes was created by overlapping the binned and MAD gene lists. B, histogram analysis of this set of 55 survival genes revealed a cell viability distribution in the range of 6 to 35%. C, survival genes were classified by shared molecular and biological functions (Mi et al., 2007). This classification scheme revealed enriched protein classes, including proteases, kinases, transferases, transcription regulators, RAS family proteins, and signaling molecules. The largest class of genes was proteases (12 genes).
Candidate survival genes
Our siRNA screen identified 55 survival genes that significantly induced glioma cell death when suppressed. Classification based on shared molecular and biological function was used to subdivide these genes by shared molecular and biological functions, and the subdivisions were further ordered by increasing cell viability. Cell viabilities were calculated by normalizing data from siRNA targeting wells to in-plate negative controls.
Survival Genes.
The set of 55 survival genes was classified by shared molecular and biological functionality (Table 1) (Mi et al., 2007). This classification scheme revealed enriched protein classes among the survival genes, including proteases, kinases, and transferases (Fig. 1C). It is noteworthy that 12 of 55 survival genes (22%) were components of the 20S and 26S proteasome complexes. These 12 proteasome components and several other survival genes were reassayed using the pooled targeting siRNAs (Fig. 2, A and B). Among the most growth inhibitory siRNAs were RAN (93.3%), DDX39 (92.9%), K-ALPHA-1 (91.9%), PSMC3 (91.2%), PSMD14 (88.2%), and PSMB2 (85.2%). The therapeutic target genome used in our study contained siRNAs targeting 26 proteasome components, and because of the high representation of proteasome components in the survival gene list (12 of 55 genes), we re-assayed siRNAs targeting the 14 remaining nonsurvival gene proteasome components (Fig. 2C). Only PSMB3, PSMC4, and PSMD7 siRNA induced >60% decrease in cell viability.

Replicate validation of screening siRNAs. Replicate validation of screening siRNAs demonstrated reproducible cytotoxicity. A, suppression of cell viability was confirmed by replicate transfection with the 12 survival gene proteasome components. siRNAs targeting PSMC3, PSMD14, and PSMB2 induced the most significant cytotoxicity. Student's t test indicated significant differences from cells treated with scrambled siRNA (p < 0.05). B, replicate validation confirmed cytotoxicity of various nonproteasome targeting survival gene siRNAs. Transfection with siRNA targeting RAN, DDX39, and K-ALPHA-1 induced the most significant cytotoxicity. C, reassaying the remaining 14 nonsurvival gene proteasome component siRNAs did not result in significant cytotoxicity, except for PSMD7. Each experiment was performed in triplicate, and error bars represent S.E.M. Student's t test indicated significant differences from cells treated with scrambled siRNA (p < 0.05).
To further provide clinical significance to our list of 55 candidate survival genes, we focused our efforts on survival genes that were reportedly overexpressed in primary or secondary GBM tissue samples from the Oncomine cancer profiling gene database or previous reports (Parsons et al., 2008) and that were known or potential targets of small molecule inhibitors. Of interest were a number of proteasome components and other genes overexpressed in GBM (Supplementary Table S7). These included 10 of 12 survival gene proteasome components as well as targets that have been previously implicated in gliomagenesis, cell proliferation, and cancer invasion, including AKT3, and CLCN3.
Validation of Survival Genes.
We validated the survival genes by reassaying several candidate siRNAs using the unpooled siRNA sequences. Our validation criteria demanded that at least two of three unpooled siRNA sequences suppressed cell viability. The survival genes PSMA3, PSMB4, PSMD14, PSMC3, RAN, and DDX39 were chosen for this validation (Supplementary Table S7). PSMA3, PSMD14, PSMC3, and RAN revalidated with all three individual sequences; PSMB4 revalidated with two sequences; DDX39 revalidated with only one sequence, suggesting a possible off-target effect (Supplementary Fig. S2).
Suppression of Cell Viability by PSMB4 Silencing.
PSMB4 was chosen for further validation, because of its role as a proteasome component, the potential clinical interest of proteasome inhibitors, and its reported overexpression in GBM. To further validate the on-target mechanism of the PSMB4 siRNA, we demonstrated that transfection with unpooled sequences A, B, but not C, induced protein knockdown as did pooled A, B, and C sequences (Fig. 3A). This result was consistent with cell viability data for sequences A (p < 0.0001), B (p = 0.0002), C (p = 0.87), and pooled (p < 0.0001), where sequences A, B, and pooled resulted in significant suppression of cell viability, whereas sequence C did not. Furthermore, cell viability was measured at 24-h time points over a 96-h period to determine the effect of siRNA transfection of the pooled and unpooled sequences (Fig. 3B). Protein knockdown was also observed at 48 and 72 h (Fig. 3C) and seemed to occur before induction of significant cell death.

Suppression of cell viability by PSMB4 silencing. PSMB4 was chosen as a representative proteasome target for further validation. A, transfection with individual siRNA sequences A, B, but not C demonstrated protein knockdown and suppression of cell viability as did the pooled A, B, and C sequences. Student's t test indicated significant differences from control cells (p < 0.05). B, cell viability after PSMB4 transfection was measured at 24-h time intervals over a 96-h period. There was minimal cytotoxicity with sequences A, B, C, and pooled (A, B, and C) at 0 to 48 h, whereas significant cytotoxicity was observed with sequences A, B, and pooled (A, B, and C) at 72 to 96 h. C, protein knockdown was observed at 48 and 72 h and seemed to occur before induction of cell death. The values below the Western blots are the quantification of band density normalized to β-tubulin from three independent experiments. Asterisks indicate a significant difference from cells treated with scrambled siRNA using a Student's t test (p < 0.05).
We assessed the expression of PSMB4 in a panel of glioma cell lines versus HUVEC and HA control cell lines (Fig. 4A). The glioma cell lines SG388, which was a low passage institutionally derived glioma cell line, T98G, U373, U87, LN-Z308, LN-Z428, and A172 on average expressed increased levels of PSMB4 compared with HUVEC and HA cells. It was notable that T98G cells had almost 2-fold more PSMB4 than either HUVEC or HA cells.

Proteasome inhibition in cell lines. A, immunoblotting was used to detect relative amounts of PSMB4 protein in glioma and control cell lines. The molecular mass of PSMB4 has been reported to be 55 kDa. B, the glioma cell lines T98G, U373, U87, LN-Z308, LN-Z428, and A172, breast adenocarcinoma cell line MCF7, and lung adenocarcinoma epithelial cell line A549 were transfected with PSMB4 siRNA, and cell viability was measured at 96 h. LN-Z308 and LN-Z428 were most resistant to cell death, whereas A549 and A172 were most sensitive. Student's t test indicated the growth inhibition for all cells was significantly different from control cells (p < 0.05). C, the proteasome inhibitor MG-132 inhibited cancer cell viability. Cell viability of T98G, U87, U373, LN-Z308, LN-Z428, and A172 glioma cells 48 h after addition of increasing concentrations of MG-132 was measured with CellTiter-Blue and corroborated with cell count data. Viability is shown as a percentage of viability of cells treated with vehicle (0.5% DMSO). Values are mean ± S.E.M. from at least three independent experiments. IC50 values for cell lines are as follow: 140.7 nM (T98G), 323.6 (U87), 288.4 (U373), 972.8 (LN-Z308), 374.1 (LN-Z428), and 242 (A172). The values below the Western blots are the quantification of band density normalized to β-tubulin from three independent experiments.
We also measured PSMB1, PSMB2, and PSMB5 protein levels in all nine cell lines (Supplemental Fig. 2). PSMB1 was not markedly elevated in any tumor line compared with HUVEC. Only LN-Z428 cells had elevated PSMB2 and PSMB5 compared with HUVEC and astrocytes. Therefore, PSMB4 seemed to have a somewhat different expression profile compared with these other proteasomal subunits.
We next assessed the generality of this cytotoxic effect in a series of glioma and nonglioma cell lines (Fig. 4B). Of these eight cell lines, LN-Z308 (38.4%) and LN-Z428 (34.5%) were most resistant to cell death with this siRNA, whereas A549 (14.3%) and A172 (14.7%) were most sensitive at 96 h. We next examined the mRNA levels of PSMB4 and two other β subunits, PSMB2 and PSMB5, in cells after PSMB4 siRNA to assess the specificity of the siRNA depletion. As anticipated, we found that PSMB4 mRNA was markedly decreased 48, 72, and 96 h after transfection with PSMB4 siRNA (Fig. 5A). We observed no decrease in PSMB2 or PSMB5 mRNA levels. It is noteworthy that PSMB1, PSMB2, and PSMB5 protein levels decreased in T98G cells 48, 72, and 96 h after transfection with PSMB4 siRNA and a new higher molecular mass band appeared for each β subunit (Fig. 5B). These bands migrate as the previously published precursor forms (Nandi et al., 1997; Hirano et al., 2008). We also probed whether or not PSMB4 siRNA induce an apoptotic-like process using (PARP) cleavage. As indicated in the Fig. 5C, PARP was clearly cleaved at 72 and 96 h, with some PARP cleavage at 48 h.

PSMB1, PSMB2, and PSMB5 mRNA and protein levels in T98G cells after PSMB4 siRNA transfection. A, mRNA levels of PSMB4, PSMB2, and PSMB5 were determined in T98G cells transfected with either scramble siRNA or PSMB4 siRNA. PSMB4, but not PSMB2 and PSMB5, mRNA level was substantially decreased 48 h after PSMB4 siRNA transfection. B, Western blotting for PSMB1, PSMB2, and PSMB5 expression in T98G cells transfected with scramble siRNA or PSMB4 siRNA. Treatment with siRNA against PSMB4 for 48 h resulted in a loss of all three subunits with the appearance of a higher molecular weight immunoreactive band. C, Western blotting analysis of PARP in T98G cells transfected with scramble or PSMB4 siRNA. PARP cleavage was apparent 48 h after the transfection with PSMB4 siRNA with prominent PARP protein at 72 and 96 h.
Proteasome Inhibitor Sensitization.
We used the prototypic proteasome inhibitor MG-132 to pharmacologically evaluate the essential functionality of the proteasome in glioma cells. The glioma cell lines were treated with increasing concentrations of the compound for 48 h, and cell viability was calculated as a percentage of cells treated with vehicle (0.5% DMSO). All cell lines were sensitive to MG-132 in the nanomolar range, and the 50% inhibitory concentrations (IC50) ranged from 140 to 973 nM (Fig. 4C). It is interesting that LNZ-308 cells were rather resistant to both MG-132 and PSMB4 siRNA growth inhibition, whereas T98G cells seemed more sensitive to both MG-132 and PSMB4 siRNA.
Protein-Protein Interaction Networks.
We analyzed our survival gene data with a knowledge-based protein-protein interaction network to identify critical biological networks and processes. This analysis revealed the most enriched cellular functions: dermatological disease, infectious disease, embryonic development, cellular compromise, cell-to-cell signaling and interaction, cellular function and maintenance, inflammatory response, nervous system development, cell morphology, gastrointestinal disease, and cancer, ordered according to statistical significance (Supplementary Table S9). Twenty of the survival genes were classified as genes previously implicated in cancer, including the survival genes AKT3 and CLCN3.
We then mapped clusters of survival genes to functional networks. Within these large networks, genes clustered around specific centers. In a network that consisted of genes related to dermatological diseases, infectious disease, and embryonic development, the network of genes centered on the proteasome complex and nuclear factor-κB (NF-κB) (Fig. 6).
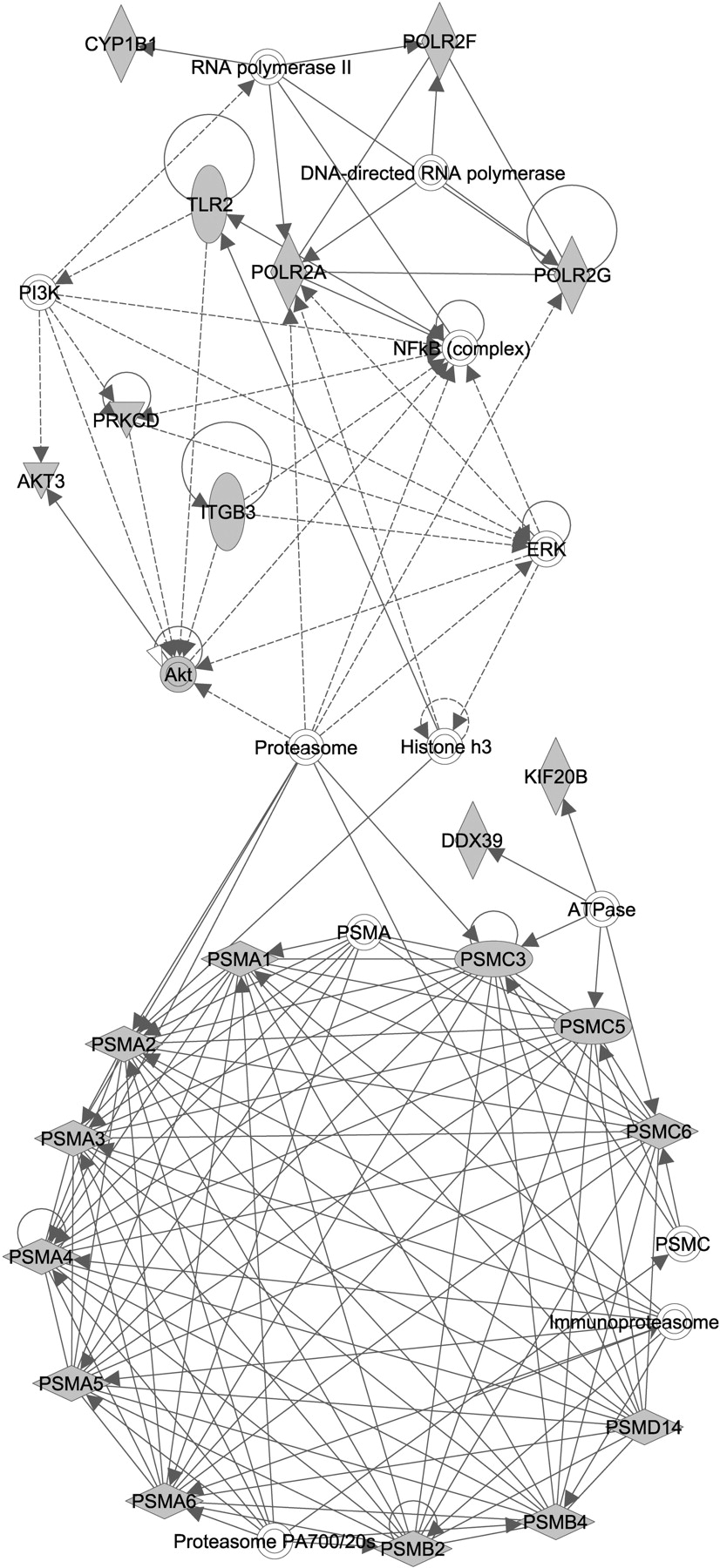
Mapping of survival genes onto a protein-protein interaction network. Functional analysis of survival genes was performed with IPA. The genes are represented as nodes, and edges connecting two nodes represent a biological relationship that is supported by at least one published reference or the IPA knowledge base. Shaded nodes represent survival genes. The network score refers to the negative exponent of the p-value calculation. In a network that comprises genes related to dermatological diseases, infectious disease, and embryonic development, the network of genes centered on the proteasome complex and NF-κB (network score: 53).
We also organized the set of survival genes into the most highly represented cellular pathways (Supplementary Fig. S3). These pathways included protein ubiquitination, purine metabolism, nucleotide excision repair, pyrimidine metabolism, and NF-κB, in order of statistical significance. Not surprisingly, the survival genes comprised several constituents of the protein ubiquitination and NF-κB pathways.
Discussion
Identifying the genes that are essential for cell survival may facilitate the discovery of opportunistic molecular targets by uncovering the molecular weaknesses in cancer cell biology. Large-scale siRNA screening can facilitate this effort by providing a platform to systematically assess the loss-of-function phenotypes associated with protein knockdown of thousands of genes. We developed a semiautomated high-throughput siRNA screen to identify survival genes in the T98G glioma cell line and employed a therapeutic target genome to focus our efforts on targets that may be candidates for small molecule inhibition. We identified 55 survival genes through the use of two orthogonal statistical methods.
Identification of several known cancer targets provided confidence that our approach was sufficiently robust to identify novel regulators of cell survival. AKT has been implicated in GBM (Gallia et al., 2009) and is a known regulator of cell proliferation, differentiation, apoptosis, and tumorigenesis. AKT3 is a gene in the AKT family (Yang et al., 2004) and has been identified as a survival gene. Furthermore, we identified the chloride channel 3 (CLCN3) as an essential gene, and inhibition with chlorotoxin has been previously explored as a treatment for glioma (Soroceanu et al., 1998).
High-throughput screening, however, is inevitably associated with false-positive and false-negative results. Our motivation was not to capture all of the essential genes for cell survival, but rather to use a rigorous algorithm to identify a few genes that could be further validated for future targeting. We speculate there are several reasons why some well known cancer survival genes were not identified by our screen. First, the therapeutic target genome comprises targets that are candidates for small-molecule inhibition, and some of the putative cancer cell survival genes, such as constituents of the Rb1 and epidermal growth factor receptor/phosphatidylinositol 3-kinase/mammalian target of rapamycin pathways, are not all targeted by this library. In addition, our goal was to implement an unbiased screen to uncover previously unknown regulators of cell survival. Furthermore, inherent limitations of this siRNA technique may also result in false-negative results. Protein knockdown depends on target protein and/or mRNA transcript stability, turnover, and overall abundance. Cancer cells may be “addicted” to specific oncogenic pathways that comprise well known cancer cell survival genes, and siRNA-induced protein knockdown may not be sufficient to suppress protein levels below the threshold for inducing cell death. Thus, these targets would not be identified by this screening strategy.
A major challenge in analyzing the large data set generated by genome-scale screening is the need to develop and implement rigorous statistical algorithms. We constructed an analysis method that combined screening reproducibility with magnitude of effect, and we ensured a high stringency threshold by employing two orthogonal statistical strategies. First, a binning method was applied to rank-ordered cell viabilities to select only those hits that consistently reproduced in the top 2.5% of genes. This method ensured that only the most cytotoxic siRNAs were selected. Second, we adapted and applied an outlier detection (MAD) method to the screening data. Outliers shift the mean and variance of the observations so that the widely used Z-scores and other mean-variance-based outlier detection methods are not suitable to detect these values (Iglewicz and Hoaglin, 1993; Zhang et al., 1999). Thus, we employed the MAD analysis to remove such outliers before calculating the target specific cell viability. Together, these two orthogonal analysis strategies facilitated the selection of a small number of high-confidence hits from the genome-scale siRNA library, although the hit threshold can be dynamically regulated depending on the desired number of hits.
Of the 55 survival genes we identified in this study, 22% were constituents of the proteasome complex, and we selected PSMB4 to further illustrate the validity of the proteasome as a target for survival inhibition. The proteasome is a multicatalytic complex that degrades most intracellular proteins, including proteins involved in cell cycle regulation and apoptosis (Voorhees et al., 2003). It is noteworthy that studies have reported a selective susceptibility of transformed cells to proteasome inhibition (Voorhees et al., 2003). For instance, transformed fibroblasts were 40 times more susceptible to proteasome inhibition than primary fibroblasts (Orlowski et al., 1998). Although the molecular mechanisms of this differential susceptibility are still unknown, possible explanations include increased susceptibility of actively proliferating cells and the de-regulation of the ubiquitin-proteasome pathway in transformed cells (Voorhees et al., 2003). Future research efforts will focus on the potential selective susceptibility of glioma cells to proteasome inhibition.
In our initial screen, systematic interrogation of 26 proteasome components with siRNA conferred significant toxicity in 12 components, and decrease in PSMB4 protein occurred before induction of significant cell death, which supported our hypothesis that cell death occurred as a result of PSMB4 knockdown. PSMB4 siRNA transfection in a panel of glioma and nonglioma cell lines demonstrated the generality of this cytotoxic effect. Depletion of many of the remaining 14 proteasome components also resulted in significant growth inhibition. siRNA against PSMB5, which is a target for the proteasome inhibitor bortezomib, did not produce as large a decrease in growth inhibition as siRNA against PSMB4 or other β subunits. One explanation for the lack of a large growth inhibitor effect to PSMB5 siRNA could be could be due to poor protein suppression with this siRNA. It is also interesting that PSMB4 siRNA caused a marked reduction in PSMB1, PSMB2, and PSMB5 protein levels. Previous results (Hirano et al., 2008) reveal that RNA interference against β subunits can result in an accumulation of intermediate forms. The proteasome subunits can stabilize each other another during assembly, and the loss of other mature β subunits might be due to destabilizing of the β-ring assembly pathway, which could contribute to metabolic stress and loss of viability. Collectively, our results suggest the presence of a subnetwork of essential proteasome components that may be most essential for cell survival, proteasome structure or function, or may have the most rapid protein turnover.
We demonstrated the cytotoxic effect of the prototypic proteasome inhibitor MG132 in a panel of glioma cell lines, and previous studies have reported a similar effect with the proteasome inhibitor bortezomib in various cell types (Fribley et al., 2004; Yin et al., 2005; Poulaki et al., 2007). Bortezomib is clinically valuable for multiple myeloma and mantle cell lymphoma and has demonstrated antitumor activity in the National Cancer Institute tumor cell line screen, in GBM cell lines (Yin et al., 2005), and in several xenograft models (Voorhees et al., 2003). Nonetheless, it is generally believed that limited penetration through an intact blood-brain barrier restricts its use in the treatment of glioma. Our results suggest that glioma treatment might be enhanced with the development of second-generation proteasome inhibitors that can penetrate the blood-brain barrier.
Another challenge in analyzing large genomic data sets is the identification of functional groups within the gene set that can identify related groups of genes in pathways that may not be readily connected from the raw data. These network analyses have provided further confirmation for our observation of the key role of the proteasome complex in cell survival. Given the numerous substrates that are regulated by proteasome degradation, it is perhaps not surprising that other survival genes would interact with this complex. For instance, NF-κB is a transcriptional factor that is activated in response to cellular stress and regulates the expression of genes involved in cell proliferation and cell death. In general, the proteasome regulates cellular levels of the inhibitor of NF-κB, although NF-κB activation can be disrupted by proteasome inhibition, thus inducing apoptosis (Jung and Dritschilo, 2001). This previously reported role of the proteasome in NF-κB activation lends further support for the molecular connectivity of this network.
Biological functions represented by these survival genes included genes implicated in dermatologic diseases, gastrointestinal diseases, and developmental processes. Previous dermatologic studies have shown that the ubiquitin-proteasome pathway regulates levels of the retinoic acid receptor in human keratinocytes (Boudjelal et al., 2000) and that topical proteasome inhibitors could be clinically valuable for the treatment of inflammatory disorders (Arbiser et al., 2005). In addition, others have found that pathways altered in GBM are also altered in colorectal cancers and may represent processes that underlie tumorigenesis (Lin et al., 2007; Parsons et al., 2008). Recent studies have also highlighted the significance of developmental processes in gliomagenesis (Bredel et al., 2005), and we have identified embryonic development and nervous system development as significant biological functions in our survival genes. Identification of these developmental processes in our screen suggests that genes implicated in tumor development and gliomagenesis may also be essential for glioma cell survival. These functional network analyses have facilitated the investigation of the molecular connectivity of genes central to cell survival, and future research will scrutinize the mechanisms of cytotoxicity.
We have catalogued the genes that are most essential for cell survival using a high-throughput screening approach and sophisticated statistical algorithms. This study provides a broad understanding of the core genes and pathways that are generally essential for cell survival and represents a first attempt to annotate the essential genes in a glioma cell-based system. Although the identified genes may not represent glioblastoma-specific chemosensitivity nodes, future research efforts using in vivo systems will reveal the selectively toxic effects of targeting these genes in GBM and other cancers.
In conclusion, siRNA is a powerful tool that provides an unbiased approach to the systematic interrogation of loss-of-function cellular phenotypes. We implemented this approach in a genome-wide siRNA screen for therapeutic targets and identified several genes that positively regulate cell survival. It is noteworthy that the proteasome complex seems to play a central role in cell survival in vitro; to our knowledge, this is the first study using a systematic siRNA-based screen of glioma cells and the first siRNA-based interrogation of the proteasome in glioma. Discovery of novel genes that contribute to cell survival validates the utility of genome-wide genetic analysis of tumors and opens new paradigms of brain tumor research.
Footnotes
↵
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.This work was supported in part by the National Institutes of Health National Institute of Neurological Disorders and Stroke [Grant P01-NS40923]; the National Institutes of Health National Cancer Institute [Grant P01-CA78039]; and the Doris Duke Charitable Foundation.
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.109.058024
-
ABBREVIATIONS:
- siRNA
- short interfering RNA
- GBM
- glioblastoma multiforme
- HA
- human astrocyte
- HUVEC
- human umbilical vein endothelial cell
- DMSO
- dimethyl sulfoxide
- PBS
- phosphate-buffered saline
- PSMB4
- proteasome subunit β
- PARP
- poly (ADP-ribose) polymerase
- PCR
- polymerase chain reaction
- MAD
- median of the absolute deviation
- NF-κB
- nuclear factor κ-light-chain-enhancer of activated B cells
- IPA
- Ingenuity Pathways Analysis.
- Received May 27, 2009.
- Accepted September 24, 2009.

- © 2009 The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}