


Abstract
Until recently, the signaling events elicited in vascular smooth muscle cells by angiotensin II (Ang II) were considered to be rapid, short-lived, and divided into separate linear pathways, where intracellular targets of the phospholipase C-diacylglycerol-Ca2+ axis were distinct from those of the tyrosine kinase- and mitogen-activated protein kinase- dependent pathways. However, these major intracellular signaling cascades do not function independently and are actively engaged in cross-talk. Downstream signals from the Ang II-bound receptors converge to elicit complex and multiple responses. The exact adapter proteins or “go-between” molecules that link the multiple intracellular pathways await clarification. Ang II induces a multitude of actions in various tissues, and the signaling events following occupancy and activation of angiotensin receptors are tightly controlled and extremely complex. Alterations of these highly regulated signaling pathways in vascular smooth cells may be pivotal in structural and functional abnormalities that underlie vascular pathological processes in cardiovascular diseases such as hypertension, atherosclerosis, and post-interventional restenosis.
I. Introduction
The vascular wall is an active, pliable and integrated organ made up of cellular (endothelial cells, vascular smooth muscle cells, and fibroblasts) and noncellular (extracellular matrix) components. It is not a static organ; the components dynamically change shape, increase, decrease, or reorganize, in response to physiological and pathological stimuli (Dubey, 1997). In the intact arterial media, smooth muscle cells and matrix are responsible for structural and functional characteristics of the vessel wall, including contraction-relaxation, growth, development, remodeling, and repair, and for the pathogenesis of cardiovascular disease, such as atherosclerosis, restenosis and hypertension (Mulvany and Aalkjaer, 1990; Schiffrin, 1992; Katoh and Periasamy, 1996; Bornfeldt, 1996). Many local and systemic factors regulate vascular smooth muscle cell function, including vasoactive peptides, such as Ang2 II and endothelin-1 (ET-1), that stimulate vasoconstriction and growth and vasorelaxing factors, such as nitric oxide, prostacyclin, and C-type natriuretic peptide that induce vasodilation by increasing levels of cyclic nucleotides (Rubanyi, 1991; Lüscher, 1993; Lüscher and Barton, 1997; Stein and Levin, 1998).
Ang II is a multifunctional peptide that has numerous actions on vascular smooth muscle—it modulates vasomotor tone, it regulates cell growth and apoptosis, it influences cell migration and extracellular matrix deposition, it is proinflammatory, and it stimulates production of other growth factors [e.g., platelet-derived growth factor (PDGF)] and vasoconstrictors (e.g., ET-1). Accordingly, Ang II plays a fundamental role in controlling the functional and structural integrity of the arterial wall and may be important in physiological processes regulating blood pressure and in pathological mechanisms underlying vascular diseases. The multiple actions of Ang II are mediated via specific, highly complex intracellular signaling pathways that are stimulated following an initial binding of the peptide to its cell-surface receptors (Matsusaka and Ichikawa, 1997). The term “intracellular signaling pathway” includes the interconnected molecular cascades that transmit information from the cell membrane receptor to the intracellular proteins that regulate cell activities such as contraction, cell growth, mitogenesis, apoptosis, differentiation, migration, and other specialized functions. Identification of such signal transduction processes is essential for understanding mechanisms that regulate vascular smooth muscle cell function, both physiologically and pathophysiologically. This review focuses on Ang II-mediated signaling in vascular smooth muscle cells and implications of altered Ang II-induced signal transduction in vascular pathological processes, concentrating specifically on hypertension. The molecular and cellular mechanisms of Ang II in cardiac and renal diseases have recently been reviewed and will not be discussed in detail here (Kim and Iwao, 2000).
II. Physiological Actions of Angiotensin II in Vascular Smooth Muscle Cells
A. The Renin Angiotensin System—Production of Angiotensin II
Ang II, an octapeptide hormone, is the active component of the renin-angiotensin system (RAS). It regulates blood pressure, plasma volume via aldosterone-regulated sodium excretion, sympathetic nervous activity, and thirst responses. It also plays a fundamental role in pathological adaptation, as manifested in myocardial remodeling after myocardial infarction and in vascular remodeling in hypertension. Ang II is produced systemically via the classical or renal RAS, and locally via tissue RAS. In the classical RAS, circulating renal-derived renin cleaves hepatic-derived angiotensinogen at the N terminus to form the decapeptide, angiotensin I, which is converted by the dipeptidyl carboxypeptidase, angiotensin-converting enzyme (ACE), in the lungs, to the active Ang II (Skeggs et al., 1967; Dorer et al., 1972; Phillips et al., 1993; Inagami, 1998) (Fig. 1). Ang I can also be processed into the heptapeptide Ang-(1-7) by three tissue endopeptidases, neutral endopeptidase (NEP) 24.11, NEP 24.15, and NEP 24.26 (Ferrario et al., 1997). Ang II is degraded by aminopeptidases to Ang III and Ang IV (Fig. 1).

Scheme of the classical renin-angiotensin system. Circulating renal-derived renin cleaves hepatic-derived angiotensinogen to form the decapeptide angiotensin I (Ang I). Ang I is converted by ACE in the lungs and tissue to active angiotensin II (Ang II), which is further metabolized to angiotensin III, angiotensin IV, and Ang II (1-7). Several non-ACEs, such as chymase, carboxypeptidase and cathepsin G, may also cleave Ang I to Ang II.
The RAS was originally regarded as a circulating system. However, many of its components are localized in tissues indicating the existence of a local tissue RAS as well (Dzau, 1989; Danser, 1996). ACE exists in plasma (as the circulating hormone), in the interstitium and intracellularly. Tissue ACE is present in all major organs, heart, brain, blood vessels, adrenals, kidney, liver, and reproductive organs (Hollenberg, 1998), and is already functional in utero (Schutz et al., 1996; Esther et al., 1997). Tissue ACE activity seems to peak during the phase of major organ development and declines thereafter (Esther et al., 1997). All components of the RAS, except renin, have been demonstrated to be produced in the vasculature. ACE is found in high concentrations in the adventitia, as well as in cultured vascular smooth muscle and endothelial cells (Dzau, 1989; Ekker et al., 1989;Naftilan, 1994). Angiotensinogen mRNA and protein have been detected in vascular smooth muscle, endothelium, and perivascular fat (Naftilan et al., 1991; Naftilan, 1994; Morgan et al., 1996). Since vascular renin is absent, local generation of Ang II in the interstitium is regulated by tissue ACE that is probably dependent on circulating renin (Fig.2). Although the function of tissue ACE is currently unclear, it may contribute to regulation of regional blood flow as recently demonstrated in the human forearm where in situ generated Ang II is more important for vasoconstriction than circulating Ang II (Saris et al., 2000).

Scheme of the tissue renin-angiotensin system. Angiotensinogen, ACE, and angiotensin receptors have been demonstrated in endothelial and vascular smooth muscle cells, as well as in perivascular fat. Tissue-derived angiotensinogen is converted to Ang I by renal-derived renin that is adsorbed from the circulation. Ang I is cleaved to Ang II by tissue ACE. endoth, endothelium; other abbreviations as in Fig. 1.
In addition to ACE-dependent pathways of Ang II formation, non-ACE pathways, which could be particularly important in pathological states, have been demonstrated. Chymotrypsin-like serine protease (chymase) may represent an important pathway for conversion of Ang I to Ang II in the human heart (Urata et al., 1990, 1996) and kidney (Hollenberg, 1998). Functional chymase and a non-ACE pathway have also been demonstrated in human vascular tissue (Hollenberg et al., 1998; Takai et al., 1998) and in dog carotid artery (Shiota et al., 1999).
B. Angiotensin Receptors
In mammalian cells, Ang II mediates its effects via at least two high-affinity plasma membrane receptors, AT1 and AT2. Both receptor subtypes have been cloned and pharmacologically characterized (Murphy et al., 1991; Sasaki et al., 1991; Kambayashi et al., 1993; Mukoyama et al., 1993). Two other Ang receptors have been described, AT3 and AT4 subtypes. The AT3receptor subtype, initially described in the neuro 2A neuroblastoma cell line (Chaki and Inagami, 1992) is peptide-specific recognizing mainly Ang II. This subtype does not bind nonpeptide ligands such as losartan (selective AT1 receptor antagonist) or PD123319 (selective AT2 receptor antagonist), and has only been observed in cell lines. The AT4receptor, which is distributed in heart, lung, kidney, brain, and liver, binds Ang IV (Swanson et al., 1992) but not losartan or PD123319. Since the pharmacology of AT3 and AT4 receptors has not been fully characterized, these receptors are not yet included in a definitive classification of mammalian AT receptors as defined by the International Union of Pharmacology Nomenclature Subcommittee for Angiotensin Receptors (de Gasparo, 1995).
The AT1 receptor belongs to the seven membrane-spanning G protein-coupled receptor family and typically activates phospholipase C through the heterotrimeric Gq protein (de Gasparo et al., 1995; Inagami, 1995) (Table1). Human AT1receptor gene is mapped to chromosome 3. To date, AT1 receptors have been shown to mediate most of the physiological actions of Ang II, and this subtype is predominant in the control of Ang II-induced vascular functions (Sadoshima, 1998). In the vasculature, AT1 receptors are present at high levels in smooth muscle cells and relatively low levels in the adventitia and are undetectable in the endothelium (Zhuo et al., 1998;Allen et al., 2000). Two AT1 receptor subtypes have been described in rodents, AT1A and AT1B, with greater than 95% amino acid sequence identity (Iwai and Inagami, 1992). AT1A and AT1B receptor genes in rats are mapped to chromosome 17 and 2, respectively. Based on the cDNA sequence, the AT1 receptor is composed of 359 amino acids (Sandberg, 1994). It is a glycoprotein and contains extracellular glycosylation sites at the amino terminus (Asn4) and the second extracellular loop (Asp176 and Asn188) (Desarnaud et al., 1993). The transmembrane domain at the amino-terminal extension and segments in the first and third extracellular loops are responsible for G protein interactions with the receptor (Hjorth et al., 1994). Internalization of G protein-coupled receptors involves receptor phosphorylation, which may be mediated, in part via caveola (Berk and Corson, 1997; Ishizaka et al., 1998). Although G protein-coupled receptors do not contain intrinsic kinase activity, they are phosphorylated on serine and threonine residues by members of the G protein receptor kinase (GRK) family. AT1 receptors are phosphorylated both in the basal state and in response to Ang II stimulation (Kai et al., 1994). Threonine and serine residues between Thr332 and Ser338 of the cytoplasmic tail are essential for receptor internalization (Hunyady et al., 1994). The AT1 receptor is also phosphorylated at tyrosine residues. Potential tyrosine phosphorylation sites within the AT1 receptor include amino acids 302, 312, 319, and 339 within the carboxyl terminus (Berk and Corson, 1997). Tyrosine at position 319 is important as it is part of the motif Tyr-Ile-Pro-Pro, which is analogous to a Src homology 2 (SH2) binding motif in the PDGF receptor (Tyr-Ile-Pro) and in the epidermal growth factor (EGF) receptor (Tyr-Leu-Pro-Pro) (Fantl et al., 1993). In EGF and PDGF receptors, these motifs are target sequences for tyrosine phosphorylation. Various tyrosine kinases, including Janus kinases (JAK and TYK), Src family kinases, and focal adhesion kinase (FAK) can tyrosine phosphorylate AT1 receptors.
Characteristics of AT1 and AT2 angiotensin receptor subtypes
The second major isoform of the Ang receptor, AT2, is normally expressed at high levels in fetal tissues and decreases rapidly after birth (Nahmias and Strosberg, 1995). The AT2 receptor gene is localized as a single copy on the X chromosome. In adults, AT2receptor expression is detectable in the pancreas, heart, kidney, adrenals, brain, and vasculature (Viswanathan and Saavedra, 1994; Touyz et al., 1999a). In the vasculature, AT2 receptors predominate in the adventitia and are detectable in the media (Zhuo et al., 1998). AT2 receptors are also expressed in several cell lines, including PC12W, R3T3, and N1E115 (Inagami, 1995). The AT2 receptor is a seven transmembrane-type, G protein-coupled receptor, comprising 363 amino acids. It has low amino acid sequence homology (∼32%) with AT1A or AT1B receptors (Mukoyama et al., 1993). Although the exact signaling pathways and the functional roles of AT2 receptors are unclear, these receptors, which appear to be regulated by intracellular cations, particularly Na+ (Tamura et al., 1999), may antagonize, under physiological conditions, AT1-mediated effects (Ciuffo et al., 1998; Yamada et al., 1998) by inhibiting cell growth, and by inducing apoptosis and vasodilation (Hayashida et al., 1996;Horiuchi, 1997a,b; Gallinat et al., 2000; Unger, 1999; Siragy, 2000). The exact role of AT2 receptors in cardiovascular disease remains to be defined.
C. Vascular Actions of Angiotensin II
Ang II promotes its effects by acting directly through Ang II receptors present on vascular cells, indirectly through the release of other factors, and possibly via cross-talk with intracellular signaling pathways of other vasoactive agents and growth factors. Although the principal function of smooth muscle cells is vasoconstriction, it has become evident that vascular smooth muscle cells have important synthetic properties during development and vascular remodeling (Table2) and are the major source of extracellular matrix components of the vascular media (Katoh and Periasamy, 1996). During blood vessel development, immature smooth muscle cells are in a dynamic state of growth and differentiation characterized by proliferation and migration (Glukhova et al., 1991). In the adult vessel, they become quiescent and assume a fibroblast-like appearance, and become filled with contractile fibers (Gordon et al., 1990). Although mature smooth muscle cells remain quiescent until injury or insult occurs, they undergo physiological hypertrophy in response to increased load (Bucher et al., 1982; Katoh and Periasamy, 1996). Ang II plays a role in these developmental processes, acting via AT1 and AT2 receptors, which are differentially expressed in vascular smooth muscle cells during normal development and during pathological processes. In vascular disease, smooth muscle cells undergo hyperplasia and/or hypertrophy as an adaptive or reactive response (Table 2). (Geisterfer et al., 1988; Berk et al., 1989; Paquet et al., 1990; Stouffer and Owens, 1992; Dubey, 1997; Touyz and Schiffrin, 1997a; Touyz et al., 1999b) and may be critical in vascular remodeling associated with hypertension, atherosclerosis, or neointimal formation. Both Ang II receptor subtypes appear to be necessary for a complete vascular smooth muscle cell response to injury (Zahradka et al., 1998).
Physiological and pathophysiological effects of angiotensin II on the vasculature
Integrated vascular responses to Ang II are the result of combined AT1- and AT2-mediated actions, as well as effects of bioactive end products of the RAS, such as Ang-(1-7). Whereas Ang II induces vasoconstriction, growth, migration, production of extracellular matrix components, and inflammation via AT1 receptors, it promotes apoptosis, and inhibits proliferation and hypertrophy via AT2 receptors (Allen et al., 2000; Siragy, 2000). Ang-(1-7) has been described as a naturally occurring competitive inhibitor of Ang II, as it has potent vasodepressor and antihypertensive effects. It can stimulate release of vasopressin, act as an excitatory neurotransmitter, augment synthesis and release of vasodilator prostaglandins, potentiate the actions of bradykinin and release nitric oxide (Ferrario et al., 1997). In addition, ACE inhibitors augment circulating levels of the vasodilator peptide, which may contribute to the antihypertensive effects associated with ACE inhibitors (Chappel et al., 1998; Iyer et al., 1998). The receptor mediating the vascular actions of Ang-(1-7) has been tentatively characterized as a non-AT1/AT2 subtype (Ferrario et al., 1997). Although the exact role of this peptide in the physiological and pathophysiological regulation of vascular function awaits clarification, its potential to antagonize AT1-mediated actions suggests that Ang-(1-7) could modulate vascular tone by promoting vasodilation.
D. Angiotensin II-Dependent Signaling Pathways
Ang II elicits complex highly regulated cascades of intracellular signal transduction that lead to short-term vascular effects, such as contraction, and to long-term biological effects, such as cell growth, migration, extracellular matrix deposition, and inflammation. Ligand-receptor binding on the external cell membrane surface induces the interaction between the receptor and effector protein on the internal cell membrane surface via G proteins (heterotrimeric proteins comprised of α, β and γ subunits). Intracellular signaling via the AT2 receptor subtype will not be discussed in detail here, as progress in Ang II type 2 receptor research in the cardiovascular system has recently been reviewed (Csikos et al., 1998;Horiuchi et al., 1999; Unger, 1999). Unless otherwise indicated, signaling events described in the present review are mediated via AT1 receptors. AT1receptors are coupled to multiple, distinct signal transduction processes, leading to diverse biological actions. The signaling processes are multiphasic with distinct temporal characteristics (Fig.3). Immediate, early, and late signaling events occur within seconds, minutes, and hours, respectively (Fig. 3). Ang II-induced phospholipase C (PLC) phosphorylation and Src activation occur within seconds and constitute immediate signaling events, activation of phospholipase A2(PLA2), phospholipase D (PLD), tyrosine kinases and mitogen-activated protein kinases (MAPKs) occurs within minutes and are early signaling processes, whereas generation of oxidative stress, proto-oncogene expression, and protein synthesis, which occur within hours, make up late signaling events.

Diagram demonstrating the multiphasic nature of Ang II-mediated signaling events in vascular smooth muscle cells. Binding of Ang II to the AT1 receptor (AT1 rec) stimulates activation of PLC and Src within seconds and constitutes the immediate signaling events. Activation of PLA2, PLD, tyrosine kinases, and MAP kinases occurs within minutes and are the early signaling processes. Generation of reactive oxygen species, proto-oncogene expression, and protein synthesis occurs within hours and makes up the late signaling events.
E. Immediate Signaling Events Stimulated by Angiotensin II
Ang II-elicited vascular contraction is rapid and utilizes various signaling mechanisms that occur within seconds of Ang II binding to its receptor. These immediate signal transduction processes include: a) G protein-mediated activation of PLC, leading to phosphatidylinositol hydrolysis and formation of inositol trisphosphate (IP3) and diacylglycerol accumulation (DAG); b) increase in cytosolic free calcium concentration ([Ca2+]i) by increasing Ca2+ influx and mobilizing intracellular Ca2+; c) activation of protein kinase C (PKC); d) changes in intracellular pH (alkalinization) via stimulation of the Na+/H+ exchanger; e) changes in intracellular free concentrations of Na+([Na+]i) and Mg2+([Mg2+]i); and f) activation of the Src family of kinases (Fig.4).

Immediate signaling events induced by Ang II stimulation in vascular smooth muscle cells. Angiotensin receptor (AT1) binding leads to G protein-coupled activation of PLC, resulting in phosphatidylinositol hydrolysis and formation of IP3 and DAG accumulation. IP3 mobilizes Ca2+ from sarcoplasmic reticular stores, and DAG activates PKC, which in turn activates the Na+/H+exchanger. These events result in increased intracellular free Ca2+ concentration ([Ca2+]i) and intracellular alkalinization. Ang II also activates the Na+-dependent Mg2+ exchanger that induces Mg2+ efflux and Na+ influx leading to increased intracellular free Na+ concentration ([Na+]i) and decreased intracellular free Mg2+ concentration ([Mg2+]i). These signaling events stimulate actin-myosin interaction resulting in vascular smooth muscle cell contraction. Src family kinases, which are also activated by Ang II within seconds, are major upstream regulators of signaling pathways associated with cell growth. Src-dependent pathways may also modulate [Ca2+]i a possible pathway that has not yet been fully elucidated (dashed line).
1. Stimulation of Phospholipase C and Phosphatidylinositol Hydrolysis.
One of the earliest detectable events resulting from Ang II stimulation of vascular smooth muscle cells is a rapid, PLC-dependent hydrolysis of phosphatidylinositol-4,5-bisphosphate (PtdInsP2) to yield water soluble IP3 and membrane bound DAG (Alexander, 1985;Griendling et al., 1985; Berk et al., 1987a; Griendling et al., 1989). PLC is a family of at least three related genes: PLC-β, PLC-γ, and PLC-δ (Rhee and Choi, 1992). PLC-β isoforms are regulated by α and βγ subunits of G proteins (Smrcka et al., 1991), whereas PLC-γ isoforms are regulated by tyrosine phosphorylation (Rhee, 1991; Homma et al., 1993; Marrero et al., 1995b). PLC-δ regulation is unclear, but may involve intracellular Ca2+. PLC-β1, PLC-γ1, and PLC-δ1 have been identified in vascular smooth muscle cells (Marrero et al., 1994;Ushio-Fukai et al., 1998b). The AT1 receptor sequentially couples to PLC-β1 via a heterotrimeric G protein and to PLC-γ1 via a tyrosine kinase (Ushio-Fukai et al., 1998b;Venema et al., 1998). The initial AT1receptor-PLC-β1 coupling is mediated by Gαq/11βγ and Gα12βγ. The βγ dimer acts as a signal transducer for activation of PLC (Touhara et al., 1995; Ushio-Fukai et al., 1998b). Both PLC-β1 and PLC-γ isoforms play a role in IP3 formation. PLC-β1 appears to be important in the rapid generation of IP3 (within 15 s), whereas PLC-γ seems to play a role in the later phase of IP3 formation (Ushio-Fukai et al., 1998b). Ang II-stimulated IP3 generation may also be mediated, in part, via tyrosine kinase-dependent pathways (Goutsouliak and Rabkin, 1997). Ang II induces a dose-dependent increase in phosphatidylinositol turnover resulting in rapid transient IP3 formation (Griendling et al., 1989) and biphasic and sustained DAG generation (Alexander et al., 1985;Griendling et al., 1985). Losartan, the selective AT1 receptor blocker, inhibits Ang II-induced hydrolysis of PtdInsP2, indicating that Ang II stimulation of the PLC pathway is mediated exclusively via AT1 receptors. IP3stimulates release of Ca2+ from sarcoplasmic/endoplasmic reticular stores and DAG, with cofactors phosphatidylserine and Ca2+, activates PKC. Ang II-elicited IP3 signal slightly precedes a rapid increase in cytoplasmic free calcium concentration ([Ca2+]i), which is in large part independent of calcium influx. These events correlate temporally with initiation of contraction in isolated vascular smooth muscle cells, as well as in intact small resistance arteries, and most likely constitute the early signaling pathway for initiation of the calcium-dependent, calmodulin-activated phosphorylation of the myosin light chain, which leads to cellular contraction (Lassegue et al., 1993; Walsh et al., 1995; Savineau and Marthan, 1997; Touyz and Schiffrin, 1997a; Touyz et al., 1999c). DAG can also be formed by the PLD-mediated hydrolysis of other phospholipids such as phosphatidylcholine and phosphatidylethanolamine.
2. Increased Intracellular Free Calcium Concentration.
Ang II-stimulated Ca2+signaling is complex and occurs via multiple pathways to elicit an integrated Ca2+ signal. Ang II typically mediates a biphasic [Ca2+]iresponse comprising a rapid initial transient phase and a sustained plateau phase (Dostal, 1990; Touyz et al., 1994; Assender et al., 1997;Touyz and Schiffrin, 1997b). Both AT1A and AT1B receptors have been shown to mediate calcium signaling in rodent vascular smooth muscle cells (Zhu et al., 1998b). The first [Ca2+]i transient is generated primarily by IP3-induced mobilization of intracellular Ca2+ and to a lesser extent by Ca2+-induced Ca2+ release (Touyz and Schiffrin, 1997b). The second [Ca2+]i phase, which appears to contribute to the sustained Ang II-induced vasoconstriction, is dependent on external Ca2+ and is the result of transmembrane Ca2+ influx (Rembold, 1992; Ruan and Arendshorst, 1996a; Inscho et al., 1997; Iverson and Arendshorst, 1998; Touyz et al., 1999c). Exact mechanisms whereby Ang II stimulates Ca2+ influx are unclear but may involve voltage-dependent calcium channels, which are directly or indirectly activated by Ang II, Ca2+-permeable, nonspecific dihydropyridine-insensitive cation channels, receptor-gated Ca2+ channels, Ca2+-activated Ca2+ release channels, and activation of the Na+/Ca2+ exchanger (Arnaudeau et al., 1996; Lu et al., 1996). In addition to IP3-mediated mobilization of intracellular Ca2+ and influx of extracellular Ca2+, tyrosine kinase-dependent increases in [Ca2+]i have been demonstrated in vascular smooth muscle cells (Hughes and Bolton, 1995;Touyz and Schiffrin, 1996a; Di Salvo et al., 1998).
3. Activation of Protein Kinase C.
Ang II-induced DAG production, together with Ca2+ and phosphatidylserine, activate PKC, a serine/threonine kinase that is a member of a multigene family consisting of at least 11 isoenzymes (Hug and Sarre, 1993; Newton, 1997). Ang II stimulates the translocation of cytosolic PKC to the plasma membrane where the activated enzyme phosphorylates specific proteins associated with vascular function (Walsh et al., 1996; Damron et al., 1998). PKC is implicated in Ang II-induced vascular contraction as well as in vascular smooth muscle cell growth (Rasmussen et al., 1987; Ruan and Arendshorst, 1996b; Orjii and Keiser, 1997; Kiron and Loutzenhiser, 1998; Bauer, 1999). These effects are mediated via activation of the Na+/H+ exchanger leading to intracellular alkalinization, an important modulator of actin-myosin interaction, and of contraction (Aalkjaer and Peng, 1997). In addition, Ang II-stimulated PKC induces its actions through phosphorylation of tyrosine kinases, such as proline-rich tyrosine kinase (PYK2) (Sabri et al., 1998), p130Cas (Sayeski et al., 1998), and Src family tyrosine kinases (Zou et al., 1998), and by stimulating MAP kinase signaling pathways (Zou et al., 1996; Wilkie et al., 1997; Kudoh, 1997; Li et al., 1998a). The PKC isoform that activates ERK-1 and ERK-2 (extracellular signal-regulated kinases) in vascular smooth muscle cells has been identified as PKC-ζ (Liao et al., 1997). Some studies failed to demonstrate that Ang II effects are PKC-dependent and others reported only a partial dependence on PKC (Berk et al., 1987b, 1989; Assender et al., 1997). Thus both PKC-dependent and -independent mechanisms are involved in Ang II-stimulated vascular contraction and growth. In addition to its second messenger function, PKC has been implicated in the rapid-agonist-induced desensitization of AT1receptors (Balmforth et al., 1997).
Some of the PKC-induced actions are mediated via the recently characterized protein kinase D (PKD), a serine/threonine kinase that is rapidly and potently activated by Ang II (Abedi et al., 1998). PKD could be an important mediator for the biological function(s) of one or more PKC isoforms in vascular smooth muscle cells, and/or may represent a component of a novel Ang II-stimulated PKC-independent signaling pathway.
4. Stimulation of Na+/H+Exchange.
Ang II elicits a biphasic change in intracellular pH (pHi), comprising an initial acidification followed by a sustained alkalinization (Griendling et al., 1989; Touyz and Schiffrin, 1997a; Touyz et al., 1999d). The rapid acidification is associated with Ca2+-ATPase-regulated Ca2+ mobilization (Berk et al., 1987b). Ang II-stimulated alkalinization is entirely dependent on activation of the Na+/H+ exchanger (Berk et al., 1987b; Touyz and Schiffrin, 1997a; Touyz et al., 1999d), which is modulated by PKC-dependent and PKC-independent mechanisms (Berk et al., 1987b). MAPKs also play a role in Ang II-stimulated activation of the Na+/H+ exchanger. ERK-1/ERK-2 and p38 activate the Na+/H+ exchanger in vascular smooth muscle cells (Kusuhara, 1998; Touyz et al., 1999d) and p90rsk has been identified as a putative potent Na+/H+ kinase (Takahashi et al., 1997a). Activation of the Na+/H+ exchanger and akalinization induce vasoconstriction in various vascular beds by increasing [Na+]i and [Ca2+]i and by sensitizing the contractile machinery to Ca2+(Grinstein et al., 1989; Carr et al., 1995; Ye, 1996; Tepel et al., 1998b; Touyz et al., 1999). In addition, increased intracellular pHi is a potent stimulus for DNA synthesis (Sachinidis et al., 1996). Thus alkalinization is an important mechanism whereby Ang II modulates vascular smooth muscle function by stimulating both contraction and growth.
5. Angiotensin II Increases Intracellular Free Concentrations of Na+ and Decreases Intracellular Free Concentrations of Mg2+.
In addition to increasing [Ca2+]i and pHi, Ang II raises [Na+]i and reduces [Mg2+]i in a concentration-dependent fashion in vascular smooth muscle cells (Johnson et al., 1991; Ye et al., 1996; Touyz and Schiffrin, 1999). These effects are rapid and maximal responses occur within 40 to 60 s (Touyz and Schiffrin, 1998). [Na+]i is regulated by the Na+/H+ exchanger, the Na+/Ca2+ exchanger, the Na+/K+ ATPase pump, and Na+ channels (Shigekawa et al., 1996; Juhaszova and Blaustein, 1997; Cox et al., 1998). The cellular mechanisms regulating [Mg2+]i are unknown, but we and others have shown that a putative Na+/Mg2+ exchanger regulates [Mg2+]i by inducing Mg2+ efflux and by stimulating Na+ influx (Touyz and Schiffrin, 1996b; Touyz and Schiffrin, 1999a; Murphy, 2000). Ang II-stimulated increase in [Na+]i and reduction in [Mg2+]i influence vascular smooth muscle contraction directly or indirectly by modulating [Ca2+]i.
6. Activation of Src Family Kinases.
The Src family of protein tyrosine kinases that characteristically interact with transmembrane tyrosine kinase receptors, also interact functionally with G protein-coupled receptors, such as AT1(Paxton et al., 1994; Marrero et al., 1995b; Parsons and Parsons, 1997;Thomas and Brugge, 1997; Ishida et al., 1998). To date, at least 14 Src-related kinases have been identified, of which the 60-kDa c-Src is the best characterized (Thomas and Brugge, 1997). The Src family kinases are subdivided into three groups based on their pattern of expression. Src, Fyn, and Yes are expressed ubiquitously, Blk, Fgr, Hck, Lck, and Lyn are found primarily in hematopoietic cells and Frk-related kinases (Frk/Rak and Iyk/Bsk) are expressed predominantly in epithelial-derived cells (Thomas and Brugge, 1997). Src family kinases share a high degree of structural similarity, with common domain architecture and regulatory mechanisms. They consist of one or more amino-terminal acylation sites (required for membrane localization), a unique domain (which defines the individual members), an SH3 domain, an SH2 domain, a catalytic domain, and a carboxyl-terminal noncatalytic domain. Regulation of Src activity is complex. Phosphorylation of Tyr527 by Csk inhibits Src activity, whereas dephosphorylation of this residue activates Src. Activation may also occur by autophosphorylation of Tyr419 in the catalytic domain, by displacement of the intramolecular interactions of the SH2 or SH3 domains by high-affinity ligands or modification of certain residues (Erpel and Courtneidge, 1995). Src family kinases are activated in response to various stimuli in many cell types and have been suggested to play an important role in signal transduction pathways that control growth and cellular architecture.
Ang II rapidly phosphorylates c-Src with maximal activation occurring within 60 s measured by either autophosphorylation or kinase activity toward enolase (Ishida et al., 1995, 1998; Marrero et al., 1995b; Touyz et al., 1999e). Src plays an important role in Ang II-induced phosphorylation of PLC-γ and IP3formation. We reported that Ang II-stimulated [Ca2+]i responses in human vascular smooth muscle cells are mediated, in part, via Src-dependent mechanisms (Touyz et al., 1999e). Src, intracellular Ca2+, and PKC regulate Ang II-induced phosphorylation of p130Cas, a signaling molecule involved in integrin-mediated cell adhesion (Sayeski et al., 1998). Src has also been associated with Ang II-induced activation of PYK2 (Dikic et al., 1996; Sabri et al., 1998) and with phosphorylation of ERKs (Ishida et al., 1998), as well as activation of other downstream proteins including pp120, p125Fak, paxillin, Jak2, signal transducers and activators of transcription (STAT)-1, Gα, caveolin, and the adapter protein, Shc (Li et al., 1996b).
F. Early Signaling Events Mediated by Angiotensin II
In addition to rapid signaling events associated with contraction, the AT1 receptor couples to multiple intracellular transduction pathways that are linked to long-term regulation of vascular smooth muscle cell function, such as growth, migration, deposition of extracellular matrix, and production of growth factors. These processes are initiated by signaling pathways that are stimulated by Ang II within minutes and include: a) phosphorylation of tyrosine kinases; b) activation of MAPKs; c) activation of PLA2 and arachidonic acid metabolism; d) activation of PLD; and e) modulation of cyclic nucleotides (Fig.5).
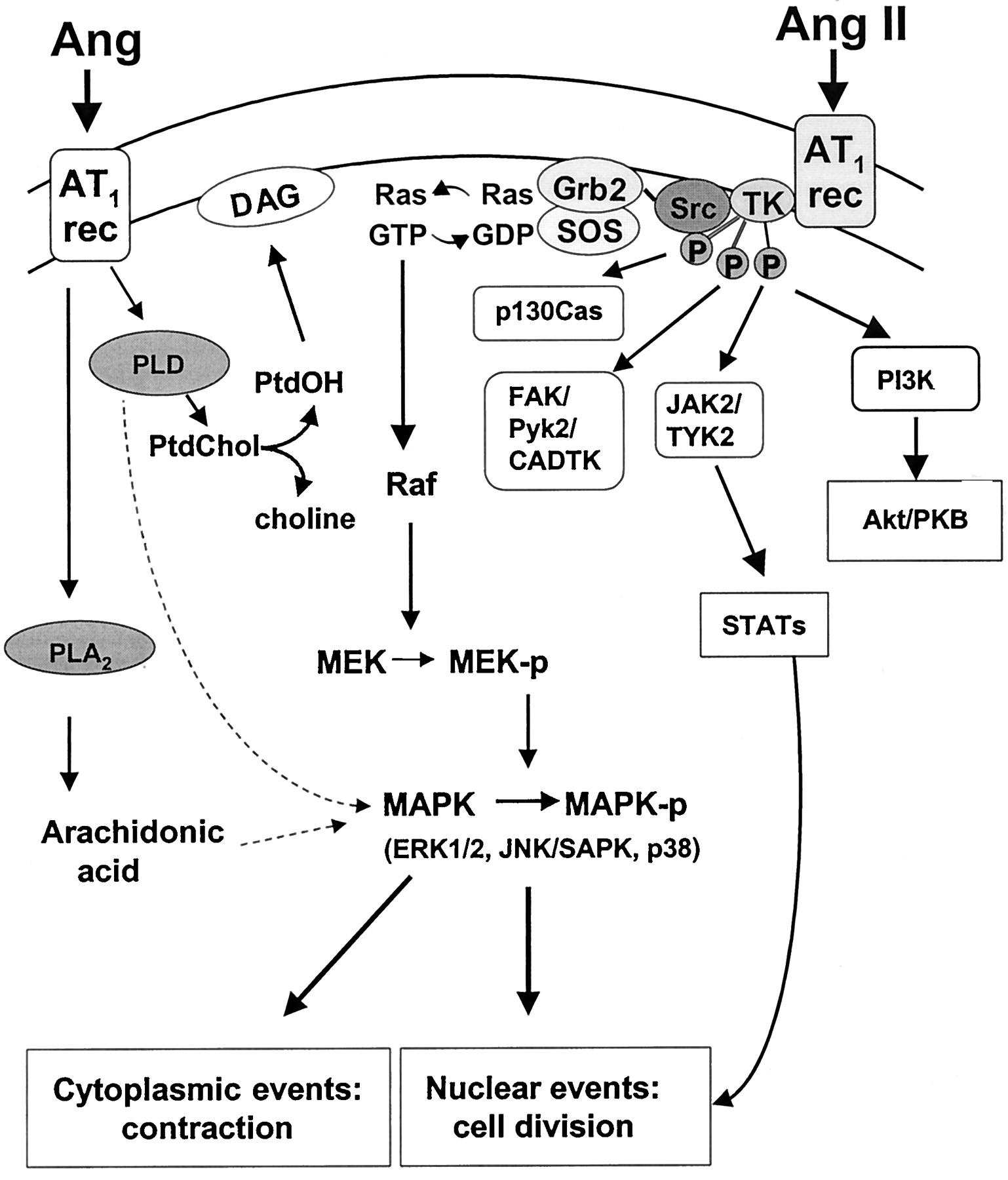
Early signaling events mediated by Ang II in vascular smooth muscle cells. Ang II phosphorylates multiple tyrosine kinases (TK) such as Janus family kinases (JAK/TYK), focal adhesion kinases (FAK and Pyk2), p130Cas and phosphatidylinositol 3-kinase (PI3K), within minutes of stimulation. Activated tyrosine kinases phosphorylate many downstream targets including the mitogen-activated protein kinase cascade (detailed in Figs. 7 and 8). Src associates with the adapter protein complex, Shc-GRB2-Sos that induces guanine nucleotide exchange on the small G protein Ras-GDP/GTP. Activated Ras-GTP interacts with Raf (MAPK kinase kinase) resulting in phosphorylation of two serine residues present in MEK (MAPK/ERK kinase) which, in turn, phosphorylates MAPKs, including ERK1/2, JNK/SAPK, and p38. Ang II also activates PLD, a major source of DAG, and phospholipase A2, which induces arachidonic acid production. PLD- and PLA2-dependent signaling pathways may also activate MAPKs. Ptd, phosphatidylcholine; PtdOH, phosphatidic acid.
1. Activation of Tyrosine Kinases.
Ang II stimulates phosphorylation of a tyrosine residue of many vascular smooth muscle cell proteins. These include the AT1 receptor itself, PLC-γ1 and Src family kinases (activated within seconds), as well as JAK and TYK, FAK, Pyk2, p130Cas (a Crk-associated substrate), and phosphatidylinositol 3-kinase (PI3K), all of which are activated within minutes (Fig.6). The role of tyrosine kinases in Ang II-mediated signal transduction pathways in cardiovascular cells was extensively reviewed in 1997 (Marrero et al., 1995a; Berk et al., 1997;Berk and Corson, 1997; Dostal et al., 1997; Griendling et al., 1997). Only recent developments relating to Ang II signaling and tyrosine kinases will be discussed in detail here.

Tyrosine kinase pathways stimulated by Ang II in vascular smooth muscle cells. Ang II rapidly activates Src, which regulates PLC-γ- and ERK-dependent signaling pathways. Ang II binding to the AT1 receptor induces the physical association and activation of JAK2/TYK2 (Janus kinases) as indicated by dashed line. JAK2/TYK2 phosphorylates STAT proteins that are translocated to the nucleus where they activate gene transcription. Ang II also activates FAK, which possesses sites favored for phosphorylation by Src. FAK associates with paxillin and talin that associate with actin. The link between AT1 receptor and FAK is unknown, but the Rho family of GTPases are potential candidates. Pyk2 and CADTK are activated by Ang II through Ca2+-dependent pathways. Activated Pyk2 regulates Src and ERK-dependent signaling cascades. p130casis transiently activated by Ang II, possibly via a Ca2+-dependent pathway. Phosphorylated p130casmay be important in the regulation of α-actin expression. PI3K activation by Ang II leads to Akt/PKB activation, which in turn stimulates cell survival pathways and activation of p70S6K. p70S6K, p70 S6-kinase.
a. Janus family kinases.
Similar to classical cytokine receptors, the AT1 receptor stimulates tyrosine phosphorylation of the Janus family kinases (Jak1, Jak2, Jak3, and Tyk2) (Ihle, 1995; Dostal et al., 1997). In vascular smooth muscle cells, Ang II binding to the AT1 receptor induces the physical association and activation of Jak2. Jak2 must be catalytically active to form a complex with the AT1 receptor, and this process appears to be regulated by an Ang II-mediated autophosphorylation event (Ali et al., 1998). JAK proteins are key mediators of mRNA expression and are characterized as “early growth response genes”. JAK phosphorylates STAT proteins that are translocated to the nucleus, where they activate gene transcription (Horvath and Darnell, 1997) (Fig. 6). In cardiovascular cells, Jak2 and Tyk2 are phosphorylated within 5 min of Ang II stimulation (Marrero et al., 1995a; Dostal et al., 1997). STAT1 and STAT2 phosphorylation in response to Ang II is maximal by ∼15 min, while STAT5 is activated within 30 to 60 min, and STAT3 phosphorylation is only detectable after ∼60 min (Marrero et al., 1995a; Kodama et al., 1998; McWhinney et al., 1998). Electroporation of antibodies against STAT1 and STAT3 abolished vascular smooth muscle cell proliferative responses to Ang II but not to other growth factors, implicating an essential role of STAT proteins in Ang II-induced cell proliferation (Marrero et al., 1997). The JAK-STAT signaling pathway activates early growth response genes and may be a mechanism whereby Ang II influences vascular and cardiac growth, remodeling, and repair (Berk and Corson, 1997; Hefti et al., 1997).
b. Focal adhesion kinase and proline-rich tyrosine kinase 2.
Ang II promotes cell migration and induces changes in cell shape and volume by activating FAK-dependent signaling pathways (Howe et al., 1998). Similar to integrin receptors, the AT1 receptor also activates FAK (Leduc and Meloche, 1995). Focal adhesion complexes, specialized sites of cell adhesion, act as supramolecular structures for the assembly of signal transduction mediators. The best characterized tyrosine kinase localized to focal adhesion complexes is a 125-kDa protein, FAK (Guan, 1997). FAK is autophosphorylated at Tyr397 in resting substrate-attached cells, and it possesses sites favored for phosphorylation by Src (Calalb et al., 1995). FAK associates with paxillin and talin, and both FAK and paxillin can bind to the cytoplasmic tail of integrins independently (Chen et al., 1995a; Leduc and Meloche, 1995) (Fig. 6). FAK is abundant in developing blood vessels, and elevation of its phosphotyrosine content in vascular smooth muscle cells is a rapid response to Ang II (Polte et al., 1994;Okuda et al., 1995). Ang II-induced activation of FAK causes its translocation to sites of focal adhesion with the extracellular matrix and phosphorylation of paxillin and talin, which may be involved in the regulation of cell morphology and movement. The link between the AT1 receptor and FAK is unknown, but the Rho family of GTPases are potential candidates (Rozengurt, 1995;Aspenstrom, 1999).
A novel p125FAK protein, calcium-dependent tyrosine kinase (CADTK), has recently been detected in rat aortic smooth muscle cells. CADTK is the rat homolog of Pyk2 (Yu et al., 1996). This nonreceptor tyrosine kinase is rapidly tyrosine-phosphorylated by Ang II, and appears to be associated with the cytoskeleton (Brinson et al., 1998). CADTK is localized to and activated by an actin cytoskeleton-dependent mechanism that is regulated in a Ca2+ and PKC-dependent manner, independently of FAK (Brinson et al., 1998). CADTK and FAK exhibit different modes of activation. Activation of CADTK is highly correlated with the stimulation of c-Jun N-terminal kinase (JNK) activity, rather than with ERK activity, as is the case for FAK (Yu et al., 1996).
Another FAK family member, Pyk2 (Lev et al., 1995), also called cell adhesion kinase-β (Sasaki et al., 1995), related adhesion focal tyrosine kinase (Avraham et al., 1995) and CADTK (Yu et al., 1996;Guan, 1997), is activated by G protein-coupled receptors, including the AT1 receptor, as well as by PKC stimulation and increased intracellular Ca2+ (Marasawa, 1998b; Murasawa et al., 1998b; Eguchi et al., 1999a). The AT1 receptor uses Ca2+-dependent PYK2 to activate c-Src, required for Pyk2-mediated ERK activation (Eguchi et al., 1999a). Since Pyk2 is a candidate to both regulate c-Src and to link G protein-coupled vasoconstrictor receptors with protein tyrosine kinase-mediated contractile, migratory, and growth responses, it may be a potential point of convergence between Ca2+-dependent signaling pathways and protein tyrosine kinase pathways in vascular smooth muscle cells (Dikic et al., 1996). In endothelial cells the balance of Pyk2 tyrosine phosphorylation in response to Ang II is controlled by Yes kinase and by a tyrosine phosphatase SHP-2 (Tang et al., 2000).
c. p130Cas.
p130Cas is an Ang II-activated tyrosine kinase that plays a role in cytoskeletal rearrangement. This protein serves as an adapter molecule because it contains proline-rich domains, an SH3 domain, and binding motifs for the SH2 domains of Crk and Src (Fig. 6). p130Casis important for integrin-mediated cell adhesion, by recruitment of cytoskeletal signaling molecules such as FAK, paxillin, and tensin to the focal adhesions (Rozengurt, 1995; Carey et al., 1998). In cultured vascular smooth muscle cells, Ang II induces a transient increase in p130Cas tyrosine phosphorylation, that peaks at ∼20 min after the addition of Ang II (Sayeski et al., 1998). Some investigators have found this phosphorylation to be dependent on Ca2+, c-Src, and PKC, and that it requires an intact cytoskeletal network (Sayeski et al., 1998). Other studies reported that Ang II-induced activation of p130Cas is Ca2+- and PKC-independent (Takahashi et al., 1998). Although the exact functional significance of Ang II-induced activation of p130Cas is unclear, it might regulate α-actin expression, cellular proliferation, migration, and cell adhesion (Nojima et al., 1995; Carey, 1998; Nakamura et al., 1998). p130Cas has recently been demonstrated to play a critical role in cardiovascular development and actin filament assembly. Mice lacking p130Cas died in utero showing marked venous congestion and growth retardation (Honda et al., 1998). Histologically, the heart was poorly developed and blood vessels were prominently dilated (Honda et al., 1998). Thus, p130Cas plays an essential role in arterial and cardiac development, and accordingly in remodeling in cardiovascular disease.
d. Phosphatidylinositol 3-kinase.
PI3Ks, a large family of intracellular signal transducers that phosphorylate inositol lipids at the 3′ position of the inositol ring to generate the 3-phosphoinositides PI(3)P, PI(3,4)P2 and PI(3,4,5)P3, are heterodimeric proteins composed of 85- and 110-kDa subunits (Leevers et al., 1999). These kinases influence cell survival, metabolism, cytoskeletal reorganization, and membrane trafficking and have recently been identified to play an important role in the regulation of vascular smooth muscle cell growth (Saward and Zahradka, 1997; Leevers et al., 1999). PI3K, characteristically associated with tyrosine kinase receptors, is also activated by AT1 receptors (Saward and Zahradka, 1997). In vascular smooth muscle cells, Ang II stimulates activity, phosphorylation, and migration of PI3K, and induces translocation of the p85 subunit from the perinuclear area to foci throughout the cytoplasm and the cytoskeletal apparatus (Saward and Zahradka, 1997). The action of Ang II peaks at 15 min and returns to control levels by 30 min. PI3K inhibition by wortmannin and LY294002 completely blocks Ang II-stimulated hyperplasia in cultured rat cells, suggesting the important regulatory role of this nonreceptor tyrosine kinase in vascular smooth muscle cell growth (Saward and Zahradaka, 1997). Several molecular targets for PI3K have been identified, including centaurin, the actin-binding protein profilin, phosphoinositide-dependent kinases, the atypical PKCs, PLCγ, Rac1, and JNK and the protein Ser/Thr kinase Akt/protein kinase B (PKB) (Wymann and Pirola, 1998). Akt/PKB has recently been identified as an important PI3K downstream target in Ang II-activated vascular smooth muscle cells (Takahashi et al., 1999). It regulates protein synthesis by activating p70 S6-kinase (p70S6K) (Eguchi et al., 1999b), and it modulates Ang II-mediated Ca2+ responses in aortic cells by stimulating Ca2+ channel currents (Seki et al., 1999). Akt/PKB has also been implicated to protect vascular smooth muscle cells from apoptosis and to promote cell survival by influencing Bcl-2 and c-Myc expression and by inhibiting caspases (Coffer et al., 1998). Mechanisms whereby the AT1 receptor mediates activation of PI3K-dependent Akt/PKB are unclear, but redox-sensitive pathways and c-Src may be important (Thomas, 1997; Ushio-Fukai et al., 1999b). Although the exact role of PI3K in Ang II signaling in vascular smooth muscle cells has not yet been established, it is possible that this complex pathway may control the balance between mitogenesis and apoptosis, a fundamental process in the regulation of vascular structure in health and disease.
2. Mitogen-Activated Protein Kinase Pathways.
MAP kinases constitute a superfamily of serine/threonine protein kinases involved in the regulation of a number of intracellular pathways. Mammalian MAPKs are grouped into six major subfamilies: a) ERK-1/ERK-2; b) JNK/stress-activated protein kinases (SAPK); c) p38; d) ERK-6, p38-like MAPK; e) ERK-3; and f) ERK-5 (also called Big MAP kinase 1) (Robinson and Cobb, 1997) (Fig. 7). MAP kinase-dependent signaling pathways have been associated with cellular growth and apoptosis, with cellular differentiation and transformation and with vascular contraction (Mii et al., 1996; Force and Bonventre, 1998; Touyz et al., 1999b,c). The ERKs are activated in response to growth and differentiation factors, whereas JNKs and p38 are usually activated in response to inflammatory cytokines and cellular stress (Robinson and Cobb, 1997; Force and Bonventre, 1998; Morinville et al., 1998; New and Han, 1998; Ip and Davis, 1998). Ang II activates the three major members of the MAP kinase family, ERKs, JNKs, and p38 (Leduc and Meloche, 1995; Kudoh et al., 1997; Touyz et al., 1999d). MAP kinase pathways comprise a three-component protein kinase cascade consisting of a serine/threonine protein kinase (MAPKKK), which phosphorylates and activates a dual-specificity protein kinase (MAPKK), which in turn phosphorylates and activates another protein kinase (MAPK) (Cobb and Goldsmith, 1995; Robinson and Cobb, 1997). In the Ras/Raf/MEK/ERK pathway, Raf corresponds to MAPKKK, MEK corresponds to MAPKK, and ERK corresponds to MAPK (Fig. 7).

Schematic diagram of the currently known mammalian MAP kinase signaling pathways. Individual components are discussed in the text. The question marks denote the signaling components that remain to be elucidated. PAK, p21-activated protein kinase; TAK, TGF-β-activated kinase; TAO, thousand and one amino acid kinase; BMK, big MAP kinase
a. Upstream events.
Activation of ERKs requires dual phosphorylation on threonine and tyrosine residues found within the motif Thr-Glu-Tyr, that is mediated by MEK (Fig.8). MEK in turn is regulated by serine (and probably tyrosine) phosphorylation by the kinase c-Raf-1, although Raf-independent pathways for ERK activation have also been demonstrated (Chao et al., 1994; Force and Bonventre, 1998). Raf is regulated by phosphorylation of Raf-1 kinase, as well as by recruitment to the plasma membrane by the small molecular weight guanine-nucleotide-binding protein, p21ras(Robinson and Cobb, 1997). The regulation of p21ras is complex, involving various adapter proteins and guanine-nucleotide exchange factors (Touhara et al., 1995;Schieffer et al., 1996a). Ligand binding to tyrosine kinase receptors stimulates autophosphorylation of the receptor, which then binds the SH2 domain of the adapter protein, Grb2. Grb2 is complexed to the guanine nucleotide factor, mammalian son-of-sevenless (Sos), that then stimulates the exchange of GDP for GTP on p21ras(Wang and McWhirter, 1994; Marshall, 1996). Tyrosine kinase receptors therefore utilize tyrosine phosphorylation to connect receptor activation to the p21ras cascade. G protein-coupled receptors, such as AT1, lack intrinsic tyrosine kinase activity but also activate p21ras (Sadoshima and Izumo, 1997; Zou et al., 1998). Although the exact mechanisms of AT1-activation of p21rasare unclear, activation might occur via G protein βγ subunits, by a receptor-associated tyrosine kinase or by tyrosine phosphorylation of a linker protein, such as Shc (Crespo et al., 1994; Apostolidis and Weiss, 1997; Berk and Corson, 1997; Schieffer et al., 1997). Activity of ERKs is modulated by MAP kinase phosphatase (MKP-1), a dual-specificity protein tyrosine phosphatase that exhibits catalytic activity toward phosphotyrosine and phosphothreonine on MAP kinases. In vascular smooth muscle cells, MKP-1 (the human homolog is CL100, 97% identity), dephosphorylates and inactivates ERK, JNK/SAPK, and p38 MAP kinase (Liu et al., 1995; Bokemeyer et al., 1998). Termination of ERK activation may also be mediated through a feedback loop, implicating Ras/Raf-mediated suppression of MAP kinase activation (Hughes et al., 1997).
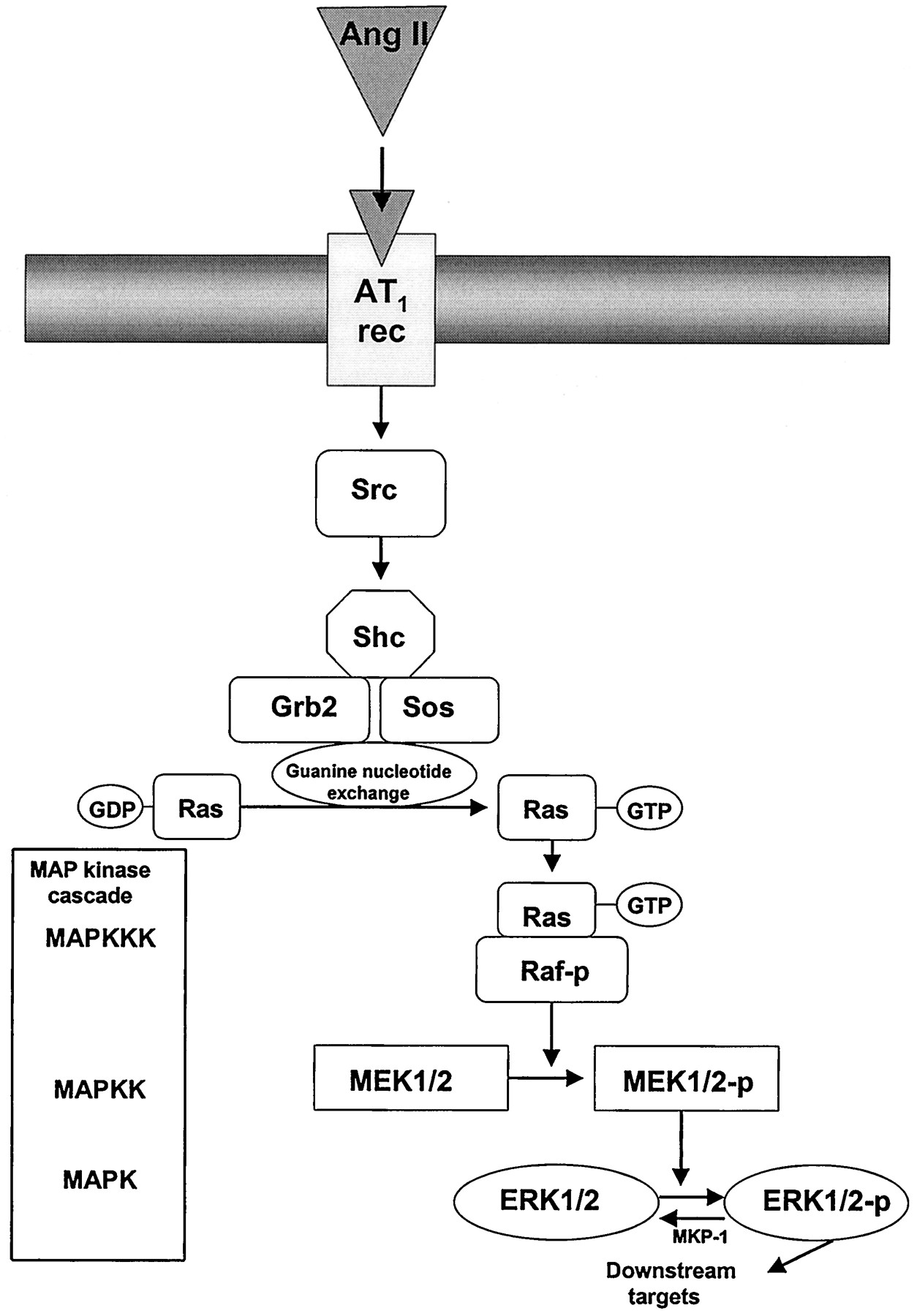
Upstream regulators of Ang II-stimulated extracellular signal-regulated kinase (ERK)-dependent signaling pathways in vascular smooth muscle cells. The ERK phosphorylation cascade is initiated by Ang II-binding to AT1 receptors that induces Shc-Grb2-Sos formation (tyrosine phosphorylation of Shc), which in turn facilitates guanine nucleotide exchange on the small G protein Ras-GDP/GTP. Activated Ras-GTP interacts with the Ser/Thr kinase Raf (MAPK kinase kinase (MAPKKK)) which translocates to the cell membrane. Activation of Raf leads to phosphorylation of two serine residues present in MEK (MAPK/ERK kinase), which in turn phosphorylates Thr/Tyr and activates MAPK, present as a 44- (ERK-1) and a 42-kDa (ERK-2) isoform. Phosphorylated ERK has diverse intracellular protein targets, which it phosphorylates and activates (Fig. 9). Dephosphorylation of ERK is accomplished by activation of MAP kinase phosphatase-1 (MKP-1).
b. Downstream events.
Events downstream to MAP kinase activation are numerous and heterogeneous and include PLA2, cytoskeletal proteins, the MAPK-activated protein kinase 2 (MAPKAPK-2), and the p90rskprotein kinase, which can move to the nucleus and activate transcription factors (Morinville et al., 1998) (Fig.9). Once phosphorylated ERKs translocate to the nucleus to phosphorylate transcription factors and thereby regulate gene expression of cell cycle-related proteins (Treisman, 1996). Both ERK-1/ERK-2 and JNK/SAPK lead to ternary complex formation at the serum response element that is present on many gene promoters, and to increased transcriptional activity (Whitmarsh et al., 1995). Alternatively, phosphorylation of the translation regulator protein, PHAS-I (phosphorylated heat- and acid-stable protein) promotes the dissociation of the PHAS-I-eukaryotic initiation factor (eIF)-4E complex, normally tightly bound when PHAS-I is relatively underphosphorylated, releasing eIF-4E that will facilitate initiation of translation in the nucleus (Brunn et al., 1997). In vascular smooth muscle cells, another downstream target of ERK is the serine/threonine protein kinase pp90rsk, which phosphorylates the S6 ribosomal protein and stimulates protein synthesis (Berk and Corson, 1997). ERK-1/ERK-2 activation ultimately results in enhanced proto-oncogene expression, and activation of the AP-1 transcription factor and probably regulates cell cycle progression as well as protein synthesis in vascular smooth muscle cells (Watson et al., 1993). Ang II may also induce protein synthesis by an ERK-independent pathway in part via activation of the 70-kDa S6 kinase (Giasson and Meloche, 1995). Other downstream targets of MAP kinases include cyclooxygenase-2, the contractile regulatory protein h-caldesmon, the high-molecular weight form of caldesmon, myelin basic protein, microtubule-associated protein, Ca2+ channels, and the Na+/H+ exchanger (Adam et al., 1995; Bornfeldt et al., 1997; Kusuhara, 1998). The functional outcome of MAP kinase activation probably depends in part on the availability of downstream substrates.

Downstream effectors of activated ERK. Once phosphorylated, ERK activates various intracellular proteins. These substrates include: 1) proteins involved in transcriptional activation such as Elk-1, TAL 1, RNA polymerase II; 2) proteins involved in protein translation such as PHAS I; 3) structural proteins such as myelin basic protein (MBP), microtubule-associated protein (MAP) and caldesmon; and 4) secondary enzymes such as PLA2, S6 kinase, Ca2+ channels, Na+/H+exchanger, and MAP kinase-activated protein kinase (MAPKAPK2). Activation of these downstream proteins regulates cellular functions associated with cell growth and contraction.
c. Angiotensin II and the mitogen-activated protein kinase pathway in cardiovascular cells.
Ang II activates the MAP kinase signaling cascade at various intracellular levels. It induces tyrosine and threonine phosphorylation of ERK-1/ERK-2, JNK/SAPK, and p38 in cultured vascular smooth muscle cells, as well as in intact arteries (Schieffer et al., 1996a,b; Epstein et al., 1997; Touyz et al., 1999c,d). It stimulates phosphorylation of Ras, Raf, and Shc, and it increases activity of MEK kinase and MEK (Eguchi et al., 1996; Liao et al., 1996; Sadoshima and Izumo, 1996; Schieffer et al., 1996a;Griendling and Ushio-Fukai, 1997; Touyz et al., 1999c). In addition, Ang II increases activation of vascular Src and PYK2, potential links between the AT receptor and Ang II-induced ERK signaling in vascular smooth muscle cells (Ishida et al., 1998; Murasawa et al., 1998b; Eguchi et al., 1999a). MAP kinase activation by Ang II is transient, with a peak at 3 to 5 min. Activity remains elevated at suprabasal levels for at least 60 min (Eguchi et al., 1996;Touyz et al., 1999b). Ang II stimulates ERK-dependent pathways via AT1 receptors (Flesch et al., 1995; Booz and Baker, 1996; Kudoh, 1997; Touyz et al., 1999c) and is associated with increased expression of the early response genes c-fos,c-myc, and c-jun (Naftilan et al., 1989; Lyall et al., 1992), DNA synthesis, cell growth and differentiation, and cytoskeletal organization (Seewald et al., 1998; Touyz et al., 1999b). Both Ras/Raf-dependent and -independent pathways have been implicated in Ang II-stimulated MAP kinase activation and protein synthesis in cultured vascular smooth muscle cells (Liao et al., 1996; Takahashi, 1997a,b).
In addition to ERKs, Ang II activates JNK/SAPKs, which regulate vascular smooth muscle cell growth by promoting apoptosis or by inhibiting growth (Kudoh, 1997; Wen et al., 1997; Ip and Davis, 1998;Schmitz et al., 1998). Ang II phosphorylates JNK/SAPK via p21-activated kinase (αPAK), which is dependent on intracellular Ca2+ mobilization and on PKC activation (Schmitz et al., 1998). Following phosphorylation, the isoforms JNK-1 and JNK-2 translocate to the nucleus to activate a number of transcription factors, such as c-Jun, ATF-2, and Elk-1 (Ip and Davis, 1998). Ang II appears to activate vascular smooth muscle cell ERK-1/ERK-2 and JNK/SAPK via different signaling pathways. ERK phosphorylation occurs via a Ca2+-dependent or -independent pathway that involves c-Src and the atypical PKC isoform PKC-ξ (Liao et al., 1997), whereas JNK/SAPK activation occurs via a Ca2+-dependent pathway that involves a tyrosine kinase other than Src and a novel PKC isoform (Schmitz et al., 1998). Furthermore, whereas Ang II-induced phosphorylation peaks within 5 min, kinase activation is maximal at about 30 min (Kusuhara, 1998). The exact functional effects of Ang II-induced signaling of ERK-1/ERK-2 and JNK/SAPK in vascular smooth muscle cells are ill-defined, but regulation of cell growth may be important as Ang II-activated ERKs and JNK/SAPKs have opposite growth effects, with ERKs facilitative and JNK/SAPK inhibitory. These signaling processes and associated cellular functions are potentially important in enhanced vascular contractility, hyperplasia, and/or hypertrophy in hypertension (Schelling et al., 1991).
Recent studies demonstrated that Ang II also phosphorylates vascular p38 MAP kinase, which plays an important role in inflammatory responses, apoptosis and inhibition of cell growth (Kusuhara, 1998; New and Han, 1998; Ushio-Fukai et al., 1998b). In the cardiovascular system, the p38 pathway has been implicated in cardiac ischemia, ischemia/reperfusion injury, cardiac hypertrophy, progression of atherosclerosis, and arterial remodeling in hypertension (New and Han, 1998). The specific upstream and downstream regulators of Ang II-activated p38 in vascular smooth muscle cells are unclear, but p38 could be a negative regulator of ERK-1/ERK-2 (Kusuhara et al., 1998). p38 has been implicated to be an essential component of the redox-sensitive signaling pathways in Ang II-activated vascular smooth muscle cells (Ushio-Fukai et al., 1998b).
Inactivation of Ang II-stimulated MAP kinases occurs via MKP-1-induced dephosphorylation of both tyrosine and threonine on MAP kinases. Inhibition of MKP-1 results in sustained activation of MAP kinase in response to Ang II, suggesting that this enzyme is primarily responsible for the termination of the MAP kinase signal (Duff et al., 1993, 1995). In vascular smooth muscle cells, Ang II modulates MKP-1 activity. MKP-1 expression is stimulated by Ang II, and activities of MKP-1, as well as tyrosine phosphatase (PTP-1C), and Ser/Thr phosphatase PP2A, are increased by Ang II (Kambayashi, 1993; Bedecs et al., 1997; Horiuchi et al., 1997a). These effects appear to be mediated via the AT2 receptor subtype, which has been associated with inhibition of cell growth and apoptosis (Bedecs et al., 1997; Horiuchi, 1997a,b; Fischer et al., 1998). Accordingly, AT1 receptors induce growth via stimulation of ERK-dependent signaling pathways, whereas AT2 receptors oppose these effects by stimulating MKP-1 activity to inhibit ERK activity, and to arrest the cell growth signal. Termination of Ang II-stimulated MAP kinase activity may also involve activation of protein kinase A (PKA), which inhibits the phosphorylation of Raf-1 (Cook and McCormick, 1993).
3. Activation of Phospholipase A2 and Arachidonic Acid Metabolism.
Ang II stimulates PLA2activity, which is responsible for the release of arachidonic acid from cell membrane phospholipids (Bonventre, 1992; Rao et al., 1994). Released arachidonic acid is processed by cyclooxygenases, lipoxygenases, or cytochrome P450 oxygenases to many different eicosanoids in vascular and renal tissues (Fig. 5). Cyclooxygenases catalyze the formation of prostaglandin (PG) PGH2, subsequently converted to thromboxane (TXA) by thromboxane synthase, to PGI2 (or prostacyclin) by prostacyclin synthase, or to PGE2, PGD2 or PGF2α, by different enzymes (Smith et al., 1991). Lipoxygenases catalyze the formation of 5-, 12-, or 15-HPETEs, that then undergo spontaneous or peroxidase-catalyzed reduction to the corresponding HETEs, and in the case of 5-HPETE to leukotrienes (Yamamoto, 1992). Cytochrome 450 oxygenases catalyze arachidonic acid epoxidation to epoxyeicosatrieenoic acids, ω and ω-1 hydroxylation to 20- and 19-HETE, and allylic oxidation to other HETEs (Harder et al., 1995; Dennis, 1997).
PLA2-derived eicosanoids influence vascular and renal mechanisms important in blood pressure regulation (Nasjletti, 1997). Vascular PLA2 activity in response to Ang II is evident within minutes and is sustained for at least 30 min after Ang II stimulation (Rao et al., 1994). In vascular smooth muscle cells and endothelial cells, these effects are mediated via AT1 receptors (Pueyo et al., 1996; Freeman et al., 1998), whereas in neonatal rat cardiac myocytes, neuronal cells, and renal proximal tubule epithelial cells, Ang II-induced activation of PLA2 occurs via AT2receptors (Rogers and Lokuta, 1994; Lokuta et al., 1994; Dulin et al., 1998; Zhu et al., 1998b). Ang II-elicited activation of vascular PLA2 is dependent on [Ca2+]i, Ca2+-calmodulin-dependent protein kinase II (CaM kinase II), and MAP kinases (Muthalif et al., 1998a,b). Activated PLA2 and its metabolites in turn activate Ras/MAP kinase-dependent signaling pathways, amplifying PLA2 activity and releasing additional arachidonic acid by a positive feedback mechanism (Muthalif et al., 1998a). In renal epithelial cells, Ang II activates PLA2 via an AT2-mediated Ca2+-independent mechanism (Jacobs and Douglas, 1996; Becker et al., 1997). Renal-derived arachidonate phosphorylates the adaptor protein Shc and stimulates its association with Grb2 and Sos 1 (Dulin et al., 1998). Ang II-generated eicosanoids regulate vascular contraction and growth, possibly by activating MAP kinases and redox sensitive pathways (Nasjletti, 1997; Dulin et al., 1998). Thromboxanes are involved in Ang II-induced contraction, whereas vasorelaxant prostaglandins such as PGE2 and PGI2 attenuate Ang II-mediated vasoconstriction in some vascular beds (Wilcox and Lin, 1993). Lipoxygenase-derived eicosanoids also influence Ang II-mediated actions in vascular smooth muscle cells. 12-HETE facilitates the stimulatory actions of Ang II on Ca2+ transients in cultured cells. Lipoxygenase inhibitors attenuate the vasoconstrictor action of Ang II and decrease blood pressure in SHR (Stern et al., 1993; Oyekan et al., 1997). Some of these effects elicited by Ang II-generated arachidonic acid metabolites may be mediated via modulation of the oxidative state of the cell (Zafari et al., 1996).
4. Phospholipase D activation.
PLD, which hydrolyzes phospholipids (mainly phosphatidylcholine) to generate phosphatidic acid, is a critical component in cellular signaling associated with mitogenesis (Dhalla et al., 1997; Gomez-Cambronero and Kiere, 1998). Sustained activation of PLD is a major source of prolonged second messenger generation in vascular smooth muscle cells and cardiomyocytes. Unlike PLC, which is activated within seconds by Ang II, PLD activation is detectable at about 2 min and remains elevated for up to 60 min (Lassègue, 1993). In contrast to the PLC response, PLD activation does not appear to desensitize significantly during this time period (Lassègue, 1993). Hydrolysis of phosphatidylcholine by PLD leads to the production of phosphatidic acid and subsequent generation of DAG by phosphatidic acid phosphohydrolase (Billah, 1993) (Fig. 5). DAG contributes to prolonged activation of PKC. This pathway probably represents the major cascade by which Ang II-induced activation of PKC remains sustained in vascular smooth muscle cells. Molecular mechanisms coupling AT1receptors to PLD have recently been identified. G protein βγ subunits as well as their associated Gα12subunits mediate Ang II-induced PLD activation via Src-dependent pathways in vascular smooth muscle cells (Freeman, 1995; Ushio-Fukai et al., 1999b). The small molecular weight G protein RhoA is also involved in these signaling cascades (Exton, 1997). The downstream pathways associated with Ang II-induced activation of PLD in vascular smooth muscle cells are PKC-independent (Freeman et al., 1995) but involve intracellular Ca2+ mobilization (Freeman et al., 1995) and Ca2+ influx that is tyrosine kinase-dependent (Suzuki et al., 1996). Ang II-induced PLD signaling has been implicated in cardiac hypertrophy as well as in proliferation of vascular smooth muscle cells (Morton et al., 1995; Dhalla, 1997). PLD-dependent signaling cascades also influence cardiac and vascular contraction (Xu et al., 1996b). These effects are mediated via phosphatidic acid and other PLD metabolites (Boarder, 1994; Wilkie et al., 1996; Dhalla, 1997) that influence vascular generation of superoxide anions by stimulating NADH/NADPH oxidase (Griendling et al., 1994; Gomez-Cambronero and Kiere, 1998; Ushio-Fukai et al., 1998b), that activate tyrosine kinases and Raf and that modulate intracellular Ca2+ signaling (Boarder, 1995; Eskildsen-Helmond et al., 1997; Gomez-Cambronero and Kiere, 1998). The long-term signaling pathways associated with Ang II-stimulated growth and remodeling in the cardiovascular system are dependent, in part, on PLD-mediated responses.
5. Angiotensin II Effects on Cyclic Nucleotides.
The cyclic nucleotides cAMP and cGMP are generated intracellularly within minutes by adenylate cyclase and guanylate cyclase, respectively, via a cyclasing reaction of α-phosphate and release of pyrophosphate from the substrates ATP or GTP in the presence of Mg2+. Downstream targets of cyclic nucleotides include cAMP-dependent protein kinase, cGMP-dependent protein kinase, intracellular Ca2+, and ionic channels (Bentley and Beavo, 1992). Increased cyclic nucleotide concentration leads to decreased [Ca2+]i and reduced Ca2+ sensitivity of phosphorylation in vascular smooth muscle, with resultant smooth muscle relaxation (Brophy et al., 1997; Frings, 1997). Ang II influences vascular dilation either directly, by increasing intracellular cAMP and cGMP concentrations, or indirectly, by potentiating vasodilator-induced formation of cyclic nucleotides. Ang II stimulation increases cAMP and/or cGMP production in cardiomyocytes, vascular smooth muscle cells, and mesangial cells, as well as in intact arteries (Boulanger et al., 1995; Magness et al., 1996; Siragy and Carey, 1997; Gohlke et al., 1998). These effects involve kinin-dependent mechanisms mediated via receptors of the AT2 subtype (Siragy and Carey, 1997; Gohlke et al., 1998). In rat carotid arteries Ang II increases release of nitric oxide and cGMP production via endothelial AT1receptors (Boulanger et al., 1995; Caputo, 1995). Ang II also induces vasorelaxation by enhancing the vasodilatory effect of agonists such as isoproterenol (McCumbee et al., 1996; Brizzolara-Gourdie and Webb, 1997; Mokkapatti et al., 1998). In pathological conditions, AT2 receptor stimulation is associated with reduced vascular cGMP levels (Moroi et al., 1997). The vasodilatory effects of Ang II linked to the AT2receptor oppose the vasoconstrictory actions of Ang II linked to the AT1 receptor. Cross-talk between these pathways could represent an important mechanism in the modulation of Ang II-regulated vascular tone.
G. Long-Term Effects Mediated by Angiotensin II
Ang II influences the long-term control of cellular growth, adhesion, and migration, as well as intercellular matrix deposition within the vasculature and the heart thereby influencing chronic adaptive changes in vascular remodeling, cardiac hypertrophy, as well as processes involved in atherosclerosis. Intracellular cascades underlying long-term Ang II signaling involve early activation of various kinases (discussed above) that phosphorylate downstream targets regulating chronic and sustained cellular functions. Stimulation of redox-sensitive pathways, induction of proto-oncogene expression, cross-talk with tyrosine kinase receptors, production of other growth factors and stimulation of nuclear signaling cascades ultimately result in cellular growth and differentiaition (Fig.10).
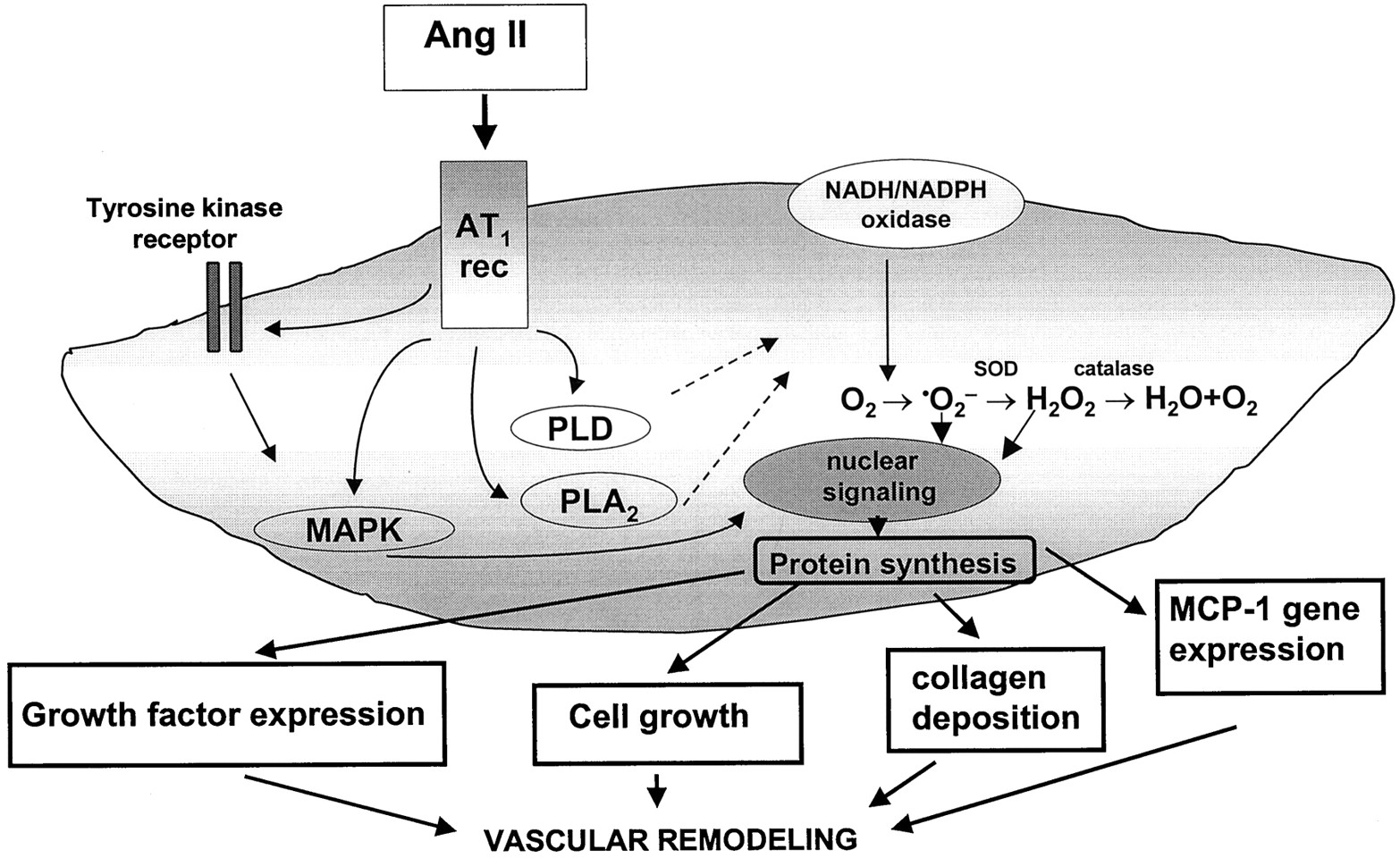
Long-term signaling events in vascular smooth muscle cells induced by Ang II stimulation. Activation of upstream regulators by Ang II, such as tyrosine kinases, MAP kinases, PLD, and PLA2 lead to activation of various signaling pathways that modulate long-term functions of vascular smooth muscle cells. These include generation of reactive oxygen species via membrane-associated NADH/NADPH oxidase, induction of proto-oncogene expression, cross-talk with tyrosine kinase receptors, stimulation of nuclear signaling cascades and production of other growth factors. The biological response of these signaling events is increased protein synthesis resulting in cell growth that contributes to vascular remodeling.⋅O⨪2, superoxide anion; H2O2, hydrogen peroxide; SOD, superoxide dismutase.
1. Generation of Reactive Oxygen Species.
Reactive oxygen species such as superoxide anions and hydrogen peroxide act as intercellular and intracellular second messengers that may play a physiological role in vascular tone and cell growth, and a pathophysiological role in inflammation, ischemia-reperfusion, hypertension, and atherosclerosis (Alexander, 1995; Irani et al., 1997;Diaz et al., 1997; Griendling and Ushio-Fukai, 1997; Finkel, 1998; Abe and Berk, 1998). Xanthine oxidase, mitochondrial oxidases and arachidonic acid are the major sources of oxidative molecules in nonvascular tissue (Finkel, 1998), whereas a nonmitochondrial, membrane-associated NADH/NADPH oxidase appears to be the most important source of superoxide anion (O⨪2) in vascular cells (Griendling et al., 1994; Rajagopalan et al., 1996; Pagano et al., 1998; Lieberthal et al., 1998). This enzyme transfers electrons from NADH or NADPH to molecular oxygen, producing superoxide anion (Fig.11). The complete molecular structure of the vascular oxidase is unknown, but it shares some features with the neutrophil oxidase. In neutrophils, NADH/NADPH oxidase consists of five subunits: a 22-kDa α-subunit (p22phox), a glycosylated 91-kDa β-subunit (gp91phox), which together make up cytochrome b558, the electron transfer element; cytosolic components p47phoxand p67phox; and a low-molecular weight G protein, rac1 or rac2 (Jones, 1994). Upon activation, the p47phox and p67phoxproteins are translocated to the membrane and associate with the cytochrome b558, creating the active oxidase. In vascular smooth muscle cells, p22phox is a critical component of the superoxide-generating NADH/NADPH oxidase system (Ushio-Fukai et al., 1996). Ang II activation of NADH/NADPH oxidase is delayed and is only detectable in vascular smooth muscle cells about 60 min after Ang II stimulation (Griendling et al., 1994;Touyz and Schiffrin, 1999b). The effect is sustained for up to 24 h, suggesting that NADH/NADPH oxidase-dependent signaling pathways probably play an important role in Ang II-mediated long-term signaling events such as cell growth. In support of this, when NADH/NADPH oxidase is inhibited by the selective inhibitor diphenylene iodinium (DPI), Ang II-stimulated protein synthesis in vascular smooth muscle cells is also inhibited (Griendling et al., 1994; Ushio-Fukai et al., 1998b). The O⨪2 that is generated by NADH/NADPH oxidase is rapidly converted by superoxide dismutase to H2O2, which is scavenged by catalase or by peroxidases (Fridovich, 1997) (Fig. 10). O⨪2 and H2O2 can undergo further reactions with each other or with iron-containing molecules to generate the highly reactive hydroxyl radical (⋅OH) (Fridovich, 1997).

Generation of reactive oxygen species in the vasculature. Many enzyme systems stimulate production of superoxide anion (⋅O⨪2) from O2. These include NADH/NADPH oxidase, xanthine oxidase, lipoxygenase, cyclooxygenase, P450 monooxygenase, and mitochondrial oxidative phosphorylation. NADH/NADPH is a multi-subunit enzyme that is the major regulated source of reactive oxygen species in endothelial and vascular smooth muscle cells. Dismutation of ⋅O⨪2 spontaneously or enzymatically by superoxide dismutase (SOD) produces hydrogen peroxide (H2O2) that can undergo further reactions to generate the highly reactive hydroxyl radical (⋅OH). H2O2 may be metabolized by catalase or peroxidases to H2O and O2. Downstream targets of ⋅O⨪2 and H2O2 include ERK5, p38, tyrosine kinases, Src, and NF-κB.
Generation of reactive oxygen species is regulated by various cytokines and growth factors, including Ang II, which increases O⨪2 and H2O2 production in cardiac, vascular smooth muscle, endothelial, adventitial, and mesangial cells (Griendling et al., 1994; Jaimes et al., 1998; Pagano et al., 1998;Ushio-Fukai et al., 1998b; Touyz and Schiffrin, 1999b) and generation of reactive oxygen species has been implicated in the pathogenesis of Ang II-induced but not catecholamine-induced hypertension (Rajagopalan et al., 1996; Laursen, 1997). Mechanisms underlying oxidative stress-induced hypertension may be associated with degradation of endothelium-derived NO and with the potent vascular mitogenic effects of O⨪2 and H2O2 (Rao and Berk, 1992;Ushio-Fukai et al., 1996; Oskarsson and Heistad, 1997; Lu et al., 1998;McIntyre et al., 1999). Growth of vascular smooth muscle cells has an essential redox-sensitive component, which appears to be mediated in part via activation of ERK-5 (Abe et al., 1996). Reactive oxygen species stimulate hyperplasia and hypertrophy of vascular smooth muscle cells, whereas antioxidants inhibit growth, trigger apoptosis, and attenuate the response to growth factors and hypertrophic agents (Boscoboinik et al., 1991; Rao and Berk, 1992; Puri et al., 1995; Tsai et al., 1996). Ang II-mediated oxidative stress has recently been shown to stimulate endothelial vascular cell adhesion molecule-1, important in cell-cell interactions, and possibly in processes associated with atherosclerosis (Pueyo et al., 2000). The signaling pathway linking Ang II-stimulated generation of H2O2 to vascular growth has recently been identified as p38 MAP kinase (Ushio-Fukai et al., 1998b). Although ERK-5 is a redox-sensitive kinase, Ang II does not appear to mediate its oxidative stress-induced growth effects via this MAP kinase (Abe et al., 1996). Another redox-sensitive cascade whereby Ang II influences cell growth is through phosphorylation of the cell survival protein kinase Akt/PKB (Ushio-Fukai et al., 1999b).
2. Angiotensin II-Induced Expression of Proto-Oncogenes and Growth Factors.
Long-term control of Ang II-regulated cellular growth, adhesion, migration, fibrosis, and collagen deposition within the vasculature involves protein synthesis (Fig. 10). Ang II induces the expression of several proto-oncogenes in human and rat vascular smooth muscle cells, including c-fos, c-jun, c-myc, erg-1, VL-30, proto-oncogene/activator protein 1 complex (Lyall et al., 1992; Grohé et al., 1994; Duff et al., 1995; Pollack, 1995; Puri et al., 1995; Patel et al., 1996). Ang II increases expression of vascular c-fos in a PKC- and Ca2+-dependent manner via multiple regulatory mechanisms (Garcia-Sainz et al., 1995; Chen et al., 1996). The c-fos promoter contains a cAMP/calcium response element (CRE), a serum response element (SRE), and a sis-inducing factor element (SIE) (Bhat et al., 1994). These promoter elements are regulated by various proteins activated by Ang II, including cAMP and PKA, which regulate CRE, MAPK-stimulated phosphorylation of p62TCF and PKC, which regulate SRE, and STATs, which regulate SIE (Marrero et al., 1995a). Stimulation of early response genes by Ang II is associated with increased gene expression and production of growth factors, such as PDGF, EGF, transforming growth factor-β (TGF-β), insulin-like growth factor-1 (IGF-1), basic fibroblast growth factor (bFGF) and platelet activating factor (PAF) (Dubey, 1997; Force and Bonventre, 1998), vasoconstrictor agents, such as ET-1 (Itoh et al., 1993), adhesion molecules such as ICAM-1, VCAM-1, and E-selectin, and integrins ανβ3 and β5 (Kim et al., 1996; Krejcy et al., 1996;Grafe et al., 1997; Hsueh et al., 1998), and finally, chemotactic factors such as tumor necrosis factor-α (TNF-α) and monocyte chemoattractant protein-1 (MCP-1) (Chen et al., 1998).
Ang II is a powerful mitogen for many cell types and a potent competence and/or progression factor, stimulating transition from the Go-Gi phase in the cell cycle, which leads to increased DNA synthesis in certain conditions and to mitogenesis in combination with other growth factors (Owens et al., 1981; Gibbons et al., 1992; Jahan et al., 1996). Ang II induces hypertrophy and/or hyperplasia of vascular cells, both in vivo (Li et al., 1998a; Levy, 1998; Schiffrin et al., 2000) and in vitro (Touyz and Schiffrin, 1997a,b) and is a potent stimulus for collagen and fibronectin production (Kaiura et al., 2000). These effects may be direct, via activation of ERK-1/ERK-2-dependent pathways and by activation of 70-kDa S6 kinase, an ERK-independent pathway, or indirectly, by increasing the local production of TGF-β, PDGF, bFGF, ET-1, IL-6, PAF, IGF-1, heparin-binding EGF, and osteopontin (Gomez-Garre et al., 1996; Dubey, 1997; Mangiarua et al., 1997; Schott et al., 1997; Border and Noble, 1998). TGF-β and PDGF may play pivotal roles in the vascular growth effects of Ang II. In human and rat vascular smooth muscle cells, Ang II up-regulates TGF-β mRNA levels and increases production of TGF-β through the AP-1 complex (Liu et al., 1997; Morishita et al., 1998; Fukai, 2000). Ang II-stimulated hyperplasia is significantly increased in the presence of TGF-β antibodies, whereas in their absence, Ang II induces hypertrophy of vascular smooth muscle cells (Itoh et al., 1993; Dubey, 1997). TGF-β also contributes to the fibrogenic and migratory actions of Ang II. In vascular smooth muscle and mesangial cells, Ang II time and dose dependently increase TGF-β mRNA, which is associated with increases in mRNAs for matrix proteins biglycan, fibronectin, and collagen type 1 (Border and Noble, 1998). In the presence of neutralizing antibody to TGF-β, matrix protein production is almost completely blocked, indicating that Ang II-stimulated increases in extracellular matrix production are mediated in large part by TGF-β (Kagami et al., 1994). In human and rat vascular smooth muscle cells, Ang II induces a bimodal migratory effect where both migratory and antimigratory pathways are activated. Ang II directly stimulates migration in a concentration-dependent manner whereas autocrine release of TGF-β1 induced by Ang II has an antimigratory action (Liu et al., 1997).
PDGF-A mRNA expression and PDGF-A secretion associated with increased expression of c-fos and c-myc and augmented cell growth, are enhanced by Ang II (Naftilan et al., 1989; Linesman et al., 1995; Mangiarua et al., 1997). Interestingly, Ang II exerts a transient inhibitory effect on PDGF-BB-induced DNA synthesis, and reduces vascular smooth muscle cell proliferation (Dahlfors et al., 1998). The interaction between Ang II and PDGF is complex, as the growth response induced by Ang II-mediated PDGF is dependent on the form of PDGF produced. The homodimer AA is a less potent mitogen than its AB or BB counterparts. Other growth factors that also play a role in Ang II-stimulated vascular growth include IGF, EGF, and bFGF. Ang II increases IGF-1 receptor mRNA levels and IGF-1 receptor gene transcription in vascular smooth muscle cells via PKC-independent pathways (Du et al., 1996). In addition, Ang II stimulates tyrosine phosphorylation and activation of insulin receptor substrate 1 and protein-tyrosine phosphatase 1D, suggesting the presence of a convergent intracellular signaling cascade that is stimulated by IGF-1 and Ang II (Ali et al., 1997). In large but not in small arteries, Ang II stimulates smooth muscle cells replication dependent on mediation by bFGF (Su et al., 1998). This differential response may be important in vessel wall remodeling in atherosclerosis and following balloon injury. In rat aortic vascular smooth muscle cells, Ang II increased FGF-2 but not FGF-1 mRNA levels (Peifley and Winkles, 1998). EGF also induces growth actions of Ang II, but these effects appear to be mediated via Ang II-induced transactivation of the EGF receptor by a PKC-independent Ca2+/calmodulin-dependent pathway (Murasawa et al., 1998a). Besides growth factors, mechanical stretch and collagen potentiate the mitogenic activity of Ang II in vascular smooth muscle cells. This synergy is blocked by antibodies to PDGF-BB and TGF-β (Hsueh et al., 1995; Li et al., 1998b,c). Ang II also stimulates production of chemotactic factors that may be important in vascular inflammatory processes associated with cardiovascular diseases. Ang II stimulates monocyte chemoattractant protein-1 gene expression in rat vascular smooth muscle cells (Chen et al., 1998) and in renal tissue, Ang II increases expression of TNF mRNA and enhances production of TNF 5-fold (Ferreri et al., 1998).
Ang II controls growth by inhibiting cellular proliferation and hypertrophy and/or by inducing apoptosis (deBlois et al., 1997; Diep et al., 1999). In vascular smooth muscle cells, Ang II inhibits apoptosis via AT1 receptors, whereas it induces apoptosis via AT2 receptors (Pollman et al., 1996; Yamada et al., 1998; Horiuchi et al., 1999; Lemay et al., 2000). The AT2-mediated proapoptotic effects of Ang II have been demonstrated in vascular smooth muscle cells, neonatal cardiomyocytes, PC12W cells, R3T3 mouse fibroblasts, and human umbilical vein endothelial cells (Hayashida et al., 1996; Yamada et al., 1996; Dimmeler et al., 1997; Horiuchi et al., 1997a). The exact signaling pathways associated with these actions have not yet been fully identified, but inhibition of ERK activity, by MKP-1, may result in inactivation of Bcl-2, activation of caspases and the induction of apoptosis (Dimmeler et al., 1997; Horiuchi et al., 1997a;Yamada et al., 1998). The role of AT2-mediated actions has recently been extensively reviewed (Matsubara et al., 1998;Horiuchi et al., 1999) and the reader is referred to these reviews for more detailed information.
Ang II influences the architecture and integrity of the vascular wall by modulating cell growth and regulating extracellular matrix composition. It increases expression and production of fibronectin, collagen type 1, tenascin, glycosaminoglycans, chondroitin/dermatan sulfates, and proteoglycans, major constituents of the extracellular matrix in the vessel wall (Hsueh et al., 1995; Dubey et al., 1997). In vascular smooth muscle cells, mesangial cells and endothelial cells, Ang II increases levels and activity of plasminogen activator inhibitor-1 (PAI-1), influencing fibrinolysis, extracellular matrix turnover, and degradation and regulation of cell migration (Feener et al., 1995; Oikawa et al., 1997; Wilson et al., 1997; Yoshizumi et al., 1998). Some of these effects have been linked to the AT4 receptor subtype (Kerins et al., 1995). However this remains to be clarified. Ang II also stimulates activity of matrix metalloproteinases (Singhal et al., 1995) responsible for extracellular matrix degradation. Accordingly, Ang II influences vascular structure by stimulating synthesis of structural components of the extracellular matrix (Egido, 1996) and by increasing production of factors that degrade the extracellular matrix proteins (Oikawa et al., 1997; Wilson et al., 1997; Yoshizumi et al., 1998).
H. Why the Special Role for Angiotensin II Signaling in Vascular Smooth Muscle Cells?
In vivo, Ang II does not act alone and many vasoactive agents that signal through G protein-coupled receptors, such as ET-1, AVP, catecholamines, and serotonin, influence vascular smooth muscle cell function. Each agonist binds to its specific Gq-linked receptor to elicit a signaling response that translates into a functional event, such as contraction, hypertrophy or proliferation. Although these agonists mediate effects through similar signal transduction pathways the relative importance of each is probably related to unique processes associated with receptor expression, ligand-receptor interactions, receptor phosphorylation, G protein coupling to second messengers and cytosolic proteins, cross-talk between signaling pathways, termination of signaling events and receptor internalization (Fig.12). Other important characteristics that differentiate cellular responses to agonists that signal through similar pathways relate to: 1) underlying mechanisms generating the signal; 2) kinetics of the signaling event; and 3) magnitude of the signal. For example, in vascular smooth muscle cells, Ang II and ET-1 both increase [Ca2+]i. However, the underlying processes and kinetics are different (Fig.13). Whereas Ang II elicits a potent biphasic response that is generated primarily by mobilization of Ca2+ from intracellular stores, ET-1 increases [Ca2+]i mainly by stimulating influx through Ca2+ channels (Dostal et al., 1990; Douglas and Ohlstein, 1997). Furthermore Ang II-elicited [Ca2+]i responses and associated vascular smooth muscle cell contraction are relatively rapid, whereas ET-1 actions are more sustained. The kinetics of ERK activation by Ang II and ET-1 are also different. Maximal ERK phosphorylation by Ang II occurs within 5 min, whereas ET-1-stimulated ERK activation peaks later (Eguchi et al., 1996; Douglas and Ohlstein, 1997; Touyz et al., 1999c). These differences could be due to differential regulation of ERK by the two peptides and may explain, in part, why Ang II has a potent mitogenic effect, whereas ET-1 requires the presence of co-mitogens to elicit its growth action. Thus activation of common signaling pathways by different agonists may manifest as diverse functional responses (Fig. 12).
Scheme demonstrating mechanisms whereby different ligands that signal through Gq-coupled receptors can elicit diverse cellular responses. The specificity of response in a given cell is achieved by differential expression of the receptors that activate the signaling event and the nature of the intracellular signal. As indicated, Gq receptor coupling elicits common signals (A and B). However, the intensity of the signal, the kinetics and the cross-talk between signals differ between the cells. These differential processes result in integrated cellular responses, specific for each ligand. Rec, receptor; ↑, increased intensity of signal; ↓, decreased intensity of signal.

Diagram demonstrating common signaling pathways in vascular smooth muscle cells that are differentially regulated by Ang II and ET-1. Although both vasoactive agents signal through Gq-coupled receptors, the cellular responses differ. Ang II increases [Ca2+]i primarily via IP3-mediated effects, whereas Ca2+ influx is more important in ET-1-stimulated cells. ERK is potently and rapidly phosphorylated by Ang II, whereas responses to ET-1 are less pronounced and delayed. These differences could contribute, in part, to the diverse functional responses. Rec, receptor; VSMC, vascular smooth muscle cell; ↑, increased response; dashed lines indicate main pathway.
Of the many G protein-coupled receptors, those linked to Ang II seem to be one of the most important in vascular smooth muscle cell regulation. This is supported by in vivo studies that demonstrate that ACE inhibitors and AT1 receptor blockers attenuate Ang II-mediated signal transduction and decrease vascular smooth muscle cell functional and growth responses. Exact reasons for the apparent selective importance of Ang II are unclear but may be due to the ability of Ang II to amplify its vascular responses via other agonists. Ang II stimulates production of growth factors and vasoactive peptides, such as PDGF and ET-1, respectively, as well as transactivates multiple receptors, such as IGF, PDGF, and EGF, thereby amplifying vascular smooth muscle cell signaling responses to Ang II. Selective activation of multiphasic signaling pathways that cross-talk with other cascades, together with the phenotype of the stimulated vascular smooth muscle cell determines whether the cell undergoes contraction, proliferation, hypertrophy, and/or migration in response to Ang II. Another distinguishing feature of Ang II is the down-regulation of Ang II responsiveness (tachyphylaxis, desensitization) to repeated applications of Ang II. In vascular smooth muscle cells, Ang II down-regulates its own receptor, decreases the amount and coupling to Gq and increases G protein receptor kinase 5 (GRK5) mRNA and protein expression, which reduces efficiency of coupling between the receptor and G protein. The net effect of these processes is attenuation of responsiveness to Ang II. Although tachyphylaxis is a phenomenon common to many vasoactive agents, it is particularly potent for Ang II (Harada et al., 1999). AT receptor internalization and creation of a signaling domain specific for Ang II further contribute to unique signaling events associated with this peptide. These special qualities and the ability to stimulate production of agonists that signal through other Gq-linked receptors suggest that Ang II is an important primary regulator of vascular smooth muscle cell function. The role of G protein signaling in vascular smooth muscle cells is probably not exclusive for Ang II. However, most of our current knowledge on signal transduction pathways in vascular smooth muscle cells has been described for Ang II. As we learn more about signaling processes for other vasoactive agents it may become evident that many G protein-coupled receptors could be equally important in vascular smooth muscle cell regulation.
II. Altered Angiotensin II Signaling in Vascular Smooth Muscle Cells in Cardiovascular Diseases—Special Reference to Hypertension
A. Introduction
Ang II is an exceptional peptide that generates signaling events to elicit pleiotropic effects in vascular smooth muscle cells. Not only does it stimulate classic G protein-coupled phospholipases to induce contraction, but it also activates many tyrosine kinase pathways that are characteristically associated with growth, inflammatory, migratory, and fibrotic responses. These data suggest that Ang II is crucial in maintaining the structural and functional integrity of the vessel wall and that it plays an important role in cardiovascular diseases associated with vascular smooth muscle cell contraction and growth such as hypertension and restenosis. In addition, Ang II induces vascular wall adhesion molecule-1 expression and contributes to atherogenesis by activation of VCAM-1 through proteasome dependent, NF-κB-like transcriptional mechanisms (Kranzhofer et al., 1999; Tummala et al., 1999). In clinical trials with angiotensin-converting enzyme inhibitors and AT1 receptor blockers demonstrating improved morbidity and mortality in hypertension, congestive cardiac failure, and myocardial infarction, support the significance of Ang II in the pathogenesis of cardiovascular disease. We focus here on hypertension and the signal transduction mechanisms whereby Ang II influences vascular smooth muscle cell responses underlying vascular functional and structural alterations associated with blood pressure elevation. Ang II also plays an important pathophysiological role in cardiac and renal disease, but will not be discussed here. The reader is referred to a recent review on this topic (Kim and Iwao, 2000).
B. Vascular Changes
The primary hemodynamic characteristic of essential hypertension is increased peripheral vascular resistance that is associated with structural, mechanical, and functional alterations in the peripheral vasculature (Korner et al., 1989; Folkow, 1990). The major structural changes include reduced vessel lumen diameter and media thickening (vascular remodeling) (Mulvany and Aalkjaer, 1990; Schiffrin, 1992;Mulvany et al., 1996; Laurant et al., 1997; Rizzoni, 1998a; Sharifi and Schiffrin, 1998; Williams, 1998; Intengan et al., 1999). At the cellular level, there is hyperplasia, hypertrophy, elongation of vascular smooth muscle cells, reorganization of the cells around the lumen of the artery, and/or altered extracellular matrix composition, resulting in a smaller lumen and outer diameter (Mulvany et al., 1985;Lee, 1987; Korsgaard et al., 1993; Owens and Schwartz, 1993; Nag, 1996;Gibbons, 1998; Sharifi et al., 1998; Tsoporis et al., 1998; Intengan et al., 1999). Some studies failed to demonstrate hyperplasia or hypertrophy of vascular smooth muscle cells in small arteries from hypertensive patients and spontaneously hypertensive rats (SHR), a popular rat model of human essential hypertension. Vascular remodeling accordingly was attributed to changes in extracellular matrix content and to rearrangement of vascular smooth muscle cells (Korsgaard et al., 1993; Nag, 1996; Intengan et al., 1999). In intramyocardial arteries in SHR, the volume and number of arterial smooth muscle cells is significantly increased (Amann et al., 1995) and in Ang II-induced hypertensive rats, arterial smooth muscle cell thickness is increased without a change in the number of cell layers (Simon et al., 1998). In prehypertensive SHR, structural changes of the small muscular arteries are associated with an increase in the media volume, increased number of smooth muscle cell layers, and elongation of vascular smooth muscle cells (Dickhout and Lee, 1997; Lee and Dickhout, 1998). Mesenteric resistance arteries from SHR have a significantly increased number of cell layers, which is normalized when rats are treated for 8 weeks with ACE inhibitors or AT1 receptor blockers (Rizzoni et al., 1998b). Other studies showed that increased media thickness results from greater collagen deposition rather than increased smooth muscle cell number (Sharifi et al., 1998). These conflicting data indicate that cellular processes underlying media thickening are complex, and exact mechanisms contributing to arterial remodeling in hypertension are not yet well understood.
Functional changes accompany structural changes in small arteries in hypertension, which contribute to enhanced vasoconstrictor responses and to elevation of vascular tone. Functional alterations that increase peripheral resistance include enhanced vascular reactivity to vasoconstrictor agents or impaired relaxation and reflect changes in excitation-contraction coupling and/or electrical properties of cells (Dominiczak and Bohr, 1989; Schiffrin, 1992; Schiffrin et al., 1993;Touyz et al., 1994, 1999d; Chen et al., 1995b; Schiffrin et al., 1996;Feldman and Gros, 1998). Excess systemic or local production of vasoconstrictor agents or growth factors, abnormal agonist-receptor interactions, increased cell membrane permeability, defective transplasmalemmal ion transport, and altered transduction of intracellular signaling pathways in vascular smooth muscle cells may contribute to the pathological vascular changes that characterize hypertension (Touyz and Schiffrin, 1993b,c).
Among the many vasoactive agonists implicated in vascular hyperresponsiveness in hypertension, Ang II appears to be one of the most important. Whereas responses to ET-1, vasopressin, and norepinephrine have been reported to be decreased, unchanged, or rarely, increased, vascular reactivity to Ang II has, for the most part, been found to be enhanced in experimental and human hypertension (Bodin et al., 1993; Schiffrin et al., 1993; Touyz et al., 1994, 1999d;van Geel et al., 2000). The significance of Ang II in the pathogenesis of hypertension is supported by experimental and clinical studies demonstrating that ACE inhibitors and AT1receptor blockers not only lower blood pressure, but also regress arterial and cardiac remodeling and normalize mechanisms that regulate intracellular second messengers (Touyz and Schiffrin, 1993a; Schiffrin et al., 1994, 2000; Schiffrin, 1996; Schiffrin and Deng, 1995; Li et al., 1997; Ennis et al., 1998; Li et al., 1998a; Rizzoni et al., 1998a;Sharifi et al., 1998; Benetos et al., 2000; Zhan et al., 2000). Many alterations in signal transduction have been described in cardiovascular cells in hypertension (Touyz and Schiffrin, 1993b,c;Witte and Lemmer, 1996). The present review concentrates specifically on changes in Ang II-mediated intracellular signaling in vascular smooth muscle cells in hypertension. Other agents, and particularly vasoactive peptides such as ET-1, may also be important in vascular pathological processes and complications of hypertension (Schiffrin et al., 1997; Schiffrin, 1998) but will not be discussed here and the reader is referred to recent reviews (Schiffrin et al., 1997; Schiffrin and Touyz, 1998; Barton and Luscher, 1999).
C. Vascular Angiotensin Receptors
Altered Ang II-mediated signal transduction in hypertension may occur at, or beyond, the level of the cell membrane receptor. Recent studies demonstrated enhanced mRNA expression for AT1 and AT2 receptors in aortic vessels from adult SHR compared with age-matched normotensive Wistar-Kyoto rats (WKY rats) (Otsuka et al., 1998a). We recently reported that AT2 receptor mRNA and protein expression are augmented in mesenteric arteries from young SHR compared to age-matched controls (Touyz et al., 1999a). Differential regulation of AT2 receptors has also been demonstrated in cultured aortic smooth muscle cells from SHR and in kidneys from Ang II-induced hypertensive rats (Ishiki et al., 1996; Wang et al., 1999). Binding studies demonstrate that AT1 receptor density is greater in the adrenal cortex, outer medulla of the kidney, and heart from SHR compared with WKY rats (Song et al., 1995; Touyz et al., 1996). However in the vasculature, Ang receptor density and affinity do not seem to be significantly different between adult SHR and WKY rats (Schiffrin et al., 1984; Cortes et al., 1996), suggesting that up-regulation of Ang II-mediated vascular signaling events in SHR probably occur primarily at the post-receptor level. It has also been suggested that hyperresponsiveness to Ang II in hypertension could be due to altered desensitization of AT1 receptors. mRNA and protein expression of GRK5, a member of the G protein-coupled receptor kinase family that phosphorylates and participates in the desensitization of Ang receptors, is significantly increased in aorta of Ang II-induced hypertensive rats compared with normotensive controls (Ishizaka et al., 1997; Feldman and Gros, 1998).
D. Short-Term Signaling Events
1. Angiotensin II Stimulation of the Phospholipase C-IP3-Diacylglycerol Pathway Is Augmented.
In vascular smooth muscle cells from young and adult SHR, Ang II-stimulated PLC-mediated signaling is increased (Fig.14). These events may be fundamental in the pathological vascular changes and target organ sequelae that characterize hypertension. PLC activity, IP3generation, and DAG production as well as the second messengers [Ca2+]i and pHi are significantly augmented in response to Ang II in cells from SHR compared with WKY rats (Bendhack et al., 1992;Kato et al., 1992; Osanai and Dunn, 1992; Redon and Batlle, 1994; Touyz et al., 1994, 1999d; Baines et al., 1996). Intracellular Ca2+ overload and alkalinization are partially due to increased Ca2+ influx and mobilization and to enhanced activity to the Na+/H+ exchanger (Roufogalis et al., 1997; Touyz and Schiffrin, 1997b; Ennis et al., 1998). Altered Ang II-induced [Ca2+]i handling in hypertension may also be due to increased TGF-β-stimulated Ang II-induced transplasmalemmal Ca2+ influx (Zhu et al., 1995a). ACE inhibition and AT1 receptor blockers, but not AT2 receptor antagonists, normalize Ca2+ and pHiregulatory mechanisms in experimental and human hypertension, suggesting that AT1-mediated processes play a role in modified Ang II-stimulated second messenger responses (Touyz and Schiffrin, 1993b; Ennis et al., 1998). Activity of vascular smooth muscle cells from SHR shows a greater dependence on Ang II-mediated Ca2+ mobilization than cells from WKY rats (Lucchesi, 1996). This Ca2+-dependent MAP kinase activation in SHR vascular smooth muscle has been defined as a hypertensive signal transduction phenotype (Lucchesi, 1996). [Ca2+]i elevation and alkalinization are major determinants of vascular contraction and growth and could be critical in Ang II-induced vascular hyperreactivity and dysfunction in hypertension (Grinstein et al., 1989; Rembold, 1993).
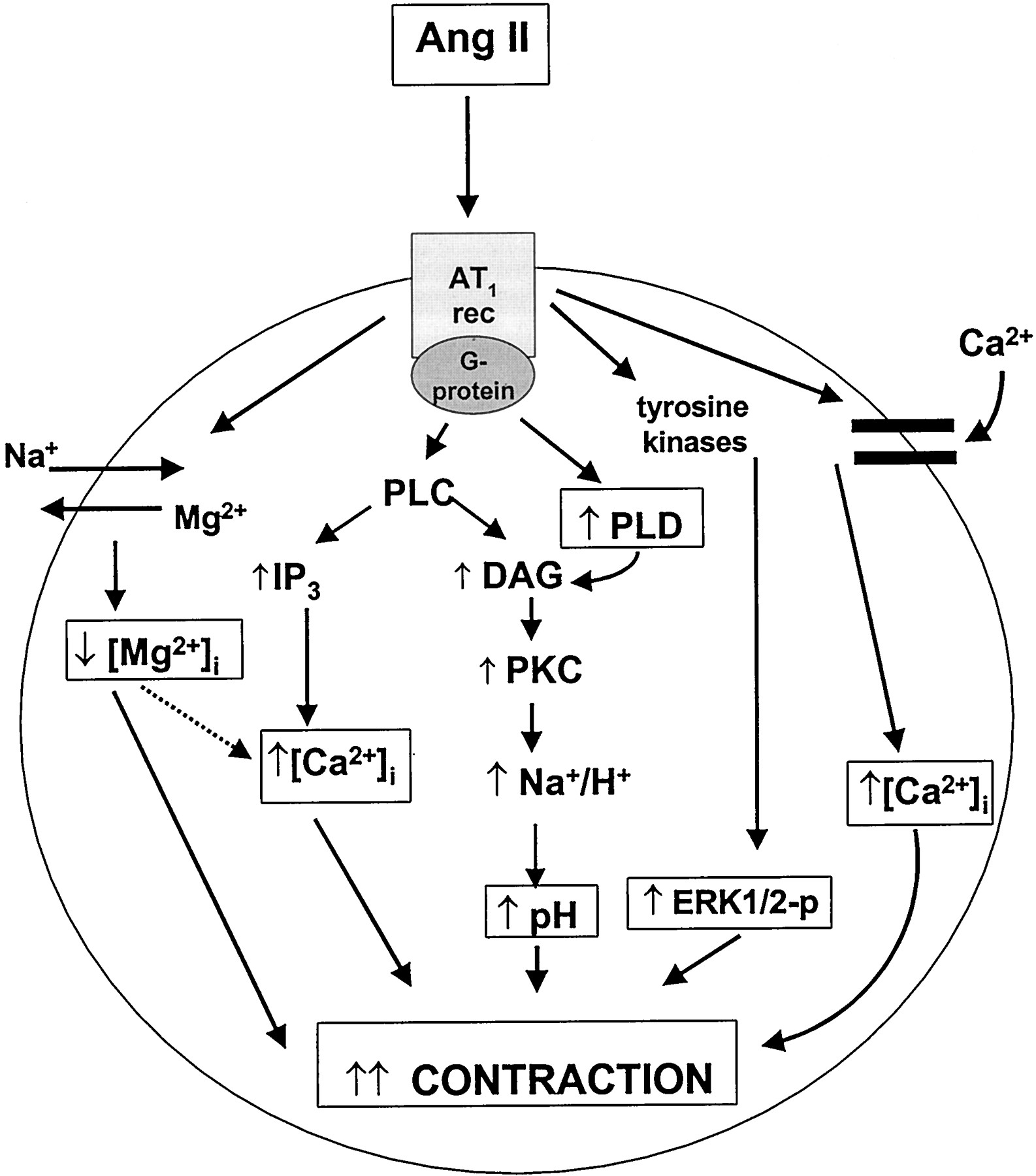
Alterations in some of the short-term Ang II-stimulated signaling events in hypertension. In hypertension, PLC activity, IP3 generation, and DAG production are increased in response to Ang II stimulation. Increased IP3 results in augmented mobilization of intracellular Ca2+ with resultant increased [Ca2+]i. Increased Ang II-stimulated Ca2+ influx also contributes to elevated [Ca2+]i in hypertension. Generation of DAG, due to increased activation of PLC and PLD, leads to increased protein kinase C activity, which activates the Na+/H+exchanger resulting in intracellular alkalinization. Activation of the Na+-dependent Mg2+ exchanger induces Mg2+ efflux resulting in decreased intracellular free Mg2+ concentration ([Mg2+]i) contributing to increased [Ca2+]i. These processes lead to enhanced contraction. Increased activation of ERK-dependent signaling pathways contributes to the sustained phase of contraction. ↑, increase; ↓, decrease; dashed line, indirect effect.
2. Angiotensin II-Stimulated Effects on Vascular [Mg2+]i and [Na+]i.
Magnesium, the second most abundant intracellular cation is an important modulator of vascular [Ca2+]i (Fig. 14). Total and free concentrations of intracellular Mg2+ are significantly reduced in various cell types in experimental and human hypertension (Touyz et al., 1992; Touyz and Schiffrin, 1993a, 1998;Resnick et al., 1997). Mechanisms that regulate [Mg2+]i in hypertension are unknown, but we recently reported that the magnitude of Ang II-induced reduction in [Mg2+]i is increased in vascular smooth muscle cells from SHR (Touyz and Schiffrin, 1999a). This augmentation was associated with alterations in Na+-dependent Mg2+exchange, which was linked to increased activation of the Na+/H+ exchanger and increased [Na+]i (Touyz and Schiffrin, 1999a). Because of the Ca2+-antagonistic properties of Mg2+, reduced [Mg2+]i, both basal and in response to agonists, may contribute to increased [Ca2+]i and enhanced contractile responsiveness to Ang II in hypertension (Zhu et al., 1995b; Yoshimura et al., 1997).
Ang II increases [Na+]iin a concentration-dependent manner that is augmented in hypertension (Touyz and Schiffrin, 1996, 1999a). This is associated with increased Na+/Ca2+exchange, increased Na+ influx, increased activation of the Na+/H+exchanger, alterations in activity of the Na+/K+ ATPase pump, and the presence of a Na+ pump inhibitor (Shigekawa et al., 1996; Juhaszova and Blaustein, 1997; Cox et al., 1998). In addition, altered regulation of the Na+/Mg2+ exchanger contributes to enhanced Ang II-stimulated [Na+]i responses (Touyz and Schiffrin, 1999a). Elevated [Na+]i influences vascular smooth muscle growth and contraction (Henrion et al., 1997; Gu et al., 1998). In SHR increased [Na+]i responses have been shown to convert hypertrophy to hyperplasia synergistically with activated PKC via MAP kinase-dependent signaling pathways (Osanai et al., 1996). These responses could contribute to changes in vascular smooth muscle cell phenotype that lead to vascular remodeling in hypertension.
3. Vascular Eicosanoids, Angiotensin II, and Hypertension.
The role of eicosanoids in Ang II-dependent hypertension has recently been reviewed (Nasjletti, 1997). Eicosanoids have the potential to act as either prohypertensive or antihypertensive agents. Ang II-induced hypertension in rats is accompanied by increased vascular production of TXA2 and of lipoxygenase-derived metabolites that have the ability to inhibit prostacyclin synthase (Nasjletti, 1997). As a result of these alterations, the activity of pressor mechanisms mediated by TXA2 and/or PGH2 is augmented. Thromboxane synthase inhibitors, TXA2/PGH2 receptor blockers and inhibitors of lipoxygenase lower blood pressure in Ang II-treated rats, supporting the role of eicosanoids in this model of hypertension (Nasjletti, 1997). Eicosanoids also influence blood pressure elevation in genetically hypertensive rats (Kunimoto et al., 1998). Enhanced Ang II-stimulated vascular reactivity in de-endothelialized small mesenteric arteries is associated with alterations in metabolism of cyclooxygenase products in SHR (Cortes et al., 1996). Treatment with inhibitors of thromboxane synthase and of lipoxygenase significantly reduced blood pressure in SHR (Stern et al., 1993; Keen et al., 1997).
4. Angiotensin II Increases Activity of Phospholipase D.
Some of the altered prolonged signaling events mediated by Ang II in hypertension have been attributed to increased activation of PLD (Fig.12). The magnitude of increase in PLD activity and the rate of activation in response to Ang II, as well as the heptapeptide Ang-(2-8), is greater in aortic vascular smooth muscle cells from SHR compared with cells from WKY rats (Freeman, 1995). This effect appears to be [Ca2+]i-dependent. Increased Ang II-stimulated activation of PLD contributes to enhanced vasoconstriction via DAG-PKC-dependent pathways, and to increased cell growth via phosphatidic acid (Dhalla, 1997; Gomez-Cambronero and Kiere, 1998). PLD and its metabolites also activate NADH/NADPH to generate superoxide anions; which are important modulators of cell growth and vascular remodeling in hypertension (Gomez-Cambronero and Kiere, 1998;Touyz and Schiffrin, 1999b).
5. Cyclic Nucleotides and Angiotensin II.
Augmented Ang II-induced vasoconstriction in hypertension is related, in part, to changes in cyclic nucleotide signaling. In cultured preglomerular microvascular smooth muscle cells, Ang II enhances cAMP responses to β-adrenoceptor agonists via a PKC-dependent mechanism, resulting in vasodilation and attenuation of Ang II-stimulated contraction (Mokkapatti et al., 1998). In hypertension, this buffering mechanism is altered leading to blunted vasodilation and increased vascular contractility. Similar findings have been reported in the renal vasculature of SHR in the early phases of blood pressure elevation. In renal resistance arteries of 8-week-old SHR, exaggerated vascular reactivity to Ang II was found to be due to defective cAMP generation in the presence of a normally operating PKC pathway (Ruan and Arendshorst, 1996b). Changes in cGMP regulation have also been demonstrated to play a role in enhanced responsiveness to Ang II in SHR. Basal and stimulated cGMP responses are significantly lower in vascular smooth muscle cells from SHR compared with WKY rats (Baines et al., 1996). In balloon-injured rat aorta, AT2receptor stimulation results in reduced basal cGMP levels (Moroi et al., 1997). Modified cross-talk between constrictor and dilator signaling pathways may contribute to Ang II-mediated vascular hyperresponsiveness in some vascular beds in hypertension.
E. Long-Term Signaling Events
1. Angiotensin II-Induced Generation of Reactive Oxygen Species.
There is increasing evidence that vascular oxidative stress plays a pathogenic role in hypertension (Touyz, 2000) (Fig.15). Ang II increases production of reactive oxygen species in vascular smooth muscle, endothelial, adventitial, and mesangial cells (Harrison, 1997; Pagano et al., 1997; Jaimes et al., 1998; Touyz and Schiffrin, 1999b). In Ang II-dependent models of hypertension, vascular production of superoxide anions is increased (Laursen et al., 1997; Aizawa et al., 2000; Zalba et al., 2000). This effect is mediated via Ang II-stimulated activation of vascular NADH/NADPH oxidase (Rajagopalan et al., 1996). In Ang II-induced hypertensive rats, treatment with liposome-encapsulated superoxide dismutase reduced production of vascular reactive oxygen species, decreased blood pressure by ∼50 mm Hg, and enhanced responses to vasodilators, both in vivo and in vitro (Laursen, 1997). NADH/NADPH oxidase-generated reactive oxygen species also contribute to Ang II-mediated vascular hypertrophy in hypertension. Both O⨪ and H2O2 are potent mitogens that elicit effects via p38 MAP kinase, ERK-5 and NF-κB (Abe and Berk, 1998; Ushio-Fukai et al., 1998a). Inhibition of NADH/NADPH oxidase inhibits Ang II-induced vascular smooth muscle cell hypertrophy (Ushio-Fukai et al., 1996; Touyz and Schiffrin, 1999b), supporting a potential role of reactive oxygen species as inducers of increased vascular growth in hypertension.

Alterations in some of the long-term Ang II-stimulated signaling events in hypertension. Increased activation of tyrosine kinase- and MAP kinase-dependent signaling pathways lead to increased nuclear signaling events resulting in enhanced protein synthesis. In the vasculature in hypertension, increased protein synthesis is associated with enhanced cell growth, mitogenesis, greater extracellular matrix (ECM) deposition, and increased growth factor production leading to increased media thickness and vascular remodeling. Augmented Ang II-stimulated generation of superoxide anion (⋅O⨪2) and hydrogen peroxide (H2O2), potent mitogens, may also contribute to increased mitogenesis in hypertension. JAK, Janus kinase.
2. Angiotensin II, Tyrosine Kinases, and Hypertension.
In cardiac, renal, and vascular tissue from hypertensive rats, basal and Ang II-stimulated activation of tyrosine kinases and ERKs is increased (Wilkie et al., 1997; Hamaguchi et al., 1998; Izumi et al., 1998; Touyz et al., 1999d) (Fig. 15). Of the tyrosine kinases stimulated by Ang II, JAK/STAT has been most extensively studied in hypertension. In acute pressure overload in the rat, cardiac JAK/STAT is activated (Pan et al., 1997). In this model, where pressure overload was produced by abdominal aortic constriction in Wistar rats, Ang II activated both Tyk2 and JAK2 (Pan et al., 1997). These effects were completely blocked by ACE inhibitors and AT1 receptor antagonists (Pan et al., 1997). Activation of the JAK/STAT pathway is associated with Ang II-induced vascular and cardiac remodeling in hypertension as well as with inflammatory processes that underlie atherosclerosis (Dostal et al., 1997; Ghatpande et al., 1999). These actions are mediated by increased phosphorylation of STATs. In Ang II-induced hypertension, activated STATs bind to the SIE of the gene promoter leading to enhanced expression of vascular smooth muscle cell growth-related early response genes, such as c-fos, c-myc, and α2-macroglobulin (Pollack, 1995; Chen et al., 1996; Xu et al., 1996a; Dostal et al., 1997). In vivo studies demonstrate that renal c-fos mRNA expression in response to AT1 stimulation is augmented in SHR compared with WKY rats (Otsuka et al., 1998a,b). Although many tyrosine kinases, such as Src family kinases, Fak, receptor tyrosine kinases, PI3K, and Pyk2 are phosphorylated by Ang II, the role of these kinases in the pathogenesis of vascular damage and cardiovascular diseases has not yet been elucidated.
3. Angiotensin II-Mediated Mitogen-Activated Protein Kinase Signaling Is Increased.
Of the growth-signaling pathways activated by Ang II, MAP kinase-dependent cascades have been most extensively studied in hypertension (Fig. 15). Ang II-stimulated activation of ERK-1/ERK-2 is augmented in vascular smooth muscle cells from aorta and mesenteric arteries of SHR (Wilkie et al., 1997; Touyz et al., 1999c,d). In aortic vessels and cardiac tissue from SHRSP and in Ang II-induced hypertension, activities of both JNK/SAPK and ERKs are increased compared with normotensive controls (Kim et al., 1997;Izumi et al., 1998). These effects appear to be specific, as there is no significant increase in ERK or JNK/SAPK activity in noncardiovascular tissue, such as liver, stomach, spleen, or lung of hypertensive rats (Kim et al., 1997; Kim and Iwao, 2000). Alterations in MAP kinase function in hypertension include a more rapid inactivation of MAP kinase after Ang II stimulation and a greater dependence of MAP kinase phosphorylation on intracellular Ca2+ mobilization (Lucchesi et al., 1996). Mechansims underlying these enhanced Ang II-induced MAP kinase responses in hypertension are related to amplification at the level of sequential PKC and tyrosine kinase steps (Wilkie et al., 1997;Hamaguchi et al., 1998). In cardiac hypertrophy associated with Ang II-induced hypertension, both cardiac ERK and JNK/SAPK activities are increased, but JNK/SAPK activation occurs in a more sensitive manner than ERK activation (Yano et al., 1998). The differential regulation suggests that JNK/SAPK may be critical in Ang II-induced cardiac hypertrophy (Yano et al., 1998), whereas in vascular hypertrophy, ERK-1/ERK-2-dependent signaling pathways may be more important (Dubey, 1997). This is further supported by studies of balloon-injured vessels, in which activity of ERK-1/ERK-2 was greater than that of JNK/SAPK (Kim et al., 1998). In vivo studies in SHRSP demonstrate that ACE inhibitors and AT1 receptor blockers significantly reduces ERK activity, implicating a role for AT1receptors in enhanced ERK activation in hypertension (Hamaguchi et al., 1999; Kim and Iwao, 2000).
Increased activity of MAP kinases contributes not only to augmented growth responses, but also to increased vascular contractility in hypertension. We reported that Ang II-induced contractile and associated [Ca2+]isignaling responses are significantly enhanced in vascular smooth muscle cells from resistance arteries of SHR, and that these phenomena are dependent on hyperactivation of ERK-1/ERK-2 (Touyz et al., 1999d). The second phase of contraction, which was sustained and of greater magnitude in vessels from SHR than WKY rats, was particularly sensitive to MAP kinase phosphorylation (Touyz et al., 1999d). Similar findings have been demonstrated in intact arteries (Epstein et al., 1997).
Altered regulation of vascular MAP kinase activity in hypertension is related to modifications in the balance between MKP-1 (as well as related phosphatases) and MAP kinase levels/activity. Normally, activity of MAP kinases is regulated by reversible phosphorylation of tyrosine and threonine residues by protein phosphatases (Hunter, 1995). Ang II induces MKP-1 expression in the vasculature, which is secondary to induction of MAP kinase (Xu et al., 1997). In vascular smooth muscle cells from SHR, increased MAP kinase activity appears to be due to increased tyrosine phosphorylation because of reduced dephosphorylation resulting from impaired growth factor-mediated MKP-1 gene expression that attenuates MAP kinase signaling via feedback inhibition (Begum et al., 1998). Thus dysregulation of the balance between MAP kinase and MKP-1 levels/activity could contribute to increased vascular smooth muscle cell growth in hypertension.
4. Indirect Effects of Angiotensin II on the Vasculature.
Some Ang II actions on vascular signaling in hypertension are mediated via indirect mechanisms through other vasoactive agents such as ET-1, growth factors such as TGF-β1 and PDGF-A, or cytokines such as TNF (Fig. 15). Enhanced Ang II-elicited contractile responsiveness in aorta from SHR is mediated by an ET-1 component that is especially important at suppressor Ang II concentrations (Balakrishnan et al., 1996). Ang II-stimulated ET-1 production also regulates vascular structural changes in hypertension. In rats infused with Ang II, ET-1 expression in vascular smooth muscle cells is increased (Moreau et al., 1997) and arterial remodeling in Ang II-induced hypertensive rats is completely reversed when rats are treated with an ET receptor blocker (Barton et al., 1998). However the role of endogenous Ang II on ET-1 production is still unclear, since ET receptor blockade has little blood pressure-lowering effect in Ang II-dependent models of hypertension (Li and Schiffrin, 1995). Also in renin-independent hypertension vascular ET-1 gene expression is enhanced (Schiffrin et al., 1996; Sventek et al., 1996). Furthermore in ren 2 transgenic rats, the ET receptor blocker, SB 2099670, had no significant effect on blood pressure (Gardiner et al., 1995). Hence the exact contribution of ET-1 to mediation of Ang II effects in vivo is unclear.
Exaggerated growth of vascular smooth muscle cells and vascular remodeling in SHR has also been attributed, in part, to abnormal regulation of growth factors and their receptors by Ang II. Expression of TGF-β1 and PDGF-A mRNAs and TGF-β receptor is greater in vascular smooth muscle cells from SHR than in cells from WKY rats (Hahn et al., 1991; Hamet et al., 1991; Fukuda et al., 1995; Parker et al., 1998). Abnormal regulation of TGF-β receptors in hypertension appears to be related to locally generated Ang II (Fukuda et al., 1998). Ang II-induced effects are also mediated via cytokines and other tyrosine kinase receptor-linked agonists. In Ang II-dependent hypertension, renal levels of the cytokine TNF and the constrictor prostaglandin PGE2 are higher than those of normotensive controls. Anti-TNF antiserum exacerbates Ang II-mediated increase in blood pressure (Ferreri, 1997). These data suggest that TNF and PGE2 modulate pressor actions of Ang II in hypertension, and that signaling pathways typically associated with inflammation are also involved in Ang II-associated vascular dysfunction and structural remodeling in hypertension.
F. Mechanisms Underlying Enhanced Angiotensin II Vascular Responsiveness
The significance of Ang II-dependent effects in the cardiovascular system in hypertension is evidenced by clinical studies demonstrating that pharmacological interruption of the RAS not only normalizes enhanced Ang II-induced signaling responses but improves endothelial function, regresses cardiac and vascular structural changes, and reduces blood pressure (Rizzoni et al., 1998a; Schiffrin et al., 2000). Blockade of other systems linked to G protein-coupled receptors, such as ET-1, fails to effectively improve vascular remodeling and only modestly reduces blood pressure in genetically hypertensive rats and in patients with essential hypertension (Barton and Luscher, 1999). These data suggest that Ang plays an important and specific role in the pathogenesis of hypertension and that altered regulation of Ang II at the cellular and molecular level could be fundamental in the pathological processes associated with the development and maintenance of blood pressure elevation. Both in vitro studies in cultured cells and data from whole animal experiments indicate that Ang II signaling in hypertension is up-regulated. Exact reasons for this are unclear, but it appears that augmented Ang II signaling is a post-receptor phenomenon. This is based on studies demonstrating that Ang II receptor status, determined by binding studies, mRNA and protein expression, is not significantly altered in vascular smooth muscle cells in hypertension (Cortes et al., 1996; Schiffrin, 1996).
Since multiple Ang II-mediated signaling pathways and downstream effectors are up-regulated in hypertension, it seems likely that the primary abnormality occurs very early in the signaling process. This may be at the level of AT1 receptor-G protein interraction, or at the level of a common upstream signaling molecule. Possible post-receptor mechanisms underlying these events include increased phosphorylation of the AT1 receptor, altered receptor-G protein coupling, impaired receptor-mediated activation of upstream signaling molecules and dysregulation of second messengers. Defective AT1 receptor internalization and termination of the signaling event could also contribute to sustained and augmented responses. Although our current knowledge of the precise mechanisms responsible for Ang II hyperresponsiveness is limited, it is apparent that genetic factors play a role. This is based on studies demonstrating that Ang II-induced signaling events remain up-regulated in serially passaged cultured cells and in immortalized lymphocytes from hypertensive rats and humans. The need to identify primary causes responsible for altered Ang II signaling in hypertension is clinically relevant, as targeting specific abnormally regulated molecules in the signaling cascade has therapeutic potential. With the availability today of highly selective pharmacological inhibitors, molecular tools, and genetically manipulated animal models, hopefully it won't be too long before we are able to elucidate in greater detail the fundamental origin responsible for abnormal Ang II signaling in hypertension.
IV. Conclusions
Ang II influences arterial tone and remodeling in hypertension by stimulating vascular smooth muscle cell contraction, augmenting cell growth, increasing deposition of extracellular matrix, inhibiting apoptosis, inducing cell migration, and promoting inflammation. Recent data showing that ACE inhibitors or AT1 receptor antagonist treatment of essential hypertensive patients regresses structural and functional abnormalities of the vascular wall (Schiffrin, 1996, 1998; Schiffrin et al., 2000) suggest that Ang II plays a critical role in abnormal behavior of vascular cells in hypertension. Mechanisms underlying these cellular effects seem to occur at the post-receptor level and appear to be associated with hyperactivity of Ang II-stimulated G protein-coupled phospholipases, tyrosine kinase-, and MAP kinase-dependent pathways, as well as with oxidative stress. Interactions between these cascades is highly complex, and dysregulation at any level could manifest as pathological functional sequelae and structural vascular changes in hypertension. The impact and significance of altered Ang II-induced intracellular signaling in the vasculature in hypertension is becoming more evident. However, although there has been significant progress in the last few years in the elucidation of aberrations in Ang II-induced signal transduction in hypertension, we still know very little about the processes that underlie these phenomena and at what point some pathways become more important than others. With molecular and pharmacological tools that allow manipulation of specific signal transduction molecules, identification of distinct abnormalities in intracellular signaling in hypertension should be possible. This will further our understanding of the role of Ang II in the vascular pathophysiological processes that are associated with hypertension and other cardiovascular diseases.
Acknowledgment
This study was supported by funds from the Medical Research Council of Canada, Heart and Stroke Foundation of Canada, Canadian Hypertension Society, and the fonds de la recherche en santé du Quebec.
Footnotes
-
↵1 Address for correspondence: R. M. Touyz, M.D., Ph.D., Clinical Research Institute of Montreal, 110 Pine Ave. West, Montreal, Quebec H2W 1R7 Canada. E-mail: touyzr{at}ircm.qc.ca
-
c-Jun N-terminal kinase; PKB, protein kinase B; SAPK, stress-activated protein kinase; MKP, MAP kinase phosphatase; PHAS-I, phosphorylated heat- and acid-stable protein; eIF, eukaryotic initiation factor; PKA, protein kinase A; PG, prostaglandin; TXA, thromboxane; HPETE, hydroperoxyeicosatetraenoic acid; HETE, hydroxyeicosatetraenoic acid; CRE, cAMP/calcium response element; SRE, seum response element; SIE, sis-inducing factor element ; TGF-β, transforming growth factor-β; IGF-1, insulin-like growth factor-1; bFGF, basic fibroblast growth factor; PAF, platelet-activating factor; TNF-α, tumor necrosis factor-α; MCP-1, monocyte chemoattractant protein-1; SHR, spontaneously hypertensive rats; WKY, Wistar-Kyoto; MEK, MAPK/ERK kinase.
Abbreviations
- Ang
- angiotensin
- ET-1
- endothelin-1
- PDGF
- platelet-derived growth factor
- RAS
- renin-angiotensin system
- ACE
- angiotensin-converting enzyme
- NEP
- neutral endopeptidase
- GRK
- G protein receptor kinase
- SH2
- Src homology 2
- EGF
- epidermal growth factor
- FAK
- focal adhesion kinase
- PLC
- phospholipase C
- PLA2
- phospholipase A2
- PLD
- phospholipase D
- MAPK
- mitogen-activated protein kinase
- PKC
- protein kinase C
- DAG
- diacylglycerol
- PtdInsP2
- phosphatidylinositol-4,5-bisphosphate
- PYK
- proline-rich tyrosine kinase
- PKD
- protein kinase D
- ERK
- extracellular signal-regulated kinase
- STAT
- signal transducers and activators of transcription
- PI3K
- phosphatidylinositol 3-kinase
- CADTK
- calcium-dependent tyrosine kinase
- The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
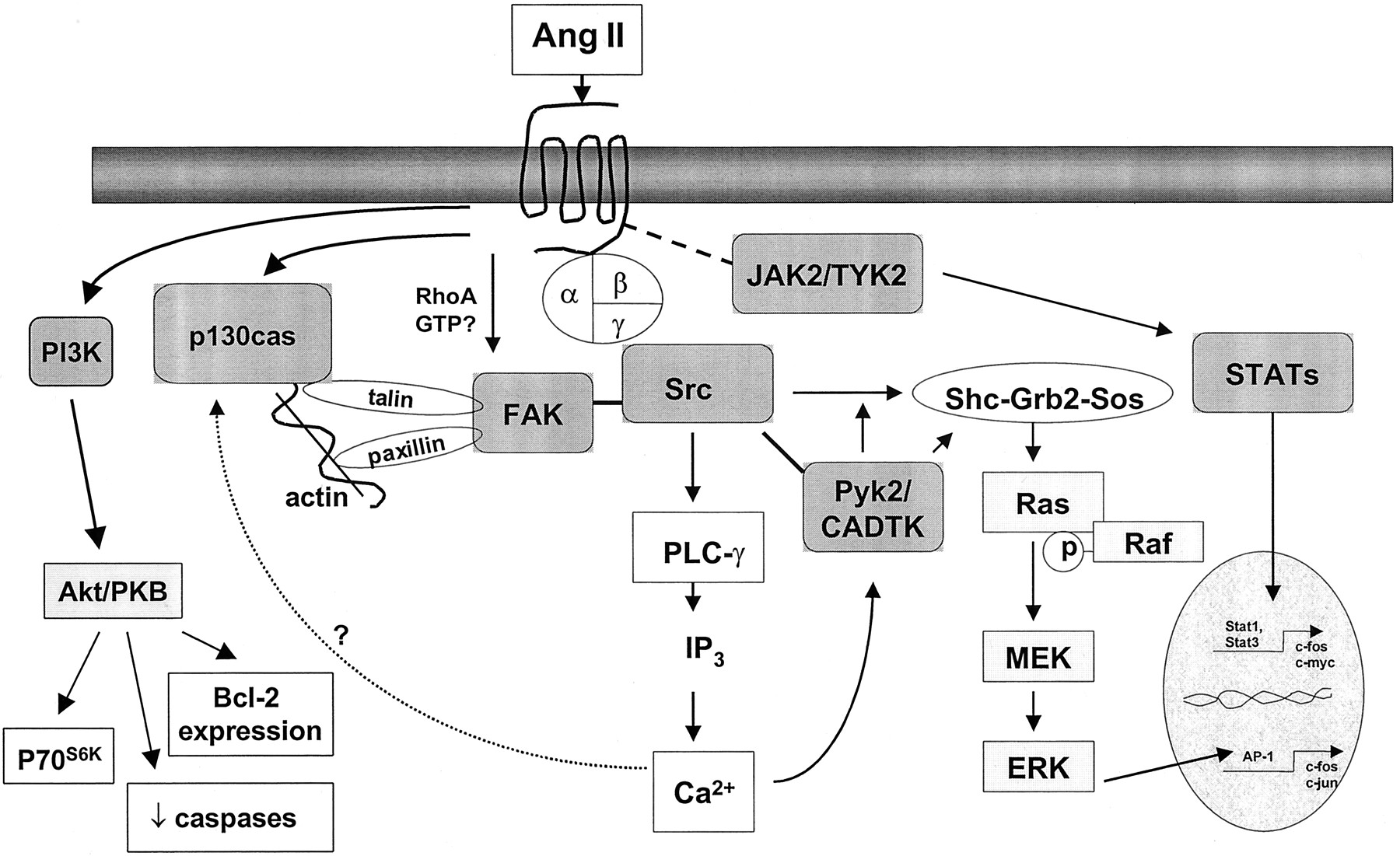{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}