


Abstract
In recent years, the focus of interest on the role of the renin-angiotensin system (RAS) in the pathophysiology of hypertension and organ injury has changed to a major emphasis on the role of the local RAS in specific tissues. In the kidney, all of the RAS components are present and intrarenal angiotensin II (Ang II) is formed by independent multiple mechanisms. Proximal tubular angiotensinogen, collecting duct renin, and tubular angiotensin II type 1 (AT1) receptors are positively augmented by intrarenal Ang II. In addition to the classic RAS pathways, prorenin receptors and chymase are also involved in local Ang II formation in the kidney. Moreover, circulating Ang II is actively internalized into proximal tubular cells by AT1 receptor-dependent mechanisms. Consequently, Ang II is compartmentalized in the renal interstitial fluid and the proximal tubular compartments with much higher concentrations than those existing in the circulation. Recent evidence has also revealed that inappropriate activation of the intrarenal RAS is an important contributor to the pathogenesis of hypertension and renal injury. Thus, it is necessary to understand the mechanisms responsible for independent regulation of the intrarenal RAS. In this review, we will briefly summarize our current understanding of independent regulation of the intrarenal RAS and discuss how inappropriate activation of this system contributes to the development and maintenance of hypertension and renal injury. We will also discuss the impact of antihypertensive agents in preventing the progressive increases in the intrarenal RAS during the development of hypertension and renal injury.
I. Introduction
The critical role of the circulating RAS1 in the regulation of arterial pressure and sodium homeostasis has been recognized for many years. Ang II is the most powerful biologically active product of the RAS, although there are other bioactive Ang peptides, including Ang III, Ang IV, and Ang 1-7. Ang II directly constricts vascular smooth muscle cells, enhances myocardial contractility, stimulates aldosterone production, stimulates release of catecholamines from the adrenal medulla and sympathetic nerve endings, increases sympathetic nervous system activity, and stimulates thirst and salt appetite. Ang II also regulates sodium transport by epithelial cells in intestine and kidney. There has also been a growing appreciation of the organ-specific roles exerted by Ang II acting as a paracrine factor (Navar et al., 1996; Paul et al., 2006). In addition to its physiological roles, locally produced Ang II induces inflammation, cell growth, mitogenesis, apoptosis, migration, and differentiation, regulates the gene expression of bioactive substances, and activates multiple intracellular signaling pathways, all of which might contribute to tissue injury. Clinical and preclinical studies on the effects of pharmacological investigations with ACEIs and ARBs support the notion that Ang II exerts a cardinal role in the pathogenesis of hypertension and renal injury via activation of AT1 receptors when inappropriately activated (Timmermans et al., 1993; Navar et al., 2000). Importantly, because the kidney plays a crucial role in the development of hypertension, hypertension is both a cause and consequence of renal disease (Navar, 1997, 2005; Paul et al., 2006). Accordingly, the Seventh Report of the Joint National Committee (JNC7), the European Society of Hypertension/European Society of Cardiology (2003 ESH-ESC), and the Japanese Society of Hypertension (JSH2004) recommended that ACEIs and ARBs be used in concert with diuretics as first-line therapy to reduce blood pressure in patients with hypertension and renal disease (Chobanian et al., 2003; Cifkova et al., 2003; Ikeda et al., 2006).
Recent attention has been focused on findings that local Ang II levels are differentially regulated in the kidney. Because there often is not clear evidence for markedly elevated circulating renin or Ang II concentrations, identification of local RAS activity is essential for understanding the mechanisms mediating pathophysiological functions. In particular, the Ang II contents in renal tissues are much higher than can be explained on the basis of equilibration with the circulating concentrations (Navar et al., 1997, 1999a,b; Navar and Nishiyama, 2004). Furthermore, the demonstration of much higher concentrations of Ang II in specific regions and compartments within the kidney indicates selective local regulation of intrarenal Ang II (Navar and Nishiyama, 2001, 2004; Ichihara et al., 2004b; Pendergrass et al., 2006). Thus, it is now apparent that intrarenal Ang II levels are regulated in a manner distinct from circulating Ang II concentrations. It has also been revealed that Ang II produced locally in the kidney exerts an important regulatory influence on renal hemodynamics and functions as a paracrine factor (Navar et al., 2000; Paul et al., 2006). Further studies demonstrate that reduced renal function and its structural changes are associated with inappropriate activation of the intrarenal Ang II, leading to the development of hypertension and renal injury (Navar et al., 2003; Navar, 2005).
In this review, we will briefly summarize the paracrine roles of intrarenal Ang II and review recent findings related to its independent regulation with special emphasis on roles in the pathogenesis of hypertension and renal injury. We will also discuss evidence regarding the effects of pharmacological intervention with antihypertensive agents on intrarenal Ang II. The molecular mechanisms responsible for Ang II-induced cell injury have been reviewed by Kim and Iwao (2000) and Touyz and Schiffrin (2000) and will not be discussed in detail in this review.
II. Physiological Actions of Angiotensin II in the Kidney
A. Role of Angiotensin II in the Regulation of Renal Hemodynamics
Exogenous administration of Ang II elicits dose-dependent decreases in renal blood flow and glomerular filtration rate (Yamamoto et al., 2001; Paul et al., 2006). Although there is agreement that Ang II exerts substantial direct effects on the renal microvasculature and glomerular mesangium, there remains controversy regarding the intensity of actions at various sites and the relative contribution of systemically and intrarenally formed Ang II to the overall regulation of renal hemodynamics. The observation that Ang II increases the filtration fraction has frequently been used to support the notion that Ang II predominantly constricts the postglomerular arterioles (Schor et al., 1980; Heller and Horacek, 1986; Alberola et al., 1994). It should be emphasized, however, that this misconception is based on the failure to recognize that an increase in filtration fraction can occur as a consequence of parallel increases in both pre- and postglomerular arteriolar resistances (Navar and Rosivall, 1984; Rosivall et al., 1984; Carmines et al., 1987). Indeed, in vivo micropuncture studies in rats have clearly demonstrated that Ang II elicits reductions in single nephron glomerular filtration rate and glomerular plasma flow and increases in both afferent and efferent arteriolar resistance (Blantz et al., 1976; Baylis and Brenner, 1978; Schor et al., 1980; Rosivall and Navar, 1983). The decreases in glomerular filtration rate are also attributed to the effects of Ang II to reduce the glomerular filtration coefficient, which is thought to be due to changes in contractility of mesangial cells (Blantz et al., 1976; Baylis and Brenner, 1978; Schor et al., 1980; Paul et al., 2006). Because both AT1 and AT2 receptors are expressed in mesangial cells (Sharma et al., 1998), these may influence the glomerular filtration coefficient. However, the exact mechanism by which Ang II regulates the glomerular filtration coefficient remains to be clarified.
Although it was originally reported that Ang II did not constrict isolated rabbit afferent arterioles, there are many reports demonstrating that Ang II constricts both afferent and efferent arterioles (Carmines et al., 1986; Mitchell and Navar, 1988; Loutzenhiser et al., 1991; Ichihara et al., 1997; Yamamoto et al., 2001). Ito et al. (1991, 1993), and Yoshida et al. (1994) showed that inhibition of nitric oxide synthesis markedly augmented the afferent arteriolar responses to Ang II, indicating that high levels of nitric oxide may be present in the dissected afferent arterioles perfused with cell-free solutions. Studies using the in vitro blood-perfused juxtamedullary nephron preparation (Carmines et al., 1986; Ichihara et al., 1997), renal tissue transplantation into hamster cheek pouch (Click et al., 1979), and hydronephrotic rat kidneys (Steinhausen et al., 1987; Dietrich et al., 1991; Loutzenhiser et al., 1991; Inman et al., 1995) also showed similar results. Yamamoto et al. (2001) used an intravital tapered-tip lens-probe video-microscopy system and demonstrated that intrarenal infusion of Ang II constricts both afferent and efferent arterioles in anesthetized dogs. These collective observations indicate that, rather than predominantly constricting efferent arterioles, Ang II elicits vasoconstrictor actions on both pre- and postglomerular resistance vessels; however, the experimental circumstances may influence the reactivity of the afferent more than of the efferent arterioles.
It should be recognized that Ang II elicits the glomerular hemodynamic changes described above without causing significant proteinuria. In both animals and humans, acute Ang II infusion sufficient to change renal hemodynamics does not elicit proteinuria (Loon et al., 1989; Pagtalunan et al., 1995). These observations are in agreement with the prediction based on the mathematical modeling that alterations in glomerular pressure can cause less change in macromolecule filtration if the capillary wall structure is not altered (Bohrer et al., 1977). However, sustained elevation of intrarenal Ang II induces proteinuria accompanied by progressive injury of the glomerular filtration barrier, which is composed of the glomerular endothelium, glomerular basement membrane, and podocytes (glomerular visceral epithelial cell) (Miller et al., 1991; Hoffmann et al., 2004; Whaley-Connell et al., 2006). Locally produced Ang II directly induces podocyte injury via activation of AT1 receptors, independent of hemodynamic changes (Durvasula et al., 2004; Liang et al., 2006; Liebau et al., 2006). Therefore, pharmacological interventions of these effects of Ang II are useful for reducing proteinuria in patients with renal injury.
The overall renal hemodynamic responses to Ang II blockade with ACEIs and ARBs have been quite variable because of the counteracting influences of the associated decreases in systemic arterial pressure. If arterial pressure remains within the renal autoregulatory range, renal blood flow is generally increased by Ang II blockade (Navar et al., 1996; Paul et al., 2006); however, the glomerular filtration rate responses have been much more variable, either increased (Kimbrough et al., 1977; Rosivall et al., 1986; Tamaki et al., 1993), unchanged (Omoro et al., 2000), or decreased (Hall et al., 1979b). In vivo micropuncture studies showed that Ang II blockade increases single nephron filtration rate as well as single nephron plasma flow when arterial pressure is not markedly reduced (Kon et al., 1993; Cervenka et al., 1998; Cervenka and Navar, 1999; Paul et al., 2006). Similarly, intrarenal infusion of subpressor doses of ARBs significantly increased both whole kidney renal blood flow and glomerular filtration rate (Nishiyama et al., 1992; Tamaki et al., 1993), suggesting that Ang II blockade increases the glomerular filtration coefficient. Most clinical studies also show that the glomerular filtration rate remains stable when Ang II blockade is instituted (Andersen et al., 2000; Fridman et al., 2000; Agodoa et al., 2001). The most direct way to explain increases in renal blood flow without changes in glomerular filtration rate is by combined decreases in both pre- and postglomerular arteriolar resistance. In some studies, glomerular filtration rate has been shown to be increased slightly in response to treatment with ACEIs and ARBs (Fridman et al., 1998; Pechère-Bertschi et al., 1998). However, a significant reduction in the glomerular filtration rate has often been seen in patients with renal disease (Hansen et al., 1995; Apperloo et al., 1997). Decreases in arterial pressure in response to Ang II blockade are pronounced during sodium-depleted states (Navar et al., 1996; Paul et al., 2006). Usually, in hypertensive patients with renal disease, ACEIs and ARBs are often added to other drugs, including diuretics, under the conditions where intake of sodium is restricted. Thus, it seems likely that Ang II blockade with ACEIs and ARBs causes a marked reduction in blood pressure, leading to decreases in glomerular filtration rate when extracellular fluid volume is low. In addition, in patients with established glomerular disease, it may be difficult to maintain the glomerular filtration rate by sufficient increases in glomerular filtration coefficient when glomerular pressure is reduced by treatment with ACEIs and ARBs. In patients with more severe renal disease, the afferent arterioles may also become less responsive to ACEIs and ARBs.
In addition to its direct constrictor effects on glomerular arterioles and mesangium, Ang II also regulates renal hemodynamics by exerting a modulatory influence on the sensitivity of the tubuloglomerular feedback mechanism (Navar et al., 1996; Paul et al., 2006). This mechanism provides a balance between the reabsorption capabilities of the tubules and the filtered load by regulating the glomerular filtration rate (Nishiyama et al., 2004a). When flow-dependent changes in the tubular fluid solute concentration at the level of the macula densa in the terminal part of the loop of Henle are sensed, signals are transmitted to the afferent arterioles and glomerular mesangium to constrict or dilate to maintain stability of the filtered load (Navar et al., 1996; Paul et al., 2006). The tubuloglomerular feedback mechanism also participates in autoregulatory responses of renal vascular resistance and glomerular filtration rate (Nishiyama et al., 2004a; Paul et al., 2006). Although it was demonstrated that Ang II does not directly mediate the tubuloglomerular feedback response, its level of activity exerts an important modulatory influence on the sensitivity of the vascular and mesangial elements that respond to signals from the macula densa cells (Ploth, 1983; Schnermann and Briggs, 1986; Mitchell et al., 1992; Braam et al., 1995; Schnermann et al., 1997; Traynor et al., 1999). The tubuloglomerular feedback responsiveness is enhanced during either systemic or peritubular capillary infusion of exogenous Ang II (Schnermann and Briggs, 1986; Mitchell et al., 1992). Furthermore, Ang II blockade with ACEIs and ARBs markedly attenuates the tubuloglomerular feedback responsiveness as assessed by stop-flow pressure feedback responses to increases in distal nephron perfusion rate (Ploth, 1983; Braam et al., 1995). Similarly, both AT1 receptor knockout and ACE-deficient mice have markedly attenuated tubuloglomerular feedback responses to increases in distal nephron perfusion rate (Schnermann et al., 1997; Traynor et al., 1999). Collectively, these findings indicate that Ang II enhances the sensitivity of the vascular and mesangial elements that mediate tubuloglomerular feedback-induced alterations in single nephron function. These effects probably are mediated by direct actions on the vascular smooth muscle cells and mesangial cells as well as by modulating the Na+/H+ exchange activity of the macula densa cells (Peti-Peterdi and Bell, 1998; Kovács et al., 2002). A modulatory influence of Ang II on tubuloglomerular feedback responsiveness shifts the operating point of the system and allows the nephron filtration rate to be maintained at a lower distal nephron volume delivery (Navar et al., 1996; Paul et al., 2006). During conditions of elevated intrarenal Ang II levels, the modulatory influence of Ang II on tubuloglomerular feedback responsiveness is of pivotal importance in maintaining the Ang II-mediated stimulation of proximal tubular reabsorption and the consequent decrease in distal nephron volume delivery. In this manner, the interactive effects of increased Ang II levels to enhance both proximal tubular reabsorption rate and sensitivity of the tubuloglomerular feedback mechanism elicit sustained decreases in distal nephron volume delivery and, thus, urinary sodium excretion.
Enhanced preglomerular vascular tone and blunted microvascular autoregulatory responsiveness to changes in perfusion pressure are observed in Ang II-dependent hypertensive models (Ichihara et al., 1997; Inscho et al., 1999). The blunted autoregulatory responsiveness of the afferent arteriole in Ang II-dependent hypertension apparently results from chronic elevation of Ang II levels because acute exposure to 10-fold greater concentrations of Ang II does not affect autoregulatory behavior (Inscho et al., 1996). Chronic treatment with ARBs prevents the deterioration of renal autoregulatory responsiveness in Ang II-infused rats (Inscho et al., 1999). However, Ang II blockade does not affect renal autoregulatory behavior in normal animals (Navar et al., 1986; Persson et al., 1988).
B. Role of Angiotensin II in the Regulation of Tubular Function
Ang II is one of the most powerful sodium-retaining hormones in the body. The direct intrarenal actions of Ang II that contribute to increased tubular reabsorption are complex, including constriction of glomerular arterioles, which alter peritubular capillary dynamics and renal medullary blood flow, and direct actions on tubular epithelial cell transport. Although the quantitative contribution of each of these hemodynamic and tubular actions may vary in different physiological circumstances, high intrarenal Ang II levels contribute to salt and water retention through direct actions on renal tubular transport function when inappropriately stimulated (Navar and Nishiyama, 2004).
Ang II is also one of the body's most important regulators of aldosterone, which stimulates sodium reabsorption, primarily through the mineralocorticoid receptors in the connecting and cortical segments of the collecting tubule. Furthermore, Ang II directly enhances urinary concentration in the collecting tubule and collecting ducts.
Because all of the components of the RAS are found in the kidney and significant amounts of Ang II can be formed locally, considerable interest has focused on the possibility that intrarenally formed Ang II may be more important than circulating Ang II in controlling renal function. Several studies demonstrated the fact that intrarenal infusion of ARBs or ACEIs, at rates that produced no changes in plasma aldosterone concentration and minimal effects on systemic hemodynamics, increased sodium excretion (Kimbrough et al., 1977; Hall et al., 1979a; Klag et al., 1996; Cervenka et al., 1998). Intrarenal infusion of Ang I, to stimulate local formation of Ang II, also reduced sodium excretion (Rosivall and Navar, 1983). These results emphasize the contribution of intrarenally formed Ang II in regulating sodium excretion.
In addition to maintaining fluid and electrolyte homeostasis, Ang II participates in a variety of tubular functions, including induction of cellular hypertrophy and oxidative stress. Details of biological function specific for each tubular segment are described below.
1. Proximal Tubules.
Normally, the potent antinatriuretic effects of Ang II are due primarily to increased tubular reabsorption rather than to reductions in glomerular filtration rate (Hall et al., 1986; Mitchell et al., 1992). In vivo perfusion of rat proximal tubules with an ultrafiltrate-like solution containing either ACEIs or ARBs decreased the volume reabsorption, suggesting modification of proximal tubule transport by locally produced Ang II independent from the systemic RAS (Quan and Baum, 1996).
Microperfusion studies of isolated proximal tubules have shown that the Ang II effect on proximal tubule sodium transport is bimodal; Ang II at physiological concentrations (picomoles per liter) significantly stimulates proximal tubule sodium reabsorption, whereas pharmacological micromole per liter concentrations inhibit transport (Harris and Young, 1977; Schuster et al., 1984). Reabsorption of sodium by Ang II in proximal tubules is coupled with bicarbonate reabsorption, which is mediated by inhibition of adenylate cyclase (Liu and Cogan, 1989). Using in vivo microperfusion in the Munich-Wistar rat, Liu and Cogan (1987) showed that administration of luminal Ang II increased proximal tubule bicarbonate reabsorption. These findings were confirmed using electrophysiological methods in isolated perfused rabbit renal proximal tubules (Coppola and Fromter, 1994a,b). Perfusion of rabbit proximal tubules with luminal Ang II after treatment with ACEIs increased volume and bicarbonate reabsorption (Baum et al., 1997), supporting the role of intrarenally produced Ang II to stimulate proximal tubule volume and bicarbonate transport.
Molecular mechanisms of direct stimulation of fluid reabsorption by Ang II within the proximal tubule involve increased transcellular sodium and bicarbonate reabsorption via activation of apical Na+/H+ exchange, basolateral Na+-HCO3- cotransport, and basolateral Na+/K+-ATPase and via insertion of H+-ATPase into the apical membrane (Liu and Cogan, 1988; Garvin, 1991; Mitchell et al., 1992; Eiam-Ong et al., 1993; Wang and Giebisch, 1996). Stimulation of Na+-HCO3- cotransport by Ang II is mediated by diverse signaling pathways, including activation of the Src family of tyrosine kinase and the classic mitogen-activated protein kinase pathway (Espiritu et al., 2002; Robey et al., 2002).
In view of these observations that the intratubular activation of the RAS stimulated proximal fluid reabsorption, recent analysis of tissue-specific ACE knockout mice using micropuncture techniques gave unexpected results (Hashimoto et al., 2005). In this unique model, tissue ACE is deleted, but ACE is selectively expressed in the liver (Cole et al., 2003). Whereas disruption of ACE often causes low blood pressure, which complicates renal functional studies, this model is able to maintain sufficient plasma levels of ACE and subsequently normal blood pressure. Proximal tubular fluid reabsorption of these genetically altered mice was comparable with that observed in wild-type mice despite the essentially complete absence of tissue ACE. These findings are in contrast with the previous findings that an acute reduction in local Ang II formation exerted a profound inhibitory effect on fluid reabsorption. The discrepancy may lie in the chronicity of Ang II blockade, and chronic ACE deficiency is apparently associated with compensatory events that normalize fluid reabsorption along the proximal tubule. It is also possible that proximal tubular Ang II is formed through alternative pathways not requiring ACE.
In addition to regulation of fluid and electrolyte balance, Ang II plays an important role in hypertrophy of proximal tubular cells. In rat proximal tubular epithelial cells, Ang II induces cellular hypertrophy and activates relevant downstream signal transduction pathways (Wolf et al., 1993; Hannken et al., 1998, 2000; Guo et al., 2004). The Ang II-induced tubular cell hypertrophy is inhibited by ARBs, suggesting that the AT1 receptor contributes to the tubular cell hypertrophy (Chatterjee et al., 1997). Cells undergoing hypertrophy are arrested in the G1 phase of the cell cycle, and p27Kip1, an inhibitor of cyclin-dependent kinases, is required for Ang II-induced hypertrophy of proximal tubular cells (Wolf and Stahl, 1996; Terada et al., 1999; Wolf et al., 2001, 2003).
Transfection of AT1 receptors into a renal proximal tubular cell line LLCPKcl4, which does not express endogenous Ang II receptors, increased protein synthesis without DNA synthesis in response to Ang II, as indicated by increased [3H]leucine incorporation without increases in [3H]thymidine incorporation (Burns and Harris, 1995). The stimulation of protein synthesis and cell hypertrophy without increasing cell number was mediated by activation of the epidermal growth factor receptor (Chen et al., 2006). Recent studies also demonstrated involvement of connective tissue growth factor in mediating Ang II-induced tubular cell hypertrophy (Liu et al., 2006). In cultured proximal tubular cells, Ang II stimulated the expression of connective tissue growth factor and increased the total protein content as well as cell size, which were markedly inhibited by cotreatment with an antisense oligonucelotide for connective tissue growth factor.
With blockade of transforming growth factor-β receptor, Ang II-mediated hypertrophy can be converted into cell proliferation. Rats that received Ang II infusion had an increased number of proliferating cell nuclear antigen- and transferase dUTP nick-end labeling-positive cells in proximal tubules with a possible involvement of AT2 receptors (Cao et al., 2000), suggesting that Ang II also triggers both proliferation and apoptosis in tubular epithelial cells under certain circumstances.
A role for Ang II in induction of oxidative stress in the kidney has been extensively studied. Treatment of Wistar-Kyoto rats with subcutaneous Ang II infusions from osmotic minipumps induced oxidative stress in association with increased expression of the p22phox component of NADPH oxidase and decreased expression of extracellular superoxide dismutase in the renal cortex (Welch et al., 2005). These effects were mediated via AT1 receptors and were offset by protective effects of AT2 receptors (Chabrashvili et al., 2003). Measurement of PO2 in the lumen of proximal tubules and distal tubules gave low values, which can be ascribed to inefficient utilization of O2 due to oxidative stress. Therefore, it is likely that Ang II induced oxidative stress in both proximal and distal tubules.
Another function of renal proximal tubule cells regulated by Ang II is endocytosis of urinary protein components. Ang II at physiological concentrations as low as 1 nM increased albumin endocytosis through AT2 receptors located on the luminal side and triggered the activation of protein kinase B in a porcine proximal tubular cell line (Caruso-Neves et al., 2005). This report clearly indicates that Ang II is also involved in the regulation of endocytosis of urinary protein in renal proximal tubule cells under physiological conditions.
2. Distal Tubules.
Ang II infusion increased distal fractional sodium reabsorption (Olsen et al., 1985). Intravenous infusion of Ang II stimulated distal bicarbonate reabsorption during microperfusion experiments (Levine et al., 1994). Ang II also regulated distal bicarbonate reabsorption during modifications of food intake in the rat (Levine et al., 1996). Studies of separate perfusions of early and late segments of cortical distal tubule showed that Ang II stimulated early distal bicarbonate reabsorption, whereas the late distal effect was mostly on amiloride-sensitive sodium reabsorption, i.e., on sodium channels (Wang and Giebisch, 1996). Ang II acts to stimulate Na+/H+ exchange in both early and late distal segments via activation of AT1 receptors and the vacuolar H+-ATPase in late distal segments (Barreto-Chaves and Mello-Aires, 1996). Experiments performed in distal tubules of nephrectomized rats indicated that AT1 receptor blockade caused marked reduction of synthesis and insertion of apical H+-ATPase in A-type intercalated cells (Levine et al., 2000). The effects of Ang II on sodium reabsorption in distal tubular segments further enhance and amplify the effects in proximal tubules, leading to much greater overall efficiency of sodium conservation.
3. Collecting Ducts.
In the proximal and distal tubules, Na+ serves as a counterion for H+ secretion. Thus, Ang II augments H+ secretion and Na+ absorption in these tubular segments. Despite a dramatic up-regulation of H+ secretion in the proximal and distal tubules by Ang II, infusion of Ang II does not produce a metabolic alkalosis, suggesting a compensatory regulation of acid secretion in other segments of the nephron. To support this notion, Ang II decreased H+ secretion in the perfused rat outer medullary collecting ducts (Weiner et al., 1995; Wall et al., 2003). This can be explained by a reduction in H+-ATPase activity (Tojo et al., 1994; Valles and Manucha, 2000). However, different results regarding the effect of Ang II on H+ secretion have been reported. Whereas selective aldosterone deficiency created by adrenalectomy with glucocorticoid replacement resulted in down-regulation in the expression of the H+-ATPase B1 subunit in medullary collecting ducts, Ang II increased the expression of the B1 subunit of H+-ATPase in the medullary collecting ducts and thus may up-regulate H+ secretion in this tubular segment of these animals (Valles et al., 2005).
Ang II also plays an important role in regulation of the sodium channel in the collecting ducts via a mechanism that is not dependent on circulating aldosterone. In isolated perfused rabbit cortical collecting ducts, Ang II directly stimulated apical membrane epithelial sodium channel activity (Peti-Peterdi et al., 2002). With low-salt diets, associated with activation of the RAS, the expression of the α-epithelial sodium channel was markedly decreased in AT1a receptor knockout mice (Brooks et al., 2002).
The inner medullary collecting ducts are responsible for the final concentration of the urine. Mice with gene deletion of the AT1a receptor exhibit defects in urinary concentrating ability (Oliverio et al., 2000). The effects of RAS activation in the inner medullary collecting ducts may be mediated by stimulation of urea transport, which maintains the medullary interstitial osmotic gradient. In rat terminal inner medullary collecting ducts, low concentrations of basolateral Ang II increases vasopressin-stimulated urea permeability and induces phosphorylation of the urea transporter (Kato et al., 2000). These data suggest that Ang II stimulates the urinary concentrating mechanism, leading to increased water reabsorption.
In addition to direct effects, Ang II regulates function of the collecting ducts via aldosterone. Ang II stimulates the zona glomerulosa of the adrenal cortex to produce the sodium-retaining hormone, aldosterone. Aldosterone stimulates ionic transport in the principal cells by increasing the number of open sodium and potassium channels in the luminal membrane and the activity of Na+/K+-ATPase pump in the basolateral membrane. Thus, aldosterone promotes sodium chloride reabsorption and potassium secretion in the principal cells of the cortical collecting tubular segment of the nephron. It further stimulates H+ secretion in the intercalated cells of the cortex and tubular cells in the outer medulla (Navar et al., 1996; Paul et al., 2006).
III. Regulation of Circulating Renin-Angiotensin System—Classic Renin-Angiotensin System Pathways
Ang II is produced systemically via the classic RAS. An aspartyl protease, renin, in the plasma is released primarily from the juxtaglomerular cells on the afferent arterioles of the kidney (Hackenthal et al., 1990; Schnermann et al., 1997). Although circulating active renin and prorenin are released mainly from the kidney, other tissues also secrete prorenin into the circulation, and prorenin can be converted to renin by limited proteolysis such as that with trypsin activation in the circulation (Sealey et al., 1986). Angiotensinogen is primarily formed and constitutively secreted by hepatic cells into the circulation, thus allowing systemic formation of Ang II throughout the circulation (Brasier and Li, 1996). On release into the circulation, renin cleaves angiotensinogen at the N terminus to form the decapeptide, Ang I (Navar et al., 1997). The circulating concentrations of angiotensinogen are abundant, being more than 1000 times greater than the plasma Ang I and Ang II concentrations (Navar and Nishiyama, 2001). Although some species variation exists, changes in renin activity thus determine the rate of Ang I formation in the plasma from the huge stores of circulating angiotensinogen (Ichihara et al., 2004b; Paul et al., 2006). Figure 1 shows the representative plasma angiotensinogen concentrations measured in anesthetized rats and expressed as nanomoles per liter; the Ang I and Ang II concentrations are expressed as picomoles per liter, indicating that the active Ang II concentration in the plasma is a small fraction of the available Ang II in the form of angiotensinogen. Therefore, even small relative changes in the rates of Ang I and Ang II generation may make large absolute differences in the circulating concentrations. As is well known, renin is synthesized and stored in substantial quantities in the granules of juxtaglomerular cells and is released in response to various stimuli (Schweda and Kurtz, 2004; Paul et al., 2006). Thus, large changes in plasma renin levels can occur rapidly, leading to changes in Ang I generation. The concentrations of angiotensinogen in the plasma are close to the Michaelis-Menten constant of the proteolytic activity of renin such that changes in substrate concentrations can also influence the Ang I generation rate; however, changes in angiotensinogen synthesis occur slowly and thus are less responsible for the dynamic regulation of plasma Ang I and Ang II than renin (Deschepper, 1994; Brasier and Li, 1996). Ang I is easily converted to Ang II, due not only to the circulating dipeptidyl carboxypeptidase, ACE, but also to the widespread presence of ACE on endothelial cells of many vascular beds including the lung (Navar et al., 1997; Ichihara et al., 2004b; Paul et al., 2006). Although other pathways for Ang II formation have been identified in certain tissues (Fig. 1), the circulating levels of Ang II reflect primarily the consequences of the renin and ACE enzymatic cascade on angiotensinogen (Erdös, 1990; Johnston, 1994). The resultant increases in plasma Ang II exert powerful actions throughout the body through activation of AT1 receptors (Timmermans et al., 1993; Paul et al., 2006). Several angiotensinases and peptidases are then able to metabolize Ang II further (Reudelhuber, 2005). It is recognized that several of the smaller peptides, including Ang III, Ang IV, and Ang 1-7, have biological activity, but their plasma levels (except for Ang1-7) are much lower than those of Ang II (Haulica et al., 2005; Pendergrass et al., 2006).

Brief scheme of the systemic RAS. The representative plasma concentrations for angiotensinogen, Ang I, and Ang II in anesthetized rats are shown. The fine details of regulatory function of the RAS products may differ depending on the environment. tPA, tissue plasminogen activator.
IV. Mechanisms Responsible for Independent Regulation of Intrarenal Renin-Angiotensin System
The RAS has been acknowledged as an endocrine, paracrine, autocrine, and intracrine system (Navar et al., 2002; Re, 2003; Kobori et al., 2006; Re and Cook, 2006; Suzaki et al., 2006b), and, thus, it has been difficult to delineate the quantitative contributions of systemically delivered versus locally formed Ang peptides to the levels existing in any given tissue. Emerging evidence suggests that local formation is of major significance in the regulation of the Ang levels in many organs and tissues. For example, there is substantial evidence that the Ang peptide levels in the brain are regulated in an autonomous manner (Baltatu et al., 2000). Although every organ system in the body has elements of the RAS, the kidney is unique in having every component of the RAS with compartmentalization in the tubular and interstitial networks as well as intracellular accumulation. Recent attention has been focused on the existence of unique RASs in various organ systems. Various studies have demonstrated the importance of the tissue RAS in the brain, heart, adrenal glands, and vasculature as well as in the kidney (Mitchell and Navar, 1995; Navar et al., 2006). In this regard, the kidneys, as well as the adrenal glands, are unique in terms of the tissue concentrations of Ang II, which are much greater than can be explained by the concentrations delivered by the arterial blood flow (Ingert et al., 2002a). There is substantial evidence that the major fraction of Ang II present in renal tissues is generated locally from angiotensinogen delivered to the kidney as well as from angiotensinogen locally produced by proximal tubule cells. Ang I delivered to the kidney can also be converted to Ang II (Rosivall and Navar, 1983; Komlosi et al., 2003). Renin secreted by the juxtaglomerular apparatus cells and delivered to the renal interstitium and vascular compartment also provides a pathway for the local generation of Ang I (Hackenthal et al., 1990; Schnermann et al., 1997). ACE is abundant in the rat kidney and has been located in the proximal and distal tubules, the collecting ducts, and renal endothelial cells (Casarini et al., 1997). Therefore, all of the components necessary to generate intrarenal Ang II are present along the nephron.
A. Angiotensinogen
Although most of the circulating angiotensinogen is produced and secreted by the liver, the kidneys also produce angiotensinogen (Kobori et al., 2006). Intrarenal angiotensinogen mRNA and protein have been localized to proximal tubule cells, indicating that the intratubular Ang II could be derived from locally formed and secreted angiotensinogen (Darby and Sernia, 1995). The angiotensinogen produced in proximal tubule cells seems to be secreted directly into the tubular lumen in addition to producing its metabolites intracellularly and secreting them into the tubule lumen (Lantelme et al., 2002). Proximal tubule angiotensinogen concentrations in anesthetized rats have been reported in the range of 300 to 600 nM, which greatly exceed the free Ang I and Ang II tubular fluid concentrations (Navar et al., 2001). Because of its molecular size, it seems unlikely that much of the plasma angiotensinogen filters across the glomerular membrane, further supporting the concept that proximal tubule cells secrete angiotensinogen directly into the tubule (Rohrwasser et al., 1999). To determine whether circulating angiotensinogen is a source of urinary angiotensinogen, Kobori et al. (2003b) infused human angiotensinogen into normotensive rats; however, circulating angiotensinogen was not detectable in the urine. The failure to detect human angiotensinogen in the urine indicates limited glomerular permeability and/or tubular degradation. These findings support the hypothesis that urinary angiotensinogen originates from the angiotensinogen that is formed and secreted by the proximal tubules and not from plasma in rats (Kobori et al., 2003b). Formation of Ang I and Ang II in the tubular lumen subsequent to angiotensinogen secretion may be possible because some renin is filtered and/or secreted from juxtaglomerular apparatus cells. The identification of renin in distal nephron segments may also provide a possible pathway for Ang I generation from proximally delivered angiotensinogen. Intact angiotensinogen in urine indicates its presence throughout the nephron and, to the extent that renin and ACE are available along the nephron, substrate availability supports continued Ang I generation and Ang II conversion in distal segments (Ding et al., 1997; Davisson et al., 1999). Once Ang I is formed, conversion readily occurs because there are abundant amounts of ACE associated with the proximal tubule brush border. Casarini et al. (1997) found that ACE activity is present in tubular fluid throughout the nephron except in the late distal tubule. They demonstrated that the ACE activity is higher at the initial portion of the proximal tubule but then decreases to the distal nephron and increases again in the urine. This evidence suggests proximal ACE secretion, degradation, and/or reabsorption associated with secretion in the collecting ducts. Therefore, intratubular Ang II formation may occur not only in the proximal tubule but also beyond the connecting tubule (Fig. 2). Thus, renal tissue ACE activity is critical to maintain the steady-state Ang II levels in the kidney. Indeed, Modrall et al. (2004) demonstrated that knockout mice that do not exhibit bound tissue ACE in the kidney have 80% lower intrarenal Ang II levels compared with wild-type mice. In addition to the marked reduction of intrarenal Ang II levels, this tissue ACE knockout mouse showed significant depletion of its immediate precursor Ang I in renal tissue, which supports the concept that Ang II exerts a positive feedback loop on proximal angiotensinogen (Ingelfinger et al., 1999; Kobori et al., 2001a,b, 2002).

Intrarenal RAS in proximal and distal nephron segments. In Ang II-dependent hypertension, increased proximal tubular secretion of angiotensinogen spills over into the distal nephron and increases Ang II effects on distal tubular reabsorption. AGT, angiotensinogen; PT, proximal tubules; DT, distal tubules; CD, collecting ducts.
The proximally formed angiotensinogen that is secreted into the tubular fluid flows into the distal nephron, allowing intraluminal Ang II formation to continue throughout the length of the nephron with the residual angiotensinogen appearing in the urine (Ding et al., 1997; Rohrwasser et al., 1999). Ding et al. (1997) demonstrated in mice harboring the gene for human angiotensinogen fused to the kidney-specific androgen-regulated protein promoter that human angiotensinogen was localized primarily to proximal tubule cells (see section V.A.1.c.). They found abundant human angiotensinogen in the urine but only slight traces in the systemic circulation. This finding suggests that most of the angiotensinogen formed in proximal tubule cells is destined for secretion into the lumen. Rohrwasser et al. (1999) demonstrated luminal localization of angiotensinogen in proximal tubular cells in vivo and showed, in monolayer proximal tubule cell cultures, that most of the angiotensinogen was detected near the apical membrane. They also reported that angiotensinogen was detected at low nanomoles per liter concentrations in urine from mice and human volunteers. Kobori et al. (2002) evaluated the changes in urinary angiotensinogen excretion rates in Ang II-infused rats maintained on high-salt diets to suppress basal levels and observed an approximately 4-fold increase with Ang II infusion (80 ng/min) in urinary angiotensinogen excretion rates. Angiotensinogen was measured using both Western blot analysis and radioimmunoassay determination of generated Ang I after incubation with excess renin, thus demonstrating the fact that urinary angiotensinogen contained intact active angiotensinogen. They extended these results further to show that chronic Ang II infusions to normal rats significantly increased the urinary excretion rate of angiotensinogen in a time- and dose-dependent manner that was associated with elevations in systolic blood pressure and kidney Ang II levels but not with plasma Ang II concentrations (Kobori et al., 2003b). To determine whether the increase in urinary angiotensinogen excretion was simply a nonspecific consequence of the proteinuria and hypertension, further studies were done in rats made hypertensive with DOCA salt plus a high-salt diet. Although urinary protein excretion in DOCA salt-induced volume-dependent hypertensive rats was increased to the same or to a greater extent, urinary angiotensinogen was significantly lower in volume-dependent hypertensive rats than in Ang II-dependent hypertensive rats and was not greater than in control rats. This study also demonstrated that there was a significant relationship between urinary angiotensinogen and kidney Ang II content in rats given different doses of Ang II to achieve different levels of hypertension. These results provide further evidence that urinary angiotensinogen may be a useful index of intrarenal Ang II activity (Kobori et al., 2002, 2003b, 2004) (Fig. 2). Recently, two independent groups have developed an enzyme-linked immunosorbent assay system to measure angiotensinogen directly (Lantelme et al., 2005; Suzaki et al., 2006a). Outcomes of clinical studies are expected in the near future.
B. Renin and Prorenin
Strictly speaking, renin is not a hormone; however, it can be considered as such because of its role in determining Ang I generation and because it is subject to tight control. Hence, the plasma renin concentration or activity is often used as a measure of the overall activity of the RAS. In most species, renin synthesized by the juxtaglomerular apparatus cells is the primary source of both circulating and intrarenal renin levels. However, some strains of mice also produce substantial amounts of renin in the submandibular and submaxillary glands as an expression of the duplicated renin gene, Ren2 (Catanzaro et al., 1983).
The secreted active form of renin contains 339 to 343 amino acid residues after proteolytic removal of the 43-amino acid residue at the N terminus of prorenin. Circulating active renin and prorenin are released mainly from the kidney, but other tissues also secrete prorenin into the circulation (Sealey et al., 1986). Besides serving as the precursor for active renin, it has been suggested that circulating prorenin is taken up by some tissues where it may contribute to the local synthesis of Ang peptides (Prescott et al., 2002). In the heart under normal conditions, renin is not produced and its transcript is undetectable or extremely low (Ekker et al., 1989). Nevertheless, transgenic rats expressing the Ren2 renin gene exhibit high circulating prorenin levels in the absence of the cardiac transgene, prorenin internalization into cardiomyocytes with generation of Ang, and cardiac damage (Peters et al., 2002). These effects suggest that uptake of circulating prorenin but not active renin may play an important role in cardiac hypertrophy.
Although there have been suggestions that renin itself or perhaps prorenin may directly elicit cellular effects, independent of the generation of Ang II, the well established role of renin is to act on angiotensinogen, a protein with a glycosylated weight of 52 to 64 kDa and synthesized primarily by the liver to form the decapeptide Ang I. However, the (pro)renin receptor may also initiate intracellular signaling to activate extracellular signal-regulated kinases 1/2 (Nguyen et al., 2002) and p38 mitogen-activated protein kinase (Saris et al., 2006). In the heart and kidney, the recently described renin receptor (Nguyen et al., 1996) binds renin and prorenin, leading to an increase in the catalytic efficiency of Ang I formation from angiotensinogen. It has also been reported recently that the binding of prorenin to an intrinsic prorenin-binding receptor plays a pivotal role in the development of diabetic nephropathy by a mechanism that involves the receptor-associated prorenin system (Ichihara et al., 2004a,b).
It should be recognized that juxtaglomerular apparatus cells are not the only intrarenal structures in which renin has been localized. Kidneys from rats treated chronically with ACEIs also exhibit renin immunoreactivity of the afferent arteriole extending well beyond the juxtaglomerular apparatus loci up toward the interlobular artery, suggesting that ACE inhibition induces a recruitment of cells that in the basal state were not expressing the renin gene (Gomez et al., 1988). Positive renin immunoreaction has been observed in cells of glomeruli and of proximal and distal nephron segments as well as its mRNA (Moe et al., 1993). In addition, renin mRNA and protein expression have been also reported in proximal and distal nephron segments (Rohrwasser et al., 1999; Lantelme et al., 2002; Prieto-Carrasquero et al., 2004). Using immunoblotting, Rohrwasser et al. (1999) found that renin was secreted by microdissected arcades of connecting tubule cells, indicating that renin is probably secreted into the distal tubular fluid. They also demonstrated that renin activity was observed in excreted urine (Rohrwasser et al., 2003).
C. Angiotensin-Converting Enzyme
Ang I is rapidly converted into the major effector of this system, Ang II, by ACE, which is located on endothelial cells in many vascular beds and on membranes of various other cells including brush border membranes of proximal tubules (Schulz et al., 1988; Mezzano et al., 2003a,b). The localization of ACE within the kidney in various species has been well characterized. However, there are some important differences between humans and commonly used experimental animals (Metzger et al., 1999). Indeed, Metzger et al. (1999) reported that kidneys from normal human subjects predominantly expressed ACE in the brush border of proximal tubular segments, and very little ACE expression was observed on vascular endothelial cells. ACE was not detectable in the vasculature of the glomerular tuft or even in the basolateral membranes of epithelial cells. In contrast, there was intense labeling on the endothelial cells of almost all of the renal microvasculature of rats. However, kidneys from human subjects with non-neoplastic diseases manifested increased expression on vascular endothelial cells (Metzger et al., 1999). These data indicating much lower endothelial expression in renal vascular endothelial cells in humans help explain the much lower Ang I to Ang II conversion rates that have been reported for human kidneys compared with those of other species (Danser et al., 1998). Danser et al. (1998) reported that less than 10% of arterially delivered Ang I is converted to Ang II, which is much lower than the amount reported for dogs (Rosivall et al., 1983). The reduced ACE expression on renal vascular endothelial cells in humans implies that the influence of intrarenal Ang II formed from circulating precursors may not be of major significance.
D. Angiotensin II Receptors
Most of the actions of Ang II on renal function are the consequence of activation of Ang II receptors, which are widely distributed in various regions and cell types of the kidney. Two major categories of Ang II receptors, type 1 (subtypes 1a and 1b) and type 2, have been described, pharmacologically characterized, and cloned (Murphy et al., 1991; Sasamura et al., 1992; Nakajima et al., 1993). However, most of the Ang II hypertensinogenic actions are generally attributed to the AT1 receptors (Ito et al., 1995). AT1 receptor transcript has been localized to proximal tubules, the thick ascending limb of the loop of Henle, glomeruli, arterial vasculature, vasa recta, arcuate arteries, and juxtaglomerular cells (Tufro-McReddie et al., 1993b). In rodents, there are two AT1 receptor subtypes, with type 1a being the predominant subtype in all nephron segments, whereas type 1b is more abundant than type 1a only in the glomerulus (Bouby et al., 1997). In mature kidneys, type 1a receptors have been localized to the luminal and basolateral membranes of several segments of the nephron, as well as on the renal microvasculature in both cortex and medulla, smooth muscle cells of afferent and efferent arterioles, epithelial cells of the thick ascending limb of Henle, proximal tubular apical and basolateral membranes, mesangial cells, distal tubules, collecting ducts, and macula densa cells (Paxton et al., 1993; Harrison-Bernard et al., 1997; Miyata et al., 1999). This evidence is consistent with the localization of the transcript for the AT1 receptor subtypes in all of the renal tubular and vascular segments (Miyata et al., 1999). Nevertheless, renal microvascular functional studies obtained from mice lacking the type 1a receptor gene have shown that the afferent arteriole has both type 1a and type 1b receptors, whereas the efferent arteriole only expresses type 1a receptors (Harrison-Bernard et al., 2003).
The regulation of intrarenal Ang II receptors in hypertensive conditions is complex because vascular and tubular receptors respond differently during high Ang II states (Navar et al., 2002). In general, high Ang II levels associated with a low-salt diet decrease glomerular AT1 receptor expression but increase tubular AT1 receptor levels (Cheng et al., 1995). Studies in two-kidney, one-clip Goldblatt hypertensive rats demonstrated that glomerular AT1 receptors were decreased by 2 weeks after clipping, but vascular receptors were not decreased until 16 weeks (Amiri and Garcia, 1997). However, glomerular AT1 receptor density was not increased in the one-kidney-one-clip model, although vascular AT1 receptor density was increased (Amiri et al., 1999). In the Ang II-infused rat model of hypertension, total kidney AT1 receptor mRNA levels and protein levels were not suppressed but were maintained by 2 weeks of Ang II infusion sufficient to cause marked hypertension (Harrison-Bernard et al., 1999). However, Wang et al. (1999) reported that type 1 receptor protein was reduced in both ischemic and contralateral kidneys of two-kidney, one-clip Goldblatt and two-kidney, one-wrap Grollman hypertensive models and in kidneys of Ang II-infused rats. AT2 receptors were down-regulated only in ischemic kidneys. In transgenic rats harboring the mouse Ren2 renin gene, Zhuo et al. (1999) found increased AT1 receptor binding in vascular smooth muscle of afferent and efferent arterioles, juxtaglomerular apparatus, glomerular mesangial cells, proximal tubular cells, and renomedullary interstitial cells. It was suggested that up-regulation of AT1 receptors in multiple renal cells may contribute to the pathogenesis of hypertension in these rats. Harrison-Bernard et al. (2002) extended the analysis in Ang II-infused rats with in vitro autoradiography and showed differential responses with significant decreases in glomeruli and inner stripe but not in proximal tubules. Furthermore, ACE binding was significantly increased in proximal tubules of Ang II-infused rats. Thus, vascular and glomerular AT1 receptors are down-regulated, but the proximal tubular receptors are either up-regulated or not significantly altered in Ang II-dependent hypertension.
The AT2 receptor is highly expressed in human and rodent kidney mesenchyme during fetal life and decreases dramatically after birth (Norwood et al., 2000). AT2 receptors have been localized to the glomerular epithelial cells, proximal tubules, collecting ducts, and parts of the renal vasculature of the adult rat (Miyata et al., 1999). Although the role of AT2 receptors in regulating renal function remains uncertain, it has been suggested that AT2 receptor activation counteracts AT1 receptor effects by stimulating formation of bradykinin and nitric oxide, leading to increases in interstitial fluid concentration of cyclic guanosine monophosphate (Siragy and Carey, 1999). AT2 receptor activation seems to influence proximal tubule sodium reabsorption either by a cell membrane receptor-mediated mechanism or by an interstitial nitric oxide-cyclic guanosine monophosphate pathway (Jin et al., 2001). Ang II infusion into AT2 receptor knockout mice leads to exaggerated hypertension and reductions in renal function, probably due to decreased renal interstitial fluid levels of bradykinin and cyclic guanosine monophosphate available that counteract the direct effect of Ang II (Siragy et al., 1999).
E. Intrarenal Angiotensin II
1. Interstitial and Tubular Angiotensin II.
Intrarenal Ang II is not distributed in a homogeneous fashion but is compartmentalized in both a regional and segmental manner (Navar et al., 2001). Earlier studies indicated that medullary Ang II levels are higher than the cortical levels in normal rats and increase further in Ang II-infused hypertensive rats (Navar et al., 1997). The combination of high Ang II levels in the medulla coupled with the high density of Ang II receptors suggest that Ang II exerts a major role in regulating hemodynamics and tubular function in the medulla (Harrison-Bernard et al., 2002; Pendergrass et al., 2006). The higher Ang II levels in the medulla suggest that there may be specialized Ang II-forming pathways or accumulation mechanisms in medullary tissues that are subject to local regulation. However, Ingert et al. (2002a,b) failed to confirm the fact that medullary Ang II contents are higher than cortical Ang II contents. These authors found that Ang I and Ang II levels in cortex and medulla are equivalent and respond in a similar manner to alterations in dietary salt intake.
Within the cortex, there is distribution of Ang II in the interstitial fluid, tubular fluid, and intracellular compartments. The interstitial as well as the intratubular compartments contribute to the disproportionately high total Ang II levels. Studies using microdialysis probes implanted in the renal cortex demonstrated that Ang II concentrations in interstitial fluid are much higher than the plasma concentrations, with recent results suggesting values in the range of 3 to 5 nM (Siragy et al., 1995; Siragy and Carey, 1999; Nishiyama et al., 2002a,b). Increases in renal interstitial fluid Ang II levels have been reported for two models of hypertension. Siragy and Carey (1999) found that renal interstitial Ang II is increased in the wrapped kidney of rats with two-kidney, one-wrap Grollman hypertension. Nishiyama et al. (2003b) reported that renal interstitial fluid Ang II concentrations are also increased in rats infused with Ang II for 2 weeks. Because the renal interstitial values are so much higher than can be explained on the basis of equilibration with the plasma concentrations, the data suggest local regulation of Ang II formation in the renal interstitial compartment and enhancement of interstitial Ang II production in Ang II-dependent hypertension.
Measurements of tubular fluid Ang II concentrations in anesthetized rats have not revealed significant differences among control rats and several hypertensive models (Mitchell and Navar, 1987; Wang et al., 1997; Cervenka et al., 1999). Considering that kidneys of the hypertensive rats are markedly renin depleted and exposed to elevated arterial pressures, the maintenance of high proximal tubular Ang II concentrations reflects an inappropriate maintenance of intrarenal Ang II formation levels. Nevertheless, the results so far have not demonstrated further elevations in proximal tubule Ang II concentrations above the levels found in normal anesthetized rats. In normal rats, volume expansion failed to suppress proximal tubule Ang II concentrations, but increased levels were documented after reductions in renal perfusion pressure (Boer et al., 1997).
The Ang II concentrations in tubular fluid from the other segments of the nephron remain unknown. Several studies support an important role for Ang II in regulating reabsorptive function in distal nephron and collecting duct segments, as well as in proximal tubule segments, which activate the Ang II receptors on the luminal borders (Navar et al., 1999b). Recently, a direct action of Ang II on the luminal amiloride-sensitive epithelial sodium channel was reported (Peti-Peterdi et al., 2002; Komlosi et al., 2003). These data indicate that when luminal distal nephron Ang II concentrations are augmented, they could contribute directly to the regulation of distal tubule and collecting duct sodium reabsorption.
2. Intracellular Angiotensin II.
As indicated earlier, some of the Ang II that binds to receptors is internalized via AT1 receptor-mediated endocytosis (Zou et al., 1998; Ingert et al., 2002a; Li et al., 2006; Zhuo et al., 2006). Imig et al. (1999) provided direct evidence of endosomal accumulation of Ang II in intermicrovillar clefts and endosomes of Ang II-infused hypertensive rats. It was also shown that AT1 receptor blockade with candesartan prevented the endosomal accumulation even though plasma Ang II increased, further demonstrating the importance of AT1 receptor-mediated uptake (Zhuo et al., 2002). The presence of Ang II in renal endosomes indicates that some of the internalized Ang II remains intact and contributes to the total Ang II content measured in tissue homogenates (Navar et al., 2002; Zhuo et al., 2002). As shown for proximal tubule cells, endocytosis of the Ang II-AT1 receptor complex seems to be required for the full expression of functional responses coupled to the activation of signal transduction pathways (Becker et al., 1997). In Ang II-dependent hypertension, a higher fraction of the total kidney Ang II is internalized into intracellular endosomes (light endosomes as well as intramicrovillar clefts) via an AT1 receptor mediated process (Zhuo et al., 2002). The demonstration that AT1 receptor blockade prevents the augmentation of intrarenal Ang II that occurs during chronic infusions of Ang II suggests that AT1 receptor-mediated accumulation of Ang II into an intracellular compartment is progressive and that some of the internalized Ang II is protected from degradation (Ingert et al., 2002a; Zhuo et al., 2002). This process can also occur during acute infusions of Ang II (van Kats et al., 1997). van Kats (1997) infused labeled Ang II and showed a 6- to 7-fold increase in intrarenal Ang II, which was prevented by an AT1 receptor antagonist. It was also recently reported that megalin, a multiligand receptor heavily involved in protein endocytosis, binds and mediates internalization of Ang II as well as Ang 1-7 in renal proximal tubule cells (Gonzalez-Villalobos et al., 2005, 2006). These findings suggest a role for megalin not only as a scavenger receptor but also as a regulator of local activity of the intrarenal RAS in the kidney. These observations may widen the area in which to explore the pleiotropic effects of Ang II.
There are several possible functions of the internalized Ang II. Ang II could be recycled and secreted to exert further actions by binding to Ang II receptors on the cell membranes. Ang II may also act on cytosolic receptors to stimulate the inositol 1,4,5-triphosphate pathway as has been described for vascular smooth muscle cells (Haller et al., 1996). A particularly intriguing hypothesis is that Ang II migrates to the nucleus to exert genomic effects (Chen et al., 2000). Chen et al. (2000) transfected Chinese hamster ovary cells with an AT1a receptor fused with green fluorescent protein, which allowed visualization of trafficking of the internalized ligand-receptor complex. Nuclear binding sites for Ang II in renal cells were reported by Licea et al. (2002). The nuclear receptors were primarily of the AT1 subtype as they were displaced by losartan as well as saralasin. Nuclear Ang II receptor density was not altered in Ang II-infused hypertension. Ang II increased colocalization of green fluorescent protein fluorescence with nuclear markers, suggesting migration of the receptor complex to the nucleus (Chen et al., 2000). Pendergrass et al. (2006) established a new congenic model of hypertension, the mRen2.Lewis rat, from the back-cross of the (mRen2)27 transgenic rat that expresses the mouse Ren2 gene onto the Lewis strain. Although plasma Ang II levels were not different between strains, both cortical and medullary Ang II concentrations were 60% higher in the mRen2.Lewis rats. Intracellular Ang II binding distinguished nuclear and plasma membrane receptors using the Ang II radioligand 125I-sarthran. Evaluation of intracellular Ang II receptors revealed lower cortical nuclear and medullary plasma membrane AT1 sites in mRen2.Lewis rats. The down-regulation of AT1 sites in the mRen2.Lewis rats may reflect a compensatory response to dampen the elevated levels of intrarenal Ang II (Pendergrass et al., 2006). Because Ang II exerts a positive stimulation on angiotensinogen mRNA and protein production, it is possible that the intracellular Ang II may have genomic actions to regulate angiotensinogen or renin mRNA expression in proximal tubule cells (Navar et al., 2002).
F. Alternative Enzyme Pathways
Tonin, cathepsins, and kallikreins are capable of acting on angiotensinogen to form Ang I or Ang II directly (Belova, 2000). Among these, a growing body of evidence indicates that chymase-dependent pathways play a critical role in forming Ang II from Ang I in cardiovascular tissues (Urata et al., 1993; Miyazaki and Takai, 2000, 2001). Chymase, a chymotrypsin-like serine protease, is predominantly present in the secretory granule of mast cells (Urata et al., 1993; Miyazaki and Takai, 2001). Mast cell-derived chymase can convert Ang I to Ang II in dogs, monkeys, hamsters, and humans, but not in rats and mice (Urata et al., 1993; Miyazaki and Takai, 2001). Chymase has no enzymatic activity in granules, because the optimal pH of chymase is between 7 and 9 whereas the pH in the granule is approximately 5.5 (McEuen et al., 1995). However, once the mast cells are activated in injured or inflammatory tissues, chymase is released into the extracellular matrix (pH 7.4) and immediately activated at maximum levels (Takai et al., 1996, 1999). Of note, strong chymase inhibitors, such as serine protease inhibitors, are mostly contained in the blood (Urata et al., 1993; Miyazaki and Takai, 2000, 2001). Thus, the chymase activity is inactivated in blood immediately after being released, indicating that chymase has enzymatic activity only in local tissues. In addition to mast cell-derived chymase, Guo et al. (2001) purified a novel rat chymase, rat vascular chymase, that is constitutively expressed in vascular smooth muscle cells and converts Ang I to Ang II. Rat vascular chymase expression is increased in vascular smooth muscle cells of spontaneously hypertensive rats (Guo et al., 2001). Furthermore, the conditional and targeted overexpression of rat vascular chymase in vascular smooth muscle cells induces hypertension in mice (Ju et al., 2001). Akasu et al. (1998) reported that chymase-dependent Ang II formation was more dominant than ACE-dependent Ang II formation in aorta extracts of normotensive rats. There is also Ang II-forming chymase in the pulmonary arteries of the monocrotaline-induced pulmonary hypertensive rats (Kishi et al., 2006). These data suggest that vascular chymase may play an important role in vascular Ang II formation. In the human heart, chymase is synthesized and stored in endothelial cells and mesenchymal cells and is secreted directly into the interstitium, contributing up to 80% of Ang II (Petrie et al., 2001). However, there is less information regarding the role of chymase in Ang II formation within the kidney.
ACE knockout mice have been shown to have unchanged kidney Ang II contents but 14-fold increases in chymase activity, suggesting the possible involvement of chymase and the residual ACE activity in Ang II generation in the kidney (Wei et al., 2002). Murakami et al. (1997) showed that intra-arterial infusion of [Pro11,d-Ala12]-Ang I, which is inactive but yields Ang II on digestion by chymase but not by ACE, induces renal vasoconstriction in dogs. They also revealed that the Ang II-forming activity in dog renal cortex was 20% chymase-dependent in vitro (Murakami et al., 1997). Further studies showed that intra-arterial infusion of a nonspecific chymase inhibitor, chemostatin, significantly decreased intrarenal Ang II contents in the ischemic kidney (Tokuyama et al., 2002). In the ischemic kidneys of two-kidney, one-clip rats and in kidneys of subtotal nephrectomized rats, increases in chymase activity and expression are also observed (Sadjadi et al., 2005a). Clinical studies reported increased chymase expression in rejected kidneys (Yamada et al., 2001) and kidneys of patients with renovascular hypertension (Morikawa et al., 2005) and diabetes (Huang et al., 2003; Koka et al., 2006). Collectively, these data support the potential contribution of chymase-dependent intrarenal Ang II formation to the progression of renal injury.
As described before, ACE processes the decapeptide Ang I to the octapeptide Ang II (Navar et al., 1997; Ichihara et al., 2004b; Paul et al., 2006). On the other hand, another carboxypeptidase, ACE2, cleaves only a single amino acid from the C terminals of Ang I to form the nonapeptide Ang 1-9, whereas ACE2 does not convert Ang 1-9 to Ang II (Donoghue et al., 2000; Burrell et al., 2004; Danilczyk et al., 2004; Danilczyk and Penninger, 2006; Shaltout et al., 2007). Therefore, it is possible that ACE2 regulates ACE-dependent Ang II formation by stimulating an alternative pathway for Ang I degradation. ACE2 also directly converts Ang II to Ang 1-7 (Burrell et al., 2004; Danilczyk et al., 2004; Danilczyk and Penninger, 2006; Shaltout et al., 2007). It has been suggested that Ang 1-7 acts on its own receptor, postulated to be the orphan heterotrimeric guanine nucleotide-binding protein-coupled receptor, MAS receptor (Santos et al., 2003). Recent studies demonstrated that genetic deletion of the guanine nucleotide-binding protein-coupled receptor encoded by the MAS protooncogene abolished the binding of Ang 1-7 to mouse renal cells (Santos et al., 2003). Ang 1-7 is thought to serve as an endogenous antagonist of the Ang II-induced actions mediated via AT1 receptors (Stegbauer et al., 2003; Burrell et al., 2004; Danilczyk et al., 2004; Danilczyk and Penninger, 2006; Shaltout et al., 2007). Thus, actions of ACE2 could have a substantial impact on the balance of Ang peptides found in the kidney by diverting the RAS cascade from Ang II to Ang 1-7. This helps explain the elevated Ang II levels in the ACE2 knockout mice (Crackower et al., 2002). ACE2 is abundantly expressed in renal epithelial cells including proximal tubular cells (Donoghue et al., 2000; Danilczyk and Penninger, 2006; Shaltout et al., 2007). Kidney ACE2 expression is significantly decreased in hypertensive (Zhong et al., 2004) and diabetic rats (Tikellis et al., 2003). Although the pathophysiological significance of ACE2 in renal injury remains to be established, emerging evidence suggests that ACE2 deficiency leads to increases in intrarenal Ang II levels (Wolf and Ritz, 2005).
Collectrin, a novel homolog of ACE2 has been identified in mouse, rat, and human (Tipnis et al., 2000). Both ACE2 and collectrin have tissue-restricted expression in the kidney. Collectrin is localized on the luminal surface and in the cytoplasm of collecting duct cells and its mRNA is expressed in renal collecting duct cells (Zhang et al., 2001) whereas ACE2 is present throughout the endothelium and in proximal tubular epithelial cells. Collectrin is up-regulated in the hypertrophic phase of the ablated kidney in the five-sixths nephrectomized rat model (Zhang et al., 1999a). In contrast with ACE and ACE2, collectrin does not contain dipeptidyl carboxypeptidase domains, and thus it may play a role in the hypertrophic phase through other pathways.
Other peptides with reported biological activity formed as part of the Ang cascade include Ang III and Ang IV as a consequence of action by aminopeptidases and other degrading enzymes. Although there is growing interest in the potential roles of these other Ang peptides, the bulk of the evidence continues to support the established premise that most of the vascular and transport actions attributed to the RAS that lead to vascular constriction, enhanced sodium transport, and hypertension are due to the actions of Ang II and also Ang III acting primarily on AT1 receptors (Touyz and Schiffrin, 2000). Nevertheless, Ang 1-7-, Ang IV-, and Ang II-mediated activation of AT2 receptors may exert significant counteracting or protective actions partially buffering the AT1 receptor-mediated effects under certain circumstances (Carey et al., 2000).
G. Other Factors
1. Renal Development and Aging.
Several lines of studies demonstrate the important roles of the RAS in renal development. Inadvertent use of ACEIs or ARBs during pregnancy causes structural and functional developmental abnormalities of the kidney (Pryde et al., 1993; Martinovic et al., 2001; Guron et al., 2006). In addition, mice lacking angiotensinogen (Kim et al., 1995; Niimura et al., 1995), renin (Yanai et al., 2000), ACE (Krege et al., 1995; Esther et al., 1997) and AT1a/AT1b receptors (Oliverio et al., 1998a,b) show low survival rates and diverse congenital renal abnormalities including renal vascular abnormalities, abnormal glomerulogenesis, renal papillary hyperplasia or atrophy, hydronephrosis, urinary concentrating defect, and interstitial fibrosis. Deletion of the AT2 receptor gene in mice causes a spectrum of congenital abnormalities of the kidney and urinary tract (Oshima et al., 2001). These observations reveal that the RAS has a fundamental role in renal development. Independent roles of the local RAS in developing kidney have also been indicated (Esther et al., 1997; Norwood et al., 2000). The mechanisms by which Ang II mediates tubulogenesis and nephrovascular development have been recently clarified and reviewed in detail (Yosypiv and El-Dahr, 2005; Lasaitiene et al., 2006).
Expression of angiotensinogen, renin, ACE, and AT1/AT2 receptors has been demonstrated in mesonephrons (Egerer et al., 1984; Celio et al., 1985; Wintour et al., 1996). Additionally, developing metanephrons express all components of the RAS (Gomez et al., 1989; Jung et al., 1993; Yosipiv et al., 1994; Yosipiv and El-Dahr, 1996; Norwood et al., 1997; Prieto et al., 2001). Intrarenal RAS activity is high during fetal and neonatal life but declines during postnatal maturation (Gomez et al., 1989; Yosipiv and El-Dahr, 1996). Actual Ang II contents in the kidney are much higher in newborn than in adult rats (Yosipiv and El-Dahr, 1996). Increased intrarenal renin and ACE activities may contribute to high levels of kidney Ang II levels in newborn rats (Yosipiv and El-Dahr, 1996). However, the precise mechanisms responsible for the independent regulation of the high levels of RAS activity in developing kidney remain unclear.
Renal function becomes lower with aging (Ferder et al., 2003; Basso et al., 2005). Structural and functional changes associated with aging include reductions in size, glomerular number, tubular mass, renal blood flow, and glomerular filtration rate, as well as glomerular sclerosis and tubulointerstitial fibrosis or inflammation (Ferder et al., 2003; Basso et al., 2005). Experimental findings that chronic treatment with ACEIs or ARBs attenuates most of the deleterious effects due to aging in the kidney (Inserra et al., 1996; Ferder et al., 2002) indicate the contribution of the RAS to age-dependent changes in renal function and structure. Further studies indicate that the beneficial effects of Ang II blockade on kidney aging are mediated through their antioxidative actions (Ferder et al., 1993; de Cavanagh et al., 1997, 2000). However, studies also indicate that aging is associated with reduced plasma renin activity and unchanged plasma angiotensinogen or ACE levels (Weidmann et al., 1975; Corman and Michel, 1986). In addition, there is no clear evidence that the intrarenal RAS is activated with age. Our preliminary data showed that kidney Ang II contents were not different in male Sprague-Dawley rats from 6 to 25 weeks of age (A. Nishiyama, unpublished observations). Further studies are needed to determine age-dependent changes in intrarenal Ang II levels and the expression of RAS components. Recent studies by Kunieda et al. (2006) demonstrated that Ang II induces premature senescence of human vascular smooth muscle cells via the p53/21-dependent pathway. However, the mechanisms responsible for Ang II-induced kidney aging remain unclear.
2. Gender Differences.
Gender differences in components of the RAS have been shown to play a role in the control of blood pressure (Dubey et al., 2002). It is well know that plasma renin activity is higher in men than in women regardless of age and ethnic heritage (Kaplan et al., 1976; James et al., 1986; Schunkert et al., 1997). However, the cause of this gender difference is unclear. In animal studies in Wistar-Kyoto rats, it was demonstrated that 1) renal angiotensinogen mRNA levels in males increase significantly during puberty, 2) renal angiotensinogen mRNA levels in adult females are considerably lower than those in adult males, 3) renal angiotensinogen mRNA levels in adult males are decreased by castration, and 4) renal angiotensinogen mRNA levels in adult females are increased by testosterone treatment. These data clearly indicated that androgen up-regulates rat renal angiotensinogen mRNA expression (Ellison et al., 1989; Ingelfinger et al., 1990). In contrast to the renal angiotensinogen mRNA level, circulating angiotensinogen levels are higher in women than in men (Clauser et al., 1989). Moreover, premenopausal women have slightly higher angiotensinogen levels than postmenopausal women, and estrogen replacement therapy or contraceptive medication both increase angiotensinogen in the circulation (Schunkert et al., 1997). An estrogen-response element in the angiotensinogen gene promoter markedly stimulates angiotensinogen synthesis in the liver and explains these findings (Clauser et al., 1989). Whereas angiotensinogen is up-regulated by estrogen, renin, ACE, and AT1 receptors are down-regulated by estrogen (Fischer et al., 2002; Harrison-Bernard et al., 2003). These data may account for the gender differences in the control of blood pressure (Dubey et al., 2002).
V. Augmentation of the Intrarenal Renin-Angiotensin System during Progression of Hypertension and Renal Injury
Several clinical studies have demonstrated significant renoprotection through blockade of the RAS compared with the effects other antihypertensive drugs, suggesting a crucial role of the intrarenal RAS activation in human kidney diseases. Covering all of the prospective trials and meta-analysis of the RAS blockade in patients with renal injury is beyond the scope of this review. Despite the enthusiasm for ACEIs and ARBs in patients with kidney disease, direct evidence of augmentation of the intrarenal RAS in human is relatively sparse. In human subjects, direct measurements of the intrarenal RAS components, microperfusion studies, or micropuncture investigations are not available. However, accumulating evidence including functional investigations and studies of human biopsy samples emphasize augmentation of the intrarenal RAS in human patients. Therefore, we will summarize evidence for a crucial role of the intrarenal RAS activation in renal injury in experimental studies first. Then, up-to-date clinical findings regarding significant renoprotective effects of RAS blockade will follow.
A. Animal Studies
1. Angiotensin II-Dependent Hypertensive Models.
a. Angiotensin II-infused hypertensive animals.
It is recognized that the tissue RAS exerts particularly important roles in several pathophysiological conditions (Dzau and Re, 1994). Chronic infusion of low doses of Ang II provides a useful experimental model of Ang II-dependent hypertension and develops in association with progressive enhancement of intrarenal Ang II (Mitchell and Navar, 1995). Ang II-infused rats have increases in renal angiotensinogen mRNA (Schunkert et al., 1992; Kobori et al., 2001b) and protein (Kobori et al., 2001a) and an enhanced urinary excretion rate of angiotensinogen (Kobori et al., 2002). Moreover, AT1 receptor blockade prevents the enhancement of intrarenal angiotensinogen that occurs in Ang II-infused hypertensive rats. These data suggest that the augmentation of intrarenal angiotensinogen in Ang II-dependent hypertension depends on activation of AT1 receptors and that the enhanced urinary excretion rate of angiotensinogen during Ang II infusion is blocked by AT1 receptor blockade (Kobori et al., 2003b).
In Ang II-dependent hypertension, AT1 receptor blockade increased plasma Ang II concentrations; however, it markedly limited the enhanced kidney Ang II contents elicited by chronic Ang II infusions (Kobori et al., 2004). This dissociation between plasma Ang II and intrarenal Ang II demonstrates a differential regulation of Ang II in the kidney and in the circulation. This dissociation between plasma Ang II and intrarenal Ang II has also been observed in other hypertensive models. In the Page cellophane-wrapped kidney model, the cellophane-wrapped group had progressive increments in blood pressure and Ang II content in the kidney; the plasma levels of Ang II were similar in the cellophane-wrapped group and the sham-operated animals and unchanged from baseline (Vanegas et al., 2005).
Renin is synthesized primarily by the juxtaglomerular apparatus (Hackenthal et al., 1990). However, renin mRNA and protein have been detected in proximal and connecting tubules and in collecting duct cells of human, rat, and mouse kidneys as well as in extrarenal tissues (Deschepper et al., 1986; Yanagawa et al., 1991; Moe et al., 1993; Rohrwasser et al., 1999). Whereas regulation of renin synthesis and secretion from juxtaglomerular apparatus cells has been extensively studied (Hackenthal et al., 1990), very little is known about the regulation of tubular renin (Henrich et al., 1996; Rohrwasser et al., 1999; Lantelme et al., 2002). It is well established that the intravenous infusion of Ang II decreases plasma renin activity or renin secretion, suggesting a negative feedback loop of renin on juxtaglomerular apparatus by Ang II (Blair-West et al., 1971; Shade et al., 1973). However, a recent study demonstrated that chronic Ang II infusions to normal rats significantly increased renin mRNA and protein levels in principal cells of connecting ducts and collecting tubules (Prieto-Carrasquero et al., 2004). This augmentation depends on activation of AT1 receptors (Prieto-Carrasquero et al., 2005b). Although plasma renin activity and juxtaglomerular apparatus renin are markedly suppressed in Ang II-induced hypertension, increased distal nephron renin associated with increased proximal tubular angiotensinogen production and spillover into the distal nephron segments may collectively contribute to elevated and sustained intratubular Ang I and Ang II formation in this hypertensive model (Kobori et al., 2004; Prieto-Carrasquero et al., 2005b).
The above-mentioned studies established the fact that there is a quantitative relationship between urinary angiotensinogen and intrarenal angiotensinogen and/or Ang II production and that there is both augmented angiotensinogen and distal nephron renin, leading to increased Ang II-mediated sodium reabsorption in distal nephron segments of Ang II-infused hypertensive rats (Kobori et al., 2001a,b, 2002, 2003b, 2004; Prieto-Carrasquero et al., 2004, 2005b). Ang II also directly stimulates epithelial sodium channel activity in cortical collecting duct cells (Peti-Peterdi et al., 2002), and there is intraluminal conversion of Ang I to Ang II in cortical collecting ducts (Komlosi et al., 2003). Thus, renin in distal nephron segments may synergistically contribute to the Ang II stimulatory effect on distal tubular renin and could help explain the marked stimulation of sodium reabsorption and suppression of the pressure-natriuresis relationship observed in Ang II-infused hypertensive rats (Wang et al., 2000). Therefore, the concomitant increases in proximal tubular angiotensinogen and distal nephron renin may play a crucial role in the sustained high intrarenal Ang II levels and hence contribute to the progressive high blood pressure observed in Ang II-dependent hypertension.
The importance of angiotensinogen and renin in the tubular cells in inducing of systemic hypertension was also reported in a transgenic mouse model (Lavoie et al., 2004). Lavoie et al. (2004) produced double transgenic female mice that express renal proximal tubule-specific, androgen-responsive, human renin and human angiotensinogen genes. Double transgenic female mice had normal baseline mean arterial blood pressure, which increased after a 2-week treatment with a testosterone pellet and returned to the baseline after elimination of the testosterone pellet. They showed that dual production of renin and angiotensinogen in the renal proximal tubules results in a systemic increase in arterial pressure, suggesting the pathophysiological significance of the tissue RAS in the renal proximal tubules. Transgenic animal models are also discussed in section V.A.1.c.
b. Renovascular hypertensive animals.
The RAS has been implicated in the pathogenesis of renovascular hypertension; however, plasma levels of Ang II are not significantly elevated in renovascular hypertension (Brown et al., 1965). To mimic clinical renovascular hypertension, a two-kidney, one-clip Goldblatt hypertensive rat model has been used extensively.
The development of hypertension in this model is accompanied by significant increases in intrarenal Ang II levels in both the clipped, ischemic kidney and the contralateral, nonischemic kidney (Guan et al., 1992; Sadjadi et al., 2002). The contralateral, nonischemic kidney plays a pivotal role in the maintenance of hypertension in renovascular hypertension by increasing sodium and water reabsorption to augment blood pressure (Huang et al., 1985), so the observation of increased intrarenal Ang II levels in that kidney is particularly relevant to the pathogenesis of renovascular hypertension.
Previous studies demonstrated that small, incremental increases in plasma Ang II are capable of inducing substantial increases in intrarenal Ang II that far exceed plasma Ang II levels (Von Thun et al., 1994; Zou et al., 1996a,b). Because intrarenal ACE activity is increased in the nonischemic kidney of two-kidney, one-clip rats (Guan et al., 1992), increases in plasma Ang II augment intrarenal Ang II in the nonischemic kidney by an ACE-dependent mechanism (Sadjadi et al., 2005b). Sadjadi et al (2005b) also reported that ACE-independent Ang II production by chymase is up-regulated in the ischemic kidney in renovascular hypertension. Blood pressure-independent up-regulation of collecting duct renin in two-kidney, one-clip Goldblatt hypertensive rats has also been recently reported (Prieto-Carrasquero et al., 2005a). These data indicate that many factors are involved in the mechanism of the development of hypertension in the two-kidney, one-clip Goldblatt hypertensive rat model.
c. Transgenic animals.
Several transgenic animal models have contributed to our understanding of the role and the regulation of the intrarenal RAS in hypertension. Smithies (1997) and Smithies and Kin (1994) generated an angiotensinogen gene titration model in mice. Experimental analysis of complex quantitative genetic traits, such as essential hypertension, should be greatly facilitated by being able to manipulate the expression of a gene in living animals without altering the nucleotide sequence, chromosomal location, or regulatory elements of the gene. To explore this possibility, they used targeted gene disruption and duplication to generate mice that are genetically identical except for having from zero to four functional copies of the gene coding for angiotensinogen. The two-copy animals have two normal copies of the angiotensinogen gene, whereas the one-copy and three-copy animals have one normal copy with the other either disrupted or duplicated by gene targeting. The duplicated pair of genes was generated by a special form of gap-repair gene targeting that tandemly duplicates the whole of a gene together with 5′- and 3′-flanking regions. They found progressively and significantly higher levels of the gene product in the animals having increasing numbers of gene copies: the one-copy animals have steady-state plasma angiotensinogen levels approximately 35% of normal, and the three-copy animals have levels approximately 124% of normal. Varying gene copy numbers by targeting consequently offers a promising approach to quantitative genetics. These data have clearly indicated the involvement of angiotensinogen in the development of hypertension due to activation of the RAS.
The development of a transgenic mouse model with tissue-specific targeted expression of the human angiotensinogen gene provides a unique opportunity to evaluate the specific role of proximal tubule angiotensinogen in the regulation of intrarenal Ang II concentrations. Sigmund and associates (Ding et al., 1997) generated an interesting transgenic mouse model in which the expression of human angiotensinogen in the kidney is regulated by the kidney-specific androgen-regulated protein promoter. Because the RAS is species-specific, they bred these human renin-transgenic mice with the mice expressing human angiotensinogen under the same promoter to produce offspring that expressed both transgenes. Renal expression of the transgene in female mice was undetectable under basal conditions but could be strongly induced by administration of testosterone. Testosterone treatment did not cause a transcriptional induction in any other tissues examined. In situ hybridization demonstrated that expression of human angiotensinogen and kidney-specific androgen-regulated protein mRNA in males and testosterone-treated females was restricted to proximal tubule epithelial cells in the renal cortex. Although there was no detectable human angiotensinogen protein in plasma, it was evident in the urine, consistent with a pathway of synthesis in proximal tubule cells and release into the tubular lumen. Double transgenic female mice had a normal baseline mean arterial blood pressure, which increased by 15 mm Hg after 2 weeks of testosterone treatment and returned to baseline after elimination of the testosterone pellet. The change in arterial pressure paralleled the change in plasma testosterone. There was no mean arterial blood pressure change in testosterone-treated control littermates. These results demonstrated that dual production of angiotensinogen and renin in the renal proximal tubules can result in an increase in arterial systemic pressure. These data support a role for a tissue-specific RAS in the renal proximal tubules that contributes to the regulation of systemic blood pressure. Sigmund and associates (Davisson et al., 1999) also showed that male and testosterone-treated female kidney-specific androgen-regulated protein-fused human angiotensinogen plus human renin double transgenic mice express human angiotensinogen mRNA in the kidney by Northern blot analysis and exhibit high mean arterial pressure via a carotid arterial catheter. Plasma Ang II concentrations measured by enzyme-linked immunosorbent assay were not increased in those mice compared with the control mice (Davisson et al., 1999). These results provide evidence for the potential importance of the intrarenal RAS in blood pressure regulation by showing that expression of angiotensinogen specifically in the kidney leads to chronic hypertension independently from the systemic RAS.
Luft and his associates (Bohlender et al., 1997, 2000; Dehmel et al., 1998) developed a model of spontaneously high human renin hypertension in the rat by producing two transgenic strains, one for human angiotensinogen with the endogenous promoter and one for human renin with the endogenous promoter. Because of the species specificity of the RAS, Ang II synthesis and cardiovascular physiology in these animals is unaffected by the human transgene. Therefore, neither transgenic strain was hypertensive. These strains were then crossed, producing a double transgenic strain. The double transgenic rats, both males and females, developed severe hypertension and died after a mean of 55 days if untreated. The rats had significantly higher levels of human plasma renin concentration, plasma renin activity, rat angiotensinogen concentration, and human angiotensinogen concentration compared with the control rats. Angiotensinogen transgene expression by RNase protection assay was ubiquitously present but most prominent in liver. Renin transgene expression was high in kidney but absent in liver. The rats featured severe cardiac hypertrophy, with increased cross-section of cardiomyocytes but little myocardial fibrosis. The kidneys showed atrophic tubules, thickened vessel walls, and increased interstitium. Both the ACEI, lisinopril, and the specific human renin inhibitor, remikiren, lowered blood pressure to normal values. The fact that human renin can be studied in the rat is a unique feature of this model. This animal model can be used for studies on species-specific interactions of RAS proteins and for the analysis of protein expression patterns. Furthermore, it provides a system for testing drugs modulating human RAS activity, such as renin-inhibitory pharmaceuticals for use in antihypertensive therapy (Bohlender et al., 1997, 2000; Dehmel et al., 1998).
Mullins and associates (Kantachuvesiri et al., 2001) created a transgenic rat line [strain name: TGR-(Cyp1a1Ren2)] that allows the induction of Ang II-dependent malignant hypertension. This transgenic rat line was generated by inserting the mouse Ren2 renin gene, fused to the cytochrome P450 1a1 promoter, into the genome of the Fischer 344 rat (Kantachuvesiri et al., 2001). Cytochrome P450 1a1, which catalyzes the oxidation of a wide range of endogenous lipophilic compounds and xenobiotics, is not constitutively expressed but is highly inducible on exposure to various aryl hydrocarbons such as indole-3-carbinol. Induction of cytochrome P450 1a1 is mediated by the aryl hydrocarbon receptor, which is a basic helix-loop-helix-transcription factor that binds to specific DNA elements in the cytochrome P450 1a1 promoter. Rats transgenic for the cytochrome P450 1a1-Ren2 construct do not constitutively express the Ren2 renin gene. Rather, the Ren2 gene is expressed, primarily in the liver, only on induction of the cytochrome P-450 1a1 promoter by aryl hydrocarbons such as indole-3-carbinol (Kantachuvesiri et al., 2001). In this transgenic rat model, induction of the cytochrome P450 1a1 promoter by dietary administration of indole-3-carbinol results in a fixed level of expression of the Ren2 renin gene and in the development of Ang II-dependent hypertension (Kantachuvesiri et al., 2001). At a dose of 0.3%, dietary indole-3-carbinol induces malignant hypertension characterized by loss of body weight, polyuria, lethargy, and piloerection. This model allows the induction of Ang II-dependent malignant hypertension using a benign and naturally occurring dietary supplement without the need for surgical intervention, dietary salt manipulation, or administration of steroids (Mitchell and Mullins, 2005).
2. Other Hypertensive Models.
a. Dahl salt-sensitive rats.
Previous studies indicated a clear linkage between salt-sensitive hypertension and a polymorphism of the angiotensinogen gene (Kamitani et al., 1994; Ishikawa et al., 2001; Katsuya et al., 2003). Various epidemiological studies have shown a correlation of dietary salt intake with the prevalence and progression of hypertension (Haddy and Pamnani, 1995). Although the degree of salt sensitivity is variable, some individuals are particularly prone to development of hypertension in response to an increased dietary salt intake. Subjects with essential hypertension have a higher frequency of salt sensitivity than is found in the normotensive population (Luke, 1993).
Dahl salt-sensitive rats have been used as a model of human salt-sensitive hypertension because salt loading exaggerates the development of hypertension in strains that are genetically predisposed to hypertension (Iwai et al., 1973). Mature Dahl salt-sensitive rats are reported to have low plasma renin activity that has been interpreted as being indicative of an overall suppression of the RAS (Iwai et al., 1973); however, few studies of angiotensinogen have been carried out in these rats. Although generally considered to be characterized by low activity of the circulating RAS, several studies indicate that treatment with ACEIs or ARBs reduces cardiac and/or renal dysfunction in Dahl salt-sensitive rats fed with a high-salt diet (Hayakawa et al., 1997; Kodama et al., 1997; Otsuka et al., 1998; Hayashida et al., 2001; Sakata et al., 2001; Nishikimi et al., 2002). These findings suggest that the local RAS may be inappropriately activated and may contribute to the development of hypertension in this animal model. This concept has received further support by findings that there is an inappropriate response in intrarenal angiotensinogen in Dahl salt-sensitive rats fed with a high-salt diet (Kobori et al., 2003a). Dahl salt-resistant rats and Dahl salt-sensitive rats were maintained on a high-salt or a low-salt diet. Systolic blood pressure was unaltered in Dahl salt-resistant rats; however, systolic blood pressure was significantly increased in Dahl salt-sensitive rats fed with a high-salt diet compared with Dahl salt-sensitive rats fed with a low-salt diet. The high-salt diet suppressed plasma renin activity in both strains. Plasma angiotensinogen levels were also suppressed by the high-salt diet in both strains. However, kidney angiotensinogen levels were significantly increased in Dahl salt-sensitive rats fed with a high-salt diet compared with Dahl salt-sensitive rats fed with a low-salt diet, Dahl salt-resistant rats fed with a high-salt diet, and Dahl salt-resistant rats fed with a low-salt diet. These data indicate that Dahl salt-sensitive rats fed with a high-salt diet have an inappropriate and paradoxical augmentation of intrarenal angiotensinogen (Kobori et al., 2003a).
This result could also be related to an exaggerated effect of the high-salt diet to elicit oxidative stress in the Dahl salt-sensitive rats. The inappropriate augmentation of intrarenal angiotensinogen in Dahl salt-sensitive rats by a high-salt diet may be caused by augmented production of reactive oxygen species. Systolic blood pressure was significantly increased in Dahl salt-sensitive rats fed with a high-salt diet compared with Dahl salt-sensitive rats fed with a low-salt diet. Treatment with a superoxide dismutase mimetic, tempol, or treatment with a nonspecific vasodilator, hydralazine, attenuated the hypertension to an equivalent extent. Urinary excretion of thiobarbituric acid-reactive substances, a marker of oxidative stress, was significantly increased in Dahl salt-sensitive rats fed with a high-salt diet compared with Dahl salt-sensitive rats fed with a low-salt diet. Tempol treatment prevented this effect, but hydralazine treatment only partially prevented the effect. Kidney angiotensinogen levels were significantly increased in Dahl salt-sensitive rats fed with a high-salt diet compared with Dahl salt-sensitive rats fed with a low-salt diet. Tempol but not hydralazine treatment prevented the intrarenal angiotensinogen augmentation. The evidence suggests that reactive oxygen species-dependent activation of intrarenal angiotensinogen plays an important role in the development of the hypertension in Dahl salt-sensitive rats fed with a high-salt diet (Kobori and Nishiyama, 2004).
b. Spontaneously hypertensive rats.
Previous studies also indicated a clear linkage between genetic hypertension and a polymorphism of the angiotensinogen gene (Nakajima et al., 2004; Rudnichi et al., 2004). Spontaneously hypertensive rats have been used as a model of genetic hypertension (Okamoto et al., 1966). Although generally considered to be characterized by low activity of the circulating RAS (Vincent et al., 1976; Kuriyama et al., 1982), several studies indicated that treatment with ACEIs and/or ARBs reduces cardiac and/or renal dysfunction in spontaneously hypertensive rats (Nakamura et al., 2001; Teng et al., 2002; Pu et al., 2003), suggesting that the intrarenal RAS may be inappropriately activated and in turn contribute to the development of hypertension and hypertension-induced renal damage in this animal model.
To determine whether augmented intrarenal angiotensinogen contributes to the enhanced renal Ang II and associated tissue injury, spontaneously hypertensive rats and Wistar-Kyoto rats were maintained on a normal diet and sacrificed at either 7 or 14 weeks of age. Two groups of spontaneously hypertensive rats received either an ARB (olmesartan) or triple therapy (hydralazine, reserpine, and hydrochlorothiazide) during weeks 7 through 14. Systolic blood pressure and renal Ang II were significantly increased in spontaneously hypertensive rats at 14 weeks of age compared with Wistar-Kyoto rats at 7 weeks of age, Wistar-Kyoto rats at 14 weeks of age, and spontaneously hypertensive rats at 7 weeks of age; furthermore, ARB treatment prevented these increases. However, although triple therapy prevented the development of hypertension in spontaneously hypertensive rats, this combination therapy failed to decrease renal Ang II. Using urine samples or fixed renal sections, the degree of renal injury was quantified to determine urinary excretion rate of total protein, glomerular sclerosis, interstitial expansion, monocyte/macrophage infiltration in interstitium or glomeruli, and renal arterial proliferation. Angiotensinogen mRNA and protein levels in kidney cortex and all parameters of renal damage were changed in parallel, and ARB treatment also prevented these increases. In contrast, triple therapy failed to prevent these increases. These results indicate that spontaneously hypertensive rats have enhanced intrarenal angiotensinogen production that contributes to increased Ang II levels, leading to the development of hypertension and thus renal injury in this strain (Kobori et al., 2005).
3. Diabetic Animals.
As observed in human subjects, the intrarenal RAS is generally activated in diabetic animals despite suppression of the systemic RAS. Whereas measurements of Ang II in whole kidney in diabetes models sometimes produced inconsistent results, from no change (Kennefick et al., 1996; Vora et al., 1997; Campbell et al., 1999) to even decreases in levels (Vallon et al., 1995), this may reflect the fact that whole-kidney measurements do not accurately reflect Ang II concentrations in the glomerulus or tubules.
Several studies have demonstrated that, despite normal or suppressed plasma renin activity, the intrarenal content of renin is increased in experimental diabetes models (Correa-Rotter et al., 1992; Jaffa et al., 1992; Burns and Harris, 1995; Carey and Siragy, 2003). Studies using Otsuka Long-Evans Tokushima Fatty rats, a model of type 2 diabetic nephropathy, showed increased activity of ACE in the kidney, although serum ACE activity was lower than that in control animals (Taniguchi et al., 2002). Increases in kidney Ang II levels were also observed in Otsuka Long-Evans Tokushima Fatty rats (Nagai et al., 2005). The Zucker obese rat model of type 2 diabetic nephropathy with hypertension is also associated with an increase in intrarenal Ang II. Treatment with an ACEI prevented increases in intrarenal Ang II and reversed manifestations of diabetic nephropathy (Sharma et al., 2006). The BioBreeding spontaneously diabetic rats showed an initial increase in renin gene expression, although there was a progressive decrease in renin gene expression and in the number of cells containing renin as the duration of diabetes lengthened (Everett et al., 1992)
Streptozotocin destroys pancreatic β cells and induces type 1 diabetes mellitus. In streptozotocin-induced diabetic rats, an increase in renal renin mRNA and protein content was demonstrated (Anderson et al., 1993). In early streptozotocin-induced diabetes mellitus, proximal tubule renin mRNA was significantly up-regulated, and this process was reversed with insulin therapy (Choi et al., 1997; Zimpelmann et al., 2000). Streptozotocin-induced diabetic animals also showed an increase in intrarenal angiotensinogen mRNA (Anderson et al., 1993; Zimpelmann et al., 2000). Recent studies using a unique microdialysis technique demonstrated increased levels of Ang II in renal interstitial fluid at week 3 through week 12 in streptozotocin-induced diabetic rats (Siragy et al., 2003).
High extracellular glucose stimulates angiotensinogen expression in cultured proximal tubular cells (Zhang et al., 1999b,c; Chen et al., 2001; Hsieh et al., 2003). High extracellular glucose was shown to increase angiotensinogen gene expression via reactive oxygen species in the rat proximal tubule, providing evidence of activation of the intrarenal RAS in diabetes mellitus (Hsieh et al., 2002). Glucose-induced angiotensinogen gene expression and Ang II secretion was also mediated via a glucose-responsive element on the angiotensinogen promoter in cultured tubular cells (Zhang et al., 2002). Modulation of angiotensinogen expression by high glucose or insulin in an immortalized rat proximal tubular cell line was mediated by heterogeneous nuclear ribonucleoprotein F, which binds to the insulin-responsive element in the rat angiotensinogen gene promoter (Wei et al., 2005).
To support intraglomerular activation of the RAS in diabetic nephropathy, Ang II was produced by primary cultures of rat mesangial cells and glucose increased Ang II synthesis by these cells (Singh et al., 1999). Angiotensinogen and Ang II levels were also increased significantly by 2.2- and 1.9-fold, respectively, in streptozotocin-induced diabetic rat glomerular extracts compared with nondiabetic controls (Singh et al., 2005).
Intrarenal activation of the RAS is associated with local oxidative stress. Oxidative stress in the kidney of diabetic animals estimated by the electron spin resonance imaging technique (Sonta et al., 2005), high-performance liquid chromatography (Nangaku et al., 2003), and an immunohistochemical method (Fan et al., 2004) was ameliorated by ARB treatment.
A crucial role of the intrarenal RAS activation in the pathogenesis of diabetic nephropathy was highlighted by induction of diabetes mellitus in a transgenic rat model (mRen2)27, in which renin was overexpressed in juxtaglomerular cells and proximal tubules. Administration of streptozotocin induced development of type 1 diabetes mellitus and rapid onset of diabetic nephropathy with severe glomerulosclerosis (Kelly et al., 1998). In contrast, only mild glomerulosclerosis was seen in nondiabetic (mRen2)27 rats or in diabetic spontaneously hypertensive rats. All of these changes were attenuated by ACE inhibition or by AT1 receptor blockade (Kelly et al., 2000).
4. Other Kidney Disease Models.
Activation of the RAS was also demonstrated in a variety of other kidney disease models. Immune complex nephritis is a form of glomerulonephritis that is mediated by a type III hypersensitivity reaction in which immune complex deposition or formation is thought to be the cause of the disease process. Immune complex nephritis involves poststreptococcal glomerulonephritis, membranous glomerulonephritis, minimal change glomerulonephritis, mesangiocapillary glomerulonephritis, IgA nephropathy, and systemic lupus erythematosus glomerulonephritis. In the renal cortex of a normotensive model of immune complex nephritis in rats, there was a significant increase in ACE activity (Ruiz-Ortega et al., 1995). ACEI administration to this model decreased proteinuria and glomerular and tubulointerstitial lesions, probably modulating local activation of the RAS.
Albumin, the predominant protein in the glomerular filtrate, is taken up by proximal tubular cells via receptor-mediated endocytosis. The albumin overload is a well known model of renal tubulointerstitial diseases representing an outstanding platform for in vivo investigation of the impact of excess protein load on proximal tubular cells in a primarily nonhemodynamic and nonimmunologic setting. ACE was up-regulated in proximal tubular epithelium with an increment in the gene expression of angiotensinogen in the renal cortex in a bovine serum albumin overload model (Largo et al., 1999). This model was also associated with up-regulation of renal Ang II expression via nuclear factor κβ-dependent mechanism (Takase et al., 2005). Local renal ACE activity and the Ang II concentration were also elevated in models of focal segmental glomerulosclerosis induced by puromycin, an aminonucleoside antibiotic produced by Streptomyces alboniger (Zhou et al., 2000) or Adriamycin, an anthracycline antibiotic commonly used in the treatment of a wide range of cancers (Li et al., 1999).
Unilateral ureteral obstruction, a model of experimental hydronephrosis, is characterized by tubulointerstitial fibrosis in the obstructed kidney. Unilateral ureteral obstruction leads to increases in renal renin content, ACE activity, and the Ang II concentration in the experimental kidney during the first 24 h (Pimentel et al., 1995). Intraparenchymal injection of recombinant adenovirus expressing angiotensinogen antisense into the cortex of obstructed kidneys induced reduction of transforming growth factor-β1 expression and fibrosis (Shin et al., 2005). In the setting of unilateral ureteral obstruction, Ang II stimulated the synthesis of transforming growth factor-β in tubular epithelial cells and also mediated collagen type IV production in the obstructed kidney (Ishidoya et al., 1995). These experiments suggest a pathogenic role of the intrarenal RAS in the fibrosis of unilateral ureteral obstruction.
Cyclosporine A has markedly improved the success of solid organ transplantation since its introduction in 1976. However, the effect of this immunosuppressive drug on renal graft longevity remains an intractable problem, due largely to its most prominent side effect, chronic cyclosporin A nephropathy. This disease has many complex mechanisms leading to a sustained afferent arteriole constriction, cytotoxicity, and elevated deposition of extracellular matrix. Thus, the disease is characterized histologically by tubular damage, afferent arteriolopathy, and striped tubulointerstitial fibrosis, all of which are associated with renal dysfunction. In the rat model of cyclosporin A nephropathy, renin expression in the juxtaglomerular apparatus was enhanced (Tufro-McReddie et al., 1993a; Young et al., 1995; Mazzali et al., 2001). Immunohistochemical studies of a model of chronic cyclosporin A toxicity in rats revealed intrarenal deposits of Ang II, which increased with time (del Moral et al., 1997). Increased expression of Ang II was observed in the outer medulla and medullary rays in rats treated with cyclosporin A (Ramírez et al., 2000).
To emphasize a pathogenic role of local activation of the RAS, in vitro studies showed that cyclosporin A stimulated the number of AT1 receptors and increased the intracellular level of calcium, a downstream intracellular signal of Ang II, in subcultured mouse medullary thick ascending limb cells (Wu et al., 2003). Chronic cyclosporin A infusion into rats increased plasma and kidney Ang II levels. AT1 receptor blockade markedly increased plasma Ang II levels and decreased kidney Ang II levels (Fig. 3). These studies support clinical observations of activation of the intrarenal RAS in human patients with cyclosporin A nephropathy.
Okada et al. (2002) used subtotally nephrectomized mice that are transgenic for transforming growth factor-β1 as a model of renal fibrosis. They found enhanced expression of AT1a receptor in the tubular epithelial cells and the interstitial fibroblast-like cells, whereas angiotensinogen was increased in the interstitial fibroblast-like cells and reduced in the tubular epithelial cells.
Although the response to Ang II may be variable immediately after a renal ischemic event in ischemia-reperfusion acute renal failure, there was reduced responsiveness to the constrictor actions of intra-arterially administered Ang II (McGiff and Itskovitz, 1964), suggesting that increased endogenous levels of Ang II reduced response to exogenous Ang II via a reduction in AT1 receptors. Acute renal ischemia also induced an increase in tissue levels of renin and Ang II at 24 h (Kontogiannis and Burns, 1998; Allred et al., 2000).
The vascular hypersensitivity to renal nerve stimulation and paradoxical vasoconstriction to renal perfusion pressure reduction was observed in a model of norepinephrine-induced acute renal failure in rats (Robinette and Conger, 1990). Blocking studies by saralasin showed that they are the result of intrarenal Ang II acceleration of neurotransmitter release to adrenergic nerve activity.
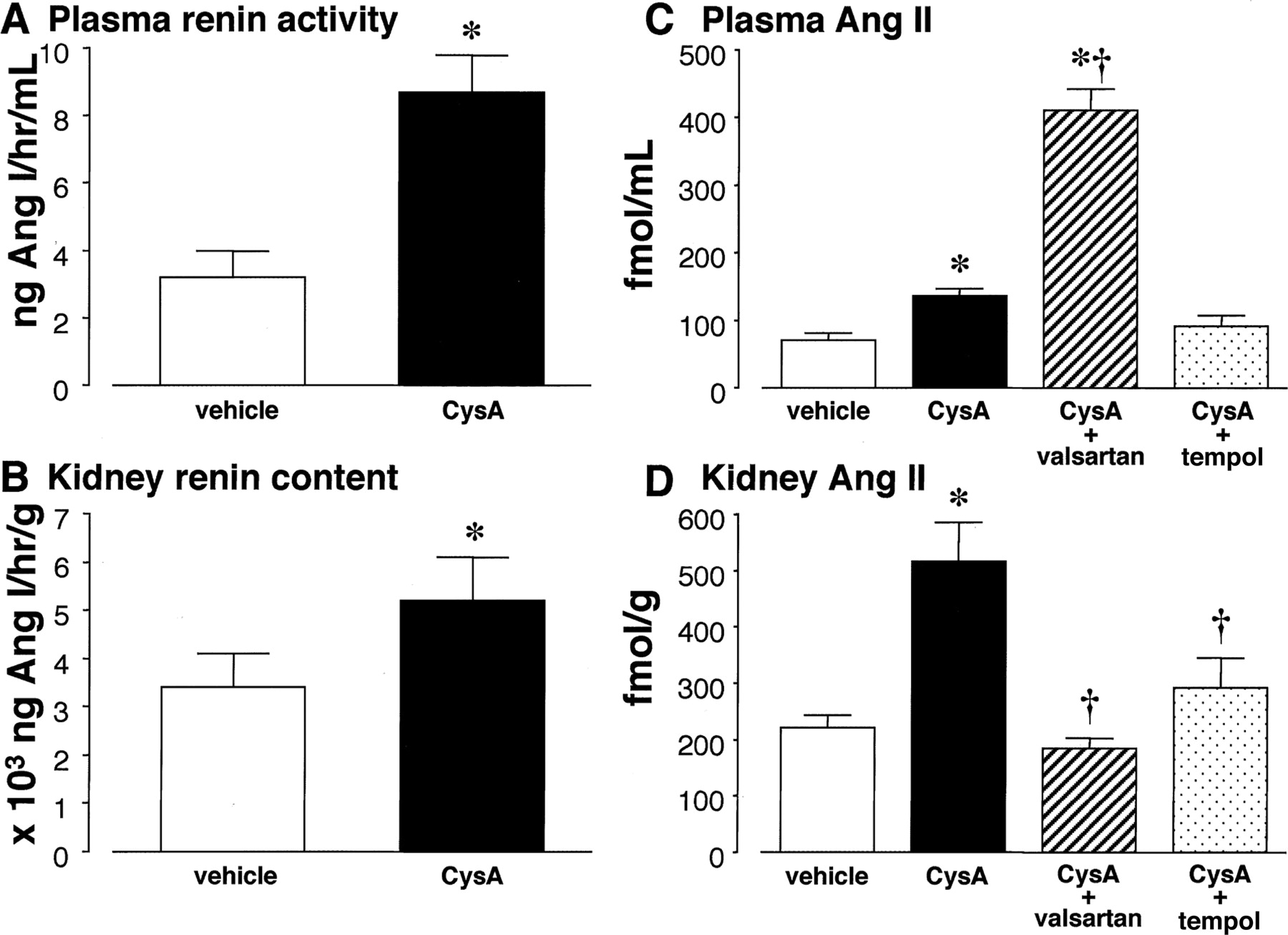
Plasma renin activity (A), kidney renin contents (B), plasma Ang II concentrations (C), and kidney Ang II contents (D). Cyclosporin A (CysA)-treated rats showed significantly higher plasma renin activity and kidney renin contents compared with vehicle-treated rats (A and B, respectively). Similarly, plasma Ang II concentration and kidney Ang II contents were higher in CysA-treated rats than in vehicle-treated rats (C and D, respectively). AT1 receptor blockade with valsartan markedly increased plasma Ang II levels in CysA-treated rats (C). In contrast, kidney Ang II contents were significantly decreased by treatment with valsartan (D). Tempol did not alter plasma Ang II levels (C) but significantly decreased kidney Ang II contents in CysA-treated rats (D). *, P < 0.05 versus vehicle-treated rats. From Nishiyama et al. (2003a) with modifications. Published with permission from Lippincott Williams & Wilkins.
Hypertonic glycerol injection is one of the most frequently used models of experimental acute renal failure. The mechanism of glycerol-induced acute renal failure is mediated by a reversible renal ischemia. Previous studies showed attenuation of intrarenal Ang II generation due to the inability of intrarenal converting enzyme during glycerol-induced acute renal failure (Baranowski and Westen-felder, 1988). However, recent studies demonstrated increased Ang II staining in the tubulointerstitium of the renal cortex at later time points from day 5 to 60 after glycerol injection (de Jesus Soares et al., 2005).
Activation of the intrarenal RAS also occurs in various conditions of altered vascular volume. Schunkert et al. (1993) found higher levels of renal angiotensinogen, renin, and Ang II in rats with severe congestive heart failure. The total renal Ang II content was 50-fold higher than that in plasma and correlated with renal angiotensinogen mRNA levels, consistent with intrarenal generation of Ang II. Acute changes in extracellular fluid volume regulated the endogenous RAS in the proximal tubule, independent from the systemic system (Quan and Baum, 1998). Perfusion of proximal tubules in vivo with an ultrafiltrate-like solution was performed to determine the rate of proximal tubule volume transport in both volume-contracted and volume-expanded animals. The proportion of transport inhibited by addition of luminal ARB was 2-fold greater in volume-contracted than in volume-expanded rats, demonstrating that the effect of endogenously produced Ang II on proximal tubule transport is higher after volume contraction than after volume expansion. These studies support the existence of a functional intrarenal RAS that may serve to modulate fluid and solute transport under altered vascular volume states.
Uninephrectomy activates the intrarenal RAS of the contralateral kidney. Tank et al. (1996) found a 7.3-fold rise in glomerular renin mRNA and a 3.8-fold rise in proximal tubule renin mRNA in the remaining kidney after uninephrectomy. The activity of the ACE in vesicles obtained from luminal membranes of proximal tubular cells was significantly larger after uninephrectomy (Amorena et al., 2001). Angiotensinogen expression in proximal tubules was significantly increased at both the mRNA and protein levels in response to uninephrectomy with no concurrent changes of either renin or angiotensinogen in the circulation (Gociman et al., 2004). Renal excretion of angiotensinogen was also increased with other maneuvers that elevate urinary volume (Kobori et al., 2002; Lantelme et al., 2002). Thus, regulation of the proximal tubule RAS may serve as an adaptive response to uninephrectomy.
While 5/6 nephrectomy is a representative model of chronic kidney failure, two different methods are used to reduce kidney mass: the ligation-infarction model involves uninephrectomy plus infarction of approximately two-thirds of the other kidney and surgical excision or polectomy model involves uninephrectomy plus surgical excision of both poles of the other kidney. Activation of the RAS is more marked in the ligation model than in the polectomy model. When the two models were compared, plasma renin activity, plasma aldosterone levels, and the renin concentration in the scar region of the kidneys were higher in the infarction group than in the polectomy group (Ibrahim and Hostetter, 1998). Ang II levels in the peri-infarct portions of the remnant kidney was already high early after renal ablation (at 2 weeks) in the ligation model (Mackie et al., 2001). Ang II-positive cells remained markedly increased in the renal cortical interstitium of a remnant kidney rat until the end of the observation period, 120 days after ligation (Gonçalves et al., 2004). Most of the Ang II-positive cells appeared in association with peri-infarct areas. In another study, renin and Ang II expression were noted predominantly in renal tubular epithelial cells of the ligation model in association with overexpression of transforming growth factor-β1 at 12 weeks (Gilbert et al., 1999). Mechanical strain increased Ang II production and AT1 receptors expression in cultured podocytes, whereas AT1 receptor staining was increased in a podocyte distribution in the 5/6 remnant kidney (Durvasula et al., 2004). These results suggest that mechanical strain up-regulates local Ang II in podocytes, thereby resulting in a progressive reduction in podocyte number.
5. Cardiovascular Implications of Renal-Specific Regulation of the Renin-Angiotensin System.
Inhibition of the RAS has become a gold standard of modern heart failure and myocardial infarction therapy. When cardiac output declines, subsequent reduction of renal blood flow stimulates baroreceptors of the renal vessels and enhances renin secretion. Examination of rats with stable compensated heart failure after experimental myocardial infarction showed up-regulation of the intrarenal RAS without changes in components of the circulating RAS (Schunkert et al., 1992). Activation of the RAS results in increased sodium and water retention, initially leading to the adaptive restoration of the effective arterial volume. However, the hyperactivation of the RAS leads to the elevation of peripheral vascular resistance, resulting in a vicious cycle and further decline in cardiac performance, eventually leading to cardiac remodeling (Makaritsis et al., 2006; Bhatia et al., 2005). In a multicenter study of patients with reduced cardiac output, RAS activity was significantly increased in most patients with heart failure, and plasma renin activity correlated poorly with the survival of these patients (Francis et al., 1990).
Although a pathogenic role of the RAS in the pathophysiology of cardiovascular diseases is obvious, the cardiovascular implications of renal-specific regulation of the RAS have been a matter of debate since it has been difficult to differentiate the effects of intracardiac Ang II generation from actions by plasma-borne Ang II (Paul et al., 2006). Sachetelli et al. (2006) used kidney androgen-regulated promoter and activated the intrarenal RAS by the overproduction of angiotensinogen alone in the kidney. This transgenic mouse line showed an increase in systemic blood pressure in the absence of activation of the circulating RAS or other tissue RAS. Recently Crowley et al. (2006) have given an important insight on a crucial role of the intrarenal RAS by using a kidney cross-transplantation strategy to separate the actions of AT1 receptor pools in the kidney from those in systemic tissues (Crowley et al., 2006). For the systemic knockout group, AT1a receptor-deficient recipients were transplanted with kidneys from wild-type donors; these animals lack AT1a receptors in all tissues except the kidney. Kidney knockout animals are wild-type recipients of AT1a receptor-deficient kidneys, thus lacking expression of AT1a receptors only in the kidney, but with normal expression of receptors in all systemic, nonrenal tissues. Continuous Ang II infusion induced systemic hypertension and cardiac fibrosis in the systemic knockout animals. Conversely, in the kidney knockout group, the absence of AT1 receptors from the kidney alone is sufficient to protect these animals from Ang II-dependent hypertension and vascular pathology. These results indicated that vascular injury and fibrosis in the heart were consequences of elevated blood pressure rather than local actions of cardiac AT1 receptors and that AT1 receptors expressed in the kidney are the primary determinants of hypertension and end-organ damage in Ang II-dependent cardiac injury.
B. Clinical Studies
1. Hypertensive Patients.
a. Renovascular hypertension.
Local intrarenal Ang II formation was demonstrated by Admiraal et al. (1993) in studies involving infusions of tracer-labeled Ang I and Ang II to patients with renovascular hypertension. Renal venous Ang I on the affected side was three times higher than arterial Ang I, whereas on the unaffected side, renal venous Ang I was similar to arterial Ang I (Admiraal et al., 1993). Estimation of regional Ang I to Ang II conversion showed that virtually none of the de novo-produced venous Ang II in the kidney could be accounted for by conversion of arterially delivered Ang I (Admiraal et al., 1993). These results suggested local activation of the RAS in the stenotic kidney.
b. Other hypertension.
Some hypertensive patients have marked activation of the intrarenal RAS even though it is not apparent from the plasma renin activity data or from the responses in systemic arterial pressure to ARB (Navar, 2004).
Renal vascular responses to a renin inhibitor were sustained in healthy men and those with essential hypertension, in marked contrast to waning concentration and activity of the drug in the plasma compartment (Fisher et al., 1995). These results provided indirect but strong evidence for a biological influence of intrarenal Ang II formation in human subjects. The renal vascular response to infused Ang II also serves as an indirect measure of activity of the intrarenal RAS. In 249 subjects, of which 68% were hypertensive, both systolic and diastolic blood pressure negatively correlated with the renal plasma flow response to Ang II (Perlstein et al., 2004). Admiraal et al. (1990) showed the intrarenal RAS activation in patients with essential hypertension by tracer studies. Intravenous infusion of 125I-Ang I allowed estimation of de novo production of Ang I in the kidney, which corresponded to 80% of the renal venous plasma concentration of Ang I. Estimation of Ang I-to-II conversion in the kidney also demonstrated local synthesis of Ang II (Admiraal et al., 1993). To support local activation of the RAS in hypertension, in situ hybridization studies demonstrated stronger signals for renin, angiotensinogen, and ACE mRNA in mesangial and epithelial cells of kidney tissues from hypertensive patients (Lai et al., 1998).
2. Patients with Renal Injury.
a. Chronic kidney diseases.
Ang II has a number of deleterious effects in chronic kidney diseases, including stimulation of fibrogenic mediators, enhanced free radical formation, aggravation of glomerular hypertension, and induction of tubulointerstitial hypoxia. Overall, the net result of these effects is fibrogenesis and oxidative stress within the kidney.
In many parts of the world, IgA nephropathy is the most common form of glomerulonephritis. The hemodynamic studies of the acute administration of ACEIs demonstrated a decrease in filtration fraction consequent to an increase in the effective renal plasma flow, suggesting local activation of the RAS in some cases of IgA nephropathy (Coppo et al., 1993). To investigate intrarenal activation of the RAS in IgA nephropathy, Kobori et al. (2007) evaluated renal specimens from 39 patients with IgA nephropathy. Angiotensinogen was localized predominantly in proximal tubular cells, and the immunoreactivity of intrarenal angiotensinogen in IgA nephropathy was significantly increased compared with that in normal kidneys. Severe tubulointerstitial injury in IgA nephropathy was induced by expression of ACE and chymase mRNAs in atrophic tubules and infiltrating cells, and such expression correlated with the degree of tubulointerstitial damage (Chan et al., 2005). When proximal tubular cells were cultured with conditioned culture medium from human mesangial cells activated with IgA, Ang II production was up-regulated (Chan et al., 2005). These results suggest activation of the tubular RAS by a humoral factor released from mesangial cells in IgA nephropathy. Tubular expression of AT1 receptors and AT2 receptors was also increased in IgA nephropathy (Lai et al., 1998; Chan et al., 2005).
Activation of the RAS in glomeruli was demonstrated by in situ hybridization studies of renal specimens from patients with IgA nephropathy. Glomerular expression of mRNAs for RAS components such as renin, ACE, chymase, AT1 receptors, and AT2 receptors was up-regulated, which correlated with the degree of mesangial hypercellularity and expansion in IgA nephropathy (Lai et al., 1998; Miyake-Ogawa et al., 2005).
Membranous nephropathy is the most common form of nephrotic syndrome in the adult population and is also associated with intrarenal RAS activation. Immunohistochemical studies of renal biopsies from 20 patients with membranous nephropathy demonstrated elevated ACE and Ang II in tubular cells and interstitial cells (Mezzano et al., 2003b). In serial sections from patients with progressive disease, the ACE and Ang II up-regulation was associated with the tubular expression of profibrogenic factors including transforming growth factor-β and platelet-derived growth factor and with interstitial infiltration and myofibroblast activation.
b. Diabetes.
Patients with type 1 diabetes showed increases in renal vascular resistance and filtration fraction, which were reversed by administration of the ARB (Miller, 1999). Blockade of the RAS with an ACEI or ARB treatment in patients with type 1 diabetes led to concomitant increases in renal plasma flow and glomerular filtration rate (Hollenberg et al., 2003). The apparent paradox of a heightened renal hemodynamic response to blockers of the RAS in the face of low plasma renin activity was also observed in type 2 diabetes mellitus (Price et al., 1999a,b). Fliser et al. (2005) investigated the effect of chronic AT1 receptor blockade on intrarenal hemodynamics in patients with type 2 diabetes in a double-blind parallel group study. Renal plasma flow increased significantly, whereas filtration fraction and renovascular resistance decreased significantly in the ARB group. Ito and colleagues (Ogawa et al., 2006) randomly assigned 66 type 2 diabetic patients with nephropathy to either the ARB or diuretic group. Treatment with an ARB for 8 weeks reduced the levels of urinary 8-epi-prostaglandin F2-α and 8-hydroxydeoxy-guanosine, biochemical markers of oxidative stress. These studies emphasize the importance of changes in intrarenal hemodynamics and oxidative stress caused by local RAS activation in diabetic nephropathy.
Direct demonstration of activation of the intrarenal RAS was shown by in situ hybridization studies demonstrating stronger signals for renin mRNA in the expanded mesangial area of kidney tissues from diabetic patients (Lai et al., 1998). Immunohistochemical studies of the biopsies from 10 patients with type 2 diabetes mellitus and overt nephropathy also demonstrated elevated ACE and Ang II immunostaining in tubular and interstitial cells (Mezzano et al., 2003a). Elevated levels of Ang II in tubules were correlated with proteinuria and interstitial cell infiltration.
Although an ACE-dependent Ang II-generating system is a major source of intrarenal Ang II production, chymase also participates in Ang II generation. In the human diabetic kidney, ACE expression is significantly up-regulated by tubular epithelial cells, but there is also markedly increased chymase expression by both mesangial cells and vascular smooth muscle cells (Huang et al., 2003). This up-regulation of chymase correlated significantly with the increase in blood pressure and the severity of extracellular matrix deposition.
In summary, the intrarenal generation of Ang II is increased, despite suppression of the systemic RAS, in diabetic nephropathy (Burns and Harris, 1995). This increase can contribute to the progression of diabetic nephropathy via several hemodynamic, tubular, and growth-promoting actions.
c. Dialysis patients.
Mailloux (2001) recently summarized the mechanisms involved in hypertension of dialysis patients as follows: 1) inappropriate increased Ang II in relationship to volume and exchangeable sodium, 2) increased vascular sensitivity to endogenous vasopressor agents, 3) increased cardiac output in the presence of an inappropriately high peripheral vascular resistance, and 4) failure to fully suppress vasoconstrictor systems.
Reports on the systemic RAS status in dialysis patients provided variable results. Whereas some hemodialysis patients tended to have higher basal plasma renin and Ang II levels than normal subjects, others showed impaired renin release during periods of hypotension (Textor et al., 1981; Schohn et al., 1985). Although microneurographic studies demonstrated sympathetic overactivity in chronic hemodialysis patients, sympathetic nerve activity was normal in hemodialysis patients with bilateral nephrectomy, suggesting that a neurogenic signal arising in the failing kidney may induce sympathetic overactivity and subsequent activation of the RAS (Converse et al., 1992; Augustyniak et al., 2002). The decrease in serum potassium concentrations during hemodialysis paralleled the decline in plasma aldosterone and Ang II concentrations, suggesting a regulatory role of serum potassium on the RAS during hemodialysis (Elias et al., 1989; Krämer et al., 1990). Although hemodialysis does not change serum ACE activity, plasma exchange with fresh frozen plasma increased ACE activity and plasma exchange with plasma substitutes induced a dramatic decrease in serum ACE activity (Fourrier et al., 1988).
Immunohistochemical analysis of renin on paraffin sections of nephrectomy and/or autopsy specimens of patients with dialysis showed that the degree of immunoreactivity was most striking in patients with severe, dialysis-resistant hypertension, exceeding that found in renovascular hypertension and present in arterioles at a distance from the glomeruli (Faraggiana et al., 1988). Thus, the intrarenal RAS may be activated in dialysis patients with remaining kidneys.
d. Other kidney diseases.
Theoretically, the RAS is important in mediating sustained renal vasoconstriction of acute renal failure. Tubular injury, from ischemia or nephrotoxins, causes impaired proximal tubular reabsorption of sodium and chloride and results in the delivery of excessive sodium chloride to the macula densa segment of the distal tubule. Decreased sodium chloride at the macula densa stimulates the adjacent juxtaglomerular apparatus to increase local renin activity, initiating a cascade of events that leads to increased Ang II generation. In addition, volume contraction, which predisposes to the development of acute renal failure, is associated with stimulation of the RAS. However, in view of the instability of the internal biochemical environment and systemic hemodynamics, assessment of the status of the RAS in humans with acute renal failure is difficult. Although patients with acute renal failure are reported to be generally hyperreninemic (Kokot and Kuska, 1976), no direct evidence of activation of the intrarenal RAS has been demonstrated in humans.
Several studies support the notion of overactivity of the intrarenal RAS in autosomal dominant polycystic kidney disease (Wang and Strandgaard, 1997). The relative insensitivity of the renal vasculature in normotensive subjects with autosomal dominant polycystic kidney disease to exogenous Ang II infusion is consistent with down-regulation of receptors occurring due to chronically increased intrarenal Ang II levels (Watson et al., 1992). Local activation of the RAS in autosomal dominant polycystic kidney disease is supported by demonstration of immunoreactive renin in vessel walls, interstitial cells, dilated tubules, and cysts (Graham and Lindop, 1988; Torres et al., 1992). Culture of cyst epithelial cells also indicated that the tubulocystic epithelium has the potential to synthesize renin (Torres et al., 1992). In addition to renin, angiotensinogen, ACE, Ang II receptor, and Ang II peptide are present in cysts and in dilated tubules of kidneys in autosomal dominant polycystic kidney disease (Loghman-Adham et al., 2004). Of note, ACE-independent, chymase-mediated Ang II formation has been reported in the interstitium of kidney tissues in autosomal dominant polycystic kidney disease (McPherson et al., 2004). These observations suggest that an increased intrarenal RAS contributes to the development of hypertension in autosomal dominant polycystic kidney disease (Barrett et al., 1994).
Chronic allograft nephropathy is the main cause of renal transplant failure in the first decade posttransplant. Elevated plasma levels of renin and Ang II were observed in patients with worsening renal graft function, implicating this as an important risk factor for allograft injury (Bresticker et al., 1991). Immunohistochemical examination of allograft biopsy specimens in 23 renal transplant recipients diagnosed with chronic allograft nephropathy demonstrated increased renin-positive cells in the juxtaglomerular apparatus (Oka et al., 2005), suggesting a potential role of the intrarenal RAS activation in the development of chronic allograft nephropathy.
Cyclosporin A nephropathy is characterized by patchy interstitial fibrosis, usually in a striped pattern, associated with degenerative hyaline changes in the afferent arteriole walls. Accumulating evidence suggests that systemic and intrarenal RAS activation by cyclosporin A plays a pathogenic role in cyclosporin A nephropathy. Immunohistochemical analysis of renal biopsy specimens obtained from 26 children with idiopathic nephrotic syndrome who were treated with long-term moderate-dose cyclosporin A revealed immunoreactivity to renin in the vessels upstream from the juxtaglomerular apparatus in association with an increased number of renin-positive cells per glomerulus (Iijima et al., 2000). In contrast, there was a decrease in the proportion of renin-positive arterioles with cyclosporin A-arteriopathy in biopsy specimens of cyclosporin A-treated transplanted kidneys (Strøm et al., 1995). This discrepancy may be explained by the difference in severity of the disease in these studies.
VI. Effects of Pharmacological Intervention with Antihypertensive Agents on the Intrarenal Renin-Angiotensin System
Inappropriate activation of the intrarenal RAS closely contributes to the development of hypertension and renal injury. Therefore, ACEIs and ARBs effectively reduce blood pressure in hypertensive patients and are recommended as first-line therapy in those with renal disease. Other pharmacological interventions with antihypertensive agents are also indicated by several clinical and preclinical studies
A. Angiotensin-Converting Enzyme Inhibitors
Treatment with ACEIs results in decreases in both plasma and intrarenal Ang II levels (Imig et al., 1999; Komine et al., 2002). As described before, ACE is abundantly expressed in the kidney, including the vascular endothelium as well as both the luminal and basolateral membranes of both proximal and distal tubules, but its activity is more abundant in the proximal tubule brush border and fluid (Erdös, 1990; Sibony et al., 1993; Casarini et al., 1997; Harrison-Bernard et al., 2002; Mezzano et al., 2003a,b). Intrarenal ACE is involved in generating Ang II not only from systemically delivered Ang I but also from intrarenally generated Ang I in view of abundant ACE and angiotensinases found in brush border (Ward et al., 1976; Ikemoto et al., 1987; Erdös, 1990; Sibony et al., 1993; Casarini et al., 1997). Furthermore, Ang I directly added by perfusion into the peritubular capillaries is converted to Ang II and exerts afferent arteriolar vasoconstriction, decreases in single nephron glomerular filtration rate, and increases in proximal fractional reabsorption rate (Mitchell and Navar, 1987, 1988). Wilcox and Dzau (1992) showed that renal lymph Ang II levels were higher than renal venous concentration and were decreased in response to administration of captopril. Similarly, Nishiyama et al. (2002a,b) used renal microdialysis methods and demonstrated that interstitial infusion of Ang I significantly increased the renal interstitial fluid Ang II concentration, and this conversion was blocked by the addition of an ACEI to the perfusate. Imig et al. (1999) showed that intermicrovillar clefts and endosomal Ang II levels and ACE activity are significantly reduced in rats acutely treated with an ACEI. Collectively, these data indicate that ACEIs can reduce Ang II formation in several regions and compartments within the kidney.
B. Angiotensin Receptor Blockers
Blockade of AT1 receptors with ARBs results in increases in plasma Ang II levels, which are associated with increases in renin release from juxtaglomerular cells (Timmermans et al., 1993; Paul et al., 2006). However, it should be emphasized again that treatment with ARBs leads to decreases, rather than increases in kidney Ang II levels through prevention of AT1 receptor-mediated uptake of Ang II and AT1 receptor-mediated stimulation of intrarenal angiotensinogen, leading to intrarenal production of Ang II (Navar et al., 2002, 2003; Nishiyama et al., 2004b). In several experimental models of renal injury, elevated kidney Ang II levels are significantly decreased by treatment with ARBs (Anderson et al., 1994; Navar and Harrison-Bernard, 2000; Nishiyama et al., 2004b; Kobori et al., 2005; Nagai et al., 2005; Kasper et al., 2005; Fan et al., 2006). As described in section V.A.1.a., the mechanisms by which ARBs decrease intrarenal Ang II levels have been investigated in detail by studies in Ang II-infused rats (Zou et al., 1996a,b; Zou et al., 1998; Navar and Harrison-Bernard, 2000; Kobori et al., 2001a,b, 2002, 2003, 2004, 2006; Zhuo et al., 2002; Nishiyama et al., 2003a; Prieto-Carrasquero et al., 2004, 2005b). Kidney Ang II contents were significantly increased in Ang II-infused hypertensive rats (Zou et al., 1996a,b, 1998; Navar and Harrison-Bernard, 2000; Kobori et al., 2001a,b, 2002, 2003b, 2004, 2006; Zhuo et al., 2002; Nishiyama et al., 2003a; Prieto-Carrasquero et al., 2004, 2005b). Furthermore, augmentation of kidney Ang II was prevented by concurrent administration of ARBs (Zou et al., 1996a; Zhuo et al., 2002; Nishiyama et al., 2003a; Kobori et al., 2004, 2006; Prieto-Carrasquero et al., 2005b). Similarly, chronic infusion of Val5-Ang II, which has essentially the same immunoreactivity and biological activity as the endogenous isoform Ang II (Ile5-Ang II), elicited the development of hypertension and increases in total intrarenal Ang II seen with the native form of Ang II (Zou et al., 1996a, 1998). The intrarenal levels of Val5-Ang II were proportionately much higher than circulating levels, indicating that Val5-Ang II had accumulated in the kidney. In these animals, the renal content of Val5-Ang II was significantly decreased by concurrent administration of an ARB, losartan (Zou et al., 1996a, 1998). As described in section IV.E.2., Zhuo et al. (2002) showed that Ang II levels in renal cortical endosomes and intermicrovillar clefts are markedly increased in Ang II-infused hypertensive rats. Furthermore, concurrent administration of an ARB, candesartan, prevented the increases in endosomal and intermicrovillar cleft Ang II levels (Zhuo et al., 2002). These data demonstrate that there is increased AT1 receptor-mediated intracellular trafficking/accumulation of circulating and/or intrarenally formed Ang II into cortical tubular endosomes during Ang II-dependent hypertension, and this process is blocked by an ARB. In addition, treatment with an ARB, olmesartan, prevented increases in proximal tubular angiotensinogen (Kobori et al., 2004, 2006) and collecting duct renin levels (Prieto-Carrasquero et al., 2005b) seen in Ang II-infused hypertensive rats (see section IV.E.1.). Collectively, it is now clear that ARBs decrease intrarenal Ang II levels through prevention of AT1 receptor-mediated uptake of Ang II and AT1 receptor-mediated stimulation of intrarenal angiotensinogen leading to intrarenal production of Ang II when Ang II levels are inappropriately elevated (Fig. 4).
C. β-Blockers
Chronic kidney disease and end-stage renal disease are often characterized by an activated sympathetic nervous system (Converse et al., 1992; Augustyniak et al., 2002; Neumann et al., 2004). Sympathetic activation results in activation of the RAS (Reid, 1992), and renin release is inhibited by β-blockers (Assaykeen et al., 1970; Pettinger et al., 1973; O'Malley et al., 1975; Proakis et al., 1989). Although renin secretion was stimulated through intrarenal β-receptors independent of changes in systemic or renal hemodynamics or in tubular sodium reabsorption, treatment with a β-blocker, propranolol, inhibited renin secretion induced by renal nerve stimulation (Taher et al., 1976). An additive effect of β-blockade with RAS blockade to reduce microalbuminuria was observed in the analysis of a prespecified secondary endpoint of the Glycemic Effects in Diabetes Mellitus Carvedilol-Metoprolol Comparison in Hypertensives (GEMINI) trial (Bakris et al., 2005). This may explain the more intense reduction of the activity of the RAS in the juxtaglomerular apparatus as well as in the local intrarenal RAS (Ritz, 2005).
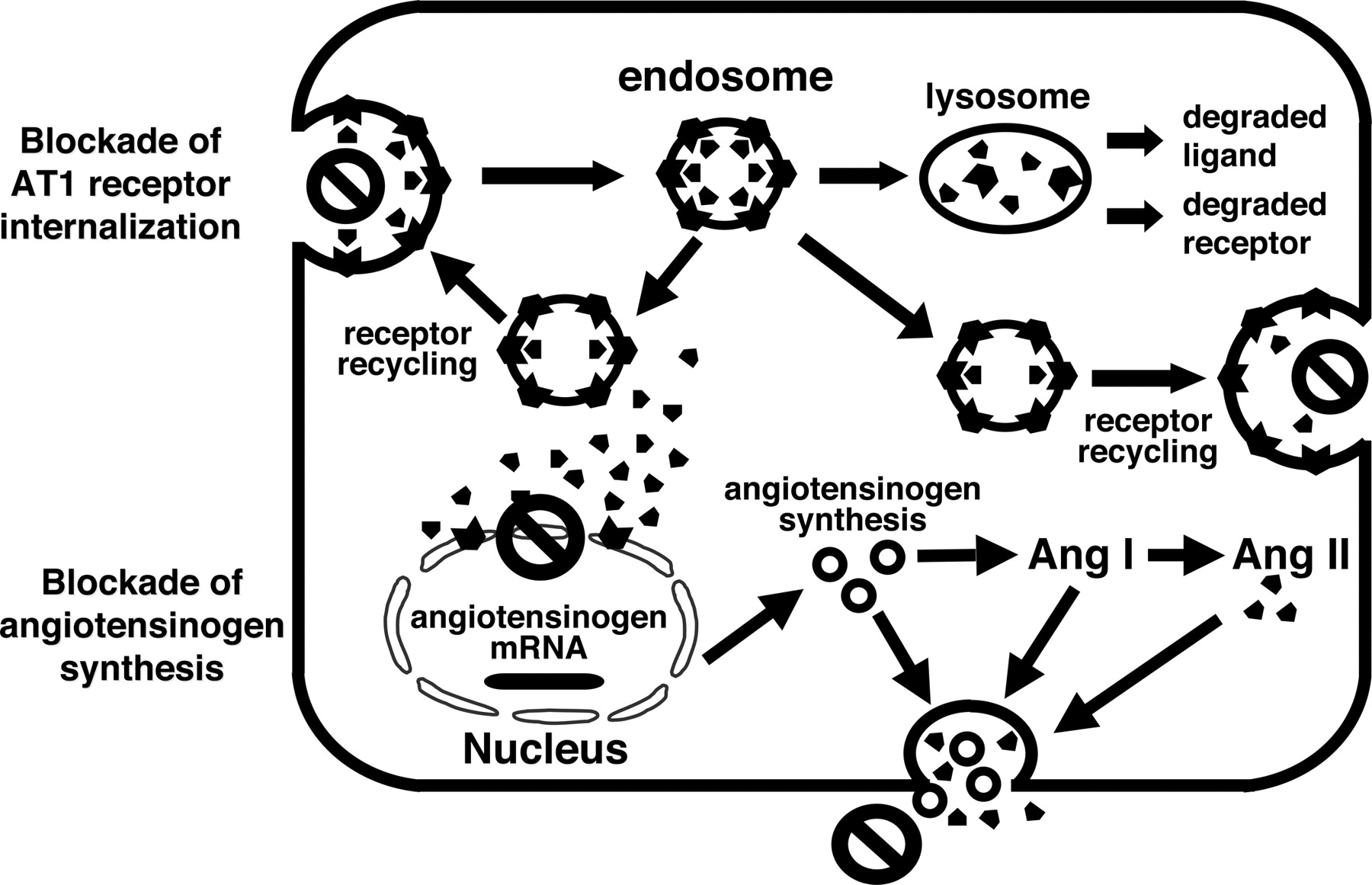
Effects of ARBs on AT1 receptor-mediated internalization of Ang II and angiotensinogen production in proximal tubular cells. We proposed that ARBs decrease intrarenal Ang II levels through prevention of AT1 receptor-mediated uptake of Ang II and intrarenal production of Ang II when Ang II levels are inappropriately elevated.
D. Calcium Blockers
Calcium blockers may theoretically activate the RAS as a response to blood pressure lowering or by decreasing calcium entry to the juxtaglomerular cell, which inhibits renin secretion directly. The effects of calcium blockers on the RAS have varied, depending on what compounds were used; however, most calcium antagonists lower blood pressure through a reduction of elevated systemic vascular resistance without clinically relevant activation of sympathetic reflexes or the RAS (Bauer and Reams, 1987; Nathan et al., 2005).
Neither the intravenous nor the oral administration of diltiazem produced a significant effect on recumbent plasma renin activity or aldosterone concentration (Ikeda et al., 1980; Aoki et al., 1983; Inouye et al., 1984; Magometschnigg et al., 1984; Mohanty et al., 1985). Short-term administration of nifedipine to normotensive subjects or hypertensive patients produced a significant increase in plasma renin activity (Aoki et al., 1976; Pedersen et al., 1979; Hiramatsu et al., 1982; Kusano et al., 1982; Leonetti et al., 1982; Kiowski et al., 1983). Results for plasma RAS components were inconsistent after treatment with nitrendipine (Thananopavarn et al., 1984; Luft et al., 1985; Pedrinelli et al., 1986), nicardipine (van Schaik et al., 1984; Elliott et al., 1985), amlodipine (Krenek et al., 2004; Lamarre-Cliche et al., 2005), or verapamil (Muiesan et al., 1982; Guthrie et al., 1983; Frohlich, 1985). Some calcium blockers such as azelnidipine may suppress the RAS although its mechanism remains unclear (Oizumi et al., 1989).
Activation of the intrarenal RAS by calcium blockers was shown in an experimental model of kidney failure. Whereas ACEIs and ARBs lowered blood pressure and proteinuria in rats subjected to renal ablation, nifedipine treatment, despite an equivalent blood pressure-lowering effect, did not reduce proteinuria. The different effects of the drugs on proteinuria were associated with differences in their effects on intrarenal peptide levels. ACEIs and ARBs reduced renal Ang II levels, whereas nifedipine increased these levels (Mackie et al., 2002).
E. Renin Inhibitors and Chymase Inhibitors
Historically, renin inhibitors have not been clinically successful because of lack of potency or bioavailability. Early renin inhibitors were peptide-like analogs of the scissile peptide bond of angiotensinogen. These compounds were poorly absorbed, rapidly degraded, and not suitable for clinical use (Wood et al., 1985; Clozel and Fischli, 1993). A novel, orally effective renin inhibitor, aliskiren, which targets the renin system at its point of activation, is now undergoing clinical investigation (Nussberger et al., 2002; Stanton et al., 2003; Gradman et al., 2005; Azizi et al., 2006). Aliskiren is a low molecular weight compound without the peptide-like backbone of the earlier compounds (Wood et al., 2003, 2005). Aliskiren monotherapy has demonstrated antihypertensive efficacy in patients with mild to moderate hypertension (Nussberger et al., 2002; Stanton et al., 2003; Gradman et al., 2005). Pilz et al. (2005) examined the effects of aliskiren in rats transgenic for human renin and angiotensinogen and showed that the antihypertensive and renoprotective effects of aliskiren are associated with decreases in renal Ang I and Ang II contents.
As described in section IV.B., studies have indicated that prorenin is activated without proteolysis by binding of the prorenin receptor to the pentameric “handle region” of the (pro)renin pro-segment, leading to an increase in the catalytic efficiency of Ang I formation from angiotensinogen (Ichihara et al., 2004a,b; Nguyen, 2006). Ichihara et al. (2004a) showed that the infusion of a decoy peptide corresponding to the handle region of prorenin in streptozotocin-induced diabetic rats resulted in decreases in kidney Ang I and Ang II contents without changing plasma Ang I and Ang II levels. Furthermore, the decoy peptide did not affect blood pressure but completely prevented renal injury in these animals (Ichihara et al., 2004a). Ichihara et al. (2006a) extended their studies in stroke-prone spontaneously hypertensive rats and showed that the inhibition of prorenin activity with a decoy peptide prevented the increases in kidney Ang II levels and markedly ameliorated renal injury. These data demonstrate that prorenin is catalytically active and contributes to Ang I and Ang II formation in the kidney during the progression of renal injury. Thus, a prorenin inhibitor will be a new therapeutic target for the treatment of renal disease.
As described in section IV.F., several studies indicated chymase-dependent Ang II formation in the kidney. However, the information regarding the effects of chymase inhibitors on intrarenal Ang II levels is limited. In chronic ischemic dog kidneys, in which intrarenal Ang II levels were elevated, acute injection of a nonspecific chymase inhibitor, chymostatin, into the renal artery significantly decreased kidney Ang II levels (Tokuyama et al., 2002). These data suggest that chymase inhibition decreases elevated kidney Ang II formation in ischemic dog kidneys. Studies by Nishiyama et al. (2005) showed that rat vascular chymase expression and chymase activity are increased in kidneys of Dahl salt-sensitive hypertensive rats. Chronic treatment with a specific chymase inhibitor, NK3201, significantly decreased intrarenal Ang II levels and elicited renoprotective effects. These data indicate that rat vascular chymase-dependent Ang II formation contributes to the pathogenesis of renal injury in Dahl salt-sensitive hypertensive rats. Treatment with a specific chymase inhibitor might therefore be a potentially useful strategy for preventing renal injury; however, clinical data are not yet available.
VII. Conclusions
Ang II exerts synergistic effects on renal microcirculatory and epithelial transport systems, which provide a powerful hypertensinogenic influence when the RAS is inappropriately stimulated. Sustained stimulation of the intrarenal RAS in a setting of elevated arterial pressure leads to enhanced fractional sodium reabsorption, renal injury, proliferation, and fibrosis. Exciting findings during the last two decades have provided insights regarding the mechanisms by which moderate increases in circulating Ang II lead to progressive increases in intrarenal Ang II. In Ang II-dependent hypertension, the intrarenal Ang II levels are increased to an extent greater than can be explained from the plasma Ang II concentrations even though there is suppression of juxtaglomerular apparatus renin and its release. The enhanced intrarenal and renal interstitial Ang II levels are the consequence of increased receptor mediated Ang II uptake by the AT1 receptor, as well as stimulation of renal angiotensinogen mRNA levels and augmentation of intrarenal angiotensinogen protein. The increased angiotensinogen production by proximal tubule cells leads to increased angiotensinogen secretion into the proximal tubular lumen leading to increased tubular formation of Ang II; increased angiotensinogen secretion leads to subsequent spillover of angiotensinogen into the distal nephron segments and the urine. In addition, the Ang II concentrations in the renal interstitial fluid are increased by chronic Ang II infusions to levels higher than can be explained from the circulating concentrations.
The enhanced intrarenal angiotensinogen mRNA levels and angiotensinogen secretion depend on AT1 receptor activation and are prevented by AT1 receptor blockade. There is a significant correlation between angiotensinogen urinary excretion rates and the intrarenal Ang II levels, indicating that urinary angiotensinogen excretion rates provide a specific index of intrarenal Ang II activity (Kobori et al., 2001a,b, 2003b, 2004). In contrast to renin suppression in juxtaglomerular apparatus, Ang II-dependent hypertension causes an augmentation of renin expression and renin content in principal cells of the collecting ducts. The increased renin produced in these principal cells may be secreted into the collecting duct lumen where it can act on the increased angiotensinogen arriving from the proximal tubule to form more Ang I. Because ACE is also present and is likewise enhanced in Ang II-dependent hypertension, there is increased Ang II formation, leading to further stimulation of distal nephron reabsorption. These effects are probably responsible for the reduced sodium excretion and the marked reduction in the slope of the pressure natriuresis relationship that occurs after chronic infusions of Ang II. The stimulation of renin in principal cells in Ang II-dependent hypertension is also blocked by AT1 receptor blockers. Thus, augmented Ang II in distal nephron segments may synergize with the elevated renal interstitial Ang II concentrations coupled with elevated aldosterone levels to contribute to further stimulation of sodium reabsorption and progression of the hypertensive response.
In addition to investigations on the many pleiotropic actions of Ang II, other emerging areas of interest are related to the discovery and characterization of the (pro)renin receptor and additional enzymes such as ACE2, that act on the peptides to provide alternative pathways that may shunt the system to provide less Ang II. The findings obtained in Ang II-dependent hypertension seem to have growing relevance to a number of conditions in other animal models and in human pathophysiological conditions. Augmented intrarenal Ang II levels have also been reported in Goldblatt hypertension, transgenic rats and mice, Dahl salt-sensitive rats, spontaneously hypertensive rats, and models of nephritis, interstitial fibrosis, and acute renal failure. Clinical studies also suggest that elevated intrarenal production of Ang II plays a key role in the pathophysiology of hypertension and diabetes as well as aging (Anderson, 1997). Accordingly, the drugs that interfere with the production of Ang II, block AT1 receptors, or block signaling pathways activated by Ang II are clearly emerging as the fist line of treatment for many hypertensive disorders as well as in diabetes and various renal diseases.
Acknowledgments
Our laboratories are supported by grants from the National Institute of Diabetes and Digestive and Kidney Diseases (R01DK072408 to H.K.), the National Center for Research Resources (P20RR017659 to H.K. and L.G.N.), the National Heart, Lung, and Blood Institute (R01HL026371 to H.K. and L.G.N.), the Health Excellence Fund from Louisiana Board of Regents (to H.K. and L.G.N.), the Japan Society for the Promotion of Science (17390246 to M.N.), the Ministry of Education, Culture, Sports, Science and Technology of Japan (17790171 to A.N.), the Salt Sciences Research (06C2 to A.N.), and Kagawa University Project Research (to A.N.).
Footnotes
-
↵1 Abbreviations: RAS, renin-angiotensin system; Ang, angiotensin; ACE, angiotensin-converting enzyme; ACEI, angiotensin-converting enzyme inhibitor; ARB, angiotensin II type 1 receptor blocker; AT1, angiotensin II type 1; AT2, angiotensin II type 2; DOCA, deoxycorticosterone acetate.
-
This article is available online at http://pharmrev.aspetjournals.org.
-
doi:10.1124/pr.59.3.3.
- The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue





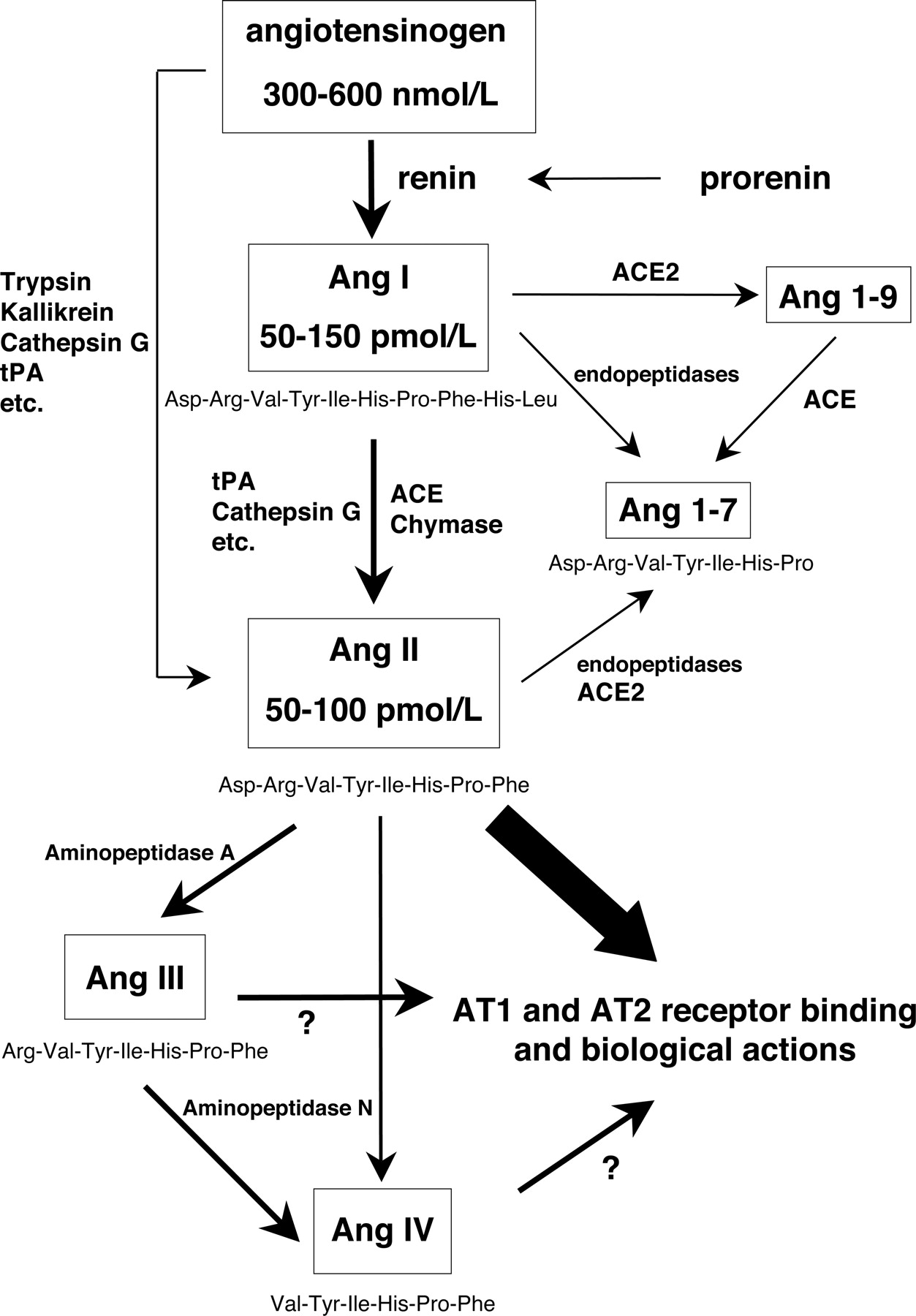{kind=link}
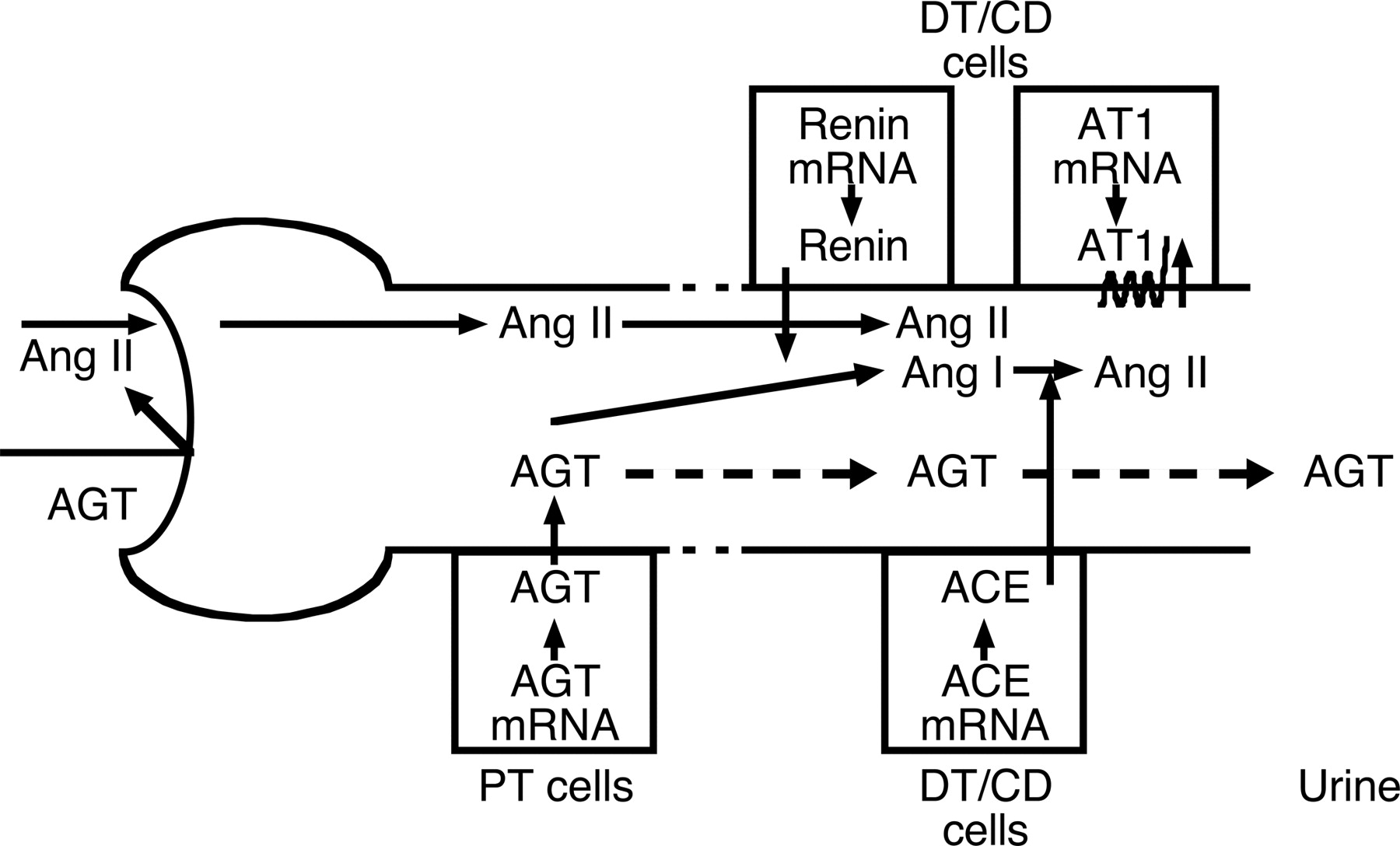{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction
- II. Physiological Actions of Angiotensin II in the Kidney
- III. Regulation of Circulating Renin-Angiotensin System—Classic Renin-Angiotensin System Pathways
- IV. Mechanisms Responsible for Independent Regulation of Intrarenal Renin-Angiotensin System
- V. Augmentation of the Intrarenal Renin-Angiotensin System during Progression of Hypertension and Renal Injury
- VI. Effects of Pharmacological Intervention with Antihypertensive Agents on the Intrarenal Renin-Angiotensin System
- VII. Conclusions
- Acknowledgments
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters