


Abstract
Adverse drug reactions (ADRs) present a serious human health problem. They are major contributors to hospitalization and mortality throughout the world (Lazarou et al., 1998; Pirmohamed et al., 2004). A small fraction (less than 5%) of ADRs can be classified as “idiosyncratic.” Idiosyncratic ADRs (IADRs) are caused by drugs with diverse pharmacological effects and occur at various times during drug therapy. Although IADRs affect a number of organs, liver toxicity occurs frequently and is the primary focus of this review. Because of the inconsistency of clinical data and the lack of experimental animal models, how IADRs arise is largely undefined. Generation of toxic drug metabolites and induction of specific immunity are frequently cited as causes of IADRs, but definitive evidence supporting either mechanism is lacking for most drugs. Among the more recent hypotheses for causation of IADRs is that inflammatory stress induced by exogenous or endogenous inflammagens is a susceptibility factor. In this review, we give a brief overview of idiosyncratic hepatotoxicity and the inflammatory response induced by bacterial lipopolysaccharide. We discuss the inflammatory stress hypothesis and use as examples two drugs that have caused IADRs in human patients: ranitidine and diclofenac. The review focuses on experimental animal models that support the inflammatory stress hypothesis and on the mechanisms of hepatotoxic response in these models. The need for design of epidemiological studies and the potential for implementation of inflammation interaction studies in preclinical toxicity screening are also discussed briefly.
- ABC, ATP-binding-cassette
- ADR, adverse drug reaction
- COX, cyclooxygenase
- P450, cytochrome P450
- DCLF, diclofenac
- FAM, famotidine
- GI, gastrointestinal
- HOCl, hypochlorous acid
- H2, histamine 2
- IADRs, idiosyncratic ADRs
- IL, interleukin
- JNK, Jun-N-terminal kinase
- KCs, Kupffer cells
- LPS, lipopolysaccharide
- MAPK, mitogen-activated protein kinase
- MIP-2, macrophage inflammatory protein-2
- MPO, myeloperoxidase
- NSAID, nonsteroidal anti-inflammatory drug
- PAI-1, plasminogen activator inhibitor-1
- PAR-1, protease-activated receptor-1
- PMNs, neutrophils
- PPARγ, peroxisome proliferator-activated receptor-γ
- ; RAN, ranitidine
- ROS, reactive oxygen species
- SECs, sinusoidal endothelial cells
- SNPs, single-nucleotide polymorphisms
- TACE, TNFα-converting enzyme
- TF, tissue factor
- TGZ, troglitazone
- TLR4, toll-like receptor 4
- TNFα, tumor necrosis factor α
- TVX, trovafloxacin
I. Drug-Induced Idiosyncratic Hepatotoxicity
A. Overview
Drug-induced toxicity is an important human health problem. A recent study in the United Kingdom found that adverse drug reactions (ADRs) 1 targeting several organs are responsible for more than 6% of hospital admissions, and the mortality rate is approximately 2% (Pirmohamed et al., 2004). Underreporting is suspected, however, and the real incidence might be greater than estimated by current methods (Bagheri et al., 2000; Sgro et al., 2002). An immediate outcome of ADRs is the withdrawal or restricted usage of otherwise efficacious drugs, leading to deficits in therapy. An example of this is felbamate (Pellock, 1999; Dieckhaus et al., 2002), which was effective in treating severe cases of epilepsy. Unfortunately, its usage was markedly reduced because of its association with aplastic anemia and hepatotoxicity in some patients. In addition to posing issues for human health, the curtailed use of a drug represents for pharmaceutical companies a loss of financial investment and of committed scientific effort and profit. Moreover, the occurrence of ADRs presents the likelihood of further financial loss from lawsuits.
Idiosyncratic ADRs that target the liver are a common cause of acute liver failure in the United States, accounting for more than 10% of cases. The results of a 5-year prospective study indicated that many dietary supplements and drugs with different pharmacological targets are associated with idiosyncratic, hepatotoxic ADRs (Chalasani et al., 2008). The reactions lead to histopathological and clinical features of acute hepatocellular necrosis, biliary injury, or a combination of the two (Zimmerman, 1993, 2000). The main culprits are anti-infective, central nervous system, musculoskeletal, and gastrointestinal drugs (Andrade et al., 2005, 2006).
ADRs can be classified as predictable (type A reactions) or idiosyncratic (type B reactions). Type A reactions are dose-dependent and occur in a relatively consistent time frame; all individuals are susceptible. A typical example is acetaminophen-induced hepatotoxicity (Larson et al., 2005; Amar and Schiff, 2007). In contrast, idiosyncratic ADRs (IADRs) occur in a minority of patients during drug therapy and are unrelated to the pharmacological action of the drug (Senior, 2008). IADRs are currently unpredictable and difficult to diagnose, and they occur at doses that do not cause toxicity in most people. They typically display a variable onset time after the beginning of drug therapy, and they have not been reproducible in animal models (Kaplowitz, 2005; Waring and Anderson, 2005; Uetrecht, 2007, 2008). These features make IADRs more insidious than type A reactions, difficult to understand, and more difficult to predict by use of current preclinical testing paradigms or during clinical trials that use relatively few volunteers. Better prediction of idiosyncratic drug-induced liver injury will require an understanding of the modes and mechanisms of the reactions.
Drug properties, genetic variation, and environmental factors probably contribute to IADRs (Kaplowitz, 2001; Boelsterli, 2003a). Two hypotheses to explain IADRs have become widely accepted in the past few decades. One of them is that the reactions are based on drug metabolism polymorphisms among patients that result in different levels of toxic drug metabolites (Williams and Park, 2003). The other argues that the reactions arise from an adaptive immune response to proteins bound to the drug or its metabolites (Park et al., 2001; Ju and Uetrecht, 2002). These two hypotheses are not mutually exclusive, in that a drug metabolism polymorphism might contribute to reactive metabolite formation and consequently to the production of hapten needed for a harmful adaptive immune response. An extension of the latter is the “danger hypothesis” (Pirmohamed et al., 2002; Séguin and Uetrecht, 2003), which suggests that, in addition to immunization and challenge, a second “danger signal” is needed to precipitate an adaptive immune response that becomes hepatotoxic. This signal might be any of a number of factors including some form of cellular stress, underlying disease, or environmental factors.
Despite the popularity of these two hypotheses, evidence for them is incomplete or lacking for the vast majority of drugs that have caused IADRs in human patients. One drug that has caused considerable concern because of its link to severe, hepatotoxic IADRs is troglitazone. The next section uses troglitazone as an example to illustrate the progress, knowledge gaps, and alternative thinking regarding mechanisms of idiosyncratic hepatotoxicity.
B. Example: Troglitazone
Troglitazone (TGZ) was initially marketed in 1997 and is one of several thiazolidinediones that have been used for treatment of type 2 diabetes. It acts pharmacologically as a peroxisome proliferator-activated receptor-γ (PPARγ) agonist and reduces insulin resistance. During clinical trials, 1.9% of patients taking the drug experienced elevated serum alanine aminotransferase activity more than 3 times the upper limit of normal, suggesting mild or moderate hepatocellular injury (Watkins and Whitcomb, 1998). After TGZ was marketed, rarer but more severe liver injury also occurred in some patients, and this serious idiosyncratic hepatotoxicity from TGZ led to its withdrawal from the market in 2000 (Graham et al., 2003). Liver biopsies from affected patients revealed predominantly hepatocellular necrosis with occasional bridging fibrosis. The time frame of hepatotoxicity occurrence varied from within a month to several years after initiation of maintenance therapy. IADRs induced by TGZ are most probably independent of its pharmacological target, because other thiazolidinediones, such as rosiglitazone and pioglitazone, also act as PPARγ agonists, yet lack the same hepatotoxic potential in patients with diabetes (Scheen, 2001a,b).
Since its withdrawal from the market, a large amount of effort has been devoted to elucidating the mechanism of TGZ-induced hepatotoxicity. A variety of hypotheses were proposed, including reactive metabolite formation and accumulation, mitochondrial dysfunction and oxidative stress, inhibition of bile salt exporter pump, and apoptosis (Chojkier, 2005; Masubuchi, 2006). The major TGZ metabolites (sulfate, glucuronide, quinone) have been identified in cultured cells, experimental animals, and human patients (Kawai et al., 1997; Loi et al., 1999; Yoshigae et al., 2000; Honma et al., 2002; Watanabe et al., 2002). However, TGZ metabolite-protein adducts have only been demonstrated in liver microsomal preparations from rats with various cytochrome P450 (P450) inducers or in “supersomes” (cDNA-expressed human P450) (He et al., 2004). Furthermore, although TGZ is cytotoxic to human HepG2 cells and to rat and human hepatocytes in vitro, inhibitors of enzymes responsible for TGZ metabolism do not protect against TGZ-induced cytotoxicity (Kostrubsky et al., 2000; Yamamoto et al., 2001; Tirmenstein et al., 2002). HepG2 cells transfected with CYP3A4 or incubated with microsomes containing cDNA-expressed CYP3A4 metabolized TGZ, leading to increased cytotoxicity (Vignati et al., 2005). However, the TGZ quinone metabolite formed by CYP3A4 is less cytotoxic than TGZ itself, both in rat hepatocytes and HepG2 cells (Tettey et al., 2001). In addition, in normal human hepatocytes, these CYP3A4-related metabolites are unlikely to be generated or to accumulate in an amount large enough to exert toxic effects. Studies investigating TGZ-induced cytotoxicity were performed with cultured cell lines or primary cells using concentrations of TGZ 1 to 2 orders of magnitude greater than those likely to occur in patients. Thus, these studies of metabolism in vitro have not provided an explanation for the infrequent TGZ hepatotoxicity that occurs in patients, and the role of reactive metabolites in TGZ cytoxicity remains unclear.
The role of metabolism in TGZ toxicity in vivo is not clear either. The hepatic expression and activity of CYP3A4 vary significantly among individuals (Eichelbaum and Burk, 2001). This might occur, in part, as a consequence of single-nucleotide polymorphisms (SNPs) in the CYP3A4 gene or its promoter. SNPs have been found in three genes important for CYP3A activity (Kuehl et al., 2001). SNPs of the CYP3A gene family members affect various ethnic groups, occurring with relatively high frequency in Europeans and Americans of European descent (Hustert et al., 2001; Kuehl et al., 2001). TGZ quinone (metabolite M3) is the putative reactive metabolite formed by CYP3A4 (Rothwell et al., 2002). However, treatment of primates with doses of TGZ sufficient to cause exposure to the TGZ M3 metabolite (i.e., up to 1.2 g/kg) did not cause hepatotoxicity (Rothwell et al., 2002).
In human patients, M3-derived reactive intermediates were found to bind covalently to microsomal protein and glutathione (Tettey et al., 2001; He et al., 2004), but the importance of these adducts to TGZ-associated hepatotoxicity has not been established. In a cohort study of 4079 patients, combined genetic polymorphisms from several genes were associated with increased susceptibility to TGZ-induced liver injury. These included CYP1A1, NAD(P)H dehydrogenase, quinone 1, glucose transporter type 1, PPARγ-892, and PPARγ-1431 (de la Iglesia FA. et al., 2003). In another study, a strong correlation was also observed in patients between TGZ-induced liver injury and the combined glutathione transferase-θ1–glutathione transferase M1 null genotype (Watanabe et al., 2003). However, none of these studies established a functional relationship between these SNPs and TGZ-induced liver injury.
Several case reports described histological evidence consistent with an adaptive immune-mediated reaction (Arioglu et al., 2000; Kohlroser et al., 2000; Murphy et al., 2000), and responses from some patients were reduced by corticosteroids (Prendergast et al., 2000; Bonkovsky et al., 2002). One of these patients developed a similar cholestatic hepatitis when switched to rosiglitazone after TGZ treatment, consistent with an adaptive immune-mediated reaction and suggesting a class effect related to the drugs' pharmacophore (Bonkovsky et al., 2002). Although eosinophils and granulomatous inflammatory infiltrates have been observed in the livers of patients with TGZ hepatotoxicity, this alone does not constitute conclusive evidence that these cells played a role in the hepatotoxic response or that an adaptive immune reaction was responsible. Animal models that recapitulate TGZ hepatotoxicity, in particular, an immune hypersensitivity component, have not emerged.
A study by Ong et al., (2007) showed that TGZ induces mild hepatocellular injury after 4 weeks of treatment of mice heterozygous for the mitochondrial antioxidant enzyme, superoxide dismutase 2. Hepatic mitochondria isolated from TGZ-treated mice exhibited enhanced oxidative stress. Furthermore, in hepatocytes isolated from untreated superoxide dismutase 2(+/−) mice, but not wild-type mice, TGZ caused a concentration-dependent increase in superoxide anion production. TGZ-induced superoxide generation was shown to induce injury to hepatocytes by activating apoptosis signal-regulating kinase 1 (Lim et al., 2008). This was the first published demonstration in an animal model that TGZ treatment could cause hepatocellular injury, even though the toxicity was mild. In agreement with other studies, wild-type animals tolerated the drug without adverse effects. The results support the general concept that subclinical stresses from prolonged drug treatment superimposed on a genetic deficiency in the same organism can lead to cell injury and organ damage. The use of the SOD heterozygous mouse as a model to study mitochondrial mechanisms of drug-induced liver injury has recently been reviewed (Boelsterli and Hsiao, 2008).
The findings of Boelsterli and colleagues suggested that healthy experimental animals are resistant to TGZ hepatotoxicity because they lack some undefined stressor required to produce hepatotoxicity (Boelsterli and Hsiao, 2008). By extension, differences in such stressors in human patients, either genetic or environmental, might constitute susceptibility factors to TGZ-induced hepatotoxicity. Moreover, the unpredictable temporal and dose relationships that characterize IADRs from TGZ and other drugs could be explained if stressor expression was episodic and its effects unnoticed in the absence of drug treatment.
C. Episodic Inflammation and the Potential for Interactions during Drug Therapy
It may not be mere coincidence that antibiotics and nonsteroidal anti-inflammatory drugs (NSAIDs) that are used in clinical conditions associated with inflammation are also the most common causes of hepatic IADRs (Hussaini and Farrington, 2007; Chalasani et al., 2008). Likewise, the presence of viral hepatitis (i.e., hepatic inflammation) is a risk factor for idiosyncratic hepatotoxicity from antiretroviral drugs in patients infected with HIV (Hussaini and Farrington, 2007). Such observations suggest the possibility that inflammatory stress during drug therapy could contribute causally to hepatotoxic IADRs.
An inflammatory response can be considered a collage of stresses originating from numerous inflammatory cells and the various mediators that they produce (section II, below). Such stresses are capable of altering tissue homeostasis and may thereby set the stage for tissue injury. Systemic or localized inflammation is commonplace and can be caused by infection, concurrent disease, or other factors. Inflammation is a feature of numerous diseases and in some cases participates in disease pathogenesis. Anti-inflammatory therapy has been successful in the treatment of arthritis, inflammatory bowel disease, asthma, and other disease entities. Furthermore, inflammation plays a key role in chronic conditions such as diabetes (Tracy, 2003), obesity (Cottam et al., 2004), cardiovascular disease (Willerson and Ridker, 2004), and cancer (Whitcomb, 2004; De Marzo et al., 2007). Adaptive immune responses (allergic reactions) to specific antigens usually have an inflammatory component. Activation of the innate immune system by exposure to exogenous bacteria, viruses, and their molecular constituents also results in inflammation. It is noteworthy that microorganisms indigenous to the gastrointestinal (GI) tract of humans are responsible for periodic inflammation. For example, liver and GI diseases are associated with increased blood levels of endotoxin (lipopolysaccharide, LPS), a cell wall component of Gram-negative bacteria (Gardiner et al., 1995), and LPS is a well characterized inducer of inflammation. Surgery can elevate plasma LPS concentration, as can changes in diet, alcohol consumption, or antibiotic treatment (Roth et al., 1997; Lepper et al., 2002). Drugs known to produce GI injury, such as NSAIDs, can cause translocation of LPS from the GI tract to blood (Kim et al., 2005; Deng et al., 2006). These and other factors result in inflammatory stress in humans that is episodic and common and can occur under numerous conditions in the absence of overt tissue injury.
Concurrent inflammation could precipitate IADRs by a variety of mechanisms. Minor hepatic injury elicited by some drugs might progress to more serious injury as damaging inflammatory mediators are generated and released. In addition to exacerbating the hepatotoxic response, inflammation might inhibit or delay liver regeneration and repair. Conversely, inflammatory stress might also initiate hepatotoxicity that could be exacerbated by certain drugs. Increased sensitivity of the liver might occur as a consequence of altered cellular signaling, accumulation and activation of extrahepatic cells, or other factors. Inflammation can inhibit expression of drug-metabolizing enzymes (e.g., P450s). This could result in accumulation of parent compound in the liver and impaired drug clearance, which could pose a significant risk for patients taking these drugs (Renton, 2005; Morgan et al., 2008). Furthermore, concurrent inflammation might modify the intrahepatic distribution of drugs by altering expression of drug transporters including the ATP-binding-cassette (ABC) transporters in hepatocytes (Renton, 2005; Petrovic et al., 2007). Thus, there are many ways in which drugs might interact with an inflammatory response to cause liver injury. The effects of inflammation on sensitivity of the liver to drug-induced injury and the diverse factors and mechanisms involved will be the focus of the remainder of this review.
D. Inflammatory Stress and Idiosyncratic Adverse Drug Reactions
As described above, episodic exposures to inflammatory stimuli are common in people, although most cause no obvious tissue injury. Moreover, they occur irregularly and often go unnoticed. These observations led us to hypothesize that an episode of inflammation during drug therapy might decrease the threshold for drug toxicity and thereby render an individual susceptible to an adverse drug reaction. This hypothesis could explain many of the characteristics of IADRs. The irregular frequency of inflammatory episodes is consistent with the erratic temporal relationship to onset of therapy. Furthermore, an inflammation-mediated increase in sensitivity to hepatic toxicity from a drug could explain the apparent lack of dose-toxicity relationship for these reactions. Accordingly, this hypothesis is an attractive explanation for the basis of some IADRs.
Small doses of LPS precipitate modest inflammatory responses in mammals, resulting in increased susceptibility to toxicity from numerous hepatotoxic chemicals (Roth et al., 1997; Ganey and Roth, 2001). For example, exposing rats to a nonhepatotoxic dose of LPS resulted in a significant decrease in the dose of allyl alcohol required to produce liver injury (Sneed et al., 1997). This increase in sensitivity depended on an LPS-stimulated inflammatory response involving resident hepatic macrophages (Kupffer cells, KCs), cyclooxygenase (COX)-2-derived eicosanoids, neutrophils (PMNs), and the coagulation cascade (Sneed et al., 1997; Ganey et al., 2001; Kinser et al., 2002, 2004). Such observations with agents that cause type A hepatotoxic reactions provide further support for the possibility that inflammation can alter the sensitivity of liver to injury and for the hypothesis that some idiosyncratic reactions might arise from drug-inflammation interaction.
Evidence is growing that mild inflammation from exposure to LPS interacts with drugs that cause human IADRs to cause liver injury in experimental animals (Table 1). For example, coadministration of nonhepatotoxic doses of LPS and the antipsychotic drug chlorpromazine to rats results in liver damage that resembles human chlorpromazine idiosyncrasy (Buchweitz et al., 2002). Trovafloxacin (TVX), a fluoroquinolone antibiotic associated with idiosyncratic reactions, also interacts with LPS, resulting in hepatotoxicity both in rats and mice (Waring et al., 2006; Shaw et al., 2007). In contrast, levofloxacin, another quinolone antibiotic without the tendency for causing idiosyncratic liver injury in people, does not share this capacity for hepatotoxic interaction with inflammatory stress in rodents. Thus, this model of drug-inflammation interaction is able to distinguish a drug that causes IADRs from one in the same pharmacological class that does not. Besides LPS, Gram-positive bacterial components (i.e., peptidoglycan and lipoteichoic acid) also interact with nontoxic doses of TVX to precipitate liver injury in mice (Shaw et al., 2008). Results of recent studies show that three other drugs associated with IADRs, sulindac, amiodarone, and halothane, also induce liver injury at otherwise nontoxic doses when coadministered with LPS (Dugan et al., 2008; Lu et al., 2009; Zou et al., 2009). A recent study indicated that activation of the innate immune response by viral RNA mimetic poly(I:C) also exaggerated halothane hepatotoxicity (Cheng et al., 2009), supporting the hypothesis that inflammation contributes to IADRs (Roth et al., 2003). Such inflammatory stress-drug interaction models in animals that mimic IADRs in human patients could provide useful tools for mechanistic study, which in turn might lead to novel biomarkers and methods to prevent or treat IADRs. Because much work to date has focused on models in which rats are treated with LPS and either ranitidine (RAN) or diclofenac (DCLF), these IADR-associated drugs will be emphasized in the remainder of this review.
Similarity in ability of drugs to cause hepatotoxic IADRs in human patients and liver injury in LPS/drug models in rodents
II. Inflammation and the Liver
A. Overview of Inflammation as a Contributor to Tissue Injury
Inflammation is traditionally defined as a local reaction of tissue to irritation, injury, or infection characterized as “redness, swelling, pain, heat and loss of function.” However, it is now viewed in terms of the activation of cells of the innate immune system, the coordinated actions of the mediators they produce, and altered inflammatory gene expression and cell signaling. Inflammation participates in host defense against microbial pathogens but also has the potential to injure tissues. Indeed, as mentioned above, it has become clear that inflammation plays a role in the pathogenesis of many diseases, can cause tissue injury by itself, and can increase sensitivity of tissues to the toxic effects of xenobiotic agents.
Inflammation encompasses not only traditional inflammatory cells (e.g., PMNs, macrophages) and the mediators they produce (e.g., cytokines/chemokines, coagulation and complement proteins, lipid mediators), but also endothelial cells and parenchymal cells in the tissue (Ganey et al., 2004). The inflammatory cells can attack and damage tissues directly by releasing toxic factors such as reactive oxygen species (ROS), proteases, etc., or they can release cytokines, eicosanoids, or other mediators that lead indirectly to damaging events. The hemostatic and complement systems are also activated in inflammatory responses and can participate in tissue injury. Various transcription factors [e.g., nuclear factor κB, activator protein-1, early growth response-1] induce the expression of gene products integral to the inflammatory response. Transcriptional regulators not only control the production of inflammatory mediators but also play an important role indirectly in the activation of various cells involved.
LPS is an important example of an inflammagen that elicits the expression of a broad range of proinflammatory genes. Monocytes/macrophages are the primary innate immune cells that orchestrate LPS-induced inflammation. LPS-binding protein presents LPS to CD14 on the plasma membrane of these and other cells. The interaction of this complex with the pattern recognition receptor, Toll-like receptor 4 (TLR4), activates a complex intracellular signaling network. TLR signaling has been well studied and is described in detail elsewhere (Guha and Mackman, 2001; Oda and Kitano, 2006). LPS activates several proinflammatory intracellular signaling cascades [e.g., the mitogen activated protein kinases (MAPKs) p38, extracellular-regulated kinase 1/2, and Jun-N-terminal kinase (JNK)1/2, and IκB kinase] and anti-inflammatory signaling cascades (e.g., PI3K-Akt signaling pathway). These intracellular signaling pathways coordinate activation of transcription factors and the induction of gene expression. The response elicited by exposure to large amounts of LPS, such as during Gram-negative bacterial sepsis, can result in damage to several organs including the liver, as well as death (Hewett and Roth, 1993). Of importance, inflammatory cells such as KCs, endothelial cells, stellate cells, and bile duct epithelial cells in the liver recognize LPS and contribute to inflammation. KCs also remove LPS from the sinusoidal blood, thereby acting as an active “filter” for bacterial products inappropriately released into the portal circulation. This can reduce the exposure of other organs to LPS, but LPS originating from the GI tract can elicit a localized, and potentially damaging inflammatory response in the liver. The next section discusses the role of some inflammatory mediators in the pathogenesis of liver injury induced by systemic endotoxemia. Particular emphasis is placed on the contribution of hepatic parenchymal and nonparenchymal cells in the inflammatory response. The involvement of these mediators in liver toxicity from selected xenobiotic agents will also be mentioned.
B. Tumor Necrosis Factor α
Tumor necrosis factor α (TNFα) is critically important in inflammatory responses. Production of this cytokine is triggered by LPS mainly in monocytes/macrophages (Michalek et al., 1980), including KCs in the liver (Hewett and Roth, 1993). Hepatic expression of TNFα mRNA increases shortly after systemic exposure to LPS, and the concentration of TNFα protein in blood rises within a few minutes (Hewett and Roth, 1993). TNFα production can be regulated at a post-transcriptional level. For example, TNFα mRNA stabilization and translation are regulated by p38 MAPK (Neininger et al., 2002; Hitti et al., 2006). Furthermore, TNFα converting enzyme (TACE) cleaves the 26-kDa membrane-bound pro-TNFα protein to generate the secreted, 17-kDa mature TNFα (Aggarwal et al., 1985; Mullberg et al., 2000). The release of TNFα from cells in vitro and in vivo can be selectively blocked by hydroxamate-based metalloprotease inhibitors that inhibit TACE activity (Gearing et al., 1994; McGeehan et al., 1994; Mohler et al., 1994).
TNFα activates two cellular receptors (TNFR1 [p55] and TNFR2 [p75]) to initiate cell death signaling, promote inflammatory mediator release, increase expression of nitric oxide synthase 2, activate the hemostatic system, and induce cell proliferation (Vassalli, 1992; Hehlgans and Pfeffer, 2005). TNFα has been identified as a critical proinflammatory cytokine of the acute inflammatory response, as well as a major component in the pathogenesis of the septic shock syndrome (Tartaglia et al., 1993; Rietschel et al., 1996). The expression of TNFα and its role in liver damage have been reviewed elsewhere (Schwabe and Brenner, 2006; Tacke et al., 2009). Indeed, TNFα infusion into the circulation leads to a sepsis-like syndrome in rats (Tracey et al., 1986). Administration of TNFα-neutralizing antibodies to baboons protects them from lethal bacteremia triggered by infusion of live Escherichia coli (Tracey et al., 1987). In these models, TNFR1 is essential in mediating TNFα signaling, because TNFR1-deficient mice are protected from septic shock induced by LPS/d-galactosamine or Staphylococcus aureus superantigen/d-galactosamine (Pfeffer et al., 1993). However, in experimental models using concanavalin A or Pseudomonas exotoxin A-induced hepatitis, TNFR2 signaling seems to be important for the host-damaging effects (Kusters et al., 1997; Schümann et al., 1998). Administration of TNFα-neutralizing antibodies or inhibition of TNFα biosynthesis significantly attenuates liver injury from large doses of LPS in rodents (Hewett and Roth, 1993; Mohler et al., 1994). Moreover, inhibitors of TACE protect against endotoxin-mediated lethality, in which TNFα plays a critical role (Mohler et al., 1994).
TNFα not only promotes tissue injury, but it also has beneficial effects. Activation of TNFR2 by endogenous TNFα is important for the development of LPS-induced resistance to bacterial infection (Echtenacher and Mannel, 2002). The cecal ligation and puncture model generates invasion of gut-derived bacteria into the blood stream and into organs, causing a sepsis-like syndrome. In this model, TNFα is important for recovery and survival from septic peritonitis (Echtenacher et al., 1990, 1995). A state of immunoparalysis characterized by a reduced production of TNFα develops after cecal ligation and puncture, which results in bacterial superinfection and subsequent lethality. TNFα administration during this phase of immunoparalysis can be beneficial or deleterious, depending on the location of TNFα activity, timing of TNFα administration, and the type of infection (Echtenacher et al., 2003). From these results, it is clear that TNFα has multiple, sometimes opposing actions, and understanding of the role of TNFα in both defense against pathogens and host damage is currently incomplete.
C. Neutrophils
PMNs are involved in producing liver injury induced by large, hepatotoxic doses of LPS (Hewett et al., 1992). After exposure of rodents to LPS, the mRNAs encoding various PMN chemoattractants, such as cytokine-induced neutrophil chemoattractant-1, interleukin (IL)-8, or KC/Gro, and macrophage inflammatory protein-2 (MIP-2) increase in the liver, and these proteins are detectable in the plasma. The cellular source of these chemokines in the livers of endotoxemic mice has not been definitively identified, although Kupffer cell depletion does not affect hepatic MIP-2 and KC/Gro expression in LPS-treated mice (Kopydlowski et al., 1999). Adhesion molecules become up-regulated on the surfaces of sinusoidal endothelial cells (SECs), PMNs, and hepatocytes (Jaeschke et al., 1996). The up-regulation of these chemokines and adhesion molecules facilitates PMN transmigration across the endothelial cell layer and promotes subsequent localization of PMNs close to hepatocytes (Butcher, 1991; Springer, 1994). This transmigration and activation process involves rolling along, binding to, and migrating across the endothelium. The migration of PMNs from the liver microvasculature is regulated by at least three distinct types of molecules interacting with their respective receptors: selectins, integrins, and chemokines. A key feature is that these ligand-receptor interactions act in sequence, not in parallel (Ley et al., 2007). Selectins initiate the rolling of PMNs across the endothelium, whereas integrins cause firm adhesion. Integrins and chemokines comprise the driving force for transmigration into the liver parenchyma (Kobayashi, 2008). This concept of sequential action has been confirmed by the observation that inhibition of any one of these steps gives essentially complete, rather than partial inhibition of PMN emigration from blood vessels.
PMNs are critical mediators of injury in models of LPS-potentiated hepatotoxicity, such as from aflatoxin B1, monocrotaline, and TVX (Barton et al., 2000; Yee et al., 2003b; Waring et al., 2006). There are several likely mechanisms by which these cells contribute to tissue damage. When activated, PMNs release numerous cytotoxic factors, including ROS and proteases (Ganey et al., 1994; Jaeschke et al., 2002), such as cathepsin G and elastase (Ho et al., 1996). ROS and proteases mediate tissue injury in several pathological conditions such as acute lung injury, endotoxemia, and ischemia-reperfusion injury (Jaeschke et al., 1990, 1991; Kawabata et al., 2002).
There is no consensus on the relative contributions of ROS and proteases to PMN-induced hepatocyte injury. PMN-mediated hepatocellular death can occur within 1 h in vivo and coincides with the appearance of intracellular oxidant stress and the formation of hypochlorite-mediated chlorotyrosine protein adducts (Jaeschke et al., 2002; Gujral et al., 2004; Hasegawa et al., 2005). A selective inhibitor of PMN NADPH oxidase, the enzyme that catalyzes production of ROS, significantly delayed liver injury in galactosamine-sensitized mice given LPS (Gujral et al., 2004). Mice deficient in glutathione peroxidase-1 also showed enhanced susceptibility to PMN-mediated liver injury (Jaeschke et al., 1999). These results suggest a crucial role for the PMN-dependent oxidative burst and ROS generation in the pathogenesis of liver injury. On the other hand, in experiments using activated PMNs cocultured with hepatocytes, protease inhibitors, but not antioxidant enzymes, prevented PMN-mediated cell injury, and cell killing could be reproduced by substituting cathepsin G or elastase for PMNs (Mavier et al., 1988; Harbrecht et al., 1993; Ganey et al., 1994).
PMNs stimulated to release proteases and ROS kill hepatocytes with a time course similar to that of proteases alone (Ganey et al., 1994). This time frame (15–20 h) is much longer than that by which hepatocellular injury develops in LPS-treated rats, suggesting, however, that PMN proteases might act in concert with other factors to damage hepatocytes in vivo. It is also possible that isolated, primary hepatocytes lack key features of hepatocytes altered by an inflammatory response in vivo (Gujral et al., 2004). Such features could include increased sensitivity of hepatocytes to PMN-derived proteases. For example, when isolated hepatocytes were exposed to PMN proteases in a hypoxic environment, damage occurred more rapidly than in an oxygen-replete environment (Luyendyk et al., 2005). Moreover, PMN protease inhibitors protected the liver from injury caused by ischemia-reperfusion, although this attenuation was accompanied by a decrease in circulating chemokines and hepatic PMN accumulation (Yamaguchi et al., 1997). Thus, PMN proteases might contribute to hepatocellular injury by mechanisms independent of direct hepatocyte killing (Yamaguchi et al., 1997, 1999; Soejima et al., 1999). It is possible that PMN proteases induce intracellular oxidative stress in hepatocytes, which could be exacerbated by glutathione peroxidase deficiency. Thus, the composite of all inflammatory mediators and additional cellular stressors are probably necessary for the complete manifestation of PMN-dependent hepatocyte killing in vivo.
D. The Hemostatic System and Hypoxia
The coagulation and fibrinolytic systems are important controllers of vascular hemostasis (Levi et al., 2003) and act to limit hemorrhage after vascular injury (Mackman, 2007). On the other hand, the coagulation cascade contributes to thrombosis (e.g., myocardial infarction, stroke, deep vein thrombosis, pulmonary embolism) and to the pathogenesis of numerous diseases including atherosclerosis, cancer, and sepsis (Levi et al., 2003; Spek, 2004; Levi, 2005). Components of the coagulation and fibrinolytic systems also participate in the liver damage induced by LPS-mediated inflammation.
The hemostatic system has been the subject of numerous excellent reviews (e.g., (Mackman et al., 2007; Rau et al., 2007), and only elements germane to the studies below will be described briefly here. LPS induces the expression of tissue factor (TF; coagulation factor 3, thromboplastin) by hematopoietic cells (e.g., monocytes/macrophages, platelets) (Pawlinski et al., 2004), and this activates the coagulation cascade in mice. The formation of a complex between TF and coagulation factor VIIa activates a cascade of coagulation factors, ultimately generating the serine protease thrombin. Thrombin cleaves circulating fibrinogen into fibrin. Fibrin, upon cross-linking and polymerization, is a major component of clots in blood vessels. The coagulation cascade is negatively regulated by several proteins, such as protein C, TF pathway inhibitor, and antithrombin. Likewise, fibrin deposition in blood vessels is tightly regulated by the fibrinolytic system. Plasminogen activators, including urokinase PA and tissue-specific PA, cleave plasminogen into its active form, plasmin. Plasmin can cleave and dissolve cross-linked fibrin. The activity of the plasminogen activators is negatively regulated by plasminogen activator inhibitor-1 (PAI-1).
Inhibition of thrombin significantly attenuated LPS-induced liver injury in rats (Hewett and Roth, 1995; Moulin et al., 2001), although the injury was independent of circulating fibrinogen (Hewett and Roth, 1995). This suggests that thrombin activity, but not fibrin clots, is required for LPS-induced liver injury. Indeed, thrombin activation of the tethered-ligand receptor, protease activated receptor-1 (PAR-1), seems to play a critical role in LPS-induced liver injury. Perfusion of livers from LPS-treated rats with thrombin or a PAR-1 agonist caused hepatocellular injury in a PMN-dependent manner (Moulin et al., 2001; Copple et al., 2003). Moreover, thrombin inhibition impaired PMN activation in LPS-treated rats (Pearson et al., 1996a,b; Copple et al., 2003). These results suggest that thrombin activation of PAR-1 is important for PMN activation in endotoxemia and that both of these events are sufficient for causing inflammatory liver injury in rats given a large, hepatotoxic dose of LPS.
Although fibrin deposition does not seem to be important for liver injury caused by large doses of LPS, it might be important in animals cotreated with smaller doses of LPS and another xenobiotic agent (Luyendyk et al., 2004; Beier et al., 2009). One of the possible consequences of fibrin deposition in liver sinusoids is tissue hypoxia caused by impaired blood flow. Hypoxia can deplete cellular ATP and interfere with intracellular pH and homeostasis of ions such as Na+ and Ca2+, which could subsequently cause cell death (Carini et al., 1997,2000). In addition, hypoxia can cause the production of ROS (Lluis et al., 2005, 2007). It can also activate numerous intracellular signaling pathways related to cell stress and stabilize hypoxia inducible factor-1α, an important transcription factor (Piret et al., 2002). Breathing a low O2 atmosphere enhanced the liver lesions in rats caused by a large, hepatotoxic dose of LPS (Shibayama, 1987). This suggests that hypoxia might interact with LPS-induced inflammatory mediators to cause hepatocellular injury in vivo.
As mentioned above, PAI-1 is the major endogenous down-regulator of fibrinolysis. PAI-1 is synthesized by a variety of cells in culture (e.g., hepatocytes, adipocytes, endothelial cells, cardiac myocytes) (Macfelda et al., 2002; Westrick and Eitzman, 2007), although the cellular source of PAI-1 in vivo is probably model- and disease-dependent. A PAI-1 pool is also found in the granules of platelets, where it is stored and can be released after vessel damage (Hoekstra et al., 2004). PAI-1 is present in three forms in the circulation: active, inactive, and latent forms. The active form converts spontaneously into the latent form with a half-life of 1 h (Hoekstra et al., 2004). The latent form is more stable and can be reconverted into the active form. PAI-1 is removed from the circulation by the liver and is also inactivated by endothelium (Hekman and Loskutoff, 1985; Owensby et al., 1991). Endothelium- or platelet-derived PAI-1 is normally complexed to vitronectin, resulting in an increased half-life of PAI-1 in the circulation.
Experiments in vitro have demonstrated that expression of PAI-1 can be induced by a variety of factors involved in inflammatory responses, including TNFα, IL-1, transforming growth factor-β, LPS, glucocorticoids, and insulin. The transcriptional induction of PAI-1 is mediated through an Sp1 element, hypoxia-responsive element, and/or Sma- and Mad-related protein 3/4 (SMAD 3/4) binding sites in the PAI-1-promoter (Fink et al., 2002; Hou et al., 2004). Up-regulation of PAI-1 is associated with disseminated intravascular coagulation and other thrombotic diseases (Padró et al., 1995, 1997). PAI-1 also plays a role in models of acute and chronic liver injury (Bergheim et al., 2006; Arteel, 2008).
E. Summary of Lipopolysaccharide-Induced Inflammatory Responses
As discussed above, LPS affects numerous inflammatory pathways through activation of TLR4. These include intracellular signaling pathways and transcription factors (e.g., nuclear factor κB, p38) and inflammatory mediators (e.g., TNFα). The coordination of these mediators leads to the activation of innate immune cells (e.g., monocytes, PMNs), coagulation, and complement systems, etc., the consequence of which can be tissue injury (Table 2). In the context of hepatotoxic LPS-drug interaction, it is possible that numerous inflammatory mediators are critical, and these mediators might vary among drugs.
Examples of inflammatory mediators important in the pathogenesis of LPS-induced liver injury
III. Mechanisms of Liver Injury in Models of Inflammation-Drug Interaction: Ranitidine and Diclofenac as Examples
Given the scarcity and inconsistency of clinical data, animal models of LPS-drug interaction could be helpful in understanding IADRs. Mechanistic studies using such models could provide biomarkers to predict these reactions. This section discusses two potential IADR animal models resulting from LPS-drug interaction: LPS-RAN and LPS-DCLF models.
A. Ranitidine
1. Ranitidine-Induced Idiosyncratic Hepatotoxicity in Human Patients.
A widely used drug associated with idiosyncratic hepatotoxicity is the histamine 2 (H2)-receptor antagonist, RAN. RAN is available over the counter for oral administration or by prescription for parenteral administration for treatment of duodenal ulcers, gastric hypersecretory diseases, and gastroesophageal reflux disease. Idiosyncratic hepatotoxicity occurs in less than 0.1% of people taking RAN (Vial et al., 1991). A summary of numerous published case reports appears in Table 3. The capacity of RAN to cause IADRs is not related to its pharmacological action, because famotidine (FAM), a H2 receptor antagonist also in long-term use, does not share this liability. Most liver reactions from RAN are mild and reversible; however, extensive liver damage and death have occurred in some individuals (Ribeiro et al., 2000). RAN hepatotoxicity typically manifests as elevations in serum markers of hepatocellular injury with more modest increases in indicators of cholestatic injury. The reactions are typical of IADRs, because the time of onset of hepatotoxicity relative to initiation of therapy varies greatly (Table 3). Rechallenge with RAN does not necessarily result in a reoccurrence of toxicity (Graham et al., 1985). Indeed, in some cases the adverse response resolved despite continued therapy (Barr and Piper, 1981). These characteristics do not seem consistent with an adaptive immune response as the cause of RAN IADRs.
Summary of published case reports of ranitidine hepatoxicity
Blank entries indicate no report in the cited reference.
RAN is minimally converted into three metabolites in both humans and rats: N-oxide, S-oxide, and desmethyl metabolites (Carey et al., 1981; Chung et al., 2000). RAN is excreted mostly unchanged in urine in humans as opposed to through bile in rats (Carey et al., 1981; Huang et al., 2005). No known RAN metabolite has been reported to be toxic to liver either in humans or rodents to our knowledge. Accordingly, it seems unlikely that IADRs caused by RAN are due to metabolic bioactivation of this drug.
Although direct evidence implicating inflammation as a contributing factor to these idiosyncratic reactions is lacking, it is interesting that several human case reports of RAN idiosyncratic hepatotoxicity mentioned prodromal signs consistent with endotoxemia or inflammation (e.g., diarrhea, fever, nausea/vomiting, and/or abdominal pain; Table 3). In several of the cases, the patients had GI ulceration, which could increase translocation of bacterial products like LPS into the portal circulation. Thus, it is possible that some RAN-induced IADRs result from episodes of mild inflammation occurring during drug therapy. However, it is important to note that these clinical observations do not constitute definitive evidence of an inflammatory response in the patients with RAN hepatotoxicity and could also be related to ensuing liver damage. Results in an animal model suggest that concurrent inflammation increases susceptibility to RAN hepatotoxicity. The discussion below will summarize this model and what is known about the inflammatory cascade that results in liver injury.
2. Inflammation-Ranitidine Interaction.
Although RAN alone is not hepatotoxic in rats, modest underlying inflammation induced by a small, nontoxic dose of LPS precipitates IADR-like liver injury from RAN (Luyendyk et al., 2003). This was not the case with FAM, which does not share RAN′s propensity to cause IADRs in human patients. This indicated that the pharmacological target (H2 receptor) cannot be solely responsible for the hepatotoxic inflammation-RAN interaction. When given to rats 2 h after a nonhepatotoxic dose of LPS, RAN caused acute, midzonal, suppurative, necrotizing hepatitis that resembled lesions in animals treated with a hepatotoxic dose of LPS alone (Luyendyk et al., 2003), suggesting that RAN might increase hepatic sensitivity to this inflammagen. The liver injury in LPS/RAN-cotreated rats began 2 to 3 h after intravenous RAN injection, was maximal at 6 h, and was sustained for at least 24 h.
Hepatic inflammatory infiltrates in LPS/RAN-treated rats comprised predominantly PMNs, suggesting the possibility of a role for these cells in the liver injury (Luyendyk et al., 2005). In addition, the expression of several genes involved in hemostasis or the response to hypoxia was greatly enhanced in LPS/RAN-cotreated rats. One of these genes encodes PAI-1 (Luyendyk et al., 2004). Because PAI-1 is an important negative regulator of fibrinolysis, its enhanced gene expression suggested that fibrin clots might be involved in the injury. Furthermore, the crucial role of TNFα and other cytokines in liver toxicity from large doses of LPS raised the possibility of the importance of inflammatory cytokines in this model. The next sections review the evidence that supports the roles of these inflammatory factors and discuss how they interact with each other to contribute to the liver injury caused by LPS/RAN cotreatment.
3. Involvement of Hemostasis, Neutrophils, and Tumor Necrosis Factor α.
a. Hemostasis.
In rats, a small, nonhepatotoxic dose of LPS alone caused a mild and transient increase in plasma thrombin-antithrombin concentration, indicating activation of coagulation, and an increase in plasma PAI-1 concentration, suggesting impaired fibrinolysis. RAN cotreatment augmented and prolonged the increases in plasma thrombin-antithrombin and PAI-1 (Luyendyk et al., 2004). Activation of the coagulation system and the production of PAI-1 in LPS/RAN-cotreated rats was more pronounced than that in LPS/FAM-cotreated rats, which did not develop liver injury (Luyendyk et al., 2006a). Consistent with enhanced coagulation and impaired fibrinolysis, significant sinusoidal fibrin deposition occurred selectively in livers of LPS/RAN-treated rats. Cotreatment with either anticoagulant heparin, the fibrinolytic agent streptokinase, or a PAI-1 inhibitor decreased the hepatocellular injury induced by LPS/RAN, suggesting a role for the hemostatic system (Luyendyk et al., 2004). Hepatic hypoxia occurred in LPS/RAN-treated rats, and heparin reduced the tissue hypoxia and fibrin deposition (Luyendyk et al., 2005). RAN seems to selectively augment coagulation and PAI-1 production triggered by LPS, and this in turn causes fibrin deposition that leads to tissue hypoxia and hepatocyte death. SEC dysfunction probably contributes to coagulation system activation and PAI-1 production in LPS/RAN-treated rats (Luyendyk et al., 2004).
b. Neutrophils.
In the LPS/RAN model, PMNs accumulate in livers early in response to LPS, and pretreatment with either a PMN-depleting antiserum or an antiserum to CD18 integrin reduced the hepatocellular injury (Luyendyk et al., 2005; Deng et al., 2007). Because the PMN antiserum selectively reduced both circulating and hepatic PMNs, and the CD18 antiserum reduced hepatic PMN activation, the protection afforded by these two antisera indicates a crucial role for PMNs in the pathogenesis.
The absence of liver injury after treatment with LPS alone, despite PMN accumulation, suggested that PMNs are not extravasated and activated in liver after exposure to the noninjurious LPS dose used in these studies. This was confirmed by the observation that LPS alone did not cause an increase in immunostaining for hypochlorous acid (HOCl) adducted to liver proteins, a marker of PMN activation (Deng et al., 2007). In contrast, LPS/RAN cotreatment did cause hepatic PMN activation. Accordingly, PMNs require a secondary signal provided by RAN treatment to be activated and cause damage. RAN itself does not activate PMNs directly; in fact, it inhibits PMN activation both in vitro and in vivo (Okajima et al., 2000, 2002). This suggests that RAN acts indirectly through other inflammatory mediators produced during LPS exposure. These mediators might be PMN chemokines (i.e., MIP-2) or hemostatic factors like PAI-1, because both can promote PMN activation (Maher et al., 1997; Lentsch et al., 1998; Li et al., 2004). In fact, PAI-1 can potentiate LPS-induced PMN activation in vitro (Kwak et al., 2006). mRNA and serum protein concentration of both MIP-2 and PAI-1 were increased to a greater extent after treatment with LPS/RAN than after LPS alone at a time before liver injury onset (Luyendyk et al., 2006b), and this increase was unique to RAN compared with FAM. This raises the possibility that MIP-2 and/or PAI-1 act as signals to activate PMNs in this model. In this regard, a PAI-1 inhibitor reduced PMN activation in LPS/RAN-cotreated rats (Deng et al., 2008).
c. Tumor Necrosis Factor α.
Because TNFα is a critical cytokine involved in liver injury from large doses of LPS, the effect of LPS/RAN treatment on TNFα production was examined. At the nontoxic doses used in this animal model, LPS rapidly induced TNFα release into the serum; the serum TNFα concentration peaked at approximately 2 h and rapidly decreased after that, returning toward basal levels by 8 h (Tukov et al., 2007). RAN cotreatment caused the serum TNFα concentration increase to last longer than in rats given LPS alone, and LPS/RAN-cotreated rats developed hepatotoxicity. In contrast, FAM neither enhanced TNFα production nor caused liver injury when administered with LPS. Thus, the prolongation of LPS-stimulated TNFα production distinguished a drug that causes human IADRs from one that does not. Some TNFα-dependent cytokines/chemokines, such as IL-6, IL-1β, and MIP-2 also had the same pattern of prolonged increase after RAN cotreatment (Tukov et al., 2007).
To explore the role of TNFα in LPS/RAN-induced hepatotoxicity, pentoxifylline or etanercept was used to reduce or neutralize TNFα, respectively. Pentoxifylline is a methylxanthine that inhibits the synthesis of TNFα (Dezube et al., 1993; Barton et al., 2001; Yee et al., 2003a), but it also has several other pharmacological effects (Banfi et al., 2004). Etanercept is a dimeric fusion protein that contains a soluble TNFα receptor capable of selectively neutralizing TNFα in serum. Treatment with either pentoxifylline or etanercept significantly reduced serum TNFα concentration and activity, respectively, and both reduced hepatocellular injury in LPS/RAN-cotreated rats (Tukov et al., 2007). These results indicate that TNFα is critically involved in LPS/RAN-induced liver injury. To investigate more specifically the role of the RAN-induced prolongation of the TNFα response, a TACE inhibitor was administered immediately before RAN so that the inhibitor did not affect the initial, LPS-induced increase in serum TNFα concentration (Deng et al., 2008). This treatment regimen decreased hepatocellular injury, suggesting that the RAN-induced prolongation of the TNFα response was important for the pathogenesis.
Many of the cytokines/chemokines that are selectively up-regulated in LPS/RAN-treated rats (Luyendyk et al., 2006b; Tukov et al., 2007) are regulated by p38 and its downstream MAPK-activated protein kinase-2 (Neininger et al., 2002; Numahata et al., 2003; Hitti et al., 2006). Thus, it seemed possible that p38 MAPK activation might be an upstream signal leading to the pathogenic cascade. Indeed, RAN, but not FAM, selectively augmented p38 activation early after LPS treatment. A p38 inhibitor given at the same time as RAN reduced the hepatotoxicity (Deng et al., 2008). This suggests that p38 activation is critical for the liver injury after RAN cotreatment of LPS-treated rats.
Despite the increase in serum TNFα, RAN did not increase TNFα mRNA in liver after LPS treatment (Deng et al., 2008). Moreover, the reduction in serum TNFα protein concentration after p38 inhibition was not accompanied by diminished hepatic TNFα mRNA. These results suggested the importance of post-transcriptional events in the up-regulation of TNFα and its regulation by p38. Indeed, p38 and MAPK-activated protein kinase-2 can regulate TNFα production in macrophages mostly by increasing mRNA translation (Neininger et al., 2002; Hitti et al., 2006). As mentioned above, an increase in TNFα protein can arise from the cleavage of cell-bound pro-TNFα by TACE (Aggarwal et al., 1985; Mullberg et al., 2000); accordingly, another possibility is that p38 activated TACE, leading to increased TNFα protein release into the circulation. LPS/RAN treatment caused greater hepatic TACE activation than LPS/vehicle treatment (Deng et al., 2008). Moreover, a p38 inhibitor (SB 239603) reduced hepatic TACE activity after LPS/RAN treatment to the same level as LPS/vehicle treatment. Furthermore, a TACE inhibitor (BMS-561392; Luo et al., 2007) similarly reduced serum TNFα concentration and liver injury. All of these results suggest that RAN prolonged TNFα production after LPS treatment through augmented p38-dependent TACE activity.
4. Interaction of Hemostasis and Neutrophils and Tumor Necrosis Factor α.
Several interactions among inflammatory factors seem to be at play in the pathogenesis of LPS/RAN-induced liver injury. Some of those for which evidence exists are depicted in Fig. 1. In LPS/RAN-cotreated rats, TNFα inhibition led to decreases in PMN chemokines such as MIP-2 and in plasma markers of coagulation activation and impaired fibrinolysis (Tukov et al., 2007). This suggests that TNFα is a proximal inflammatory mediator relative to the hemostatic system or PMNs. This is supported by the observation that inhibition of coagulation did not reduce serum TNFα concentration (unpublished results). How TNFα contributes to hemostatic system activation in this model remains unknown, but it might act on vascular endothelial cells or on macrophages in an autocrine fashion to induce TF expression (Schwager and Jungi, 1994; Bierhaus et al., 1995; Parry and Mackman, 1995). In addition, in the presence of PMNs, TNFα exposure damages SECs in vitro (Smedly et al., 1986; Takei et al., 1995), and such damage can activate the coagulation system. TNFα and IL-1 also stimulate the expression and release of PAI-1 by endothelial cells in vitro (Schleef et al., 1988). The contribution of TNFα to TF production and inhibition of fibrinolysis has been demonstrated in vivo in a model of lung hemorrhagic shock (Fan et al., 2000). Thus, TNFα could promote coagulation and impair fibrinolysis by inducing TF and/or PAI-1 expression.

Proposed mechanism of LPS/RAN-induced liver injury. RAN augments TNFα production after LPS treatment in a post-transcriptional manner by enhancing p38 activation. The increase in TNFα protein occurs through the p38-dependent activation of TACE. The prolongation of LPS-induced TNF-α production by RAN seems to be crucial for liver injury. TNFα leads to coagulation system activation and PAI-1 production, both of which cause hepatic fibrin deposition. PAI-1 might also contribute to the activation of hepatic PMNs accumulated after LPS exposure. The hypoxia resulting from hepatic fibrin deposition and perhaps other factors could act synergistically with toxic proteases released from activated PMNs to kill hepatocytes. PMN proteases are also involved in enhancing PAI-1 production and fibrin deposition. HPC, hepatic parenchymal cell; STC, hepatic stellate cell; Trans-factor(s), transcription factor(s).
TNFα prompts the accumulation of PMNs in tissues by activating endothelial cells (Vassalli, 1992; Bradham et al., 1998) and primes PMNs for activation (Schleiffenbaum and Fehr, 1990; Nagaki et al., 1991; Vassalli, 1992; Kushimoto et al., 1996). In LPS/RAN-cotreated rats, neutralization of TNFα did not affect hepatic PMN accumulation but did reduce serum MIP-2 and PAI-1 concentrations (Tukov et al., 2007). These results suggest that the signal for PMN extravasation and activation might depend on TNFα, whereas PMN accumulation does not. Heparin similarly reduced serum MIP-2 and PAI-1 concentrations but had no effect on hepatic PMN accumulation (Luyendyk et al., 2006a). Thus, TNFα-mediated activation of coagulation induces the expression of MIP-2 and PAI-1, and these mediators may activate PMNs accumulated in the liver.
Mechanisms by which these events lead to activation of PMNs are diverse. One possibility is that coagulation activation causes activation of PAR-1 on KCs, endothelial cells, and/or hepatic stellate cells (Copple et al., 2003). PAR-1 can contribute to PMN activation but in an indirect manner, because this receptor is not present on PMNs (Copple et al., 2003). Fibrin clots can also modify the accumulation and activation of PMNs. For example, fibrin(ogen) interacts with adhesion molecules on rat PMNs, and this contributes to the innate immune response (Flick et al., 2004a,b). Another possibility is that hypoxia caused by occlusive fibrin clots in liver sinusoids promotes PMN activation by altering expression of chemokines and adhesion molecules. Hypoxia can induce chemokines such as MIP-2 or adhesion molecules such as intercellular adhesion molecule-1 and P-selectin on endothelial cells and/or hepatocytes (Shreeniwas et al., 1992; Pinsky et al., 1996; Xu et al., 1999; Laurens et al., 2005). These actions would favor the adhesion, transmigration, and activation of PMNs. Indeed, PMNs isolated from humans after acute exposure to a hypoxic atmosphere released more superoxide anion and elastase compared with PMNs from people who breathed air (Tamura et al., 2002).
In addition to modulating fibrin levels, PAI-1 might have direct effects on PMN activation. As noted above, a PAI-1 inhibitor decreased PMN activation in LPS/RAN-cotreated rats, whereas it did not affect hepatic PMN accumulation or serum PMN chemokine concentration (Deng et al., 2008), suggesting a direct effect of PAI-1 on PMN activation. A recent study showed that PAI-1 directly potentiated LPS-induced PMN activation through a JNK-dependent pathway (Kwak et al., 2006). These results suggest that RAN might induce activation of PMNs accumulated in the liver after LPS exposure indirectly by augmenting PAI-1 production.
In the LPS/RAN model, both PMN antiserum and CD18 antiserum reduced hepatic fibrin deposition at 6 h after RAN treatment, even though PMN depletion did not affect fibrin deposition at an earlier time at the onset of injury (Deng et al., 2007). PMNs express functional TF upon stimulation and can promote thrombin activation and fibrin deposition (Goel and Diamond, 2003, 2004; Maugeri et al., 2006). Although either PMN depletion or administration of PMN protease inhibitor (eglin C), decreased fibrin deposition, neither influenced plasma thrombin-antithrombin concentration, suggesting that the contribution of PMNs to fibrin is independent of coagulation cascade activation (Deng et al., 2007).
PMN lysosomal proteases cathepsin B, D, and G increased PAI-1 activity in the medium of human umbilical vein endothelial cells by cleaving PAI-1 from extracellular matrix (Pintucci et al., 1992, 1993; Kimura and Yokoi-Hayashi, 1996). Accordingly, PMNs could contribute to deposition of fibrin through inhibition of fibrinolysis by increasing active PAI-1. Indeed, PMN depletion and eglin C each reduced active PAI-1 at 6 h after RAN (Deng et al., 2007). This suggests that PMNs contribute to fibrin deposition during injury progression by releasing proteases to activate PAI-1 and thereby inhibit fibrinolysis. The lack of effect of PMNs on active PAI-1 at an earlier time (i.e., 2 h) is consistent with the observation that these cells are not activated until 3 h after LPS/RAN treatment. In this regard, the shedding of PAI-1 from endothelial matrix by PMN proteases might play a more dominant role at later times (i.e., 6 h). This could represent a feed-forward mechanism to cause more PMN protease release, because PAI-1 can cause PMN activation directly as mentioned above.
PMN depletion reduced liver hypoxia 2 h after LPS/RAN treatment (Deng et al., 2007). This early contribution of PMNs to hypoxia did not depend on sinusoidal fibrin deposition, because PMN depletion did not affect liver fibrin at this time. Furthermore, PMNs that have accumulated in liver are not activated at 2 h, suggesting that enhancement of hypoxia by PMNs at this time does not require their activation and might be mediated directly by plugging of sinusoids by these cells. LPS-RAN cotreatment caused a greater degree of tissue hypoxia than LPS given alone (Luyendyk et al., 2004), although these two treatments had a similar effect on PMN accumulation (Luyendyk et al., 2005). Thus, it is possible that RAN cotreatment causes PMNs to adhere more firmly to sinusoidal endothelium and/or to undergo a shape change that results in reduced sinusoidal perfusion and consequent hypoxia.
In addition to their role in hemostasis, activated PMNs release toxic products such as ROS and proteases that can damage hepatocytes directly. As noted above, a PMN-protease inhibitor diminished LPS/RAN-induced hepatocellular injury (Deng et al., 2007). The killing of hepatocytes in vitro by PMN elastase is enhanced by a hypoxic environment (Luyendyk et al., 2005). The time course over which elastase caused hepatocyte death during hypoxia in vitro (i.e., 2 h) is consistent with the onset of liver injury in LPS/RAN-treated rats. Taken together with the evidence that fibrin deposition and hypoxia are critical to liver injury in LPS/RAN-treated rats, it seems possible that PMNs contribute to the pathogenesis by releasing proteases that kill hepatocytes in an environment made hypoxic by fibrin deposition or other events.
The proposed pathogenic mechanism of LPS/RAN-induced liver injury is summarized in Fig. 1. RAN augments TNFα production after LPS treatment in a post-transcriptional manner by enhancing p38 activation, which in turn activates TACE. The prolongation of LPS-induced TNFα production by RAN seems to be crucial for the liver injury. TNFα leads to coagulation system activation and PAI-1 production, both of which cause hepatic fibrin deposition. PAI-1 might also contribute to the activation of hepatic PMNs accumulated after LPS exposure. The hypoxia resulting from hepatic fibrin deposition acts synergistically with toxic proteases released from activated PMNs to kill hepatocytes. PMN proteases are also involved in enhancing PAI-1 production and fibrin deposition.
In the LPS/RAN model, the initial molecular target of RAN remains unknown. The lack of liver injury after cotreatment of LPS with FAM at a dose pharmacologically comparable with RAN rules out H2 receptor antagonism as the only mechanism of RAN/LPS interaction. Because RAN, but not FAM, augmented hepatic p38 activation induced by LPS, and because p38 is crucial for the downstream cascade of TNFα production, coagulation activation, and PMN activation, it is possible that activation of the p38 pathway is the point at which RAN exerts its initial effect. RAN itself did not activate p38, whereas it enhanced the p38 activation after LPS exposure. Thus, RAN might perturb one or more of the signaling molecules in the pathway of LPS-induced p38 activation.
B. Diclofenac-Induced Idiosyncratic Hepatotoxicity
NSAIDs inhibit COX-1 and/or COX-2. Many drugs in this class cause hepatic IADRs. For example, DCLF has caused rare but sometimes serious hepatotoxicity in humans (Boelsterli, 2003b). In an analysis of 180 patients with DCLF hepatotoxicity, the latent period to onset of liver injury was quite variable, ranging from less than one month to greater than a year (Banks et al., 1995). The hepatotoxicity was predominantly hepatocellular, appeared most frequently in women with osteoarthritis, and was not accompanied by signs typically associated with an adaptive immune reaction. Although the apparent incidence of severe DCLF-induced hepatic adverse reactions is quite low (one to two cases per million prescriptions or 6–18 cases/100,000 person-years), the large number of patients treated with DCLF makes the absolute number of cases impressive (Walker, 1997). In addition, severe injury leading to liver transplantation occurred in a large proportion of the reported cases of DCLF-induced hepatotoxicity (Lewis, 2003). The scarceness of liver biopsies from patients and the diverse histopathlogical presentations in available samples make it difficult to draw clues about mechanisms from human liver pathology alone (Zimmerman, 1999).
Several mechanisms of DCLF-induced hepatotoxicity have been proposed, including metabolic and kinetic factors (Seitz et al., 1998; Aithal and Day, 2007; Daly et al., 2007), oxidative stress (Cantoni et al., 2003), and mitochondrial injury (Masubuchi et al., 2002; Lim et al., 2006; Lim et al., 2008; Siu et al., 2008). However, additional studies are required to determine the relevance of these mechanisms in vivo. For instance, DCLF or its metabolites are cytotoxic to hepatocytes in vitro only at large concentrations that are not achieved in vivo (Bort et al., 1999; Masubuchi et al., 2002; Gómez-Lechón et al., 2003). In patients who developed hepatotoxicity after DCLF treatment, associations have been identified with polymorphisms in enzymes that metabolize DCLF, such as UGT2B7 and CYP2C8, and in the DCLF glucuronide transporter ABCC2 (Daly et al., 2007). These could lead to increased exposure to hydroxylated and/or glucuronidated DCLF metabolites. However, there is currently no evidence to support a causal relationship between these DCLF metabolites and hepatotoxicity in vivo, although there are reports that DCLF acyl glucuronide is directly involved in small intestinal injury in rats (Seitz and Boelsterli, 1998).
There is some evidence suggesting an adaptive immune-mediated hypersensitivity in DCLF-induced hepatotoxicity. For example, a 53-year-old female developed fulminant hepatic failure after inadvertent rechallenge with DCLF (Greaves et al., 2001). DCLF conjugated to mouse serum albumin or keyhole limpet hemocyanin is immunogenic in mice (Kretz-Rommel and Boelsterli, 1995). Furthermore, T cells isolated from DCLF-keyhole limpet hemocyanin conjugate-treated mice killed hepatocytes previously exposed in vitro to noncytotoxic concentrations of DCLF. However, there are no animal models that have reproduced hepatotoxic DCLF-induced immunoallergic reactions in the liver. In fact, the T cell popliteal lymph node reaction was attenuated if animals were treated with DCLF orally compared with injection into the footpad (Gutting et al., 2002, 2003). Because DCLF is normally given orally to human patients, this raises a question about the likelihood that immunoallergic reactions contribute to DCLF-induced IADRs. DCLF autoantibodies have been found in human patients, but their relevance to idiosyncratic hepatotoxicity is unknown (Aithal et al., 2004). Thus, the lack of a demonstrable connection between an immunoallergic response to DCLF and overt hepatotoxicity raises a question as to whether DCLF IADRs occur by this mechanism.
As observed for RAN, a small, nontoxic dose of LPS given to rats rendered a nontoxic dose of DCLF injurious to the liver (Deng et al., 2006), suggesting inflammatory stress as a susceptibility factor for DCLF-induced IADRs. This merits special attention because most NSAIDs, including DCLF, have been associated in humans with GI injury, and the latter can promote LPS and/or bacterial translocation from the intestine into the circulation. In laboratory rodents a single, small dose of DCLF (1.5 mg/kg) induced intestinal ulceration (Seitz and Boelsterli, 1998; Atchison et al., 2000). Moreover, DCLF is normally prescribed to patients with inflammatory conditions, in which inflammatory mediators are usually present. The role of inflammatory stress in DCLF hepatotoxicity is further suggested by the clinical findings that IL-4 and IL-10 polymorphisms have been found in human patients who developed hepatotoxicity after DCLF treatment (Aithal et al., 2004). These polymorphisms would lead to less IL-10 and more IL-4 production. Because IL-10 is an anti-inflammatory cytokine and IL-4 can promote PMN activation (Bober et al., 1995), these changes could result in hepatotoxic interaction between DCLF and endotoxin or other inflammagens to which patients are exposed during drug therapy.
The mechanism by which LPS-DCLF interaction causes hepatocellular injury remains unknown. Antiserum-induced PMN depletion in LPS/DCLF-cotreated rats protected against liver injury, demonstrating a role for PMNs in the pathogenesis (Deng et al., 2006). PMNs cause cytotoxicity through the release of ROS and other toxic factors as described above. It is interesting in this regard that the addition of noncytotoxic concentrations of peroxidase/H2O2 to hepatocyte cultures markedly increased DCLF cytotoxicity (Tafazoli et al., 2005). In fact, several novel reactive metabolites of DCLF appeared when the drug was incubated with PMN-derived myeloperoxidase (MPO) (Zuurbier et al., 1990; Miyamoto et al., 1997). Thus, PMNs that accumulate in liver after LPS exposure might render DCLF more cytotoxic by forming MPO-derived metabolites.
Unlike RAN, DCLF causes hepatotoxicity in rats when given alone at larger doses than were used in LPS/DCLF cotreatment studies. Liver injury from a large dose of DCLF was associated with a marked increase in bacteria in the liver, consistent with increased intestinal permeability (Deng et al., 2006). Under these conditions, prior treatment with nonabsorbable antibiotics to sterilize the GI tract attenuated liver damage. These results support the hypothesis that DCLF-mediated GI disturbance leads to increased hepatic exposure to bacterial products that can induce inflammatory stress and increase susceptibility to hepatotoxicity. Microarray analysis of gene expression changes suggested that DCLF might interact with bacteria/LPS through inducing oxidative stress, apoptotic signaling, or changes in fatty acid metabolism in liver (Deng et al., 2008). In addition, hypoxia occurred in the livers of DCLF-treated rats and rendered hepatocytes sensitive to DCLF-induced cytotoxicity in vitro (Deng et al., 2008). These results suggest that hypoxia might be a secondary factor that interacts with DCLF to kill hepatocytes.
In summary, as depicted in Fig. 2, inflammation induced by a small dose of LPS interacts with a nontoxic dose of DCLF to produce hepatocellular injury in rats through a PMN-dependent mechanism. Furthermore, results from animal models are consistent with large, toxic doses of DCLF causing hepatotoxicity through a mechanism that involves gut-derived, inflammatory stimuli that probably induce oxidative stress and apoptotic signaling or alter lipid metabolism.

Working hypothesis for DCLF-induced hepatocellular injury. DCLF could cause hepatocellular injury through different modes depending on the dose. A nontoxic dose of DCLF interacts with an independently generated inflammatory stress (e.g., from LPS translocation) to produce hepatocellular injury in rats through a PMN-dependent mechanism. Large, toxic doses of DCLF cause hepatotoxicity through a mechanism that depends on gut-derived bacteria/LPS and probably do so by inducing oxidative stress and apoptotic signaling or altering lipid metabolism. Mito, mitochondrial.
IV. Summary and Conclusions
There is evidence, especially in experimental animals, suggesting that inflammatory stress could be a susceptibility factor for IADRs. In this review, rodent models of human hepatic IADRs were discussed that involve interaction between inflammatory stress and RAN or DCLF. It would be helpful to have more information about the role of inflammatory stress in IADRs from these two drugs in humans. In the case of RAN, epidemiological study evaluating the connection between inflammation and liver injury has not been conducted. For DCLF, the type of arthritis might be important in determining sensitivity to DCLF-induced liver injury; a retrospective study showed that patients with osteoarthritis were at greater risk for DCLF-induced liver injury than those with rheumatoid arthritis (Banks et al., 1995). This difference might relate to differences in patterns of inflammation in the two conditions. Furthermore, as mentioned above, polymorphisms in genes encoding cytokines that would favor a proinflammatory state have been identified in patients developing DCLF-induced liver injury. However, epidemiological studies designed to explore the role of inflammatory stress in human IADRs caused by DCLF are lacking. Thus, in human patients, the connection between inflammatory stress and IADRs has not been established with certainty, although results in animals render this connection plausible. It seems likely from the results in animals that an acute inflammatory response (rather than chronic inflammation) during drug therapy might be needed to evoke an IADR. Because many inflammatory responses such as cytokine production are short-lived events, the lack of such connection in available clinical data might be related to the fact that clinical samples are invariably taken after hepatotoxicity develops (i.e., at a time when the peak of potentially pathological inflammatory responses has passed).
The inflammatory stress-drug interaction mechanism for IADRs is not necessarily inconsistent with other hypotheses regarding underlying causes of these reactions (Fig. 3). For example, exposure to LPS can decrease hepatic P450 expression, and could therefore result in parent drug accumulation, potentially with toxic consequences. Conversely, activated leukocytes and the products they release (ie., MPO) can metabolize drugs to reactive metabolites (Uetrecht, 1991). Inflammation might also be an important factor in adaptive immune-mediated hepatotoxicity. For example, in one model of allergic hepatitis, cotreatment with LPS markedly increased the hepatotoxicity (Mizoguchi et al., 1990). Thus, inflammation might act as a sufficient danger signal to promote hepatotoxicity after drug challenge in a sensitized individual.
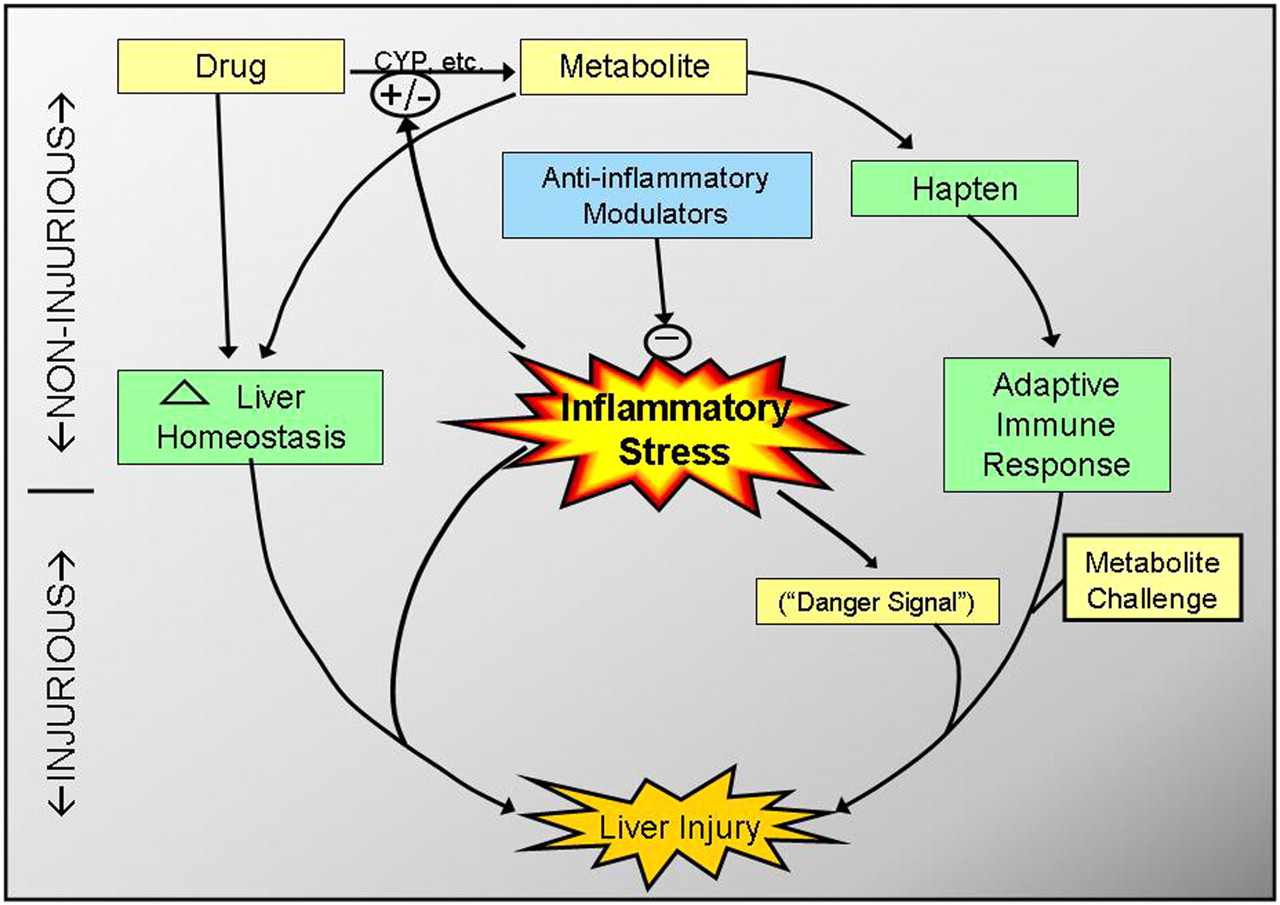
Integration of the “inflammation hypothesis” with other hypotheses for etiology of idiosyncratic hepatotoxicity. Inflammation can influence a drug's propensity to cause idiosyncratic toxicity by several modes. For example, inflammatory cytokines can increase the concentration of a drug by inhibiting P450 expression. Activated leukocytes have also been implicated in metabolism of drugs to reactive species. Modification of proteins by these metabolites could result in hapten formation and, upon rechallenge, precipitation of an adaptive immune response, which during a concurrent inflammatory stress (i.e., as a danger signal) might cause hepatotoxicity. Finally, the ability of a drug to alter hepatocellular homeostasis might render the liver sensitive to injury from normally noninjurious activation of inflammatory mediators.
In describing the “multiple determinant hypothesis,” Li and colleagues suggested that the probability of an idiosyncratic reaction occurring from a drug is related to the product of the probabilities of several factors including, but not limited to, drug exposure, environmental factors, genetic polymorphisms, altered metabolism, formation of antigens, and inadequate liver repair (Li, 2002). One important environmental factor might be exposure to inflammagens. However, one mechanism is unlikely to account for all idiosyncratic reactions, and numerous factors should be considered in future development of animal models.
It has been suggested that many drugs might cause stress to the liver at therapeutic doses, often resulting in mild injury (e.g., modest serum alanine aminotransferase increases) to which most patients adapt during continued therapy. It is hypothesized that failure to adapt in some patients could permit progression to liver damage and that this could be a mode by which IADRs occur (Watkins, 2005). If so, it could be that inflammatory stress is one of the factors that prevents adaptation and enables a liver homeostatically altered by drug exposure to progress to frank injury (Fig. 3).
One reason for continued occurrence of hepatic IADRs is that drugs with the propensity to cause these reactions are not identified under current preclinical safety testing paradigms. It is possible that this inflammation-drug interaction model could provide a basis for preclinical drug safety screening models to identify new drug candidates that might have this propensity. The concordance between IADRs in humans and inflammation-drug interaction in animals with certain known drugs (Table 1) largely supports this idea. This list is small relative to the number of drugs responsible for human hepatic IADRs, however, and the hypothesis requires validation with more drugs. Furthermore, the animal model is cumbersome as a screening tool. Nonetheless, the information gleaned regarding interaction of drugs and inflammatory mediators that result in hepatotoxicity in these animal models could be valuable for development of more streamlined, predictive models.
Another utility of these models is in the identification of early biomarkers that portend serious reactions in human patients. In the LPS/RAN rat model, several inflammatory mediators (e.g., TNFα, PAI-1, thrombin) are selectively up-regulated before liver injury, and these proved crucial for the pathogenesis. These or other inflammatory mediators might serve as early biomarkers for IADRs resulting from inflammation-drug interaction.
Acknowledgments.
This work was supported by the National Institutes of Health National Institute of Environmental Health Sciences [Grant ES004139]; National Institutes of Health National Institute of General Medicine [Grant GM075865]; and the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant DK061315].
Footnotes
-
This article is available online at http://pharmrev.aspetjournals.org.
doi:10.1124/pr.109.001727.
-
↵1 ABBREVIATIONS:
- © 2009 by The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵





{kind=link}
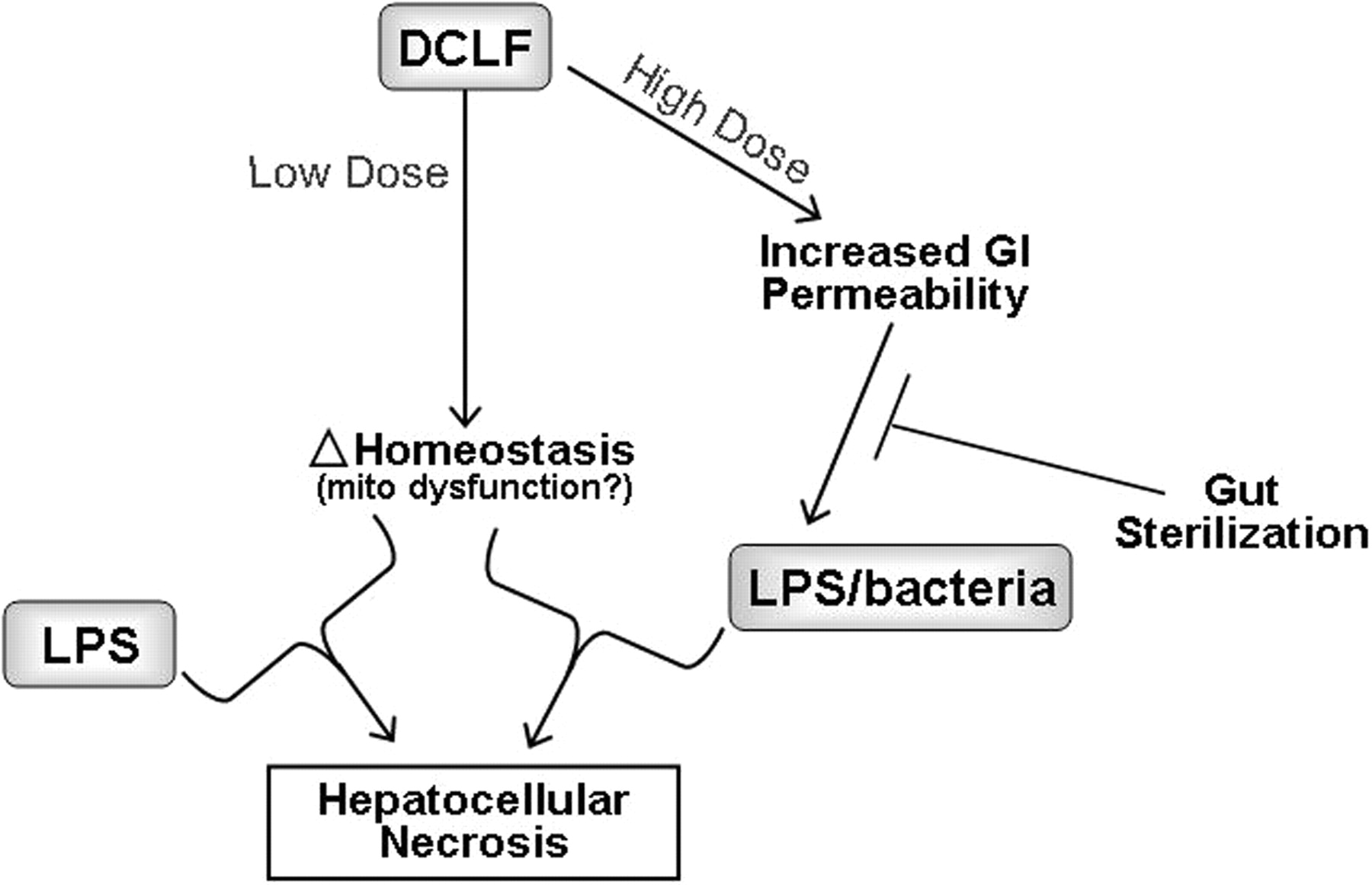{kind=link}
{kind=link}