


Abstract
For many years seven transmembrane domain G protein-coupled receptors (GPCRs) were thought to exist and function exclusively as monomeric units. However, evidence both from native cells and heterologous expression systems has demonstrated that GPCRs can both traffic and signal within higher-order complexes. As for other protein-protein interactions, conformational changes in one polypeptide, including those resulting from binding of pharmacological ligands, have the capacity to alter the conformation and therefore the response of the interacting protein(s), a process known as allosterism. For GPCRs, allosterism across homo- or heteromers, whether dimers or higher-order oligomers, represents an additional topographical landscape that must now be considered pharmacologically. Such effects may offer the opportunity for novel therapeutic approaches. Allosterism at GPCR heteromers is particularly exciting in that it offers additional scope to provide receptor subtype selectivity and tissue specificity as well as fine-tuning of receptor signal strength. Herein, we introduce the concept of allosterism at both GPCR homomers and heteromers and discuss the various questions that must be addressed before significant advances can be made in drug discovery at these GPCR complexes.
I. Introduction
The G protein-coupled receptor (GPCR1) superfamily of seven transmembrane domain-containing proteins is the largest family of integral membrane proteins encoded by the human genome, being derived from 3 to 4% of all genes (Foord et al., 2002). There are more than 400 nonolfactory GPCRs expressed in man and the GPCR superfamily can be subdivided phylogenetically into five main families: rhodopsin (also known as class A), secretin (class B), glutamate (class C), frizzled and adhesion (Fredriksson et al., 2003; Fredriksson and Schiöth, 2005). GPCRs can be traced back through evolution to plants and fungi (Fredriksson et al., 2003; Fredriksson and Schiöth, 2005) and have evolved to recognize an extraordinary diversity of ligands, including lipids, photons of light, odorants, tastants, hormones, and neurotransmitters (Perez, 2003, 2005; Lagerström and Schiöth, 2008). Although they are referred to generically as GPCRs because almost all of the family members are known to transduce at least some signals through heterotrimeric G proteins, there is a growing appreciation that GPCRs can transmit certain signals by G protein-independent means. This has resulted in the adoption of the label seven transmembrane (7TM) receptors by some commentators (Langmead and Christopoulos, 2006; Schwartz and Holst, 2006; Kenakin, 2007; Lefkowitz, 2007b).2
A. Minimal Signaling Units: G Protein-Coupled Receptors as Monomers, Homomers, and Heteromers
GPCRs were long considered to exist exclusively as monomers (refer to section VI for definitions) within the plasma membrane, where ligand binding induces a conformational change in the receptor resulting in activation of G protein and, hence, regulation of second-messenger cascades, receptor phosphorylation, desensitization, and internalization (Lefkowitz, 2004, 2007a; Hill, 2006). This view has been challenged, however, by an ever-expanding number of studies that have demonstrated GPCRs to be able to exist as dimers or even as higher-order oligomers (Milligan, 2004, 2008, 2010b; Palczewski, 2010). Although it is clear that GPCRs can (and indeed do) still function when they are purified as monomers and incorporated into reconstitution systems (Ernst et al., 2007; White et al., 2007; Whorton et al., 2007, 2008), it is equally clear that GPCRs can traffic, signal, and internalize as multimeric complexes (Gurevich and Gurevich, 2008; Milligan, 2008, 2010b) and that they have an intrinsic capacity to self-associate when present in phospholipid bilayers (Harding et al., 2009). These apparently divergent observations may reflect that, outside of the glutamate receptor subfamily, there is little evidence to indicate that covalent interactions underlie such protein-protein interactions and that a mixture of monomers and higher-order complexes may exist, with equilibria between forms defined by interaction affinities. Although homomers and heteromers are often considered to exist as dimers throughout their life cycle (Milligan, 2008), several recent and elegant studies have challenged the prevailing view that they are “born, live, and die” as dimers. Using single molecule total internal reflectance fluorescence microscopy, Hern et al., (2010) demonstrated that the average lifetime of association of a M1 muscarinic receptor homomer was ∼0.5 s and that no more than 30% of M1 receptors existed as M1-M1 homomers at any given time (Hern et al., 2010). This concept was also explored in studies on the stability of dopamine D2 receptor complexes. Although clearly able to form detectable dimers/oligomers (Han et al., 2009) these may be transient unless the interactions are captured by cross-linking between putative dimer interfaces (Fonseca and Lambert, 2009). Furthermore, the stability of dimers may vary significantly, even between closely related GPCRs. For example, via imaging of fluorescence recovery after photobleaching, the β2-adrenoceptor was shown to form stable complexes, whereas the β1-adrenoceptor interacted more transiently (Dorsch et al., 2009). There is also a complex (and potentially confusing) literature on the role of ligands in enhancing, reducing, or having little effect on GPCR dimerization. In part, this may relate to GPCR expression levels and their relative affinities of interaction. Although a series of studies has suggested that the extent of dimerization is constant over substantial ranges of GPCR expression levels (Angers et al., 2000; Maurel et al., 2008), in certain cases, ligands may modulate the extent of dimerization in a fashion that depends on receptor expression level. For example, although at high expression levels the extent of cell surface signal corresponding to dimerization of the human M3 muscarinic acetylcholine receptor was high and little affected by addition of the endogenous agonist acetylcholine or the metabolically more stable analog carbachol, at lower expression levels, the basal signal was lower, and carbachol increased this signal substantially (Alvarez-Curto et al., 2010b). By contrast, in both situations, the antagonist atropine was essentially without effect (Alvarez-Curto et al., 2010b). Although requiring many more studies and examples, this may suggest that at low expression levels, the extent of dimerization is regulated (in this case enhanced) by conformational changes associated with agonist occupancy, whereas at high expression levels, a substantial degree of constitutive interaction is present in the absence of ligands; therefore, ligand regulation is limited. However, it should be noted that differences apparently exist even within receptor subgroups. In the case of the M1 muscarinic acetylcholine receptor, the antagonist ligand pirenzepine has been suggested to enhance dimer formation (Ilien et al., 2009). This topic, although highly relevant to the current review, is beyond its scope; however, an extensive review is available (Saenz del Burgo and Milligan, 2010b).
Many such “dimeric” interactions have now been described and fit into two broad categories: GPCR homomers, in which two or more identical GPCR polypeptides combine to form a biochemically or pharmacologically distinct macromolecule, or receptor heteromers, in which two or more nonidentical (and independently functional) GPCRs exist as a complex and display behaviors distinct from those characteristic of either of the two GPCRs expressed alone (Ferré et al., 2009). Much information has been obtained from studies in which one or more protomers have been modified by mutation (Damian et al., 2006; Sartania et al., 2007; Alvarez-Curto et al., 2010b). Although we do not strictly adhere to the definitions above, for reasons of simplicity we will also refer to dimers in which such modifications have been made to the same receptor subtype as receptor homomers. An additional classification has been provided for another type of heteromer, that of the class C glutamate family GPCRs—here, the protomer subunits of these heteromers are generally not functionally active in the absence of their dimeric partner (Pin et al., 2003). Thus, these heteromers are obligate and have recently been defined as heteromeric receptors (in contrast to the case of GPCR heteromers, the constituent proteins of which are functional receptors in their own right) (Ferré et al., 2009). A database of information on GPCR dimerization/oligomerization is maintained at http://data.gpcr-okb.org/gpcr-okb/ (Skrabanek et al., 2007; Khelashvili et al., 2010).
If a GPCR is capable of signaling as a monomer, what are the potential advantages to a cell to express such receptors as homo- or heteromers? Most likely there are many advantages, not least the expanded pharmacological possibilities provided by using combinations of the gene products and ligands available to it to enhance diversity and provide improved fine tuning of response. Furthermore, receptor homo- and heteromerization has been shown to alter cell surface delivery and retention of certain GPCRs (Pin et al., 2003; Lopez-Gimenez et al., 2007; Canals et al., 2009), modulate G protein-coupling (Banères and Parello, 2003; Jastrzebska et al., 2006), cause cross-activation (Carrillo et al., 2003) or cross-inhibition (Lavoie et al., 2002; Mercier et al., 2002; Barki-Harrington et al., 2003; Lavoie and Hébert, 2003; Breit et al., 2004) of signaling, modify desensitization profiles (Pfeiffer et al., 2001) and either promote (Jordan et al., 2001; McVey et al., 2001; Pfeiffer et al., 2002, 2003; Ramsay et al., 2002; Perron et al., 2003; Stanasila et al., 2003; Xu et al., 2003) or reduce (Lavoie et al., 2002; Mercier et al., 2002; Lavoie and Hébert, 2003; Breit et al., 2004) internalization of receptor away from the cell surface. Given the wide-ranging effects reported for heteromerization between GPCR pairs, and the importance of GPCRs as the molecular targets of many therapeutic medicines, it is likely that ligands targeting receptor heteromers may offer clinical advantage. Thus, it is important to examine not only the generality of expression of such GPCR heteromers and the basis of selectivity of these interactions but also the means by which pharmacological agents alter their function, a form of allosterism.
II. Allosterism
The term allosterism is derived from the Greek word for “other.” In practice, allosterism is the process by which the interaction of a chemical or protein at one location on a protein or macromolecular complex (the “allosteric” site) influences the binding or function of the same or another chemical or protein at a topographically distinct site. When considering allosteric effects at GPCRs, the distinct site is routinely the binding site (the “orthosteric” site) of the endogenous agonist, although this does not inherently need to be the case. For example, different classes of allosteric regulators may bind to distinct, nonorthosteric sites on the receptor and modulate the action of one another. Protein-protein interactions are inherently allosteric because they result in changes in the energy landscapes of the constituent proteins. Such interactions are very common in biology, and it has been suggested that enhanced protein stability arising from such interactions is the reason for the widespread commonality of dimeric and higher-order interactions (Marianayagam et al., 2004). The prototypic example of the benefit of such allosteric interactions in enhancing biological function is the manner in which hemoglobin binds oxygen. Binding of an oxygen molecule to one protomer of the tetrameric hemoglobin complex enhances binding of subsequent molecules of oxygen to distinct binding sites. In this instance, however, each binding site, although topographically distinct, is equivalent; hence, this type of allosteric interaction is referred to as cooperativity (refer to section VI for definitions).
A. Allosterism at G Protein-Coupled Receptors
For GPCRs, the traditional view is that an allosteric binding site is not only topographically but also molecularly distinct. This derives from the expectation that GPCRs are monomers. In this situation, binding of two ligands to the same molecular site would inherently be competitive and could not occur concurrently. As such, although it has been suggested that apparent cooperativity in such studies does not inherently imply dimerization (Chabre et al., 2009), ligand binding studies not compatible with GPCRs' acting as a single class of noninteracting sites have played an important, although sometimes underappreciated and undervalued, role in providing evidence in favor of the presence of GPCR homomers (Wreggett and Wells, 1995; Chidiac et al., 1997; Armstrong and Strange, 2001; Park et al., 2002; El-Asmar et al., 2005; Pin et al., 2005; Urizar et al., 2005; Springael et al., 2006; Vivo et al., 2006; Sohy et al., 2007). Activation of GPCRs upon agonist binding is an elegant example of an allosteric transfer of energy: herein, induced conformational changes in the GPCR are detected by the interacting G protein. Thus energy, or in pharmacological terms, a “signal,” is transmitted from the extracellular milieu to the intracellular face of the plasma membrane of a cell.
More recently, it has become clear that the actions of both endogenous and synthetic ligands acting at the orthosteric site of the receptor can be altered by ligands that bind allosterically to the receptor (Fig. 1). Such allosteric modulators can alter either the affinity and/or efficacy of a ligand at the orthosteric site and do so in either a positive [positive allosteric modulator (PAM)] or negative [negative allosteric modulator (NAM)] manner. In addition, a ligand binding at an allosteric site may itself possess efficacy (an allosteric agonist) or, indeed, possess both efficacy and allosteric modulatory properties (an ago-allosteric modulator). Within this framework, it is conceptually possible although unlikely in practice that, as with a neutral antagonist, which is defined as binding to the orthosteric site without altering the signaling state of the receptor, a silent allosteric modulator could theoretically occupy an allosteric site without influencing measurable characteristics of the orthosteric ligand (May et al., 2007). Allosteric modulator effects at a single GPCR have been referred to as “on-target” allosterism (Ballesteros and Ransom, 2006). Such ligands have been discussed widely in recent years, both in an academic context and as novel ligands for therapeutic use. This reflects the fact that allosteric ligands can provide substantially greater GPCR subtype selectivity and have a theoretical maximum effect on orthosteric ligand function. Thus, in conditions in which side effects of orthosteric ligands are expected to be intolerable because receptor subtypes are widely expressed and/or mediate a wide and complex range of functions (e.g., muscarinic acetylcholine and metabotropic glutamate receptors) or the therapeutic window of orthosteric ligands may be low, allosteric ligands may become therapeutics of choice (Langmead and Christopoulos, 2006; Kenakin, 2007, 2009b; Conn et al., 2009a; Kenakin and Miller, 2010).

On-target allosteric effects on binding and function at a monomeric GPCR. A, binding of an allosteric modulator (red) to a monomeric GPCR can result in reciprocal modulation of an orthosteric ligand (yellow) binding to a nonoverlapping site on the receptor. The simulated example provided illustrates the effect of changing concentrations of a modulator (x-axis) with different α values (the cooperativity factor that reflects the influence of a modulator on affinity) on the binding of a fixed concentration of radioligand at the orthosteric site. Where α > 1, the modulator is a PAM for orthosteric binding, whereas α < 1 represents NAMs at the receptor. Thus, it is apparent how a NAM with strong negative allosteric properties can prevent binding of an orthosteric ligand at either a monomer or indeed dimer. Curves were simulated using the simple allosteric Ternary Complex Model described by Christopoulos and Kenakin (2002) using Prism 5.02 (GraphPad Software, San Diego, CA) with the following parameters for the orthosteric radioligand: KD, 1 nM; radioligand concentration, 1 nM. The modulator was assigned a KB of 10−6 M, and α was varied as indicated. B, in cases where the orthosteric ligand possesses efficacy (i.e., can generate a measurable response), represented here by fractional response (F/F0), an allosteric modulator can alter the measured potency of the orthosteric ligand. In this simulation, the arrows indicate the shift in EC50 with increasing concentrations of allosteric modulator with an α value fixed to either 20 (this would make the allosteric modulator a PAM, therefore less orthosteric agonist is required for an equivalent effect) or 0.05 (a NAM, where more orthosteric agonist is required to achieve an equivalent effect). This simulation illustrates one of the principal tenets of allosterism: despite increasing concentrations of allosteric modulator, the shift in orthosteric EC50 is saturable. If the same experiment was performed with a simple competitive antagonist, the right shift seen for the NAM would theoretically continue infinitely as the concentration of antagonist is increased. The curves were simulated as for (A) with the following parameters: EC50 = 10−6 M, KB = 10−9 M, basal = 0, Emax = 1, Hill slope = 1, and the concentration of allosteric modulator, B, was varied from 10 μM to 0.1 pM. C, in addition to modulating the binding or signaling of an orthosteric ligand, an allosteric modulator can itself possess efficacy and is thus both an allosteric modulator and an allosteric agonist, subsequently referred to as an ago-allosteric modulator. In the accompanying example, concentration-response curves were simulated for an orthosteric agonist in the presence of increasing concentrations of ago-allosteric modulator according to the operational model of allosteric modulation and allosteric agonism (Leach et al., 2007). Because the allosteric modulator possesses efficacy, increasing concentrations of the coadministered ligand increases the basal signaling in the system, yet unlike a partial agonist, this ligand is also a PAM for the signaling of the orthosteric ligand. Simulation parameters were as follows: τA = 20, τB = 1 [where τ represents the capacity of either orthosteric (A) or allosteric (B) ligands to act as agonists], KA = 10−6 M, KB = 10−7 M, α = 20, β = 10 (where β represents the allosteric effect on efficacy), slope factor = 1, basal = 0, and Emax = 1. The concentration range for ago-allosteric modulator was 3 mM to 0.1 nM. In theory, there is no reason why the same effects on orthosteric ligand binding and efficacy would not exist at a homo- or heterodimer.
There are several hallmarks of allosterism that relate to these points. First, because an allosteric modulator confers a change in conformation with respect to an orthosteric ligand, the relationship must be both reciprocal and saturable. Indeed, the so-called “ceiling effect” observed for allosteric modulators that are themselves without overt direct efficacy is one of their most favorable pharmacological attributes, because an effect should be produced only in the presence of an orthosteric (most probably the endogenous) agonist. As such, a pathological signal or “overdose” via that receptor is (theoretically) not possible. Because of the reciprocal energy exchange produced by the binding of allosteric and orthosteric ligands, another hallmark of allosterism is “probe dependence,” the observation that the existence or the extent of an allosteric effect can be dependent upon the identity of the orthosteric ligand being used as the “probe.” This characteristic makes prediction of allosteric behaviors and screening for allosteric modulators particularly challenging (Christopoulos and Kenakin, 2002; Milligan and Smith, 2007; Kenakin and Miller, 2010). This is particularly true for orphan GPCRs, because these are characterized and defined by lack of knowledge of the true endogenous orthosteric ligands and for GPCRs that have multiple endogenous “orthosteric” agonists, including, for example, the chemokine receptors. It has been argued that allosteric binding sites have escaped the evolutionary pressures that have been maintained at the orthosteric binding sites of GPCRs because the orthosteric site must continue to recognize and induce responses to the native ligand (Soudijn et al., 2004). However, other regions of the receptor subtypes are likely to have drifted in sequence more substantially, allowing the potential of selective interactions with nonendogenous, small chemical ligands that may modulate receptor function. As such, ligands that target allosteric sites of receptor subtypes can be anticipated to be substantially more selective than those at the orthosteric site. A classic example of this is in the muscarinic acetylcholine receptor family, where the endogenous ligand, acetylcholine, binds to and activates all five receptor subtypes with similar affinity/potency and where attempts to generate selective orthosteric ligands have had limited success (Christopoulos and Kenakin, 2002; Lu et al., 2002; Eglen, 2005). By contrast, a growing number of selective ligands have been identified that bind at allosteric sites of the receptor subtypes (Jakubík et al., 1997; Birdsall et al., 1999; Lazareno et al., 1999, 2004; Chan et al., 2008; Jones et al., 2008; Conn et al., 2009b). This may result, however, in a different set of challenges for drug discovery. If allosteric sites have diverged through evolution at a greater rate than orthosteric sites, this is likely to be reflected in greater differences between allosteric sites in species homologs and, therefore, to make translation from initial pharmacological studies on a heterologously expressed human GPCR to animal models of disease even more problematic. Despite these issues, two allosteric regulators, cinacalcet as a PAM at the Ca2+ sensing receptor (Nagano, 2006; Bräuner-Osborne et al., 2007) and maraviroc as a NAM at the chemokine CCR5 receptor (Dorr et al., 2005; Biswas et al., 2007), are now clinically approved medicines.
B. Homomeric and Heteromeric Allosterism
As introduced above, protein-protein interactions, including those between a receptor and an effector, are inherently allosteric in nature. Thus, it is intuitive that ligand binding and effector coupling at higher-order GPCR complexes must also be allosteric. A clear example of altered pharmacology as a result of the interactions between two different proteins is that of the receptor activity modifying protein (RAMP) family with the calcitonin and calcitonin receptor-like (CL) GPCRs. Here, interaction of a RAMP with the 7TM polypeptide is sufficient to define the pharmacological phenotype of the GPCR. For example, coexpression of RAMP1 with the CL receptor polypeptide results in a functional signaling unit that responds with high affinity to calcitonin gene-related peptide. However, when RAMP2 is expressed instead with the CL receptor, the heteromeric unit now has the pharmacological characteristics of an amylin receptor (Poyner et al., 2002; Hay et al., 2006; Sexton et al., 2009).
Allosteric effects at GPCR dimers are potentially more subtle (and far more numerous) than the example above of CL receptors. If we consider GPCR heteromers, then allosterism can occur in a number of ways (Fig. 2). First, binding at the orthosteric or an allosteric site of protomer “A” of a receptor heteromer can result in allosteric modulation within the same protomer (on-target allosterism) or allosteric modulation of ligand affinity at either an allosteric or the orthosteric site of protomer B. Second, the same series of interactions can result in changes in efficacy across the heteromer independent of, or in addition to, modulation of affinity. Additional complexity can be predicted because a GPCR polypeptide can possess more than one allosteric ligand-binding site, thus multiple on-target and off-target allosteric effects can theoretically occur simultaneously. Finally, it is conceivable that some ligands can act allosterically to influence only a subset of functions, as is the case with orthosteric ligands that display functional selectivity (also known as biased agonism, ligand-directed trafficking of receptor stimulus or pluridimensional signaling) (Galandrin et al., 2007; Kenakin, 2008). With respect to GPCR homomers, in which two copies of the same GPCR have formed a macromolecular complex, there is one difference in the definition of modulation across receptors: where the binding sites on protomer A and protomer B are identical, whether it be by definition an orthosteric or allosteric site, the interaction of the same ligand at these two sites is referred to as cooperativity (Fig. 2).

Allosteric possibilities at GPCR homomers and heteromers. Homo- or heteromerization provides the opportunity for both on-target (as seen in Fig. 1) and off-target allosterism. For simplicity, allosteric modulation of ligand binding across a dimer is illustrated (without accounting for effects on signaling), although allosterism will also occur across higher order oligomers. A, at a GPCR homodimer, an orthosteric ligand (yellow) can bind to one or both protomers, and binding of the first ligand can lead to a conformational change in the homodimer such that the affinity of the second identical protomer for the second identical copy of the ligand is altered either positively or negatively. This is referred to as cooperativity and is indicated in A by orange arrows. Cooperativity can also occur between two identical allosteric ligands (red) binding to the same site on different protomers. In addition to cooperative effects on affinity, the allosteric ligand can influence binding of the orthosteric ligand on the same protomer (on-target allosterism) and on the opposing protomer (off-target allosterism) and this modulation is reciprocal. B, for a heterodimer that, by definition, must comprise two different GPCR protomers, each of the allosteric and orthosteric binding sites is unique, and any energy transfer between them must be allosteric in nature. If we assume a single allosteric binding site for each of protomer A (blue) and protomer B (green), then up to four different ligands are capable of concurrently occupying the heterodimer and allosterically influencing each other. Note: for the purpose of illustration we have depicted the allosteric modulators binding within the transmembrane region of the protomers, although allosteric ligands are able to interact at numerous sites on a GPCR, including the extracellular loops and even the intracellular surface of the receptor.
GPCR heteromerization provides an exciting possibility for the fine-tuning of receptor signals, tissue specificity, and, potentially, the reduction of clinical side effects. Whether examining a receptor heteromer or homomer, there are clearly a number of ways in which allosterism can occur, making both the prediction of pharmacological consequences and screening for distinct outcomes difficult. Although the advent of heteromer-specific screening strategies (discussed in section V) should make identification of heteromer-selective ligands more practical, a number of measures are already available to researchers for defining or identifying allosteric interaction at dimers. For example, measures of conformational changes in protomer A in response to ligands acting at protomer B are indicative of allosterism across a dimer—such changes can be monitored as alterations in radioligand binding kinetics (El-Asmar et al., 2005; Urizar et al., 2005; Springael et al., 2006; Sohy et al., 2007) or in fluorescence (Damian et al., 2006, 2008). Allosterism can also be manifest as changes in signaling properties, whether as subtle as differences in efficacy (Ciruela et al., 2001; Parenty et al., 2008) or as profound as signal switching (George et al., 2000; Jarrahian et al., 2004; Lee et al., 2004). For example, the dopamine D1-dopamine D2 heteromer, both in transfected cell systems and in striatum, is able to activate Gαq and hence elevate intracellular [Ca2+] (Lee et al., 2004; Rashid et al., 2007), although neither partner receptor is generally associated with this signaling cascade. Likewise, certain GPCR heteromers, including the dopamine D1-D2 heteromer (Rashid et al., 2007), dopamine D2-D3 heteromer (Maggio et al., 2003, 2009; Maggio and Millan, 2010), the δ-μ-opioid heteromer (Gomes et al., 2004; Snook et al., 2006, 2008) and the δ-κ-opioid heteromer (Waldhoer et al., 2005) display selectivity to ligands not observed at the individual partner receptors. Although beguiling, experimental measures of signaling differences attributed to the presence of heteromers must be adequately controlled to exclude the influence of cross-talk downstream of monomeric or homomeric receptors, rather than representing a specific heteromer-mediated effect (Prezeau et al., 2010). For example, activation of a serotonin 5-HT2A receptor results in enhanced efficacy of morphine to promote internalization of a coexpressed μ-opioid receptor (Lopez-Gimenez et al., 2008). However, unlike ligand coregulation of internalization and recycling of coexpressed orexin OX1 and cannabinoid CB1 receptors, which does reflect heteromerization of these two GPCRs (Ellis et al., 2006), detailed studies demonstrated that the 5-HT2A receptor and μ-opioid receptor were trafficking independently, rather than within a heteromeric complex, and that the enhanced efficacy of morphine required downstream signals generated by the 5-HT2A receptor (Lopez-Gimenez et al., 2008).
A number of attempts have been made to model the interactions across dimers mathematically (Durroux, 2005; Franco et al., 2007, 2008; Rovira et al., 2010). Although probably too simplistic to be applicable in many experimental contexts [for example, the majority only include one or at most two active state conditions when receptors are now anticipated to exist in an ensemble of various conformations (Kobilka and Deupi, 2007)], they are useful theoretically because they highlight the various parameters that should be explored when considering allosterism across heteromers. For example, Franco and colleagues (Franco et al., 2007, 2008; Casadó et al., 2009) have argued that two-site agonist binding observed in many radioligand binding assays, although traditionally ascribed to the presence of both high- and low-affinity states of the receptor, could alternatively be explained by cooperative ligand binding at a receptor dimer. However, it must be noted that radioligand binding studies performed on preparations of GPCR monomers reconstituted with appropriate G proteins do recapitulate the key observations. Meanwhile, Rovira et al. (2010) have highlighted ways in which existing experimental data can be explained by functional selectivity across receptor dimers (Rovira et al., 2010). Although a discussion of the mathematical basis of these models and the assumptions required for their generation is beyond the scope of this review, interested readers are referred to several accessible articles on the topic (Durroux, 2005; Franco et al., 2008; Rovira et al., 2010).
III. Evidence for Allosterism at G Protein-Coupled Receptor Dimers
Our aim is not to provide a comprehensive list or discussion of which GPCRs have been reported to form homomers and heteromers in heterologous and native tissues, as this has been the subject of a plethora of reviews in recent years (Devi, 2000; Milligan, 2004; Terrillon and Bouvier, 2004; Pin et al., 2005, 2007; Milligan, 2006, 2009; Gurevich and Gurevich, 2008; Ferré et al., 2009; Maggio et al., 2009; Hudson et al., 2010; Khelashvili et al., 2010; Rozenfeld and Devi, 2010). Instead, we will discuss specific examples in which allosterism has been examined or identified across dimers and, where appropriate, the conclusions that may be drawn from these studies.
A. Homomers as Allosteric Complexes
Evidence for allosterism or cooperativity within homomeric protein complexes is largely derived from radioligand binding studies or from receptor complexes that are essentially heteromers, in that one of the protomers has been modified so that it can be distinguished, either biochemically or pharmacologically, from its dimeric partner. Radioligand binding experiments are one of the fundamental methods for establishing conventional “on-target” allosterism at GPCRs and have themselves provided early evidence of cooperative homomeric GPCR interactions well before the first GPCRs were even cloned (Limbird et al., 1975; Davis et al., 1977; Powell-Jones et al., 1979). More recently, negative cooperativity has been demonstrated by radioligand binding at a number of GPCR homomers, such as H2 relaxin at relaxin RXFP1 homomers (Svendsen et al., 2008b) and insulin-like peptide 3 at RXFP2 receptors (Svendsen et al., 2008a), neurotensin at neurotensin 1 receptors (White et al., 2007), secretin at secretin receptors (Gao et al., 2009), vasopressin and oxytocin at their respective receptors (Albizu et al., 2006), atypical antipsychotics clozapine and risperidone at the serotonin 5-HT2A (Brea et al., 2009), quinuclidinylbenzilate and N-methylscopolamine at muscarinic acetylcholine M2 receptors (Wreggett and Wells, 1995; Chidiac et al., 1997; Sum et al., 2001; Park et al., 2002), raclopride, spiperone, and nemonapride at dopamine D2 homomers (Armstrong and Strange, 2001; Vivo et al., 2006), and thyroid-stimulating hormone (TSH) and follicle-stimulating hormone at their respective GPCRs (Urizar et al., 2005).
In a well-controlled series of experiments examining cooperativity at the dopamine D2 receptor, Strange and colleagues (Armstrong and Strange, 2001; Vivo et al., 2006) investigated the phenomenon whereby sodium ions could alter the binding of [3H]raclopride but not N-[3H]methylspiperone (Hall et al., 1990). Armstrong and Strange (2001) demonstrated in Chinese hamster ovary (CHO) cells that both ligands bound D2 receptors orthosterically and in a competitive manner, yet [3H]raclopride seemed to recognize half the binding sites compared with those labeled by [3H]spiperone if sodium was removed from the binding buffer. Furthermore, unlabeled raclopride reduced saturation binding of [3H]spiperone in a noncompetitive manner, suggesting that the D2 receptors existed as a homomeric complex and that raclopride was a NAM both for its own binding and that of spiperone. Addition of sodium increased the affinity and Bmax of [3H]raclopride but not [3H]spiperone, whereas coincubation of [3H]spiperone with haloperidol, another member of the butyrophenone class of D2 ligands, indicated that the allosteric interaction observed was probe-dependent (Armstrong and Strange, 2001). In a later study, D2 receptors were expressed in Sf9 insect cells and allosterism was examined with radiolabeled versions of raclopride, spiperone and an additional ligand, nemonapride (Vivo et al., 2006). Again, different levels of receptor expression were reported depending upon the radioligand used, and both raclopride and nemonapride were sensitive to sodium ions. The studies were carefully controlled to account for ligand depletion [experiments were performed in either 1- or 10-ml volumes (Armstrong and Strange, 2001)], and the role of G proteins, which themselves are allosteric modulators of GPCR function—recently it has been claimed that G protein coupling and the presence or absence of guanine nucleotide can account for binding data that has otherwise been interpreted as reflecting cooperativity or allosterism across dimers (Chabre et al., 2009). However, the allosteric modulation observed at D2 was unaffected by GTP (Vivo et al., 2006), and binding differences to the various radioligands have also been reported for D2-Gαo receptor–G protein fusions (Gazi et al., 2003). Furthermore, in CHO cells expressing a vast excess of G protein in relation to D2 receptor, the agonist [3H]n-propylnorapomorphine was used to examine cooperativity and allosterism specifically at G protein-occupied homomers (Kara et al., 2010). Although ligand binding and dissociation was modulated by the addition of GTP, cooperativity was still evident, indicating that these results and those with antagonist radiolabels are not merely an artifact of asymmetrical G protein interactions across a dimer.
Differential labeling of receptor number by orthosteric ligands has also been reported for the muscarinic M2 receptor. In hamster cardiac ventricular membranes, which should contain only the M2 muscarinic receptor subtype, N-methylscopolamine was found to display strong negative allosterism toward [3H]quinuclidinylbenzilate binding, despite both ligands binding to the orthosteric site of the receptor, and the magnitude of effect was attributed to a tetrameric M2 homomer (Chidiac et al., 1997). Strong negative allosterism consistent with homomers was also reported at M2 receptors purified from pig atria, where neither ligand depletion nor failure to reach equilibrium could account for the findings (Wreggett and Wells, 1995), and a later study concluded that the negative allosteric effects were consistent with a tetramer (Park et al., 2002). The muscarinic studies discussed here also present a cautionary tale with respect to appropriate controls and alternative explanations of apparent allosterism. For example, Park et al. (2002) found that the calculated size of the homomeric complex differed depending upon the age of the membrane sample and the method of solubilization or storage, although in every case the evidence supported a complex greater than one protomer of the receptor (Park et al., 2002). Meanwhile, a number of the examples in Wreggett and Wells (1995) were later found to be incorrectly interpreted based on the surprising and disappointing discovery that the radioligand batch used contained up to 50% unlabeled precursor. However, in a thorough and commendable follow-up to this study (Sum et al., 2001), the authors were able to demonstrate that the basic concept of allosterism was still applicable to some of their findings in the original article (Wreggett and Wells, 1995) and entirely to their second article (Park et al., 2002). Thus, allosterism across homomers can be demonstrated using radioligand binding approaches similar to those used for on-target allosterism.
Non–radioligand-based approaches have also been used to effectively demonstrate communication across homomers. For example, Mesnier and Banères (2004) used detergent-isolated leukotriene B4 (LTB4) receptors (BLT1) to examine conformational changes across a receptor homomer. One protomer contained a C97A mutation such that it recognized LTB4 with 100-fold lower affinity than its wild-type counterpart and had every tryptophan except Trp234 removed so that conformational changes in the receptor could be monitored by fluorescence via 5-hydroxytryptophan labeling. By coexpressing this protomer with a wild-type receptor and purifying the resulting complex, it was possible to stimulate the wild-type protomer and monitor conformational changes in the associated mutant. As such, LTB4 binding was shown to induce a conformational change in the unbound protomer, indicating communication between the constituent receptors (Mesnier and Banères, 2004). A similar approach was employed for the purified dimeric ligand-binding domains of the metabotropic glutamate receptor, mGlu1, where glutamate binding displayed negative cooperativity and could be disrupted by prevention of dimerization (Suzuki et al., 2004). Meanwhile, functional reconstitution between two inactivated versions of the dopamine D2 receptor has recently been used to demonstrate allosteric communication across a homomer (Han et al., 2009). Dopamine D2 receptors couple endogenously to Gαi G proteins but not to Gαq, so to distinguish signaling via specific protomers within a dimer from endogenous signaling, receptor-G protein fusion constructs were generated between D2 receptors and the chimeric G protein Gqi5, which facilitates coupling to Gαi receptors but generates signaling through Gαq to ultimately increase calcium (Kostenis et al., 2005; Milligan and Kostenis, 2006). By fusing the Gqi5 element to the receptor with a short linker, the authors demonstrated that the fused monomer could not couple to its attached G protein, most likely as a result of restricted conformational movement. However, coexpression with a wild-type D2 receptor, itself unable to signal via endogenous Gαq/11, led to reconstitution of calcium signaling in response to the dopamine receptor agonist quinpirole, indicating that dimerization and allosteric communication across protomers was occurring (Han et al., 2009). Such cooperativity between protomers is in agreement with a previous study that demonstrated altered contacts at transmembrane helices 4 between two dopamine receptors within a homomer using chemical cross-linking after agonist or antagonist treatment (Guo et al., 2005).
Functional reconstitution has also been used to demonstrate allosterism across homomers in two elegant papers on glycoprotein hormone receptors. Members of this subfamily of receptors consist of a large hormone-binding domain containing numerous leucine-rich repeats and a typical rhodopsin-like 7TM region responsible for signal transduction. To examine allosterism across glycoprotein hormone receptor homomers, Urizar et al. (2005) generated chimeric [TSH receptor 7TM fused to the ligand binding domain of the luteinizing hormone/chorionic gonadotrophin (LH/CG) receptor], truncated (TSH receptor 7TM domain only), and mutant (ligand binding-competent but signaling-deficient receptor) versions of the TSH receptor. Expression of the individual receptors failed to lead to cAMP generation as a result of the absence of either TSH binding or signal transduction, yet coexpression of the nonsignaling mutant with either the truncated TSH receptor or the chimeric LH/CG-TSH receptor resulted in rescue of cAMP signaling (Urizar et al., 2005). These results suggested that allosteric communication occurred across a homodimer and were further corroborated by the demonstration of negative cooperativity between the ligand binding domains: unlabeled human CG was able to dose-dependently displace 125I-TSH from the TSH “homomer” comprising an intact TSH protomer and LH/CG-TSH receptor chimera (Urizar et al., 2005). Extending this concept in vivo, a very recent study provided the first evidence of the functional reconstitution across a homomer in a whole animal (Rivero-Müller et al., 2010). Knockout of the LH receptor in mice leads to underdeveloped external and internal genitalia, arrested testicular growth and descent, reduced gonadal sex hormone production, failure to reach sexual maturity, and infertility (Lei et al., 2001; Zhang et al., 2001). Using an LH receptor-null background, Rivero-Müller et al. (2010) introduced either a binding- or a signaling-deficient version of the LH receptor into the LH receptor gene locus, which resulted in no phenotypic change in the mice. Critically, however, cross-bred mice containing copies of both receptor mutants displayed rescued phenotypes equivalent to those of wild-type mice with intact LH receptor signaling, indicating functional complementation (Rivero-Müller et al., 2010).
B. Allosteric Interactions at Heteromers
1. Obligate Heteromeric Receptors.
Members of the class C or metabotropic glutamate group of GPCRs are the most clearly defined as dimeric or oligomeric. In part this reflects that the large extracellular domains of a number of these receptors have been generated recombinantly in large amounts, resulting in their purification and subsequent crystallization (Kunishima et al., 2000; Muto et al., 2007). Indeed, such crystals have shown direct protein-protein interactions between these domains, implying dimerization of the full-length receptors. Furthermore, because both agonist-occupied and -unoccupied forms of these domains have been crystallized, considerable insight into the mode of initial structural changes in the receptor in response to ligand binding has been gained. In the examples of the GABAB receptor and the small group of T1 taste receptors, two distinct protomers are required to produce the fully functional, pharmacologically defined receptors. At least one of the protomer subunits of these heteromers is not functionally active in the absence of its dimeric partner (Pin et al., 2003); thus, these heteromers are described as obligate. In the case of the GABAB receptor, only the GABAB1 subunit is able to bind the endogenous agonist GABA, whereas mutations of the intracellular elements of the GABAB2 subunit are sufficient to interfere with GABA-mediated G protein activation (for review, see Rovira et al., 2010). Since the cloning of the GABAB1 “receptor,” researchers had puzzled at their inability to achieve equivalent ligand affinities in some heterologous expression systems compared with native tissues (Marshall et al., 1999), a phenomenon later explained by the fact that coexpression of GABAB2 was required for expression of a fully functional receptor (Jones et al., 1998; Kaupmann et al., 1998; White et al., 1998). Furthermore, GABAB2 enhanced the affinity of agonists for GABAB1 (Kaupmann et al., 1998), providing a clear example of allosteric regulation of a receptor heteromer. Multiple levels of allosterism at the obligate GABAB heteromer were further described in an elegant study by Galvez et al. (2001). Using chimeric protomers containing the extracellular ligand binding domain of one GABAB subunit and the 7TM region of the opposing protomer, they demonstrated that the extracellular and 7TM composition of the heteromer was crucial for ligand binding and function (Galvez et al., 2001). For example, the extracellular domains of the receptor had to be heteromeric for receptor function, as chimeras expressing both GABAB1 or GABAB2 extracellular domains were nonfunctional. In contrast, G protein coupling was retained whether the 7TMs within the dimer were heteromeric or both GABAB2, although the identity of the second 7TM was clearly important allosterically as the GABAB agonists baclofen, GABA and 3-aminopropanephosphinic acid each had greater efficacy and potency if the 7TM protomers were heteromeric. In another study, direct interactions between the extracellular domains were found to allosterically modulate the effects of γ-amino butyric acid at the GABAB1 subunit (Liu et al., 2004). The examples of the T1 taste receptors are even more illuminating. Responses to savory (umami) and sweet taste sensations involve the expression of taste receptor heteromers. These two receptor heteromers share the T1R3 subunit, and its physical association with either T1R1 or T1R2 determines umami or sweet responses, respectively (Li et al., 2002; Xu et al., 2004). It is noteworthy that although the extended extracellular domain of T1R1 binds key ligands that are perceived as savory flavors, such as l-glutamate and inosine monophosphate, and the equivalent domain of T1R2 binds synthetic sweeteners, such as aspartate and the related molecule neotame (Xu et al., 2004), it is the 7TM domain of the T1R3, which is shared between the sweet and savory taste-responsive heteromers, that binds a series of taste modulators such as lactisole and cyclamate (Xu et al., 2004; Jiang et al., 2005). These act to alter the perception of the “orthosteric” ligands (Galindo-Cuspinera and Breslin, 2006) and are further clear examples of allosteric regulation within receptor heteromers. Similar aspects of the binding site(s) of allosteric modulators are also evident for the GABAB receptor. For example, 3-(3′,5′-di-tert-butyl-4′-hydroxy)phenyl-2,2-dimethylpropanol (CGP7930) is a positive allosteric modulator of the GABAB receptor (Adams and Lawrence, 2007; Pin and Prézeau, 2007), but this and related ligands appear to bind to the GABAB2 subunit, again providing evidence for allosterism across protomer partners of a heteromer.
2. Altered Ligand Binding.
Alterations in ligand binding affinity or dissociation kinetics have also been observed in cells and tissues coexpressing pairs of GPCRs that can form heteromers and provide further evidence of allosteric communication across GPCRs. Particularly when performed on cell membrane preparations, such studies can be strongly supportive of allosteric interactions between heteromers, because potential contributions of desensitization and other effects linked to protein post-translational modifications are diminished or excluded. Separate studies on the adenosine A2A receptor demonstrated that agonist binding to this polypeptide was sufficient to reduce the affinity of ligands at the partner protomer. After coexpression of the adenosine A1 and A2A receptors in HEK293 cells, Ciruela et al. (2006) found that although this did not inherently influence the affinity or binding maximum of [3H]R-PIA at the A1 protomer at equilibrium, the affinity of [3H]R-PIA for the A1 receptor polypeptide was reduced upon coincubation with the adenosine A2A agonist 4-[2-[[6-amino-9-(N-ethyl-β-d-ribofuranuronamidosyl)-9H-purin-2-yl]amino]ethyl]benzenepropanoic acid hydrochloride (CGS21680) (Ciruela et al., 2006a). Similar results were obtained in native pre- and postsynaptic neurons endogenously coexpressing the A1 and A2A receptors and potentially, therefore, the A1-A2A heteromer (Ciruela et al., 2006a).
Dopamine binding to the D2 protomer is also modulated in a manner similar to the A1 receptor when part of an adenosine A2A-dopamine D2 heteromer. A2A-D2 heteromers are found in GABAergic enkephalinergic neurons and have been demonstrated experimentally by coimmunoprecipitation, fluorescence (Förster) resonance energy transfer (FRET), and bioluminescence resonance energy transfer (BRET) in heterologous expression systems (Canals et al., 2003). Treatment with CGS21680 reduced the ability of dopamine to compete with the D2 antagonist [3H]raclopride (Ferre et al., 1991), and this was one of the key early studies consistent with expression of a GPCR heteromer. Clearly, such effects must be underpinned by further, consistent pharmacology. So, the effect of CGS21680 reflected binding to the adenosine A2A receptor element and indicated an allosteric interaction across the A2A-D2 heteromer because dopamine affinity was restored in the presence of an A2A antagonist (Ferre et al., 1991). Dopamine receptor affinity is also modulated in the proposed dopamine D1-D3 heteromer as the affinity of the D1 agonist ligand 6-chloro-2,3,4,5-tetrahydro-1-phenyl-1H-3-benzazepine hydrobromide (SKF81297) is increased by occupancy of the D3 receptor with the selective agonist R(+)-7-hydroxy-2-dipropylaminotetralin in both heterologous expression and native systems (Marcellino et al., 2008). However, the authors were unable to observe reciprocal effects of D1 receptor occupancy on agonist affinity at the D3 receptor. The authors noted that “the intramembrane interaction is not reciprocal.” However, this example illustrates a number of key points. In terms of basic chemical equilibria, allosteric effects are required to be reciprocal. However, as we have already commented on the “probe-dependence” of allosteric effects, such experiments should be performed in both directions with the same sets of ligands and many examples in the literature fail to heed this requirement. Likewise, when exploring potential α2A-adrenoceptor-β1-adrenoceptor heteromers, altered competition curves for ligands displacing the β1-adrenoceptor radioligand [3H]dihydroalprenolol were noted, affinity of tested unlabeled drugs being enhanced, decreased, or unaffected by coexpression of the α2A-adrenoceptor (Xu et al., 2003). Once more, these effects did not seem to be reciprocal, apparently violating one of the main tenets of allosterism. Of course, a further challenge in such studies is that to observe reciprocal effects, it is probably necessary that the heteromer constitutes a substantial fraction of the total receptor population. In cases in which “allosteric” effects seem to be unidirectional, it is possible that much of the protomer at which effects are observed is within a heteromer, whereas this may not be true for the partner GPCR, depending on their relative expression levels. In the case of the somatostatin sst2A-sst3 heteromer, negative allosterism seems to be so profound that, although somatostatin-14 and the sst2A ligand methyl (2S)-6-amino-2-[[(2R)-2-[[(2S)-1-[(4-nitrophenyl)amino]-1-oxo-3-phenylpropan-2-yl]carbamoylamino]hexanoyl]amino]hexanoate (L-796,778) are still able to bind to the heteromer, affinity of the sst3 ligand L-796,778, is no longer measurable (Pfeiffer et al., 2001).
A further example in which very distinct ligand binding characteristics have been recorded at a heteromer is the interaction between the serotonin 5-HT2A receptor and mGlu2 (González-Maeso et al., 2008). In membranes produced from mouse somatosensory cortex, the affinity of a series of hallucinogenic serotonergic agonists to compete with [3H]ketanserin to bind the 5-HT2A receptor were markedly higher in the presence of (−)-2-oxa-4-aminobicyclo[3.1.0]hexane-4,6-dicarboxylic acid (LY379268), a mGlu2/3 receptor agonist. Furthermore, the affinity of LY379268 and a number of other agonists at the glutamate receptor to compete for binding with the mGlu2/3 antagonist 2S-2-amino-2-(1S,2S-2-carboxycyclopropan-1-yl)-3-(xanth-9-yl)propionic acid (LY341495) was substantially lower in the presence of the hallucinogen DOI. In this example, bidirectional effects on agonist affinity are observed. Although little overlap of expression of mGlu3 and the 5-HT2A receptor was observed in brain, suggesting that the relevant heteromer was likely to be 5-HT2A-mGlu2, transfection into a heterologous cell system was required to confirm this because the mGlu2 and mGlu3 are highly similar and there is little pharmacological separation between them. Introduction of mGlu2 into cells stably expressing the 5-HT2A receptor resulted in a lower affinity for DOI, whereas mGlu3 was unable to reproduce such effects (González-Maeso et al., 2008).
Despite such elegant examples and although communication across heteromers is generally expected to result in altered ligand binding characteristics, this has not been evident in many studies on heteromers. These include β2-β3 adrenoceptor (Breit et al., 2004), β2-adrenoceptor-δ-opioid receptor (Jordan et al., 2001), β2-adrenoceptor-κ-opioid receptor (Jordan et al., 2001), μ-opioid receptor-tachykinin NK1 (Pfeiffer et al., 2003), and μ-opioid-somatostatin sst2A interactions (Pfeiffer et al., 2002). It is unclear which of the issues discussed above that could limit detection of such effects might be relevant in these examples. However, few studies have actually examined dissociation kinetics of ligands occupying each protomer in any level of detail.
With a wealth of ligands that can be radiolabeled and a substantial and relatively well understood overlap of expression patterns in white-cell populations, the chemokine receptor family has provided significant insights into allosterism in GPCR heteromers. Springael, Parmentier, and colleagues (El-Asmar et al., 2005; Springael et al., 2006; Sohy et al., 2007) used competition binding and infinite dilution of radioligand to determine the dissociation kinetics of ligands at heteromers of the closely related CCR2 and CCR5 as well as the more distantly related CCR2 and CXCR4 chemokine receptors. Using both stably transfected CHO-K1 cells and isolated lymphocytes, the affinity and dissociation kinetics of a radioligand specific to one protomer were found to be influenced by binding of ligand to the opposing protomer only when both receptors were present. For example, in cells heterologously expressing CCR2 alone, macrophage inflammatory protein 1β (MIP-1β) resulted in homologous competition with trace concentrations of 125I-MIP-1β, consistent with its role as a CCR2 agonist (El-Asmar et al., 2005). Likewise, the CCR5 agonist monocyte chemoattractant protein-1 (MCP-1) competed effectively with 125I-MCP-1 in cells expressing CCR5 alone (El-Asmar et al., 2005). Critically, neither MIP-1β nor MCP-1 displayed heterologous competition in cells expressing the nontarget chemokine receptor. However, when both receptors were expressed in CHO-K1 cells or when lymphocytes known to express both receptors were employed, both MIP-1β and MCP-1 gained the ability to displace the other radioligand (El-Asmar et al., 2005). The authors extended these observations by the use of infinite dilution experiments to demonstrate that such “negative cooperativity” reflected allosteric communication between protomers. They reported that the dissociation kinetic of the radiolabeled agonist was altered by the heterologous ligand only when both receptors were present (Springael et al., 2006). Similar results have been reported for CCR2-CXCR4 heteromers (Sohy et al., 2007) and, indeed, a number of other heteromer combinations (Springael et al., 2007).
An issue that dogs many of the studies in this area is that, at least for experiments performed using heterologous expression systems, it has been challenging to exclude effects that relate to partitioning of G proteins between individual receptor protomers (Huang et al., 2006; Tubio et al., 2010). Agonist occupancy of a GPCR is anticipated to result in enhanced interaction with a G protein, and this may sequester G protein away from the protomer that is the binding site for the radioligand probe. Because many studies have indicated that a decrease in agonist affinity or enhanced rate of dissociation is the primary effect of occupancy of the second protomer with an agonist ligand, it is interesting to note that this is also what would be expected by limiting G protein availability; as noted earlier, G proteins are themselves allosteric modulators of GPCRs.
3. Conformational Changes and Functional Reconstitution.
Measurement of changes to radioligand binding kinetics is one way of demonstrating allosterism across heteromers. However, suitable radioligands are frequently not available for many GPCRs, and in many other cases, the true endogenous orthosteric ligand(s) are of low affinity, thus limiting this approach. Furthermore, it is unwise to rely on a single strategy to reach any conclusion. Other methods, therefore, have also been employed to assess conformational changes or communication across receptor complexes. In an elegant study, Vilardaga et al. (2008) used FRET imaging to demonstrate heteromeric interactions between coexpressed forms of the α2A-adrenoceptor and the μ-opioid receptor that were modified to act as FRET donor and acceptor, respectively, by the C-terminal fusion of cyan fluorescent protein or yellow fluorescent protein (Vilardaga et al., 2008). These studies were extended by incorporation of the small fluorescein arsenical hairpin binder, which binds with high specificity to tetracysteine motifs as small as six amino acids and can act as a FRET acceptor, into the third intracellular loop of the α2A-adrenoceptor, which already had cyan fluorescent protein at the C terminus. Addition of norepinephrine to cells expressing this construct resulted in a rapid reduction in FRET signal, consistent with movement apart or reorientation of the intracellular elements of the receptor containing the FRET reporters. Most importantly, however, although morphine was without effect in these cells, after coexpression of the wild-type μ-opioid receptor, addition of morphine partially reversed the effect of norepinephrine on the α2A-adrenoceptor intramolecular FRET sensor (Vilardaga et al., 2008). That the conformational change might reflect heterologous desensitization (phosphorylation of the α2A-adrenoceptor by downstream second messengers activated by morphine) was ruled out. Furthermore, because both the α2A-adrenoceptor and the μ-opioid receptor couple selectivity to Gαi family G proteins, the authors tried to exclude the possibility that the effect resulted from a nonspecific sequestering of G proteins. So, after coexpression of the α2A-adrenoceptor intramolecular FRET sensor with wild-type adenosine A1 receptor, another Gαi-coupled GPCR, but one that does not interact with the α2A-adrenoceptor, addition of adenosine was unable to mimic the effect of morphine, suggesting this to be an unlikely explanation.
A further approach to the study of allosterism across heteromers is the use of functional reconstitution of receptor-G protein fusion constructs. Although receptor-G protein fusions have a variety of different applications (Seifert et al., 1999; Milligan, 2000, 2010a; Wurch and Pauwels, 2001; Milligan et al., 2004, 2007), the expression of a receptor-G protein fusion that lacks the ability to be activated upon ligand binding (although retaining an intact orthosteric binding site) but is potentially competent to activate the G protein, in combination with a second fusion, this time activation-competent but containing a G protein mutated in the guanine nucleotide binding site to prevent G protein activation, means that a ligand-mediated signal can be generated only if the ligand-bound protomer communicates allosterically with the partner protomer (Fig. 3A) (Milligan et al., 2004, 2007; Milligan, 2010a). This approach has been used to demonstrate allosteric communication across a number of heteromers, including κ-opioid receptor-μ-opioid receptor (Pascal and Milligan, 2005) and CXCR2-δ-opioid receptor (Parenty et al., 2008) as well as indicating that although the α1B-adrenoceptor is able to interact with the histamine H1 receptor, this occurs only with low affinity and requires supraphysiological expression levels to detect interactions (Carrillo et al., 2003). In studies on the CXCR2-δ-opioid receptor heteromer, a pertussis toxin-resistant version of Gαi2 was fused to either the chemokine CXCR2 receptor, which contained a mutation within the second intracellular loop rendering it incapable of ligand-mediated activation, or the δ-opioid receptor, where the G protein itself was mutated such that it could not exchange guanine nucleotides. By treating the cells with pertussis toxin and then performing [35S]GTPγS binding assays on isolated membranes, it was possible to measure [35S]GTPγS binding specifically at the reconstituted heteromer (Parenty et al., 2008). As such, in cells expressing only the δ-opioid receptor-Gαi2 fusion protein containing the inactivated G protein, the δ-opioid agonist [d-Ala2,d-Leu5]-enkephalin was incapable of promoting Gαi-activation, yet it regained the ability to cause G protein activation when the CXCR2 construct was coexpressed, providing clear evidence that agonist binding to one GPCR protomer can lead to activation of the G protein coupled to the partner protomer (Parenty et al., 2008). Although distinct, in that glycoprotein hormone receptors have a long N-terminal domain that is responsible for ligand binding and a 7TM domain that functions to communicate ligand binding to G protein activation, receptors of this family, including the LH receptor, can also be inactivated via either generation of binding- or signaling-deficient forms. As with the GPCR-G protein fusions, reconstitution of function can be achieved via coexpression of such pairs of LH receptors. As mentioned in section III.A, Rivero-Müller et al. (2010) extended the concept from transfected cells to in vivo activity by generating lines of mice in which one of a pair of individual inactive LH receptors constructs was knocked into the genomic locus of the LH receptor. After cross-breeding, luteinizing hormone function was restored in animals, presumably via functional complementation (Rivero-Müller et al., 2010). As indicated in section III.A, this is the first, and currently the only, example of the use of such trans-complementation in vivo and at close to physiologically normal levels of expression of a GPCR to explore the existence and importance of intermolecular cooperation/receptor dimers to function.

Experimental approaches to the identification of allosteric or heteromer-specific ligands at heteromers. A, reconstitution of a functional receptor through heterodimerization. i, protomer A is a GPCR-G protein fusion product that is able to bind ligand but contains a mutation within a conserved region in intracellular loop 2 that prevents G protein activation. ii, protomer B is also able to bind ligand and can transmit signal to the G protein, but the fused G protein is mutated such that guanine nucleotide binding is prevented, thus no signal is generated. iii, if protomers A and B are able to form a functional signaling unit (i.e., a heterodimer), it is possible for ligand binding at the functional receptor (protomer B) to result in signal rescue via the G protein of protomer A. Because signal is generated only upon functional reconstitution, the signal-to-noise ratio of the assay is high and particularly amenable to high-throughput screening. B, the use of BRET assays to examine signaling at GPCR heteromers. i and ii, BRET signal is generated by energy transfer from Renilla reniformis luciferase (Rluc, herein fused to the C terminus of Protomer B) that has oxidized the exogenously applied substrate, coelenterazine, to a fluorescent protein [such as yellow fluorescent protein (YFP), here fused to the C terminus of β-arrestin]. However, given the limited distance that the Rluc signal can travel, the YFP moiety must be within 100 Å to receive and subsequently transmit energy in the form of fluorescence. Thus, if YFP signal is generated upon stimulation of Rluc, the two proteins to which they are fused must be in close proximity, such as would be expected of a protein-protein interaction such as a homo- or heterodimer. By combining BRET with β-arrestin recruitment to an activated receptor, it is possible to monitor changes in BRET ratio upon ligand stimulation. i, for most GPCRs, ligand binding to a receptor facilitates translocation of β-arrestins from the cytoplasm to the activated receptor at the plasma membrane. In this case, protomer A is activated and recruits β-arrestin-YFP but no change in BRET signal is observed when Rluc is absent. ii, stimulation of Rluc-fused protomer B with agonist also leads to recruitment of β-arrestin-YFP to the activated receptor, yet in contrast to i, Rluc and YFP are now in close proximity and Rluc is able to excite YFP. iii, by combining the above scenarios, it is possible to determine whether two protomers are in close proximity and therefore likely to exist as a heteromer. By coexpressing β-arrestin-YFP and protomers A and B-Rluc, stimulation of protomer A will lead to β-arrestin recruitment. However, only when protomers A and B are heteromers will a change in BRET ratio be apparent.
4. Enhanced or Synergistic Signaling.
The studies on CXCR2-δ-opioid receptor heteromers also provide another pharmacological outcome specific to allosterism at heteromers: enhanced signaling of an orthosteric ligand as a result of the presence of another receptor with or without a bound ligand. When CXCR2-δ-opioid receptor heteromers were functionally reconstituted, coincubation of membranes of such cells with the CXCR2 antagonist N-(2-hydroxy-4-nitrophenyl)-N′-(2-bromophenyl)urea (SB225002) led to enhanced [35S]GTPγS incorporation in response to [d-Ala2,d-Leu5]-enkephalin agonism, despite the fact that the chemokine receptor ligand had no direct affinity for the δ-opioid receptor itself (Parenty et al., 2008). Similar observations have been reported for cross-GPCR class heteromers (e.g., between adenosine and metabotropic glutamate receptors). Found to be coexpressed in the cerebellum and primary cortical neurons and demonstrated to exist as heteromers by coimmunoprecipitation, stimulation of either A1A with R-PIA or mGlu1α with quisqualate resulted in enhanced calcium signaling if cells had been pretreated with the opposing agonist, indicating that A1A-mGlu1α heteromers display synergistic signaling as a result of allosteric interactions (Ciruela et al., 2001). This phenomenon was also observed when examining excitotoxicity at rat cortical neurons (Ciruela et al., 2001), suggesting that the allosteric effect is physiologically relevant. Synergism has also been reported at adenosine A2A-mGlu5 heteromers for extracellular signaling-regulated mitogen-activated protein kinases 1 and 2 (ERK1/2) phosphorylation and c-Fos expression in rat striatal sections as well as for motor activity induced by phencyclidine in living animals (Ferré et al., 2002). In addition to the altered binding affinity of certain agonist ligands at the serotonin 5-HT2A receptor-mGlu2 heteromer, allostery across the heteromer generates marked differences in signal generation. For example, in membranes derived from primary neural cultures, the potency of the hallucinogenic 5-HT2A receptor agonist DOI to enhance binding of [35S]GTPγS to pertussis-sensitive G proteins was greatly reduced by the coaddition of the mGlu2/3 agonist LY379268 (González-Maeso et al., 2008). It is tempting to speculate that this may be relevant to the reported clinical antipsychotic effects of mGlu2/3 agonists (Conn et al., 2009a; Moreno et al., 2009), not least because the actions of the mGlu2/3 antagonist LY341495 to increase locomotor and vertical activities are absent in serotonin 5-HT2A receptor knock-out mice, an observation consistent with the idea that the heteromer is the key therapeutic target (González-Maeso et al., 2008).
The potential contribution of heteromers to the pharmacology of other central nervous system GPCR drug targets has also been considered. Dopamine D2-dopamine D3 heteromers have been postulated to account for the obvious discrepancy between potencies of certain antiparkinsonian ligands in vivo compared with their in vitro properties (Maggio et al., 2009). When the individual receptor protomers are expressed alone, the partial agonists ropinirole and pramipexole each have markedly greater potency at D3 receptors than D2 receptors (Maggio et al., 2003). However, by using chimeric adenylate cyclase-V/VI, which was insensitive to D3 receptor stimulation and therefore acted as a measure of D2 receptor signaling alone, the authors demonstrated that each of the antiparkinsonian ligands was more potent at the D2 protomer when the receptors were coexpressed and presumably existed as D2-D3 heteromers (Maggio et al., 2003). Thus, heteromerization may explain the physiological actions of these ligands.
Two of the hallmarks of allosterism (i.e., reciprocity of allosteric effect and probe dependence) have been claimed for synergistic or enhanced cell signaling via δ-opioid–μ-opioid receptor heteromers. In a study of δ- and μ-opioid receptor heteromerization, δ-μ-opioid receptor heteromers were described in both CHO cells and in the SK-N-SH human neuroblastoma cell line (Gomes et al., 2000). Saturation binding isotherms for the μ-opioid agonist [3H]DAMGO at intact cells revealed that coincubation with the δ-opioid antagonist TIPPψ or agonist deltorphin II led to an elevation in the number of receptors recognized by the radiolabel (Bmax), whereas another δ-opioid agonist, [d-Pen2,d-Pen5]-enkephalin, did not. The effect on Bmax seemed to be reciprocal (in that treatment of cells with unlabeled DAMGO led to an equivalent increase in [3H]deltorphin II binding, although the authors used different cells to make the comparison) and specific [in that no effect on Bmax was observed in CHO cells expressing the receptors individually (Gomes et al., 2000)]. Although the similar effects of both agonist and antagonist ligands is curious and, on the basis of probe dependence, perhaps more surprising than the lack of effect of a second agonist, critically, the allosteric effect ascribed to the δ-μ-opioid receptor heteromers led to an alteration in receptor signaling in native cells also. In SK-N-SH cells, both the potency and efficacy of ERK1/2 activation by DAMGO and deltorphin II was enhanced when the partnering protomer was occupied by either TIPPψ or phenylalanyl-cyclo(cysteinyltyrosyl-tryptophyl-ornithyl-threonyl-penicillamine)threoninamide, respectively. These findings were later extended by the same group to encompass other signaling pathways in vitro and analgesia in vivo (Gomes et al., 2004). Herein, both DAMGO and morphine stimulated [35S]GTPγS binding in CHO and SK-N-SH cells and mouse spinal cord membranes, and these were enhanced (potency and efficacy) by pretreatment with a δ-opioid agonist (10 nM deltorphin II) or antagonist (10 nM TIPPψ). The ability of morphine to inhibit cAMP generation was augmented by TIPPψ, as was morphine analgesia in a murine tail-flick model of pain relief performed 30 min after intrathecal injection (Gomes et al., 2004).
5. Impaired Signaling.
A more common observation of the functional consequences of heteromerization is the cross-inhibition of signaling as a result of allosteric communication. In some cases, cross-inhibition can be directly linked to negative modulation of affinity across heteromers, as introduced earlier for the adenosine A1-adenosine A2A (Ciruela et al., 2006a) and adenosine A2A-dopamine D2 (Canals et al., 2003) heteromers, where the A2A protomer exerts an apparent dominant-negative effect on its heteromer partner. Adenosine A1 and A2A receptors are expressed on pre- and postsynaptic glutamatergic neurons, where they exert opposing effects on glutamate release. By altering the affinity of ligands at the A1 receptor upon A2A receptor occupancy, the A1-A2A heteromer has been suggested to act as a biphasic sensor of adenosine levels: because the A1 receptor has greater affinity for adenosine than the A2A protomer, low levels of adenosine will inhibit neurotransmitter release. Once adenosine concentrations are high enough to bind the A2A receptor, however, the A2A protomer will allosterically inhibit binding at the A1 protomer and concurrently stimulate glutamate release (Ciruela et al., 2006b) via a Gαi-mediated mechanism (Casadó et al., 2010). The inhibitory effect of one protomer on another is also “tunable” for the dopamine D2-D3 heteromer, where it is the concentration of individual protomer-selective drugs, rather than the endogenous ligand, that influences signaling. Aripiprazole is a recently approved antipsychotic that displays clinical efficacy in the absence of extrapyramidal side effects (Grunder et al., 2003; Maggio et al., 2009; Maggio and Millan, 2010) and is reported to be a partial agonist at both dopamine D2 and dopamine D3 receptors (Novi et al., 2007; Tadori et al., 2008). Using heterologous coexpression of the receptors with the chimeric adenylyl cyclase-V/VI introduced in the previous section, Novi et al. (2007) were able to demonstrate that aripiprazole and other atypical antipsychotics, including bifeprunox, preclamol, and N-desmethylclozapine, displayed reduced agonism when the dopamine D3 receptor was in 3-fold excess. In this setting, the ligands acted as functional antagonists of the full dopamine D2 and D3 agonist quinpirole. However, they retained partial agonism when the receptor transfection ratios were equal (Novi et al., 2007). Such inhibition of dopamine D2 activity by the D3 protomer within the dopamine D2-D3 heteromer has been suggested to account for the absence of extrapyramidal side effects of such drugs, because aripiprazole and the other partial agonists may attenuate dopamine D2-D3 heteromer postsynaptic signaling in parallel with stimulating more sensitive dopamine D2 and D3 homo-autoreceptors that prevent dopamine release (Maggio et al., 2009; Maggio and Millan, 2010).
Heteromerization can also promote uncoupling of protomers from their cognate G proteins. For example, heteromerization of β2- and β3-adrenoceptors results in uncoupling of both protomers from Gαi and subsequent ERK1/2 phosphorylation and cAMP inhibition, whereas Gαs coupling is unaltered (Breit et al., 2004). Meanwhile, at adenosine A2A-dopamine D2 heteromers, A2A stimulation results in the loss of coupling of the D2 protomer to Gαs, and reciprocal antagonism of gene expression and neuronal excitotoxicity ensues (Ferré et al., 2008). An unusual example of G protein uncoupling is provided by the melatonin MT1-GPR50 heteromer. The melatonin MT1 receptor is expressed in the brain and is responsible for the short-term inhibitory effects of melatonin on the suprachiasmatic nuclei (Jockers et al., 2008). Coexpression of the orphan 7TM polypeptide GPR50 with MT1 is sufficient to prevent high-affinity binding of 2-[125I]iodomelatonin to the MT1 protomer, reflecting the fact that the MT1-GPR50 receptor no longer couples to G protein or recruits arrestins (Levoye et al., 2006). It is noteworthy that the inhibitory effect of GPR50 was specific, because coexpression with the melatonin MT2 receptor, the β2-adrenoceptor, or the chemokine CCR5 receptor did not affect ligand binding or receptor pharmacology (Levoye et al., 2006).
An apparently clinically relevant example of impaired signaling across a heteromer was provided in an intriguing study by Barki-Harrington et al. (2003). Here, the authors demonstrated that β-blockers or angiotensin II type 1 receptor (AT1) antagonists were able to additionally cross-inhibit signaling of the opposing receptor through a potential angiotensin AT1-β2 adrenoceptor heteromer. Using isolated mouse cardiomyocytes and whole animals, the authors found that occupancy of one component of the AT1-β2 heteromer by antagonist was sufficient to prevent agonism at the opposing protomer by its cognate ligand, resulting in cross-inhibition of cardiac contractility and heart rate (Barki-Harrington et al., 2003). It is perhaps surprising that for such an intriguing study and for a potential heteromer with such a diversity of ligands, no follow-up of these observations has yet appeared.
Reciprocal inhibition of second messenger pathways in heterologous expression systems, however, has been reported widely for various potential heteromers. These include cross-inhibition of cAMP and ERK1/2 phosphorylation after coexpression of the μ-opioid and somatostatin sst2A receptors (Pfeiffer et al., 2002), and impaired ERK1/2 signaling in cells coexpressing the μ-opioid receptor and tachykinin NK1 receptor (Pfeiffer et al., 2003). It is noteworthy that interactions between the cannabinoid CB1 and orexin OX1 receptors resulted in a marked reduction in potency of the endogenous agonist orexin A to stimulate ERK/1/2 phosphorylation when the CB1 protomer was occupied by the antiobesity CB1 inverse agonist rimonabant (Ellis et al., 2006). Although more limited in extent, the orexin OX1 receptor antagonist 1-(5-(2-fluoro-phenyl)-2-methyl-thiazol-4-yl)-1-((S)-2-(5-phenyl-(1,3,4)oxadiazol-2-ylmethyl)-pyrrolidin-1-yl)-methanone (SB674042) also decreased the potency of the CB1 agonist (R)-(+)-[2,3-dihydro-5-methyl-3-[(4-morpholino) methyl]pyrrolo-[1,2,3-de]-1,4-benzoxazin-6-yl](1-naphthyl) methanone (WIN55,212-2) to promote ERK1/2 phosphorylation (Ellis et al., 2006). Inhibition of ERK1/2 signaling through the β2-adrenoceptor has been reported in two separate examples of heterodimerization. While cAMP signaling was unaffected by coexpression of β1 and β2-adrenoceptors, Lavoie et al. (2002) found that the β1-β2 adrenoceptor heteromer no longer permitted ERK1/2 phosphorylation via the β2-protomer (Lavoie et al., 2002). In the case of β2-adrenoceptor–κ opioid receptor heteromers, which are stable and unaffected by ligand stimulation (Ramsay et al., 2002), isoprenaline-mediated phosphorylation is dampened by coexpression of the κ-opioid protomer or by costimulation with the κ receptor agonist etorphine (Jordan et al., 2001). A further interesting example stemmed from the recognition that bovine adrenal medulla peptide 22 acts as an agonist at both the δ-opioid receptor and MRGPRX1 (formerly called sensory neuron-specific G-protein coupled receptor 4). These receptors play opposing roles in pain perception and are coexpressed in various dorsal root ganglia. After heterologous coexpression, these two GPCRs were shown to form a heteromer (Breit et al., 2006) and, in this setting, bovine adrenal medulla peptide 22 was no longer able to generate signals via the δ-opioid receptor protomer of the heteromer. Furthermore, in rat dorsal root ganglia neurons, bovine adrenal medulla peptide 22 acted to antagonize function of the endogenous δ-opioid receptor agonist Leu-enkephalin, suggesting that the δ-opioid receptor-MRGPRX1 heteromer might play a physiologically relevant role (Breit et al., 2006). Although such studies have yet to be reported, it would be of considerable interest to employ small interfering RNA to “knock down” levels of MRGPRX1 in isolated dorsal root ganglia and examine whether this promotes bovine adrenal medulla peptide 22 signals via the δ-opioid receptor while concurrently inhibiting signals via MRGPRX1. Such an approach has been employed successfully to provide supporting evidence for the existence and functional relevance in native cells of the melatonin MT1-GPR50 heteromer (Levoye et al., 2006).
In a similar vein, Rios et al. (2006) explored functional interactions between coexpressed cannabinoid CB1 and μ-opioid receptors. They observed that μ-opioid-mediated signaling was attenuated by a CB1 receptor agonist, that this was reciprocal in that μ-opioid receptor activation attenuated CB1 receptor signaling, and that these effects could be observed in both endogenous tissue expressing both receptors as well as cells transfected to coexpress them (Rios et al., 2006). However, although Rios et al. (2006) noted resonance energy transfer signals consistent with the presence of a cannabinoid CB1–μ-opioid receptor heteromer, it should be noted that others have observed similar pharmacology but implicated the constitutive activity of the CB1 receptor as the key driver rather than the cannabinoid CB1-μ-opioid receptor heteromer (Canals and Milligan, 2008). Likewise, although regulation of ERK1/2 phosphorylation by selective ligands in N18TG2 neuroblastoma cells, which coexpress cannabinoid CB1 and δ-opioid receptors, seemed to be produced via the same pathway and was potentially consistent with actions at a heteromer, this was not true in HEK293 cells transfected to coexpress the two GPCRs (Korzh et al., 2008). Once more, it remains unclear whether these differences might relate to sharing of G protein pools, particularly in the transfected cells (Shapira et al., 2000).
6. Altered Internalization.
Receptor-mediated signaling is a tightly regulated process and frequently follows a route of ligand binding followed by activation, phosphorylation, recruitment of arrestins, and internalization (Gurevich and Gurevich, 2006). By interacting with activated receptors, nonvisual arrestins, known as β-arrestin1 and β-arrestin2, serve two main functions in signal termination: 1) to provide steric hindrance at the GPCR-G protein interface, thereby preventing further G protein activation, and 2) to cluster GPCRs into clathrin-coated pits, whereupon they are internalized and removed from further ligand interaction (Gurevich and Gurevich, 2006) [note, however, recent studies indicating that a number of GPCRs can continue to signal after internalization (Calebiro et al., 2009, 2010; Ferrandon et al., 2009; Mullershausen et al., 2009)]. The stoichiometry of GPCR-arrestin interaction remains controversial—at least for the major form of visual arrestin, each protomer of the photon receptor rhodopsin seems to interact with its own arrestin (Hanson et al., 2007), yet the predicted interface of a GPCR dimer is more compatible with binding of only one arrestin molecule to the dimer (Liang et al., 2003). This issue is further muddied by experimental evidence that nonvisual arrestins themselves have the capacity to homo- and heterodimerize (Storez et al., 2005; Xu et al., 2008). However, given the assumption that a single arrestin might interact with a GPCR dimer, and the body of evidence that many GPCR hetero- and homomers exist throughout their life cycle as protein complexes (Terrillon and Bouvier, 2004; Bulenger et al., 2005; Milligan, 2008, 2010b), ligand-mediated recruitment of a single β-arrestin molecule to a dimer might be sufficient to induce internalization of dimers. There is certainly evidence that binding of a single ligand to dimers may be sufficient to promote internalization and this may even be true for a homomer. An interesting means to explore this involved stable coexpression of wild-type β2-adrenoceptor along with a form of this GPCR containing a point mutation (D113S) that greatly reduces the affinity/potency of isoprenaline but greatly increases the affinity/potency of the synthetic orthosteric agonist 1-(3′,4′-dihydroxyphenyl)-3-methyl-1-butanone (L158870), which does not bind to the wild-type receptor with significant affinity (Sartania et al., 2007). In cells coexpressing the two forms of the β2-adrenoceptor, both isoprenaline and L158870 were able to promote internalization of both receptor variants because they produced a homomer (Sartania et al., 2007).
Such effects can be observed for heteromers without resort to mutagenesis and medical chemistry. In the case of the β2-adrenoceptor-δ opioid heteromer, isoprenaline binding to the β2-adrenoceptor results in δ-opioid receptor cointernalization, whereas etorphine occupancy of the δ-opioid protomer triggers the reverse (Jordan et al., 2001). Similar effects have also been reported for the δ-opioid-tachykinin NK1 heteromer, where cross-phosphorylation and internalization was observed in response to either DAMGO or substance P (Pfeiffer et al., 2003). Furthermore, the α1A-adrenoceptor agonist oxymetazoline stimulated cointernalization of α1A-α1B-adrenoceptor heteromers but not control coexpressed receptors, indicating a specific interaction between these two adrenoceptors (Stanasila et al., 2003). A further example is provided by the cannabinoid CB1-orexin OX1 heteromer. When expressed alone in HEK293 cells, the OX1 receptor was found predominantly at the cell surface and underwent internalization in response to its endogenous orthosteric agonist orexin A. By contrast, CB1 receptor expression in HEK293 cells resulted in punctate vesicular localization, reflective of constitutive internalization, a phenotype that could be overcome by the presence of the CB1 antagonist/inverse agonist, rimonabant (Ellis et al., 2006). Coexpression, however, leads to heteromerization of these otherwise subcellularly separated GPCRs. In this setting the cannabinoid CB1 receptor was dominant, with the OX1 receptor adopting the internalized pattern of the CB1 receptor, and single-cell FRET imaging confirming the presence of the heteromer within intracellular vesicles (Ellis et al., 2006). Furthermore, binding of a selective ligand to either protomer was sufficient to trigger cotrafficking of the heteromer within the cell: both rimonabant and the OX1 antagonist SB674042 caused cell surface retention of the heteromer although neither ligand had direct affinity for the partner protomer (Ellis et al., 2006).
Despite these instances of cointernalization, there is clearly asymmetry across dimers, given that not all heteromers are able to undergo reciprocal internalization. For example, stimulation of the α2A-adrenoceptor with 5-bromo-6-[2-imidazolin-2-ylamino]-quinoxaline bitartrate (UK14034) resulted in cointernalization of the β1-adrenoceptor in a nonreciprocal manner (Xu et al., 2003). Meanwhile, although DAMGO is able to cause cointernalization of μ-opioid-tachykinin NK1 heteromers, it is unable to do so at the μ-opioid-sst2A heteromer despite the sst2A agonist L-796,778 demonstrating this effect (Pfeiffer et al., 2002). The β2-adrenoceptor seems to be particularly susceptible to the influence of its dimeric partner: when part of a β2-adrenoceptor-κ-opioid heteromer, the β2-adrenoceptor adopts the internalization phenotype of the κ-opioid receptor and, as such, remains at the cell surface upon isoprenaline treatment (Jordan et al., 2001). The same outcome was reported for β2-β3-adrenoceptor heteromers, where the β2-adrenoceptor adopted the noninternalizing phenotype of the β3-adrenoceptor (Breit et al., 2004), whereas β1-β2-adrenoceptor heteromerization also led to the selective loss of β2-mediated internalization (Lavoie et al., 2002). Rather than a loss of reciprocal internalization, the melatonin receptor MT1 is subject to an apparent dominant-negative effect of its heteromeric partner, the orphan 7TM receptor GPR50. Here, the MT1-GPR50 heteromer results in steric hindrance of either G protein or β-arrestin interactions, potentially as a result of the large intracellular carboxyl terminus of GPR50 (Levoye et al., 2006; Jockers et al., 2008). Finally, in contrast to many of the reciprocal or loss of internalization examples mentioned above, heteromerization can alter the kinetics of internalization without changing the overall outcome, as has been reported with sst2A-sst3 heteromers (Pfeiffer et al., 2001). The range of effects reported using the internalization endpoint is difficult to coalesce into a single mode of action and, as noted above, in many cases studies that explore such effects of different levels of receptor coexpression might provide greater insight.
7. Signal Switching and Heteromer-Specific Ligands.
Most of the above examples of allosterism at heteromers involve modulation of an existing function of one or both protomers, yet there are emerging cases in which novel pharmacology or heteromer-specific/selective ligands have been described. For example, although it has been known for some time that coadministration of agonists at the predominantly Gαi-coupled cannabinoid CB1 and dopamine D2 receptors results in a paradoxical increase in cAMP generation (Glass and Felder, 1997; Hudson et al., 2010), it has only recently been appreciated that such Gαi-to-Gαs signal switching occurs through the formation of CB1-D2 heteromers (Jarrahian et al., 2004; Kearn et al., 2005). Coexpression and δ-μ heteromerization has been shown to lead to altered rank orders of affinity for ligands selective to each protomer and the appearance of pertussis toxin insensitivity of high-affinity binding states and cAMP signaling at the δ-μ heteromer but not the individual protomers (George et al., 2000). Thus, heteromerization resulted in novel G protein coupling; the G protein was subsequently found to be Gαz (Fan et al., 2005). Signal switching has also been reported for dopamine D1-D2 heteromers, where costimulation of Gαs-coupled D1 and Gαi-coupled D2 protomers with SKF81297 and quinpirole, respectively, led to the generation of calcium transients via Gαq/11 in the absence of changes to Gαs or Gαi signaling (Lee et al., 2004). In a subsequent article by the same group, the novel Gαq/11-mediated calcium signal was demonstrated to occur in striatum and was linked to synaptic plasticity (Rashid et al., 2007). It is noteworthy that the authors also found that the ligand 3-methyl-6-chloro-7,8-hydroxy-1-[3-methylphenyl]-2,3,4,5-tetrahydro-1H-3-benzazepine (SKF83959) was able to selectively activate calcium signaling through the D1-D2 heteromer as it bound to both protomers and acted as a full D1 agonist and partial D2 agonist with functional selectivity for the Gαq/11 pathway (Rashid et al., 2007).
Waldhoer et al. (2005) have reported that 6′-guanidinonaltrindole (6′GNTI) was a selective ligand at the δ-κ-opioid heteromer, able to mediate analgesia in vivo by binding to potential heteromers within the spinal cord. 6′GNTI was initially described as a κ-opioid receptor agonist (Sharma et al., 2001) and can additionally bind to both δ- and μ-opioid receptors, albeit with limited efficacy, but was subsequently found to have highest efficacy and potency at the δ-κ-opioid heteromer (Waldhoer et al., 2005). It is noteworthy that signaling in response to 6′GNTI could be inhibited by either κ- or δ-antagonism (Waldhoer et al., 2005), suggesting that 6′GNTI might be acting as a bitopic ligand (see section VI for definition) by binding simultaneously to two pharmacophores on the δ-κ-opioid heteromer. However, this remains to be explored. Opioid heteromers have also been the targets of rational design of bitopic ligands in which two existing pharmacophores are fused together with a chemical linker (Valant et al., 2009). It has long been appreciated that coadministration of morphine (μ-agonist) and δ-opioid antagonists facilitates analgesia in the absence of the development of tolerance and dependence, the main disadvantages of long-term morphine treatment (Abdelhamid et al., 1991; Zhu et al., 1999). Thus, Daniels et al. (2005) hypothesized that a bitopic ligand containing both μ agonism and δ antagonism would result in a compound that possessed analgesia in the absence of tolerance and dependence. The authors determined empirically the optimal linker length between the μ-opioid receptor agonist oxymorphone and the δ-opioid antagonist naltrindole (also a component of 6′GNTI) and found that MDAN-21 had 50-fold greater potency than morphine in a tail-flick test of nociception in mice (Daniels et al., 2005). The same approach was employed to target various other opioid heteromers, such as δ-κ-opioid (KDN-21) (Bhushan et al., 2004) and κ-μ-opioid (KMN-21) (Zhang et al., 2009). Although the use of such ligands as therapeutics is unlikely because of their large size, they have been invaluable in proof-of-concept studies.
IV. Exploring the Expanded Pharmacological Landscape: The Challenge for Drug Discovery
There is still much to be understood about the pharmacology of heteromers, and the expanding complexity of this pharmacology makes their targeted screening problematic. In the final part of this review, we will identify some of the pressing questions in the homo- and heteromer field and discuss the implications for drug discovery.
A. How Do We Identify and Assay Dimers or, More Specifically, Heteromers?
To date, very few heteromer-specific ligands have been identified; this probably reflects the unintentional bias of many drug discovery approaches over the past 10 to 15 years. Initial high-throughput screening (HTS) campaigns based upon competition for binding with radioligands were established to detect compounds that displayed specific binding at the orthosteric site but were not designed to identify allosterism of any kind (Christopoulos and Kenakin, 2002; Rees et al., 2002; May et al., 2007; Milligan and Smith, 2007). The move to functional assays was an important advance in HTS because it enabled identification of agonists and antagonists both inexpensively and without the need for prior knowledge of the endogenous ligand (Kenakin, 2009a). However, such approaches have a number of limitations. For example, the emergence of functional selectivity as a widespread pharmacological phenomenon (Galandrin et al., 2007) means that the choice of functional endpoint measured can influence whether a ligand is identified as an agonist or an antagonist or whether it is even detected at all. A useful illustration of this point is provided by the functional selectivity of some traditional β-blockers at β2-adrenoceptors. For example, the so-called “β blocker” propranolol can be an inhibitor of hypertension (Heidenreich et al., 1999; Morgan et al., 2001), an inverse agonist for Gαs-stimulated cAMP accumulation (Chidiac et al., 1994; Azzi et al., 2003), an agonist for transcription of the cAMP response element-binding protein (Baker et al., 2003) and an agonist for ERK1/2 phosphorylation (Azzi et al., 2003; Baker et al., 2003; Galandrin et al., 2007). Thus, the choice of endpoint is a critical factor for identification of target ligands, whether searching for orthosteric site ligands, allosteric ligands, or heteromer-specific drugs.
Perhaps a bigger challenge for heteromers is identifying what constitutes a feasible, or even a legitimate, drug target. Unfortunately, the heteromer literature contains numerous instances of poorly validated “heterodimer” pairs [readers are referred to the 2007 IUPHAR nomenclature article for a discussion of appropriate criteria for the identification of a legitimate heteromer (Pin et al., 2007)], meaning that some alleged heteromers may instead represent interactions produced only after heterologous expression or, as discussed earlier, downstream integration of independently generated signals (Prezeau et al., 2010). Thus, validation of heteromers in physiologically relevant, native tissues is imperative before screening can begin, although the use of such tissues for screening itself is often impractical because of limitations in cost, scalability, and tractability.
B. How Can We Adapt High-Throughput Screening for Drug Discovery at G Protein-Coupled Receptor Heteromers?
Recent advances in heteromer-specific assay platforms have meant that drug discovery at appropriate heteromers should now be a reality for the pharmaceutical industry and some larger academic research groups. Although a description of the various strategies for heteromer ligand screening is beyond the scope of the present article and has been the subject of a recent comprehensive review (Saenz del Burgo and Milligan, 2010a), we will briefly introduce two elegant experimental approaches for the identification of novel ligands at heteromers that explicitly rely upon allosterism at heteromers but avoid, at least in part, the difficulties posed by functional selectivity (Fig. 3).
1. Receptor-G Protein Fusions and Functional Complementation.
The first technology, marketed as DimerScreen by CARA Therapeutics (Shelton, CT), relies upon the functional reconstitution of two GPCR-G protein fusions that, by themselves, are nonfunctional, as introduced in section III.B.3 above and illustrated in Fig. 3A. The added advantage of this system is that any homomers formed from either protomer in the heterologous expression system will be silent, because functional complementation is possible only for the heteromer; thus, only ligands acting at the heteromeric species will be detected by HTS. The signal-to-background is further enhanced for pertussis toxin-sensitive heteromers as incorporation of a mutation at the normal site of pertussis toxin-catalyzed ADP-ribosylation within Gαi/o subunits confers pertussis toxin resistance; thus, cultures can be treated with the toxin to further reduce background signal (Milligan, 2000; Milligan et al., 2004, 2007). It is interesting to note that although the current prevailing theory of GPCR dimer coupling to G protein is that of a 2:1 receptor/G protein stoichiometry because of the size of the dimeric interface with the G protein (Banères and Parello, 2003; Filipek et al., 2004), functional complementation in DimerScreen relies upon functional complementation between two chimeric proteins with a 1:1 stoichiometry between receptor and G protein (Milligan, 2000; Milligan et al., 2004, 2007).
2. β-Arrestin Recruitment to Unliganded Receptors by Activation of an Opposing Protomer.
An alternative approach to heteromer-specific ligand screening is based upon assays measuring β-arrestin recruitment to agonist-occupied GPCRs. This can be performed in a number of ways. One employs BRET, in which energy can be transferred from a chemically activated bioluminescent donor, usually the luciferase from Renilla reniformis (Rluc), to an acceptor fluorophore (e.g., enhanced yellow fluorescent protein), only when the two proteins are brought into close proximity. By fusing energy transfer-donor or -acceptor proteins to the C-terminal tail of a GPCR and to either β-arrestin1 or β-arrestin2, ligand-mediated recruitment of β-arrestins to an activated receptor can be monitored in real time (Milligan and Bouvier, 2005; Pfleger and Eidne, 2006; Bouvier et al., 2007; Pfleger et al., 2007; Pfleger, 2009; Alvarez-Curto et al., 2010a; Ayoub and Pfleger, 2010; Ciruela et al., 2010). Dimerix (Nedlands, WA, Australia) has modified this traditional recruitment assay to assess allosterism across heteromers by coexpressing and stimulating an unlabeled GPCR protomer in cells containing a separate BRET-labeled GPCR and β-arrestin (Fig. 3B) (Ayoub and Pfleger, 2010). Like the receptor-G protein fusion heteromer assay, this approach has a high signal-to-noise ratio because, by definition, only heteromeric, allosteric signals should generate BRET. A similar approach has been employed in the PathHunter GPCR Dimerization Assay by DiscoveRx (Fremont, CA), which measures allosterism across heteromers by enzyme fragment complementation between appropriately tagged GPCRs and β-arrestins.
β-Arrestin recruitment assays have an advantage over the receptor-G protein functional complementation approach in that they do not necessarily require knowledge of the G proteins with which a heteromer interacts; this is particularly valuable because there are situations in which novel G protein coupling is observed as a consequence of heteromerization, as was reported for the D1-D2 heteromer, discussed earlier (Rashid et al., 2007). However, the significant drawback of such β-arrestin recruitment assays is that they will always require additional validation to exclude false positives that are due to recruitment to nonheteromeric receptors because of heterologous desensitization. Furthermore, the assays will only work if one of the two protomers is able to recruit arrestins in the first place, which not all GPCRs have a propensity to do.
C. How do Homo- and Heteromers Signal?
The technologies described above are based upon the transmission of allosteric signals from a receptor to effector in trans; however, there is still much debate over the mode of signal propagation at homomers and heteromers (Carrillo et al., 2003; Damian et al., 2008) or, indeed, the minimal signaling unit of a dimer (Lambert, 2010). For example, it is unclear whether occupancy of both elements of a homomer or heteromer results in maximal efficacy or instead is less energetically favorable. This has been most effectively explored for the class C glutamate family receptors, but for the much more numerous class A receptors (e.g., the dopamine D2 receptor), a recent elegant study found that D2 homomers were able to signal asymmetrically through a two-receptor–one-G protein complex and that agonist binding to one protomer only is sufficient for maximal signaling (Han et al., 2009). It is noteworthy that cobinding of a second ligand to the opposing protomer led to allosteric modulation of the signal generated by the agonist-occupied protomer: if the second ligand was an agonist, the signal was dampened, whereas inverse agonist binding at the second protomer enhanced homomer signaling (Han et al., 2009). Thus, for the D2 receptor, it seems that the homomer couples to the G protein with greatest efficiency if one protomer is in an active conformation, whereas the second adopts an inactive structure, indicating that the conformation of one protomer can allosterically modulate the other (Fig. 4A).

Effect of ligand occupancy and intrinsic efficacy at dopamine receptor homomers and implications for bivalent ligand design. A, Han et al. (2009) examined the signaling of dopamine D2 receptor homomers using a variety of mutations at either protomer A or protomer B. The authors demonstrated that protomer B occupancy and the intrinsic efficacy of the ligand at protomer B led to allosteric modulation of the signal generated by protomer A, represented here graphically as protomer A “activation.” When both dopamine D2 protomers are unbound, they generate a basal signal illustrated by i. Agonist binding to protomer A in the absence of protomer B occupancy leads to receptor activation (iii) that is greater than when protomer B possesses constitutive activity (ii), indicating that the active state of protomer B negatively allosterically modulates the signaling of protomer A. Consistent with this observation, Han et al. (2009) were able to demonstrate that maximal signaling from protomer A is achieved when protomer B is in a completely inactive conformation, such as when an inverse agonist is bound (iv). [Adapted from Han Y, Moreira IS, Urizar E, Weinstein H, and Javitch JA (2009) Allosteric communication between protomers of dopamine class A GPCR dimers modulates activation. Nat Chem Biol 5:688–695. Copyright © 2009 the Nature Publishing Group. Used with permission.] B, extrapolation of the findings of Han et al. (2009) to the rational design of bivalent ligands highlights the importance of the intrinsic efficacy of the individual moieties attached to the linker. For example, attachment of two agonist moieties (yellow and green) would lead to moderate receptor activation, as per the example of A, ii. The signal generated by protomer A would be enhanced if the second moiety was instead a neutral antagonist (pink), reflecting the example of single occupancy in A, iii where there is no allosteric modulation of protomer A by protomer B. If maximal signal via protomer A was required, the moiety interacting with protomer B should be an inverse agonist (red), as per A, iv.
The results above agree with numerous studies over the years that have demonstrated negative cooperativity between ligands binding at dimers (Maggio et al., 1993; El-Asmar et al., 2005; Urizar et al., 2005; Albizu et al., 2006; Springael et al., 2006, 2007; Sohy et al., 2007). Furthermore, the D2-D2 homomer findings support observations from various receptor dimers, including the glutamate (Goudet et al., 2005; Hlavackova et al., 2005) and LTB4 BLT1 receptors (Damian et al., 2006, 2008), where maximal receptor signal is achieved only when one protomer is occupied by agonist or, in the case of CXCR2-δ, functionally reconstituted heteromers, where antagonism of CXCR2 by SB225002 enhances the action of δ opioid agonists (Parenty et al., 2008). However, such studies indicating asymmetry or single occupancy of homo- and heteromers are in contrast to several other reports in which co-occupancy is required. For example, cobinding of the M3 muscarinic acetylcholine receptor homomer is necessary for recruitment of β-arrestin1 to the activated receptor (Novi et al., 2004), whereas maximal ERK1/2 phosphorylation is achieved only at the δ-κ heteromer when both protomers contain ligand (Jordan and Devi, 1999). Damian et al. (2006) have argued that these observations are not inconsistent with theirs: for the BLT1 receptor, asymmetrical conformations between the homomer subunits were only observed in the presence of G protein (Mesnier and Banères, 2004). Thus, if the G protein confers asymmetry in receptor conformations, G protein-independent signaling may not be affected in the same manner.
Evidently, the occupancy requirement of homo- and heteromers is dependent upon the constitution of the dimers, and it will be important to establish this for individual cases. For example, the application of bivalent ligands for targeting multiple binding sites, whether on the same receptor (Disingrini et al., 2006; Steinfeld et al., 2007; Antony et al., 2009) or at a dimer (Waldhoer et al., 2005; Day et al., 2009), is becoming increasingly popular. With respect to dimers, the outcome of such a strategy would depend upon the ligand binding preference of individual homo- or heteromers: in the case of the D2-D2 homomer, bivalent agonist moieties would not be able to achieve full receptor activation, whereas combination of agonist and inverse agonist valencies could theoretically result in maximal signaling (Fig. 4B). How an additional protomer would influence and undoubtedly further complicate signaling through G proteins, as would be the case for higher order oligomers such as the receptor adenosine A2A–cannabinoid CB1–dopamine D2 mosaic (Carriba et al., 2008; Navarro et al., 2010) and adenosine A2A–dopamine D2–glutamate mGlu5 (Cabello et al., 2009) heteromers, is a long way from being understood.
V. Concluding Remarks
An extensive literature, not always appreciated fully at the time to reflect GPCR homo- and heteromerization, is consistent with the existence of GPCR dimers and oligomers. Furthermore, the development of interest in allosteric modulators of GPCR function as potential therapeutic entities has provided an experimental and conceptual framework in which allosteric effects of ligands at GPCR homomers and heteromers can be understood and analyzed. Much more remains to be determined. For example, it will be imperative to establish whether the binding of ligands alters the oligomerization state of receptor homo- and heteromers and whether the few currently available assay technologies that have been used to identify heteromer-selective ligands are optimal, or indeed appropriate. At a more basic and prosaic level, much needs to be explored about the effects of ligands on heteromer signaling and whether pharmacological fingerprints of GPCR heteromers observed in transfected cell systems can be translated to decode aspects of poorly understood pharmacology recorded both ex vivo and in vivo. It it still possible that many examples of GPCR heteromerization recorded in transfected cells will remain ephemeral, possibly reflecting limited current knowledge on true receptor coexpression patterns in individual native cells. However, this is an exciting time for heteromer research as more sophisticated techniques with which to address these questions are developed, and there are sufficient examples emerging from physiologically relevant systems to suggest that many examples are not mirages. Translation of these to true validated therapeutic targets will require the pharmaceutical industry to be convinced of their relevance; therefore, basic researchers must develop approaches to test hypotheses with sufficient vision that they can answer remaining key questions and doubts in an unequivocal manner.
VI. Glossary
Ago-allosteric modulator. Ligand that is both an allosteric modulator and allosteric agonist.
Allosteric agonist. Ligand that possesses efficacy and that binds to a site distinct from the orthosteric site.
Allosteric binding site. A ligand binding site distinct to the orthosteric binding site. Can be on-target or off-target.
Allosteric modulator. An exogenous or endogenous molecule that binds to a distinct and nonoverlapping site to influence binding or signaling at another, usually orthosteric, site.
Bivalent ligand. Ligand with two pharmacological moieties, often joined by a linker, which can simultaneously occupy two distinct binding pockets. Bivalent ligands can target orthosteric and/or allosteric sites on a monomer or dimer. Also known as bitopic or dualsteric ligands.
Cooperativity. The effect(s) of multiple equivalents of the same ligand binding to multiple (generally) identical sites.
Functional reconstitution. Generation of a functional dimeric unit from two otherwise nonfunctional protomers. Can exist in nature (e.g. GABAB heteromeric receptors) or be generated by mutagenesis.
Functional selectivity. Selective activation of a subset of the signaling pathways available to a receptor by a ligand.
GPCR allosterism. The reciprocated effect(s) of binding two (or more) distinct ligands at different sites on a receptor monomer, homomer or heteromer. Such effects can be positive or negative.
Heteromeric receptor. A signaling unit composed of two or more GPCR protomers that by themselves are nonfunctional.
Negative allosteric modulator. Reduces binding or activity.
Off-target allosterism. Allosteric modulation of another binding site (orthosteric or allosteric) on a distinct protein such as a dimeric partner.
On-target allosterism. Allosteric modulation of another binding site (orthosteric or allosteric) on the same protein.
Orphan receptor. A GPCR for which the endogenous ligand remains to be discovered.
Orthosteric binding site. The primary binding site of the receptor, usually where the endogenous ligand binds and elicits a signal.
Positive allosteric modulator. Enhances binding or activity.
Receptor heteromers. Two or more molecularly distinct and individually functional GPCRs that combine to form a molecular entity with distinct pharmacology.
Receptor homomers. Two or more molecularly equivalent and functional GPCRs that combine to form a molecular entity with distinct pharmacology.
Receptor monomer. Single 7TM-spanning GPCR that is capable of signal transduction.
Silent allosteric modulator. Binds to a site distinct to the orthosteric site without influencing orthosteric binding or signaling. Silent allosteric modulators are rare.
Acknowledgments.
This work was supported by the UK Medical Research Council [Grant G0900050]; the Biotechnology and Biosciences Research Council [Grant BB/E006302/1]; and a joint National Health and Medical Research Council of Australia and National Heart Foundation of Australia C.J. Martin Overseas Research Fellowship (to N.J.S.).
We thank Drs. Chris Langmead and Kirstie Bennett from Heptares Therapeutics (Hertfordshire, UK) for assistance with the equation in Fig. 1C. G.M. is a member of the scientific advisory board for Cara Therapeutics.
Footnotes
This article is available online at http://pharmrev.aspetjournals.org.
doi:10.1124/pr.110.002667.
↵2 Receptor nomenclature follows the International Union of Basic and Clinical Pharmacology guidelines as detailed by Alexander et al. (2009). Nomenclature for homo- and heterodimers and oligomers follows that of the recent recommendations by Pin et al. (2007) and Ferré et al. (2009).
↵1 Abbreviations:
- 5-HT
- 5-hydroxytryptamine (serotonin)
- 6′GNTI
- 6′-guanidinonaltrindole
- 7TM
- seven transmembrane
- AT1
- angiotensin type 1 receptor
- BLT1
- leukotriene B4 receptor
- BRET
- bioluminescence resonance energy transfer
- CB1
- cannabinoid type 1 receptor
- CCR
- chemokine receptor
- CG
- chorionic gonadotrophin
- CGP7930
- 3-(3′,5′-di-tert-butyl-4′-hydroxy)phenyl-2,2-dimethylpropanol
- CGS21680
- 4-[2-[[6-amino-9-(N-ethyle-β-d-ribofuranuronamidosyl)-H-purin-2-yl]amino]ethyl]benzenepropanoic acid hydrochloride
- CHO
- Chinese hamster ovary
- CL
- calcitonin-like receptor
- CXCR
- C-X-C chemokine receptor
- DAMGO
- [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin
- DOI
- 1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane
- ERK1/2
- extracellular signaling-regulated mitogen-activated protein kinases 1 and 2
- FRET
- fluorescence (Förster) resonance energy transfer
- GPCR
- G protein-coupled receptor
- GTPγS
- guanosine 5′-O-[γ-thio]triphosphate
- HEK
- human embryonic kidney
- HTS
- high-throughput screening
- KDN-21
- κ and δ-opioid receptor bivalent antagonist 21 linker
- KMN-21
- κ and μ-opioid receptor bivalent antagonist 21 linker
- L158870
- 1-(3′,4′-dihydroxyphenyl)-3-methyl-1-butanone
- L-796,778
- methyl (2S)-6-amino-2-[[(2R)-2-[[(2S)-1-[(4-nitrophenyl)amino]-1-oxo-3-phenylpropan-2-yl]carbamoylamino]hexanoyl]amino]hexanoate
- LH
- luteinizing hormone
- LTB4
- leukotriene B4
- LY341495
- 2S-2-amino-2-(1S,2S-2-carboxycyclopropan-1-yl)-3-(xanth-9-yl)propionic acid
- LY379268
- (−)-2-oxa-4-aminobicyclo[3.1.0]hexane-4,6-dicarboxylic acid
- MCP-1
- monocyte chemoattractant protein-1
- MDAN-21
- μ and δ-opioid receptor bivalent antagonist 21 linker
- mGlu
- metabotropic glutamate receptor
- MIP-1β
- macrophage inflammatory protein 1β
- MRGPRX1
- sensory neuron-specific G protein-coupled receptor (SNSR) 4
- MT
- melatonin receptor
- NAM
- negative allosteric modulator
- NK1
- neurokinin type 1 receptor
- OX1
- orexin 1 receptor
- PAM
- positive allosteric modulator
- RAMP
- receptor activity modifying protein
- rimonabant
- 5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-piperidin-1-ylpyrazole-3-carboxamide
- Rluc
- Renilla reniformis luciferase
- R-PIA
- (2R,3S,4R,5R)-2-(hydroxymethyl)-5-[6-[[(2S)-1-phenylpropan-2-yl]amino]purin-9-yl]oxolane-3,4-diol
- RXFP
- relaxin family peptide receptor
- SAM
- silent allosteric modulator
- SB225002
- N-(2-hydroxy-4-nitrophenyl)-N′-(2-bromophenyl)urea
- SB674042
- 1-(5-(2-fluoro-phenyl)-2-methyl-thiazol-4-yl)-1-((S)-2-(5-phenyl-(1,3,4)oxadiazol-2-ylmethyl)-pyrrolidin-1-yl)-methanone
- SKF81297
- 6-chloro-2,3,4,5-tetrahydro-1-phenyl-1H-3-benzazepine hydrobromide
- SKF83959
- 3-methyl-6-chloro-7,8-hydroxy-1-[3-methylphenyl]-2,3,4,5-tetrahydro-]H-3-benzazepine
- Sst
- somatostatin
- TIPPψ
- (2S)-2-[[(2S)-2-[[(3S)-2-[(2S)-2-amino-3-(4-hydroxyphenyl)propanoyl]-3,4-dihydro-1H-isoquinolin-3-yl]methylamino]-3-phenylpropanoyl]amino]-3-phenylpropanoic acid
- TSH
- thyroid stimulating hormone
- UK14034
- 5-bromo-6-[2-imidazolin-2-ylamino]-quinoxaline bitartrate
- WIN55,212-2
- (R)-(+)-[2,3-dihydro-5-methyl-3-[(4-morpholino)methyl]pyrrolo-[1,2,3-de]-1,4-benzoxazin-6-yl](1-naphthyl)methanone
- YFP
- yellow fluorescent protein.
- Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
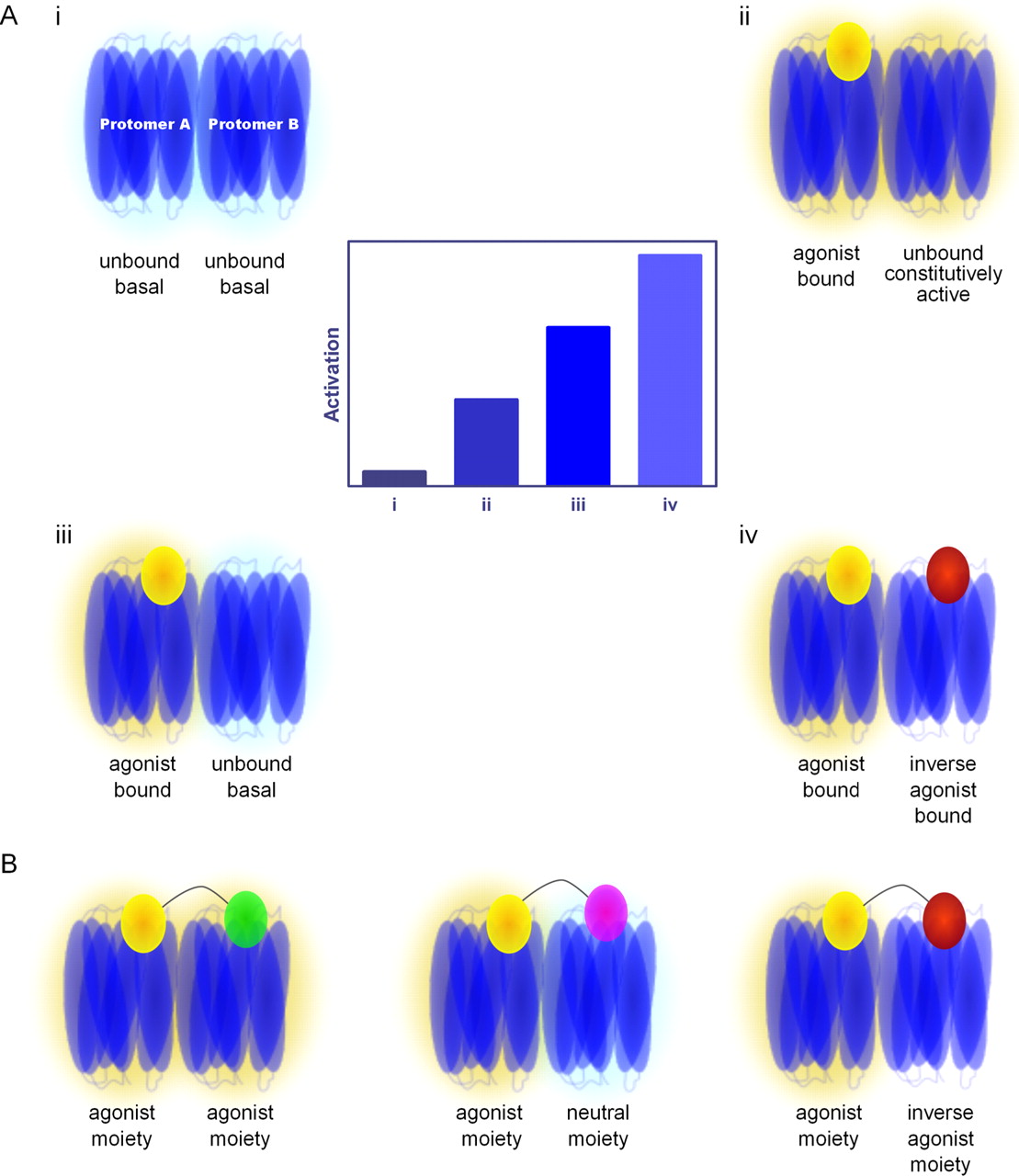{kind=link}