Article Text

Statistics from Altmetric.com
- CDK, cyclin-dependent kinase
- EGFR, epidermal growth factor receptor
- ERCP, endoscopic retrograde cholangiopancreatography
- EUS, endoluminal ultrasonography
- 5FU, 5-fluorouracil
- FNA, fine needle aspiration
- GST, glutathione S-transferase
- IPMN, intraductal papillary mucinous neoplasm
- MBTs, mechanistic based treatments
- MMP, matrix metalloproteinase
- mTOR, mammalian target of rapamycin
- PanINs, pancreatic intraepithelial neoplasia
- PTHC, percutaneous transhepatic cholangiography
- Rb, retinoblastoma
- TGF, transforming growth factor
- VEGF, vascular endothelial growth factor
Pancreatic cancer continues to pose an enormous challenge to clinicians and cancer scientists. With a more affluent world the global incidence of pancreatic cancer is rising. For the first time significant advances are now being made into the management of the disease. There is a more sophisticated approach to palliative care and the centralisation of pancreatic cancer services is leading to greater tumour resection rates. Newer adjuvant modalities are also greatly increasing the 5 year survival rates. The molecular basis of pancreatic cancer is now better understood than ever before, leading to the development of new diagnostic approaches and the introduction of mechanistic based treatments. Technical advances in imaging and great improvements in conventional and molecular pathology have led to a deeper understanding of the pathological variables of the disease. This is now an important time for making big inroads into what still remains the most lethal of the common cancers.
Pancreatic ductal adenocarcinoma remains one of the most difficult cancers to treat. It is the commonest cancer affecting the exocrine pancreas. In 2000, there were 217 000 new cases of pancreatic cancer and 213 000 deaths world wide and in Europe 60 139 new patients (10.4% of all digestive tract cancers) and 64 801 deaths.1 In 2002 there were 7152 new cases in the UK, with similar numbers in men and women.2 In the USA in 2006 there were 33 730 new cases and 32 300 deaths.3 Without active treatment, metastatic pancreatic cancer has a median survival of 3–5 months and 6–10 months for locally advanced disease, which increases to around 11–15 months with resectional surgery.4 The late presentation and aggressive tumour biology of this disease mean that only a minority (10–15%) of patients can undergo potentially curative surgery. Major advances in the past decade have included improvements in operative mortality and morbidity through the development of specialist regional centres and improved survival using systemic chemotherapy.4,5 Significant progress has been made in unravelling risk factors and key molecular events in pancreatic carcinogenesis, leading to potentially exciting new developments in diagnosis, screening of high-risk groups and mechanistic based treatments (MBTs).
MOLECULAR PATHOGENESIS AND THERAPEUTIC TARGETS IN PANCREATIC CANCER
Pancreatic precursor lesions
The ductal phenotype gives rise to three distinct cancer precursor lesions with distinct, although overlapping, gene alterations: mucinous cystic neoplasms, intraductal papillary mucinous neoplasms (IPMNs) and pancreatic intraepithelial neoplasia (PanINs) (box 1).6 PanINs are classified into early and late lesions, starting with PanIN-1A, 1B (hyperplasia) and progressing to PanIN-2 and then to PanIN-3 or carcinoma in situ (fig 1).7–9

Histological images of benign pancreatic ductal epithelial cells, progressive PanIN lesions and invasive carcinoma, with associated genetic alterations.
Box 1 Molecular pathogenesis and drug development
-
Precursor lesions PanIN 1–3 associated with specific molecular alterations.
-
Around 100 mechanistic based treatments are in early clinical trial development and there are number of large phase III trials are in progress.
-
Developmental signalling pathways such as notch/hedgehog are currently undergoing further investigation.
-
Relevant transgenic animal models are now available for molecular analysis and therapeutic studies.
-
Immunotherapies and vaccination treatments are now receiving intense evaluation.
Alterations in oncogenic molecular pathways
K-ras
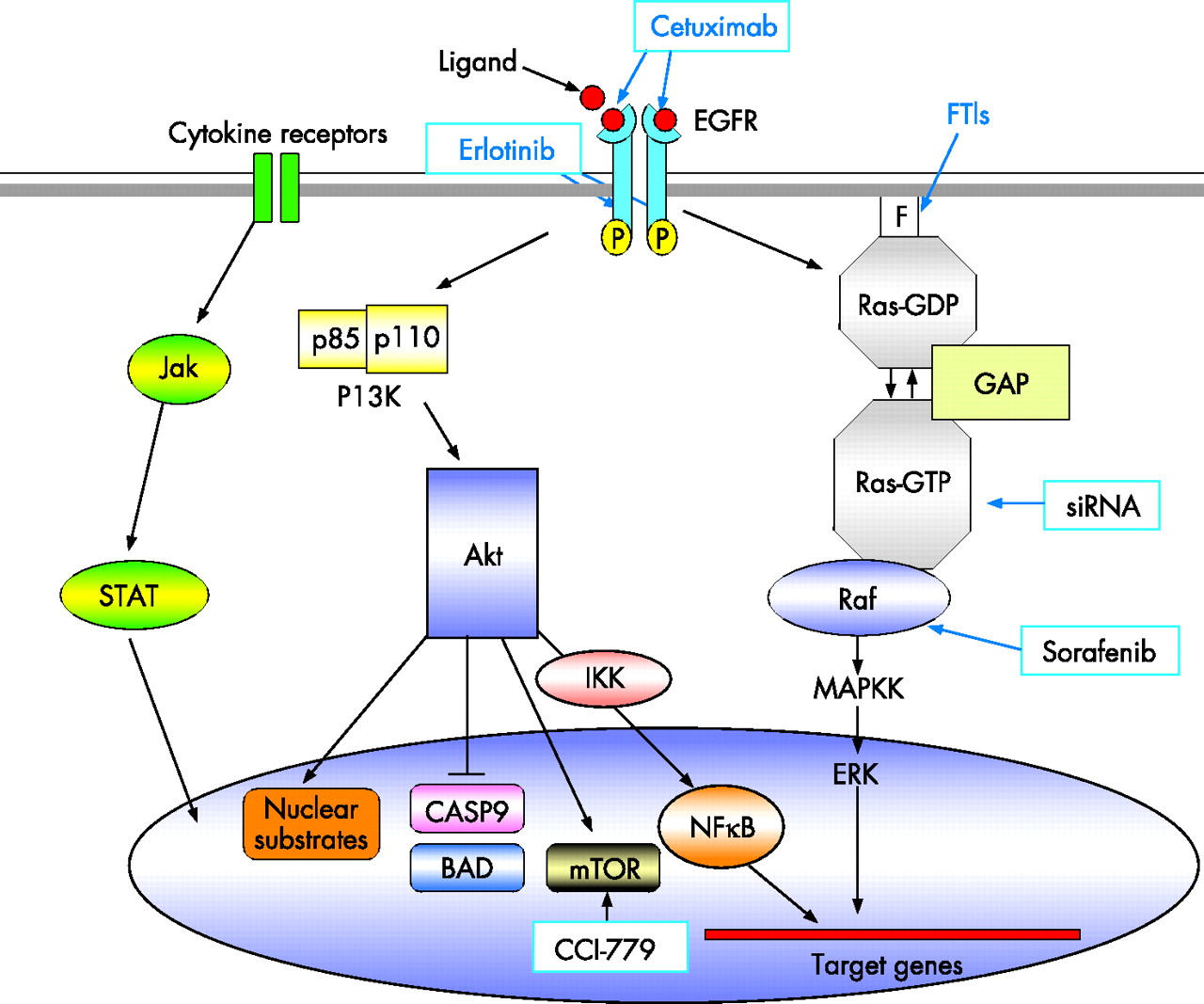
Activating mutations in K-ras, mostly codon 12 but also affecting codons 13 or 61, occur in 75–90% of pancreatic cancers.10,11 Ras is a 21 kDa membrane-bound GTP-binding protein involved in growth factor-mediated signal transduction pathways. The mutations result in a constitutively activated form of Ras in which the protein is locked in the GTP-bound state, capable of stimulating a multitude of downstream signalling cascades.12 K-ras mutations are often found in benign lesions of the pancreas13–15 as well as in early precursor lesions.6 Post-translational modification of Ras protein involves farnesylation of the C terminus, mediated by farnesyl protein transferase and is a major therapeutic target (fig 2),16,17 although farnesyl transferase inhibitors up to now have not been successful in phase III trials.18

Schematic representation of molecular oncogenic signalling pathways in pancreatic cancer. Agents targeting specific aspects of these pathways are indicated in blue boxes.
Alternative approaches that directly target K-ras are now available in the form of RNA interference19 and are showing promise both alone20 and in conjunction with radiation.21 Signalling pathways, downstream of Ras also offer therapeutic targets such as the Raf-MEK ERK pathway. Sorafenib, an inhibitor of Raf-1 kinase and vascular endothelial growth factor receptor-2 is now an FDA approved drug for the treatment of renal cell carcinoma,22 but despite being well tolerated, it is inactive in patients with advanced pancreatic cancer.23
Growth factors and their receptors
The epidermal growth factor receptor (EGFR, also known as human EGF receptor 1 or HER 1) is a major therapeutic target for pancreatic cancer. EGFR is a transmembrane glycoprotein that consists of an extracellular ligand-binding domain with cysteine-rich regions, a hydrophobic transmembrane domain, and an intracellular tyrosine kinase domain. It is a member of the ErbB family of receptor tyrosine kinases, which includes EGFR (EGFR-1), ErbB-2 (HER-2), ErbB-3, and ErbB-4. Of these, EGFR-1, ErbB-2 and ErbB-3 have all been shown to be overexpressed in pancreatic cancer.24–26 The principal natural ligands for EGFR-1, epidermal growth factor (EGF) and transforming growth factor-α (TGF-α) are also overexpressed in this disease.24,27 By binding ligands to the extracellular domain, the EGFR causes receptor homodimerisation or heterodimerisation (with other ErbB family members). This in turn leads to phosphorylation of tyrosine residues on the intracellular domain. The phosphorylated residues then provide docking sites for intracellular mediators which activate downstream signalling pathways (fig 2), including the Ras-Raf-MEK signalling pathway (transmitting growth signals), the PI3K/Akt signalling pathway (mediates cell cycle progression and survival) and the signal transducer and activator of transcription (STAT) family of proteins (mediates a variety of features conducive to cancer cell survival progression, including cell division, motility, invasion and adhesion).28
Cetuximab is a chimeric monoclonal antibody that binds to the extracellular domain of EGFR, promoting receptor internalisation and subsequent degradation without receptor phosphorylation and activation. This diminishes the available receptor for natural ligand binding and prevents activation of EGFR-associated, downstream signalling pathways.28 The first clinical trial in patients with advanced pancreatic cancer tested the combination of cetuximab and gemcitabine and showed an encouraging 1-year survival rate of 32% in a phase II trial29 and has led to the SWOG S0502 phase III trial (target 704 patients), which has completed recruitment and is due to report.30
Erlotinib (Tarceva) is an orally active small molecule that binds to the ATP binding site on the intracellular kinase domain, thus inhibiting the tyrosine kinase activity of the receptor. A recent phase III trial (569 patients) tested erlotinib in combination with gemcitabine for advanced pancreatic cancer.31 Overall survival was significantly better in the erlotinib arm than in the placebo controlled arm, with a median survival of 6.4 vs 5.9 months (p = 0.025) (hazard ratio = 0.81, 95% CI 0.67 to 0.97) and 1-year survival of 24% vs 17%, respectively, the benefit mostly restricted to patients developing a distinctive rash. Erlotinib was approved by the United States FDA in 2005, but European registration is restricted to patients with metastatic disease and does not include those with locally advanced cancer.
High levels of a number of other growth factor receptors and their ligands are also expressed in pancreatic cancer and/or PanIN lesions and represent alternative targets. These include insulin-like growth factor (IGF) and its tyrosine kinase receptor, insulin growth factor receptor 1(IGF-1R), members of the fibroblast growth factor family, the Met receptor tyrosine kinase and its ligand HGF/scatter factor and vascular endothelial growth factor (VEGF) receptors and ligands.32 VEGF promotes endothelial cell growth and survival, thus enhancing angiogenesis. VEGF expression occurs in 90% of pancreatic cancers, correlates with microvessel density and in moderate/high levels with reduced survival.33 Bevacizumab (Avastin), an anti-VEGF monoclonal antibody, showed promise in combination with gemcitabine, in a phase II trial of advanced pancreatic cancer,34 but the CALGB 80303 phase III trial was unsuccessful35 and the Avita trial, which included treatment with gemcitabine plus erlotinib with and without bevacizumab, has been closed. Multitargeted tyrosine kinase inhibitors such as, ZD6474, a dual epidermal growth factor receptor and VEGF receptor 2 small-molecule tyrosine kinase inhibitor, and sunitinib a VEGF receptor 1, 2 and 3, c-KIT, and platelet-derived growth factor receptor α and β tyrosine kinase inhibitor hold promise for pancreatic cancer treatment.36
PI3K/Akt signalling pathway
The lipid kinase phosphoinositide 3-OH kinase (PI3K)/Akt pathway (fig 2) regulates cell survival, proliferation, and resistance to apoptosis. The Akt2 gene was shown to be amplified or activated in up to 60% of pancreatic carcinomas.37–40 Akt mediates the inhibition of pro-apoptotic proteins BAD and caspase 9. Downstream of Akt, the mammalian target of rapamycin (mTOR) promotes cell survival and proliferation41 by modulating cellular signals in response to mitogenic stimuli and various nutrients, especially amino acids. The mTOR–S6K1 signalling pathway is essential for proliferation of pancreatic cancer cells in vitro,42–44 and the mTOR inhibitor, CCI-779, which demonstrates antitumour activity,45 is under investigation in early-phase trials for pancreatic cancer. Akt also activates the transcription factor nuclear factor kappa B (NFκB), which promotes survival and resistance to chemotherapy.46 Bortezomib, a proteasome inhibitor that functions, at least in part, by stabilising the IκBα protein and inhibiting NFκB activation, is currently undergoing phase I evaluation for pancreatic cancer treatment.47
Alterations in molecular pathways affecting tumour suppressor genes
p16INK4A/retinoblastoma (Rb) protein pathway
The activation of cyclin-dependent kinases (CDKs), initially in response to mitogenic signals (cyclin D-dependent kinases) and subsequently in a mitogenic-independent manner (cyclin E-dependent kinases), leads to the sequential phosphorylation of Rb, facilitating the transcription of E2F-regulated genes and consequent entry into the S-phase (fig 3).48,49 The INK4A gene product, p16INK4A, interferes with this process by binding to CDK4/CDK6, preventing the formation of active cyclin D-CDK4/CDK6 complexes. As a result, phosphorylation of Rb is suppressed, blocking entry into the S-phase. In pancreatic cancer the pRb/p16 tumour suppressor pathway appears to be abrogated, most commonly through functional inactivation of the INK4A gene. Loss of p16INK4A function occurs in 80–95% of pancreatic cancers.50–52

Mitogenic signals give rise to increased levels of cyclin D and the consequent formation of active cyclin D/cyclin-dependent kinase 4 or 6 (CDK4/CDK6) complexes leads to the phosphorylation of retinoblastoma (Rb), facilitating the transcription of E2F-regulated genes (including cyclin E) required for the S-phase. Cyclin E–CDK2 complexes further phosphorylate Rb. The tumour suppressor INK4A gene product interferes with this process by binding to CDK4/CDK6, thus preventing the formation of active cyclin D–CDK4/CDK6 complexes. The tumour suppressor, p53, is activated in response to DNA damage or other cellular stresses. MDM2 is a p53-inducible gene, the protein product of which keeps p53 levels low. The p14ARF protein inhibits MDM2, thus inducing p53. Activated p53 either initiates Rb-dependent cell cycle arrest by inducing the transcription of p21CIP1, which inhibits cyclin E-CDK2, or leads to apoptosis.
While p16INK4A inhibits cell proliferation by activating Rb, p19ARF (a gene overlapping with p16) accomplishes the same end through activation of p53 by inhibiting its MDM2-dependent proteolysis, although ARF may possess additional p53-independent functions.53,54 Around 40% of pancreatic cancers lose both the INK4A and ARF transcripts, mutations may occur in the p16 gene but not in the ARF gene, suggesting that INK4A loss alone may be a major event in the development of pancreatic cancer.55
Transcription factor p53
More than 50% of pancreatic cancer cases have mutations in the TP53 gene.50 The p53 transcription factor is normally maintained at very low levels as a result of interaction with the oncoprotein HDM2 (the human homologue of MDM2), which targets p53 for proteosomal degradation. Under conditions of cellular stress, such as genotoxic damage or oncogene activation, the HDM2–p53 interaction is inhibited, and the p53 protein is stabilised. The levels of p53 thus increase and it regulates a transcription response leading to cell cycle arrest or to apoptosis (fig 3).48 After oncogene-mediated activation, p14ARF protein inhibits MDM2, leading to the stabilisation and thus activation of p53. A recent study found that ARF was crucial for tumour suppression, while the DNA damage-induced p53 response was dispensable.56
Smad4/TGF-β pathway
Smad4, was originally isolated as a tumour suppressor gene for pancreatic cancer. Although Smad4 mutations are not particularly common in cancer, in general, pancreatic cancer is characterised by a high degree of alteration in the MADH4 locus on chromosome 18 (18q21.1) that encodes Smad4. It undergoes loss of heterozygosity in ∼90% of pancreatic cancers, and around 50% of cases have completely lost functional Smad4 protein.57,58 The loss of Smad4 has important effects on the tumour microenvironment and potentiation of invasion.59–61
The Smad4 protein is a member of the Smad family of transcription factors and has a pivotal role in mediating signal transmission of members of the TGF-β superfamily of cytokines.62 TGF-β ligands and TGF-β receptors have been found to be highly expressed in pancreatic cancer.32,63 TGF-β ligands are potent regulators of cancer cell growth, differentiation and migration. Knockdown of Smad4 was shown to lead to TGF-β-induced cell cycle arrest and migration but not to TGF-β-induced epithelial–mesenchymal transition,64 indicating that loss of Smad4 seemed to abolish TGF-β-mediated tumour suppressive functions, while maintaining at least some TGF-β-mediated tumour promoting functions. TGF-β-based therapeutic strategies in cancer are in development.62
Reactivation of developmental signalling in pancreatic cancer
Notch
The Notch pathway has an important role in directing decisions about the fate of cells in the developing pancreas and in pancreatic cancer initiation and invasion.65,66 This pathway comprises cell surface-expressed notch receptors which are activated by a number of transmembrane ligands, including Delta, Serrate, and Lag-2 of the delta and jagged families expressed on neighbouring cells, thus mediating communication between adjacent cells expressing the receptors and ligands. This signalling pathway is important for the processes of apoptosis, differentiation and proliferation. Activation leads to proteolytic intramembrane cleavage of Notch receptors, releasing their active intracellular domain, which translocates to the nucleus and binds to the transcription factor CSL (RBP-Jκ/CBF in mammals; Suppressor of Hairless (Su(H) in Drosophilia) inducing the transcription of a variety of target genes including the hairy enhancer of split (HES) family of transcriptional repressors. HES family members act to maintain cells in a precursor state. The pathway is active during embryogenesis, but not in the pancreas, while upregulation of a number of Notch target genes occurs in pre-neoplastic lesions and in invasive pancreatic cancer.67 Notch signalling promotes vascularisation in tumours68 and is a clear target for new drug development.
Hedgehog
Hedgehog signalling has a major role in the initiation and growth of pancreatic cancer.69–73 There are three Hedgehog family members or ligands, sonic hedgehog, Indian hedgehog and desert hedgehog, which are crucial for the development of the gastrointestinal tract. Together with the transmembrane proteins Smoothened and Patched, these signalling proteins/ligands closely coordinate organ development, as well as a variety of functions in adult tissues. Cyclopamine inhibits the Hedgehog pathway through direct interaction with Smoothened65,74,75 and has now entered early clinical trial development.
Sonic hedgehog may also be a feature of so called pancreatic cancer stem cells.76 The existence of cancer stem cells is based on the hypothesis that the ability of a tumour to grow and propagate is dependent on a small subset of cells (<5%) with special properties, which like normal stem cells, have a great potential for self-renewal and production of differentiated progeny.77 After a report that a clone of a distinct CD44+CD24− epithelial-specific antigen (ESA)+ could initiate human metastatic breast cancer in immunodeficient non-obese diabetic/severe combined immunodeficient mice, Li et al have recently identified a CD44+CD24+ESA+ phenotype (0.2–0.8%) isolated from primary pancreatic cancer cells with a 100-fold increased tumourigenic potential.76 Increased numbers of cells, however, were needed to generate tumours when injected into the pancreas compared with the subcutaneum.
The pancreatic tumour microenvironment
In considering the biology of any cancer, the interplay between cancer cells and the surrounding supporting host cells, known as tumour stroma, cannot be ignored. This interplay has effects on blood vessel formation, invasion, metastasis and evasion of the host immune system.78 Pancreatic cancer has a particularly intense desmoplastic stroma, which can account for a large proportion of the pancreatic tumour volume. It comprises extracellular matrix, together with a number of different host cell types, including fibroblasts, small endothelial-lined vessels, residual normal epithelia and a variety of inflammatory cells, which are both locally derived and recruited from the circulation. The biology of the pancreatic tumour microenvironment is being actively researched,79 not least, because it is a potential therapeutic site, such as for antiangiogenic strategies, as discussed above. Targeting stromal matrix, in the form, of matrix metalloproteinase (MMP) inhibitors either with or without gemcitabine, has failed to improve patients’ outcome.80,81 This might be due to the fact that there are many closely related MMPs, and current MMP inhibitors lack sufficient specificity.82
Lessons from animal models
Comprehension of transcription factor activity in the developing pancreas and elucidation of the sequence of genetic alteration in pancreatic cancer development, have been greatly advanced by the development of new genetically engineered animal models of pancreatic cancer.83,84 Targeted expression of oncogenic KRAS to pancreatic progenitor cells in mice resulted in the generation of progressive PanIN lesions, followed by low-frequency progression to invasive and metastatic adenocarcinoma.85 The development of pancreatic cancer was remarkably accelerated by the inclusion of mutations in INK4A/ARF or TP53.86 Thus it appears that activated KRAS serves to initiate PanIN formation while INK4A/ARF tumour suppressors limit the malignant conversion of these PanINs to ductal adenocarcinoma. Similarly, the concomitant expression of oncogenic KRAS and mutant p53 in the mouse pancreas led to accelerated metastatic pancreatic cancer development compared with that seen with oncogenic KRAS alone.85,86 Mutant p53 alone did not induce a cancer phenotype.86
Immunotherapy and vaccines
The limits of conventional cytotoxic drugs in pancreatic cancer have been the main driver for the development of MBTs. In parallel with this has been an explosion of preclinical development of immunotherapies, including cancer vaccination in pancreatic cancer, but the clinical results so far have proved rather disappointing.87 Telomerase overexpression occurs early in the development of pancreatic cancer and can be targeted by telomerase vaccines such as GV1001, with promising phase II results.88 These have now led to the development of a large phase III trial (TeloVac) trial that is exploring the role of simultaneous or sequential cytotoxic and vaccine treatment in advance pancreatic cancer.
AETIOLOGY AND SECONDARY SCREENING
The biggest risk factors for pancreatic cancer are increasing age, smoking,89 new onset diabetes mellitus,90 increased body mass index,89 chronic pancreatitis,91,92 hereditary pancreatitis93 and an inherited predisposition for pancreatic cancer (box 2).94,95 A variety of dietary factors are also associated with an increased risk of pancreatic cancer, all of which are amenable to intervention and comprise increased red and processed meat consumption96 and reduced intake of methionine97 and folate from food sources.98
Box 2 Risk factors for pancreatic cancer
-
Increasing age
-
Tobacco smoking
-
Inherited predisposition: at least two other family members affected
-
Hereditary pancreatitis
-
Chronic pancreatitis
-
Cancer family syndromes
-
Late-onset diabetes mellitus without diabetes risk factors
-
Increased body mass index
Tobacco smoking is associated with a twofold increase and because of the prevalence may be account for around 30% of all cases with pancreatic ductal adenocarcinoma. Chronic pancreatitis is now recognised as a risk factor, with some series finding a 15–25-fold risk.91,92 It has been observed that patients may have chronic pancreatitis for at least 20 years before the development of pancreatic cancer. These patients tend to have severe disease, increased calcification of the gland and a higher rate of complications. The risk of developing cancer is even higher with hereditary pancreatitis, with estimates of a 70-fold increase in risk.93 This is an uncommon disorder inherited as an autosomal dominant condition with an estimated 80% penetrance and an equal gender incidence, presenting in children and younger adults. The gene responsible was identified as the PRSS1 gene, and mutations have a causative role, resulting in a gain of function of the digestive enzyme trypsin.
There is an inherited component to pancreatic cancer accounting for about 10% of observed cases.94,95 Familial pancreatic cancer itself is rare and the European Registry of Hereditary Pancreatitis and Familial Pancreatic Cancer (EUROPAC) (http://www.liv.ac.uk/surgery/europac.html, accessed 22 May 2007) has been established to provide a database of these families for long-term follow-up, with the aim of identifying people at risk and developing a screening programme in the future. Diagnostic criteria are two or more first-degree relatives with pancreatic ductal adenocarcinoma or two or more second-degree relatives with pancreatic cancer, one of whom has early-onset pancreatic cancer (age <50 years at diagnosis). Overall, the observed to expected rate of pancreatic cancer is significantly raised by ninefold, rising specifically from fourfold in families with one first-degree relative, to 6.4-fold where there are two affected relatives to 32.0-fold with three relatives with pancreatic cancer.95
There is evidence that familial pancreatic cancer is an autosomal dominant condition99 and appears to demonstrate the phenomenon of anticipation with the age of onset reducing in succeeding generations.100 The main gene and causative mutation have not yet been identified, although up to 20% of families with familial pancreatic cancer have a BRCA2 mutation.101 One candidate gene is palladin,102 which encodes a component of the cytoskeleton that controls cell shape and motility, and has been identified in the susceptibility locus 4q32–34 in a large family from Seattle, USA (called family X),103 but both the susceptibility locus and the palladin gene variant described have not been confirmed in EUROPAC families.104,105
Several studies have examined the association between genetic polymorphisms and pancreatic cancer.94 Although over the whole population none of the genetic polymorphisms for two carcinogen-metabolising enzymes (cytochrome P450 1A1 (CYP1A1) and glutathione S-transferase (GST)) could be directly associated with the risk of pancreatic cancer, the combination of heavy smoking and a deletion polymorphism in GSTT1 was associated with an increased risk of pancreatic cancer among Caucasians.106 Polymorphisms of glutathione S-transferase M1 (GSTM1) and acetyltransferases (NAT1 and NAT2) enzymes may also be associated with a modest increase in susceptibility to pancreatic cancer and chronic pancreatitis.107 The UDP glucuronosyltransferase (UGT1A7) gene is predominantly expressed in the human pancreas. The low detoxification activity UGT1A7*3 allele has been identified as a new risk factor of pancreatic diseases, defining an interaction of genetic predisposition and environmentally induced oxidative injury.108
Several inherited cancer syndromes are associated with pancreatic cancer (table 1).94,101,102,109–119 The highest risk of pancreatic cancer in all of these cancer syndromes is in Peutz–Jeghers syndrome with a 120-fold lifetime risk and a 36% cumulative lifetime risk.112 Although responsible for this syndrome, germline mutations of the STK11/LKB1 gene are not involved in familial pancreatic cancer.120 Pancreatic cancer is the second most common cancer in the familial atypical multiple mole melanoma syndrome and is particularly significant in patients and families with the p16 Leiden mutation.109 Pancreatic cancer is also seen in some families with breast cancer and BRCA1 and BRCA2 mutations.110 The cumulative risk of pancreas cancer to age 75–years in BRCA2 carriers is 7%, and BRCA2 may account for as many as 5% of all cases of pancreatic cancer.94 It is evident that a number of these genes act as modifier genes on environmental and other genetic risk factors. RNASEL (encoding ribonuclease L) gene variants/mutations (Glu265X and Arg462Gln) implicated in sporadic and familial prostate cancer may also contribute to the tumourigenesis of sporadic and familial pancreatic cancer but do not directly cause pancreatic cancer.121
Hereditary cancer syndromes affecting the pancreas
Patients at high risk warrant screening,122–124 but these programmes have not been adequately assessed and at the present time secondary screening (box 3) should only be undertaken as part of an investigational study such as that organised by EUROPAC.122,123
Box 3 Secondary screening
-
All patients with an increased inherited risk of pancreatic cancer should be referred to a specialist centre offering clinical advice and genetic counselling and, where appropriate, genetic testing such as for BRCA2 mutations.
-
Primary screening for pancreatic cancer in the general population is not feasible at present.
-
Secondary screening for pancreatic cancer in high-risk cases should only be part of an investigational programme.
PATHOLOGY, STAGING AND RESECTION MARGINS (BOX 4)
Box 4 Pathological typing, staging and resection margins
-
Most pancreatic cancers are pancreatic ductal adenocarcinomas.
-
Accurate pathological typing and staging is essential to determine the most appropriate treatment and prognostic groups.
-
In 20% of cases it is not possible to distinguish the tissue of origin of pancreatic cancers: “peri-ampullary cancer”.
-
Chip-based technologies will lead to a more accurate typing of tissue origin.
-
Resection margin status needs to be clearly defined. At present, a tumour <1 mm from the margin is reported as positive.
Ductal adenocarcinoma is the most common malignant tumour of the pancreas (table 2).125–131 Characteristically, there is an intense desmoplastic reaction in the stroma surrounding these tumours. Sixty-five per cent are located within the head, 15% in the body, 10% in the tail and 10% are multifocal. Tumours of the head of the pancreas tend to present earlier with obstructive jaundice or acute pancreatitis. Tumours of the body and tail tend to present late and are associated with a worse prognosis. There are guidelines for minimum data set reporting and staging.132,133 Pancreatic ductal adenocarcinoma must be distinguished from carcinomas of the intrapancreatic bile duct, ampulla of Vater or duodenal mucosa as these tumours have a much better prognosis. In about 20% of cases it is not possible to distinguish the tissue of origin of cancers arising in the head of the pancreas, and the term “peri-ampullary cancer” is often applied. Application of chip-based DNA expression techniques will hopefully overcome this problem with the spread of molecular pathology complementing traditional histology, as even intrapancreatic bile duct cancers have genetic similarities to pancreatic ductal adenocarcinomas.134
The key factors relating to prognosis are tumour grade and diameter and lymph node status. The microscopic resection margin status is also an important survival factor, although less so within the adjuvant context.135 A positive microscopic resection margin (R1) is operationally defined as at least one cancer cell within 1 mm of any surface of the resected specimen. A positive R1 margin is unrelated to tumour diameter but rather to histological grade and lymph node status, indicating that this has more to do with the biology of the tumour than with physical factors.136
DIAGNOSIS
Advances in technology have meant that the sensitivity for detecting smaller lesions is improving, as is the identification of extrapancreatic spread (box 5).137–139
Box 5 Diagnosis
-
The use of contrast-enhanced multidetector CT is the preferred method for non-invasive staging of pancreatic cancer.
-
Other modalities such as magnetic resonance cholangiopancreatography and endoluminal ultrasonography may contribute further information but should only be used selectively.
-
Preoperative endoscopic retrograde cholangiopancreatography brushing for cytology should be undertaken in all cases undergoing endoscopic stenting.
-
Laparoscopy with laparoscopic ultrasound may be appropriate in selective cases to improve staging.
-
Tissue diagnosis should be sought in all cases deemed unresectable.
-
Transperitoneal techniques of tissue biopsy have relatively poor sensitivity and should be avoided in cases where resection is possible.
Tumour markers and proteomic signatures
The most commonly used marker in everyday practice CA19-9 has a sensitivity of 70–90% and specificity of 90%, and is better than other markers, including CA-50 and DU-PAN-2 and CEA.140 False positive results are often obtained in benign obstructive jaundice, chronic pancreatitis even in the absence of bile duct obstruction and ascites. CA19-9 is particularly useful in assessing response to prognosis and treatment in advanced cases, identifying early recurrence in resected cases and as an aid in preoperative staging.140–142 New markers, including HCGβ,143 CA72-4,143 osteopontin,144 REG4,145 RCAS1146 and MIC-1147 are under evaluation, but radically newer approaches that hold real promise are new proteomic techniques identifying unique panels of proteins associated with pancreatic cancer and protein profiles providing a distinctive pancreatic signature.148–150 Gene expression profiling may also help to categorise prognostic groups.134
Non-invasive imaging techniques
Transabdominal ultrasound can be the initial investigation and may detect tumours >2 cm in size, dilatation of the biliary and main pancreatic ducts and possible extrapancreatic spread— notably, liver metastases, with a diagnostic accuracy of 75%,151 but it is not useful in early disease, if the bile duct is not dilated and in obese patients. Therefore contrast-enhanced multidetector CT scan is the single most useful imaging procedure (using a pancreas protocol CT with 1 mm images) and can achieve diagnostic rates of 97% for pancreatic cancer.152 The accuracy for predicting an unresectable lesion is 90%, but the accuracy of predicting a resectable lesion is much less at 80–85%139,152,153 (figs 4–7). False negative results before laparotomy are mainly due to small hepatic metastases <1 cm and small peritoneal deposits. Lymph node staging is inaccurate in the absence of systematic biopsy.154

Contrast-enhanced multidetector CT scan image of a resectable pancreatic adenocarcinoma with acceptable planes of cleavage between the tumour (t) and the superior mesenteric vein (arrow) and the superior mesenteric artery (arrowhead).

Contrast-enhanced multidetector CT scan image of pancreatic tumour encasing the hepatic artery (arrow) and obliteration of the portal vein (cross) causing cavernous transformation. This patient is unresectable.

Contrast-enhanced multidetector CT scan image of pancreatic tumour encasing the superior mesenteric artery (arrow). This patient has unresectable disease.

Coronal section of multidetector CT scan demonstrating pancreatic tumour encasing the portal vein. This patient has unresectable disease.
Magnetic resonance imaging produces similar results to contrast-enhanced multislice CT and is useful for patients who cannot receive intravenous contrast.155,156 Positron emission tomography (PET) cannot differentiate inflammatory conditions from tumours accurately and the sensitivity is 71–87% with specificity of around 64–80%.157 The use of fusion CT-PET scanning adds little if anything to the use of CT alone.158 Measurement of tumour metabolism by nuclear magnetic spectroscopy holds considerable promise as a diagnostic technique but is very much in development.159
Invasive imaging techniques
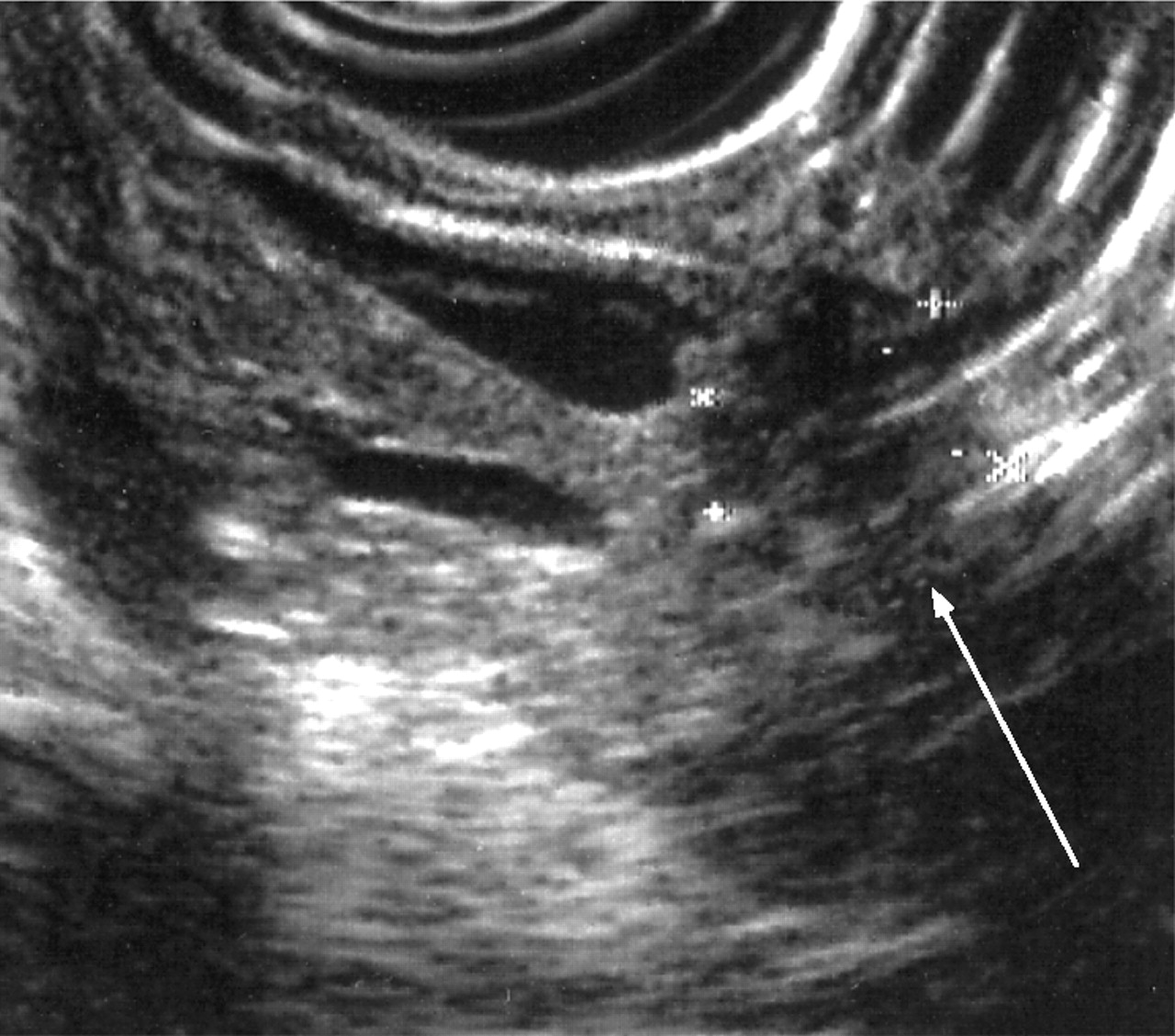
Endoluminal ultrasonography (EUS) has similar accuracy to CT in the staging of pancreatic cancer but is undoubtedly better for the detection of early pancreatic tumours as small as 2–3 mm139 (fig 8). The addition of fine needle aspiration (FNA) cytology to EUS is highly accurate for identifying malignancy in lesions identified on EUS and not seen on CT scan.139,160 The drawbacks of EUS are that distant metastases and nodal involvement cannot be accurately assessed. The sensitivity and specificity of endoscopic retrograde cholangiopancreatography (ERCP) alone are 70–82% and 88–94%, respectively, in symptomatic patients or those with suspected pancreatic cancer but should no longer be used as a pure imaging tool given the developments in magnetic resonance cholangiopancreatography and EUS.139,140,155,156,160 ERCP is used to insert biliary stents for relief of obstructive jaundice161 and to gain cytological diagnosis by sampling or brushings. These can also be obtained at percutaneous transhepatic cholangiography (PTHC).140
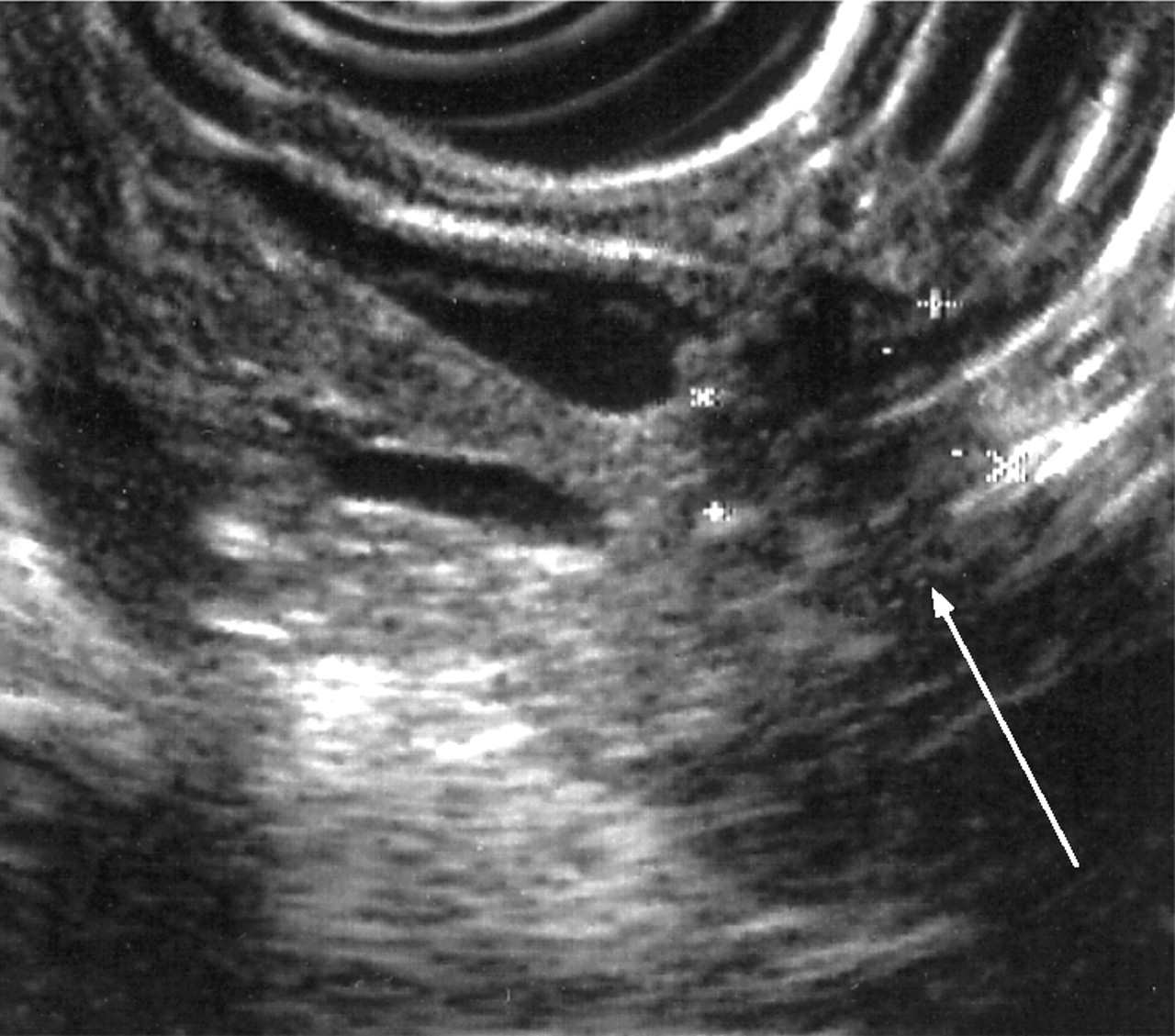
Endoluminal ultrasound demonstrating a small pancreatic cancer.
Diagnostic biopsy
Percutaneous FNA cytology has a sensitivity and specificity of 69% and 100%, respectively, for tissue diagnosis,140 but concerns have remained about intraperitoneal seeding, with an incidence of up to 16%.162 The diagnostic accuracy of EUS with FNA carries a sensitivity and specificity of >90% and ∼100%, respectively, but requires an expert team with the presence of a cytologist examining the tissue specimens in the EUS suite, repeating the procedure until the diagnosis is conclusive.163 The incidence of carcinomatosis is much less after EUS-guided biopsy than percutaneous biopsy.164 A further development is the use of EUS with an endoscopic trucut biopsy needle.165 EUS-guided biopsy is thus the preferred procedure if histological confirmation is needed in cases of advanced pancreatic cancer before chemotherapy or to diagnose small uncharacterised lesions.
Laparoscopy and laparoscopic ultrasound
Laparoscopy with laparoscopic ultrasound enables intraoperative scanning of the liver and pancreas to be performed and is highly predictive of resectability, altering the management of 15% of patients already assessed as resectable by dual-phase helical CT.166 Selective laparoscopy based on the serum level of CA19-9 is a more efficient strategy, reducing the proportion of patients undergoing laparoscopic ultrasound from 100% to around 45% while increasing the yield from 15% to 25%.167
CYSTIC TUMOURS
Pancreatic cystic neoplasms are being increasingly identified with the wider employment of high-quality abdominal imaging and comprise at least 15% of all pancreatic cystic masses (box 6).125,168,169 The three most common primary pancreatic cystic neoplasms are serous cystic neoplasm, mucinous cystic neoplasm and intraductal papillary mucinous neoplasm (IPMN). Serous cystic neoplasms predominantly affect women, are found mostly in the head of pancreas and represent 30% of primary cystic neoplasms. Mucinous cystic neoplasms also are found more often in women, but mostly in the body and tail of the pancreas, and represent 40% of primary cystic neoplasms. Unlike IPMNs the cyst does not communicate with the main pancreatic duct. IPMNs tend to affect more men than women, can involve a part or the whole of the pancreatic ductal system, affect older patients and represent 30% of primary pancreatic cysts.125,168,169
Box 6 Cystic tumours of the pancreas
-
Most non-inflammatory pancreatic cysts are malignant or premalignant: main differential diagnosis is a pancreatic pseudocyst.
-
The three main types are serous cystic neoplasms, mucinous cystic neoplasms and intraductal papillary mucinous neoplasms (IPMN).
-
Patients with pancreatic cysts have an increased risk of developing other cancers of the pancreas and also extrapancreatic cancers such as colorectal cancer.
-
Mainstays of diagnosis are CT scan, magnetic resonance cholangiopancreatography and endoluminal ultrasonography with fine needle aspiration and cyst fluid analysis (cytology, mucin, CEA and Ca19-9).
-
Serous cystadenomas are nearly always benign and may be managed conservatively and kept under radiological surveillance.
-
Side-branch IPMNs that lack malignant features may also be managed conservatively with radiological monitoring: diameter <3.5 cm, absence of nodules and thick walls, CA19-9 <25 kU/l, absence of recent-onset or worsened diabetes, absence of jaundice or of any other symptom.
-
Resection is needed for all mucinous cystic neoplasms and main duct IPMN.
Pathology
Serous cystic neoplasms consist of a well-demarcated spongy, honeycomb mass with small cysts lined by a simple cuboidal epithelium with glycogen-rich cytoplasm (fig 9) and rarely progress to serous cystadenocarcinoma. Mucinous cystic neoplasms consist of a larger often solitary cyst to begin with and may have a septum or septae contained within the cyst lined by simple mucinous columnar epithelium and there is a characteristic ovarian-type stroma (fig 10). IPMNs are classified as arising either from the main duct (fig 11) or branch duct (fig 12) and can be mixed. They are characterised by intraductal proliferation of neoplastic mucinous cells forming papillae and excessive mucous secretion. These changes lead to dilatation of the main pancreatic duct or branch duct.169,170 IPMNs arising in the branch ducts are less aggressive than those arising in the main duct, which have a high incidence of malignant lesions.171,172 There is a greatly increased risk of colorectal cancer and other extrapancreatic cancers in patients with IPMN.173,174 There is also an increased risk of developing other cancers of the pancreas.175

(A) Contrast-enhanced multidetector CT scan image of a serous cystic neoplasm, demonstrating the characteristic honeycomb appearance. (B) Honeycomb cysts of serous cystic neoplasm are lined by simple cuboidal epithelium with (glycogen-rich) clear cell cytoplasm.

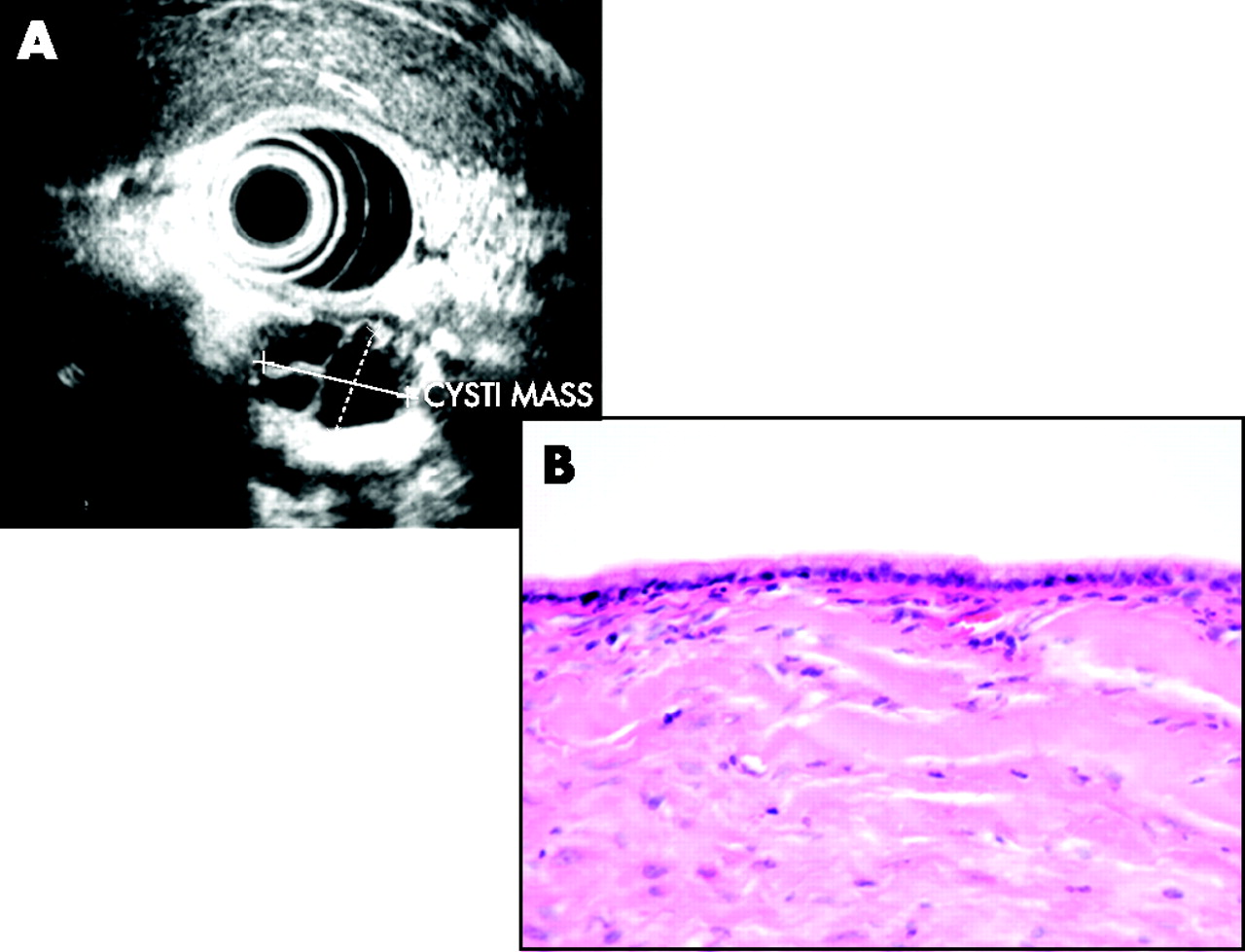
(A) Endoluminal ultrasonography showing septate mucinous cystic lesion. (B) Simple columnar mucinous epithelial lining of a mucinous cystic neoplasm.

(A) Contrast-enhanced multidetector CT scan image of a main duct intraductal papillary mucinous neoplasm, demonstrating dilatation of the whole of the main pancreatic duct. (B) High-power view of intraductal papillary mucinous neoplasm, showing minimal atypia within the lining mucinous epithelium.
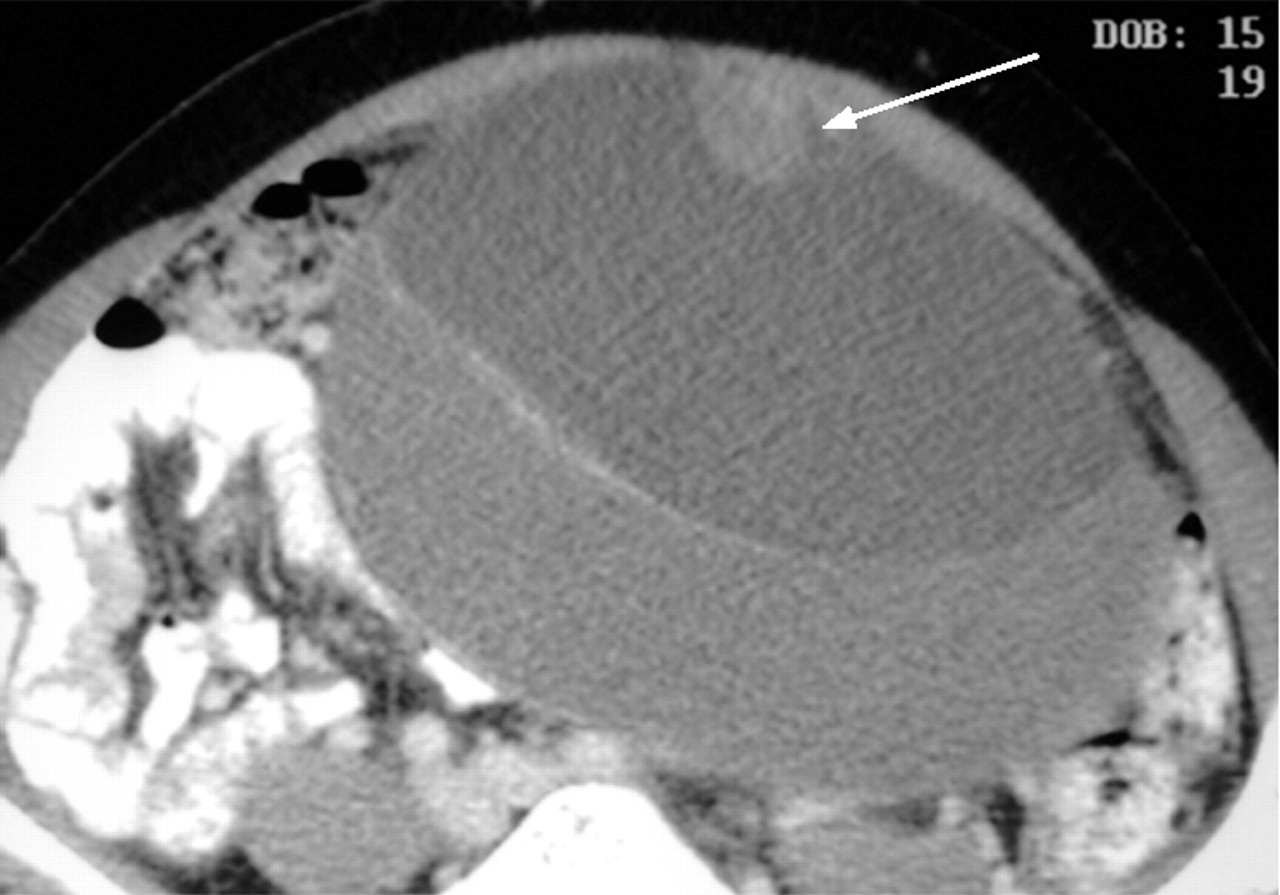
Contrast-enhanced multidetector CT scan image of a mucinous cystic neoplasm, demonstrating a papillary invagination.
Diagnosis
Serous cystic neoplasms may be polycystic or honeycomb as on cross-sectional imaging with a central scar (fig 9), and are sometimes calcified but may also appear to be solid. Mucinous cystic neoplasms may have thick irregular walls with papillary invaginations (fig 12) and sometimes peripheral calcification. The characteristic radiological feature of IPMN is side-branch cystic dilatation in communicating with the main pancreatic duct or dilation of the main pancreatic duct (figs 11 and 13) full of mucous readily seen at endoscopy or dilatation; malignant potential may be related to size (⩾3 cm) and mural nodules.172 Early lesions may be evaluated by intraductal pancreatoscopy and intraductal ultrasonography.169 Fluid by FNA from serous cystic neoplasms lacks mucin. The cyst fluid from mucinous cystic neoplasms is viscous and will stain positive for mucin with high levels of CEA or CA19-9.169

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
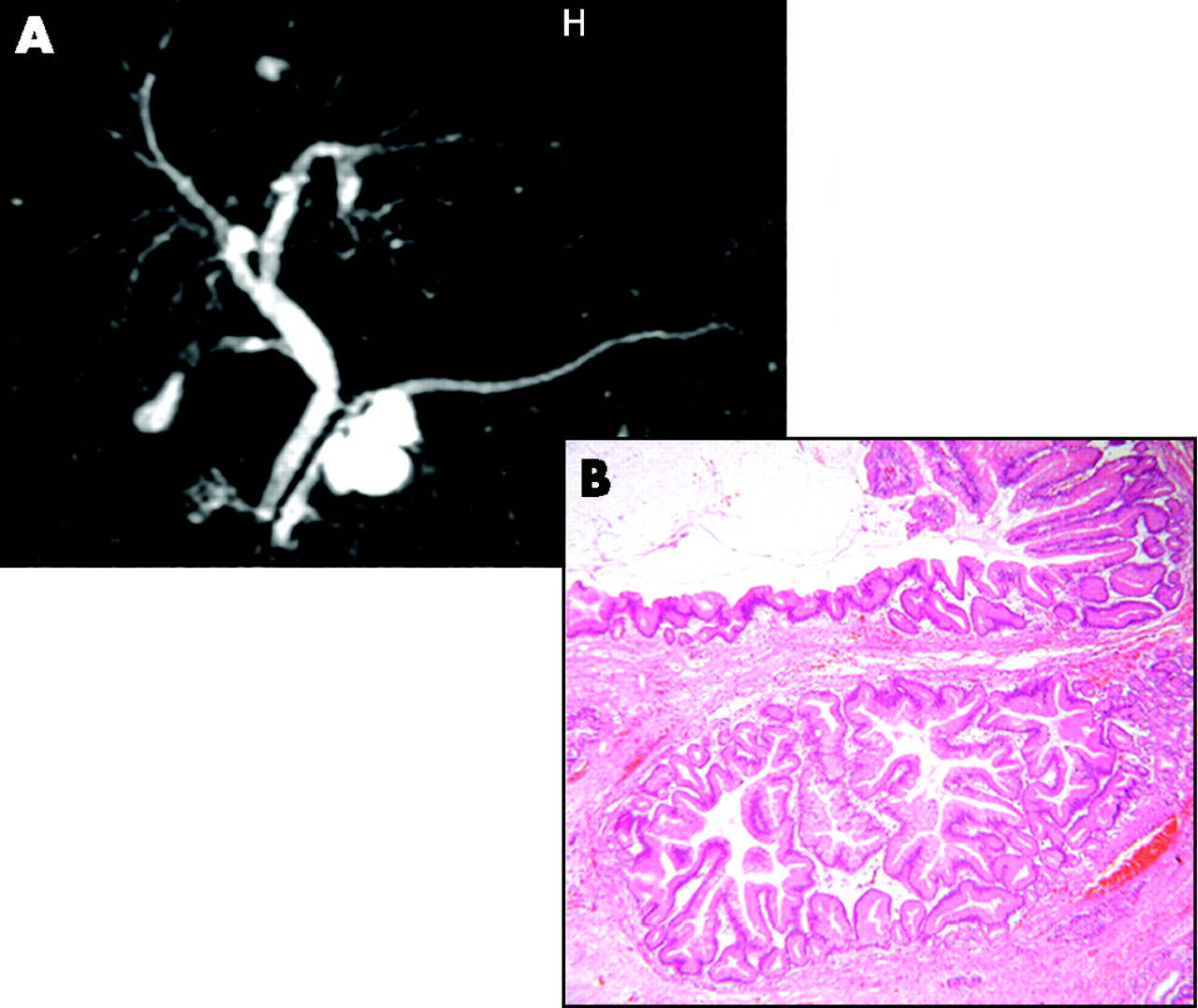
(A) Magnetic resonance imaging scan demonstrating a side-branch intraductal papillary mucinous neoplasm. (B) Low-power view of branch-type intraductal papillary mucinous neoplasm, showing papillary infoldings of lining epithelium.
Management options
If a lesion can be positively identified as a serous cystic neoplasm then a conservative approach with regular follow-up imaging is justified, particularly if the patient is frail or elderly.169,175 Mucinous cystic neoplasms should be resected if the patient is fit for major surgery owing to the high malignant potential. All main duct IPMNs should be resected if the patient is fit, combined with frozen section assessment of the main pancreatic duct resection margin; the patient should be prepared to undergo a total pancreatectomy. Patients with relatively benign features of branch duct IPMN (diameter <3.5 cm, absence of nodules and thick walls, CA 19-9 <25 kU/l, absence of recent-onset or worsened diabetes, absence of jaundice or of any other symptom) may be managed with regular follow-up imaging instead of resection in certain patients.176
TREATMENT OF PANCREATIC CANCERS
Inoperable disease
The treatment of patients who have localised advanced disease, metastases or performance status is directed at symptom control (box 7).
Box 7 Symptom control in advanced pancreatic cancer
-
The main analgesic method is the use of modern oral opiate preparations; neurolytic coeliac plexus block should be considered as complementary in selected cases.
-
Pancreatic enzyme supplements should be used to maintain weight and increase quality of life.
-
Endoscopic biliary stenting should be used in malignant biliary obstruction.
-
Metal stents should be used in patients with defined parameters (locally advanced tumour <3 cm diameter), plastic stents should be used otherwise.
-
Younger patients with relatively good performance status may undergo biliary drainage—in which case they should also undergo prophylactic gastrojejunostomy to prevent late gastric outlet obstruction (occurs in around 15%).
-
Duodenal and gastric outlet obstruction may also be treated endoscopically.
Pain
Intractable pain is a major problem and often necessitates the use of high-dose opiate analgesia. Complementary approaches include intraoperative, percutaneous CT-guided or EUS neurolytic coeliac plexus block140,177–179 and bilateral or unilateral thoracoscopic splanchnicectomy.180 In general, the results are disappointing and are particularly poor for patients with tumours in the body and tail of the pancreas. Pain control with coeliac plexus block was improved in a randomised study compared with systemic analgesia, but this was not reflected in the quality of life or survival.178
Weight loss
Weight loss initially is due to pancreatic exocrine insufficiency owing to obstruction of the main pancreatic duct as well as exclusion of bile acids from obstruction of the main bile duct. Fat maldigestion may also contribute to abdominal pain and bloating. Relief of biliary obstruction and pancreatic enzyme supplementation will alleviate these symptoms.181 Cachexia can be a marked feature of the later stages of pancreatic cancer, with no good treatment.
Biliary and duodenal obstruction
Biliary stenting using ERCP is the preferred option with the combined PTHC-endoscopy approach employed only if the former is technically not possible.140 The life of a plastic stent is about 3 months, causing recurrent jaundice. Self-expanding metal (and covered) stents have greatly reduced the risk of obstruction and acute cholangitis. Metal stents should be used for patients with a good performance status and favourable prognosis (locally advanced primary tumour <3 cm) and plastic ones for those patients with metastases and tumours ⩾3 cm in diameter.182 Expandable metal stents are being increasingly deployed endoscopically for duodenal obstruction (occurs in ∼15%), with a technical success rate of around 85%, but may be associated with serious complications, including perforation, fistula and bleeding and recurrent obstruction due to stent migration or fracture.183 Surgical bypass (open and laparoscopic) can be used to relieve jaundice using a Roux-en-Y loop hepatojejunostomy, and duodenal obstruction by gastrojejunostomy, especially in younger patients and both can be achieved laparoscopically.184,185
Chemotherapy
Pancreatic ductal adenocarcinoma is highly resistant to conventional methods of cytotoxic treatment and radiotherapy (box 8).186–189 Few chemotherapeutic agents have been shown to have reproducible response rates of more than 10%. 5-Fluorouracil (5FU) is an inhibitor of thymidylate synthetase (essential for synthesis of DNA nucleotides) and has been the most widely used in advanced pancreatic cancer, with a median survival of around 5–6 months and is better than the best supportive care.186–189 A pivotal trial in 1997 meant that the nucleoside analogue, gemcitabine, replaced 5FU as the preferred drug.190 Although the median survival improvement in favour of gemcitabine compared with 5FU was slight (5.7 vs 4.4 months), the 1-year survival rate was more encouraging (18% vs 2%), and most importantly, the toxicity was relatively mild and achieved a better clinical response (24% vs 5%, respectively).190
Box 8 Palliative therapy in advanced pancreatic cancer
-
Chemotherapy will improve survival and quality of life in patients with advanced pancreatic cancer.
-
Chemoradiotherapy and follow-on chemotherapy are no better than chemotherapy alone.
-
The best chemotherapy combination available at the present time is gemcitabine combined with either capecitabine or a platinum agent with acceptable toxicity.
-
Where possible, patients with advanced pancreatic cancer should be offered treatment with new therapeutic drugs as part of an early drug development programme or as part of a phase III randomised controlled clinical trial.
-
New agents will be expensive but will become increasingly targeted based on molecular profiling.
Capecitabine (Xeloda) is a new oral, fluoropyrimidine carbamate that is sequentially converted to 5FU by three enzymes located in the liver and in tumours, including pancreatic cancer. The Cancer Research UK GemCap trial comparing gemcitabine alone or in combination with capecitabine demonstrated significantly improved survival with this combination than with gemcitabine alone191 and is supported by other studies.189,192–194 A recent meta-analysis has demonstrated that combination gemcitabine chemotherapy is better than gemcitabine alone; the best combinations may be with capecitabine or platinum-based agents, allowing for acceptable levels of toxicity of the combinations.189
Chemoradiotherapy and follow-on chemotherapy
Radiotherapy has been widely used for the treatment of pancreatic cancer.187,188 The main drawback is the limit on the dosage owing to the close proximity of adjacent radiosensitive organs. External beam radiotherapy is routinely used with 5FU as a radiosensitising agent (chemoradiotherapy), although gemcitabine is now being evaluated as an alternative radiosensitiser. Newer techniques such as conformal radiotherapy are now being used, but these studies almost invariably employ follow-on chemotherapy once the chemoradiotherapy has been completed. A recent meta-analysis demonstrated that chemoradiotherapy is better than radiotherapy alone and that there is no survival difference between chemoradiotherapy plus follow-on chemotherapy and chemotherapy alone.188 A recent phase III study compared chemoradiotherapy and follow-on gemcitabine with gemcitabine alone in patients with locally advanced disease.195 The trial was closed prematurely because of significant toxicity in the combination arm and significantly reduced median survival in the combination arm (8.4 vs 14.3 months; p = 0.014).
Newer agents
A number of new agents and MBTs have been developed from the molecular understanding of pancreatic cancer, which are now being assessed in large phase III trials in advanced pancreatic cancer (table 3).196–201
Resectable disease
Selection and staging
Once the pancreatic cancer has been identified, the patient needs to be assessed for fitness for major surgery and the tumour staged preoperatively for resectability (box 9 and table 4). Venous resection is necessary during the course of a pancreatectomy in 5–10% of patients. Vascular reconstruction in this context results in a median and long-term survival that is similar to that of patients not needing a venous reconstruction.202 It should be emphasised, however, that routine venous resection in patients with significant venous involvement is not feasible and the results of arterial reconstruction are unacceptably poor.203 The resection rates and short- and long-term results are significantly better in high-volume centres, and major pancreas cancer surgery should only be undertaken in regional and supraregional centres.204–207
Indicators of resectability in pancreatic cancer
Box 9 Surgery in pancreatic cancer
-
Surgical resection should be confined to specialist centres; increased resection rates and survival and decreased hospital costs, morbidity and mortality.
-
Endoscopic biliary drainage before surgery does not influence surgical outcome but may assist with logistical planning.
-
Pancreatoduodenectomy with or without pylorus preservation is the most appropriate procedure.
-
Portal vein resections are needed in about 10% of resections but should not be performed routinely.
-
Arterial reconstruction cannot be supported except in exceptional circumstances.
-
Extended radical resections should not be undertaken because of increased mortality and morbidity and reduced quality of life.
-
Use of prophylactic somatostatin analogues reduces postoperative morbidity.
Surgical techniques
Preoperative endoscopic stenting does not influence surgical outcome, but it may facilitate logistical planning of staging and treatment.161,204,208 Metal stents should be avoided in patients who have tumours that may be resectable because of the tissue reaction they invoke, although resection is still technically possible. The aim of surgery is to achieve an R0 resection: complete clearance of macroscopic tumour with clear microscopic resection margins, even if there are lymph node metastases. In practical terms a large proportion of patients (at least 35%) are histologically staged as R1: complete clearance of macroscopic tumour with positive resection margins.204 R2 resections result in incomplete resection of macroscopic tumour and should be treated in the same category as patients with locally advanced pancreatic cancer as they have an equally poor prognosis. Resecting these patients may lead to longer survival than chemoradiotherapy.209
The standard operation for tumours of the head of the pancreas is the Kausch–Whipple partial pancreatoduodenectomy (KW-PPD).210 There are various methods of reconstruction involving the pancreatic anastomosis. The benefit from a pancreatogastrostomy rather than a pancreatojejunostomy is still unclear,211 and there may be no advantage for the routine use of pancreatic stents.212 The pylorus-preserving partial pancreatoduodenectomy (PP-PPD) is the most commonly used resection approach, which despite being a smaller procedure is as effective as a KW-PPD (table 5).213–215 Patients with tumours of the pancreatic body or tail undergo left pancreatectomy usually with en bloc resection of the spleen and hilar lymph nodes.204 There is no role for total pancreatectomy unless this is the only means by which an R0 resection can be achieved.204 Extended radical lymphadenectomy is associated with significantly increased morbidity without any survival benefit and is now rejected for routine practice.216–218 Involvement of para-aortic lymph nodes (Japanese Pancreas Society Lymph Node Station 16b1) is not a contraindication to resection and should probably be included as part of the routine resection procedure.219 Postoperative morbidity remains high at around 40% even in supraregional units.207 Independent risk factors are age >70 years, extended resections and main pancreatic duct diameter <3 mm.220 Postoperative complications may by reduced by the prophylactic use of octreotide.221
Adjuvant treatment
Radical resection alone will result in a 5-year survival of only 10% owing to recurrence after surgery.204 Nearly all patients develop metastatic disease, most commonly of the liver and peritoneum but also the lungs, and this may occur with or without local recurrence.187,204,222 Although chemoradiation to the area of the resection may reduce the local failure rate, survival length is the same as with systemic chemotherapy.187 After pancreatic resection, the most important independent prognostic markers are lymph node status, tumour size and tumour grade.135,136 The results from two large randomised trials show that adjuvant systemic chemotherapy will increase the 5-year survival from 9% to 12% with resection alone to 21–29% and 23% with either 5FU and folinic acid or gemcitabine, respectively (box 10).223–225 Table 6 summarises all the randomised trials of adjuvant systemic chemotherapy.168–173 The ESPAC-3(v2) trial comparing adjuvant gemcitabine and 5FU has closed to recruitment with 1030 patients, with 2-year survival as the end point. The survival benefit of adjuvant chemotherapy is maintained irrespective of the type of operation used and whether or not patients develop postoperative complications.229
Box 10 Adjuvant therapy in pancreatic cancer
-
Adjuvant 5FU-based chemotherapy significantly improves survival.
-
Adjuvant gemcitabine chemotherapy may also significantly improve survival.
-
Adjuvant chemoradiation has not been shown to improve survival in the absence of maintenance chemotherapy.
-
Adjuvant chemoradiotherapy and follow-on chemotherapy may not offer improved survival compared with chemotherapy alone—trial awaited.
-
Neoadjuvant treatments should only be administered as part of a controlled clinical trial.
Adjuvant chemoradiotherapy has been used in the USA on the basis of a small randomised trial230,231 as well as apparently improving survival as reported in a non-randomised series of patients,232,233 but these results have not been confirmed in large randomised trials,223,224,234,235 so the focus has moved to whether chemoradiotherapy and follow-on chemotherapy represents a better alternative than chemotherapy alone (table 7).223,224,230,231,234,236 The results of meta-analysis using individual patient data reject the use of chemoradiation and provide powerful evidence for systemic chemotherapy.235
The RTOG 9704 trial236 has recently reported median and 3-year survival rates. This study used background 5FU-based chemoradiotherapy together with pre- and post-chemoradiation systemic chemotherapy comprising either 5FU or gemcitabine. The original sample size was 330 patients, but this was increased to 518 patients to enable assessment of survival in patients with pancreatic head tumours. The results showed no difference in median survival or 3-year survival in all patients. There was, however, a significant improvement in survival with the gemcitabine-based treatment in patients who had tumours of the pancreatic head. These findings are in keeping with survival noted in the equivalent groups in the ESPAC-1 trial and do not show any advantage over chemotherapy alone.
The EORTC trial 40013 plans to recruit 538 patients with resectable pancreatic cancer and compare gemcitabine chemotherapy with gemcitabine followed by chemoradiotherapy. There is an initial phase II part to assess feasibility and toxicity. Neoadjuvant therapy has also been advocated to increase resection rates, reduce positive resection margins and for the early treatment of micrometastatic disease, but at present there is little evidence to support this approach and randomised trials are lacking.187,204
CONCLUSIONS
Pancreatic cancer is a formidable disease to diagnose and treat. Surgical approaches have become more standardised and are safer, with much improvement in both morbidity and mortality in specialised centres. Diagnosis has improved using conventional imaging methods, and appropriate treatment decisions can be made because of these improvements. Palliative treatment is improving, including the use of endoscopic stent placement with better but less than effective pain relief, and pancreatic enzyme supplementation. Chemotherapy regimens can prolong survival in patients with advanced disease without reducing their quality of life. At present only pancreatic resection can improve survival significantly. A further improvement in survival is achievable with adjuvant chemotherapy but not chemoradiotherapy. The molecular mechanisms responsible for pancreatic cancer point to earlier diagnosis and targeted treatments, using new genetic and biological approaches. Pancreatic cancer surgery can only be performed within a regional pancreas cancer. This is now a very encouraging phase in the diagnosis and treatment of pancreatic cancer. The information and resources now available can result in a reasoned approach to the treatment of patients with pancreatic cancer to ensure the best outcome with an optimum quality of life.
Acknowledgments
We gratefully acknowledge the provision of figures from staff at the Royal Liverpool University Hospital Trust: pathology figures are kindly supplied by Dr Fiona Campbell, consultant histopathologist, Department of Pathology and also Dr Jutta Luettges, Klinikum Saarbrücken, Germany; radiology figures are kindly supplied by Dr Jonathan Evans, consultant radiologist, Department of Radiology; and EUS figures are kindly supplied by Dr Martin Lombard, consultant gastroenterologist, Department of Gastroenterology.
REFERENCES
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
Footnotes
-
Funding: Cancer Research UK, CORE and EU Biomed Programmes 5 and 6.
-
Conflict of Interest: None.