



Abstract
Acetylcholinesterase inhibitors (AChEIs), the most developed treatment strategies for Alzheimer’s disease (AD), will be used in clinic for, at least, the next decades. Their side effects are in highly variable from drug to drug with mechanisms remaining to be fully established. The withdrawal of tacrine (Cognex) in the market makes it as an interesting case study. Here, we found tacrine could disrupt the proper trafficking of proline-rich membrane anchor-linked tetrameric acetylcholinesterase (AChE) in the endoplasmic reticulum (ER). The exposure of tacrine in cells expressing AChE, e.g., neurons, caused an accumulation of the misfolded AChE in the ER. This misfolded enzyme was not able to transport to the Golgi/plasma membrane, which subsequently induced ER stress and its downstream signaling cascade of unfolded protein response. Once the stress was overwhelming, the cooperation of ER with mitochondria increased the loss of mitochondrial membrane potential. Eventually, the tacrine-exposed cells lost homeostasis and underwent apoptosis. The ER stress and apoptosis, induced by tacrine, were proportional to the amount of AChE. Other AChEIs (rivastigmine, bis(3)-cognitin, daurisoline, and dauricine) could cause the same problem as tacrine by inducing ER stress in neuronal cells. The results provide guidance for the drug design and discovery of AChEIs for AD treatment.
SIGNIFICANCE STATEMENT Acetylcholinesterase inhibitors (AChEIs) are the most developed treatment strategies for Alzheimer’s disease (AD) and will be used in clinic for at least the next decades. This study reports that tacrine and other AChEIs disrupt the proper trafficking of acetylcholinesterase in the endoplasmic reticulum. Eventually, the apoptosis of neurons and other cells are induced. The results provide guidance for drug design and discovery of AChEIs for AD treatment.
Introduction
Alzheimer’s disease (AD) is a chronic neurodegenerative disease and accounts for 50–75% of dementias across the globe. More than 4 million new cases of dementia are reported every year with a number expected to almost double by 2030 (Prince et al., 2013), leading to a heavy burden to health care system in the society. In the brains of patients with AD, the loss of cholinergic markers, e.g., acetylcholinesterase (AChE), is one of the most consistent cholinergic alterations (Ferreira-Vieira et al., 2016). Therapeutic approaches for AD have been established to enhance cholinergic functions by using either AChE inhibitors (AChEIs) or cholinergic receptor agonists. Today, AChEI is the most sedimented strategy for AD therapy, which boosts endogenous levels of acetylcholine in the brain and thereby enhances cholinergic neurotransmission (Talesa, 2001). AChEIs can be divided into two groups according to the mechanism of action: reversible and irreversible. The toxicity of irreversible AChEI has been reported, and thus the reversible inhibitor mostly has therapeutic application (Colovic et al., 2013). Three cholinesterase inhibitors, donepezil (Aricept), rivastigmine (Exelon), and galantamine (Razadyne), are in clinical use today (Nordberg and Svensson, 1998). Tacrine (Cognex) was the first AChEI to be approved for AD treatment in 1993, but its use was abandoned because of hepatotoxicity (Watkins et al., 1994). In addition to drug development, the presence of AChE in plasma, cerebrospinal fluid, and blood cell has suggested its possible use as a biomarker reflecting the state of cholinergic system in patients with AD (Lionetto et al., 2013).
AChE is a polymorphic enzyme transcribed from a single gene of ACHE to different isoforms. The variants of AChE isoforms, i.e., AChET, AChER and AChEH, are created via alternative splicing at the 3′ end (Soreq and Seidman, 2001). The AChER variant generates a soluble monomer that is upregulated in the brain under stress (Atsmon et al., 2015); the AChEH variant produces a glycosylphosphatidylinositol-anchored dimer that is dominantly expressed in mammalian blood cells (Freitas Leal et al., 2017; Xu et al., 2018); the AChET variant, denoted here as AChE for simplicity, is the subunit mainly expressed in the brain and muscle (Tsim et al., 1988; Xie et al., 2010). The localization and oligomerization of AChE in the brain depends on the interactions of its C-terminal peptide (also called tail peptide) with proline-rich membrane anchor (PRiMA), which is an anchoring protein. The tetrameric globular (G4) form of this enzyme is produced and predominantly expressed on the surface of neuronal cells (Xie et al., 2010; Chen et al., 2011). In biosynthesis of AChE oligomer, the newly synthesized molecule does not contain enzymatic activity and is identified as an inactive precursor. A subset of polypeptide chains is assembled into catalytically active dimeric and tetrameric forms after post-translational modification (Rotundo et al., 1989). The oligomerization of AChE happens in rough endoplasmic reticulum (ER), and about 70–80% of newly synthesized AChE oligomers do not enter the Golgi apparatus, leading to a rapid degradation in the ER (Rotundo et al., 1989; Chen et al., 2011). Only a small part of AChE, catalytic active oligomers, associates with the PRiMA subunit to produce the G4 AChE in ER. The fully glycosylated AChE is produced in the Golgi apparatus and precisely targets onto plasma membrane as a functional enzyme in neurons or other cell types (Chen et al., 2011).
The newly synthesized proteins have a risk of aberrant folding and aggregation, potentially generating toxic species (Hartl et al., 2011). The unfolded or misfolded proteins could be accumulated and aggregated in ER, causing an ER stress (Schröder and Kaufman, 2005). When the stress is overwhelming, the ER cooperates with mitochondria, leading to the processes of apoptosis (Lemasters, 2005; Sano and Reed, 2013). During the process of ER stress, an evolutionarily conserved signaling, named unfolded protein response (UPR), is activated to restore cell homeostasis (Schröder and Kaufman, 2005). Protein misfolding is believed to be the primary cause of several neurodegenerative diseases, such as AD. Today, AChEIs are the best-proven efficacious agents for AD treatment. Since newer therapeutic approaches are still at a very early stage of development, AChEIs are likely to be used clinically at least for years. Interestingly, the efficacies of AChEIs for AD are usually consistent from case to case and drug to drug; however, the occurrence of side effects is highly variable (Schneider, 2000). Especially, the long-term safety of AChEIs is not known. Here, we aim to identify the role of AChEIs, e.g., tacrine, that could affect AChE protein trafficking, and subsequently we determine the potential effect of anti-AD drugs in the ER-stressed apoptosis.
Materials and Methods
Chemicals
AChEIs were purchased from Sigma-Aldrich (St. Louis, MO), and synthetic AChEIs, bis(7)-cognitin, and bis(3)-cognitin were from Prof. Yifan Han (Hong Kong PolyU) (Han et al., 2012). Reagents used for culturing cells were bought from Invitrogen Technologies (Carlsbad, CA). Thapsigargin was purchased from Abcam (Cambridge, UK). Other reagents, not mentioned, were purchased from Sigma-Aldrich (St. Louis, MO).
Cell Culture
Mouse neuroblastoma X rat glioma hybrid cells (NG108-15), human embryonic kidney fibroblast cells (HEK293T), and mouse monocytes cells (RAW 264.7) were purchased from American Type Culture Collection (Manassas, VA). These cells were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen Technologies) supplemented with100 μg/ml streptomycin, 100 U/ml penicillin, and 10% fetal bovine serum and maintained in a humid atmosphere at 5% CO2, 37°C. Cortical neurons were separated and cultured from Sprague-Dawley rats at embryonic day 18, which was previously described (Pi et al., 2004; Yu et al., 2020). In brief, Hank’s balanced salt solution (Sigma-Aldrich) supplemented with 10 mM HEPES (pH 7.4) without Ca2+ and Mg2+ and 1 mM sodium pyruvate was used to obtain cortex from the whole brain. The cortex was treated with 0.25% trypsin at 37°C and triturated for several times to get single cells. Subsequently, the dispersed cells were cultured on poly-l-lysine–coated culture plates or coverslips in neurobasal medium with 2% B27 at 37°C. After 24 hours, cytosine arabinonucleoside (Sigma-Aldrich) was added to a final concentration at 2.5 μM to inhibit nonneuronal cell division. The cortical neurons could be used at 6 days after culture. Primary osteoblasts were cultured from calvaria of Sprague-Dawley rats, as previously described (Xu et al., 2013).
AChE Enzymatic Assay
The modified method of Ellman et al. (1961) was used to determine AChE enzymatic activity. Briefly, 0.1 mmol/L tetra-isopropylpyrophosphoramide (Sigma-Aldrich) was added to each reaction for 10 minutes to inhibit butyrylcholinesterase activity. The reaction mixture contains 0.5 mmol/L 5,5-dithiobis-2-nitrobenzoic acid, 0.625 mM acetylthiocholine iodide (ATCh; Sigma-Aldrich) and 520 μl samples in 80 mM sodium phosphate (Na2HPO4) at pH 7.4. Absorbance unit per minute per gram of protein was used to express the specific enzyme activity. The absorbance was measured at 410 nm.
Subcellular Fractionation
Subcellular fractionation was carried out, as previously described (Chen et al., 2011). Briefly, the cultured NG108-15 cells were dispersed in 0.5 ml of homogenization buffer (10 mM HEPES, 1 mM EDTA, 0. 25 M sucrose, supplemented with protease inhibitors in low-salt lysis buffer, pH 7.4) and disrupted by Dounce homogenizer on ice. After centrifugation at 500g for 10 minutes, nuclei and unbroken cells were pelleted and discarded. The supernatant was centrifuged at 80,000g for 1 hour to obtain the vesicle pellet, which was resuspended in 0.4 ml homogenization buffer and layered on the top of continuous iodixanol gradients (1–20%, Sigma-Aldrich). After centrifugation by Sorvall TH 641 rotor at 200,000g for 3 hours, 1 ml fraction was collected from the top of gradient. The intracellular markers were determined by Western blot analyses with different antibodies: calnexin (1: 1,000; Sigma-Aldrich) for the ER, Na+/K+-ATPase (1:1000; Abcam) for the plasma membrane, and Golgi matrix protein 130 (1:1000; BD Biosciences, San Jose, CA) for the Golgi apparatus.
Sucrose Density Gradient Analysis
Sucrose density gradient analysis was employed to separate various molecular forms of AChE as previously described (Xie et al., 2010). The known sedimentation coefficients of alkaline phosphatase (ALP) and β-galactosidase were 6.1 S and 16 S, respectively. As internal sedimentation markers, ALP and β-galactosidase were mixed with 0.2 ml samples of cell extracts containing equal amounts of protein (200 μg). The mixture was layered onto continuous sucrose gradients (5–20%) in 12-ml polyallomer tubes. The gradient was centrifuged at 38,000 rpm in a SW41 Ti Rotor (Beckman Coulter Inc. Brea, CA) for 16 hours at 4°C. About 45 fractions were collected from the bottom to the top of tubes. ALP and β-galactosidase activities of fractions were detected to determine sedimentation coefficients of each fraction. Then, AChE activity of each fraction was assayed. According to the known sedimentation coefficients of G1 (4.3 S) and G4 (10.2 S) isoforms of AChE, relative expression of different AChE isoforms could be determined.
Real-Time Quantitative Polymerase Chain Reaction
RNAzolRT Reagent (Molecular Research Center, Cincinnati, OH) was employed to extract total RNA according to the manufacturer’s instructions. Equal amounts of RNA were reverse transcribed into cDNAs by Moloney murine leukemia virus reverse transcriptase (Thermo Fisher Scientific, Waltham, MA). The transcript level of target genes was determined by real-time quantitative polymerase chain reaction (PCR) using SYBR Green Master mix and carboxy-X-rhodamine (ROX) reference dye (Winer et al., 1999). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as housekeeping gene. Primers used were as follows: 5′-AAT CGA GTT CAT CTT TGG GCT CCC CC-3′ and 5′-CCA GTG CAC CAT GTA GGA GCT CCA-3′ for AChE; 5′ -TCT GAC TGT CCT GGT CAT CAT TTG CTA C-3′ and 5′-TCA CAC CAC CGC AGC GTT CAC-3′ for PRiMA; 5′-CAT GGT TCT CAC TAA AAT GAA AGG-3′ and 5′-GCTGGTACAGTAACAACTG-3′ for immunoglobulin heavy chain binding protein (BiP); 5′- CAT ACA CCA CCA CAC CTG AAA G-3′ and 5′- CCG TTT CCT AGT TCT TCC TTG C-3′ for CAAT/enhancer binding protein homologous protein (CHOP); 5′ -AGG TCG GTG TGA ACG GAT TTG-3′ and 5′-TGT AGA CCA TGT AGT TGA GGT CA-3′ for GAPDH. The 2−ΔΔCt method was employed to quantify the relative levels of transcript expression for target genes.
SDS-PAGE and Western Blot Analyses
The total protein from cell cultures was collected by centrifugating at 13.2 × 104 rpm for 15 minutes at 4°C in low-salt lysis buffer (Liu et al., 2020). Samples containing equal amounts of total protein were treated with direct lysis buffer and boiled at 100°C for 10 minutes (Chen et al., 2011). After the electrophoresis separation in 1× SDS-PAGE running buffer, the proteins were transferred to nitrocellulose membrane. To confirm the transfer and equal loading of samples, the membrane was stained by Ponceau S. The primary antibodies, incubated overnight at 4°C, were as follows: anti-AChE antibody (1:500; Santa Cruz Biotechnology, Santa Cruz, CA), anti–cleaved caspase 3 antibody (1:1000; Cell Signaling Technology, Danvers, MA), anti-BiP antibody (1:1000; Cell Signaling Technology), and anti–α-tubulin antibody (1:10,000; Sigma-Aldrich). The enhanced chemiluminescence method was used to visualize the immune complexes in strictly standardized conditions. The intensities of bands were quantified by Image Laboratory 6.0 (Bio-Rad, Hercules, CA). α-Tubulin served as an internal control to calculate the expression level of target proteins.
DNA Construct and Transfection
Luciferase genes, pAChE-Luc and pPRiMA-Luc, were the DNAs (∼2.2 kb) encompassing human AChE promoter and human PRiMA promoter, which were subcloned into a pGL3 vector (BD Biosciences) upstream of a luciferase gene. The cDNA encoding human wild-type AChE (AChEWT), AChE3N→3Q (three potential N-glycosylation sites were site-directed mutated from asparagine to glutamine substitution), mutant AChE (AChEMT; AChEWT residues Ser234, Glu365, and His478 were mutated to alanine) (Du et al., 2015), and mouse PRiMA were described previously; all AChE constructs were generated from human AChET subunit (Chen et al., 2011). Cultured NG108-15 and HEK293T cells were transiently transfected with the cDNA constructs by jetPRIME reagent and calcium phosphate precipitation, respectively (Chen et al., 2011). Another control plasmid having a β-galactosidase gene under a cytomegalovirus (CMV) enhancer promoter was used to determine the transfection efficiency, consistently at 20–30%. A commercial kit (Tropix Inc., Bedford, MA) was used to perform luciferase assay according to manufacturer’s protocol. The activity was measured as absorbance (up to 560 nm) per mg of protein.
Immunofluorescence Analysis
Cultured cells on glass coverslip (Marienfeld superior, Lauda-Königshofen, Germany) were fixed by 4% paraformaldehyde for 10 minutes. After several times of washing, samples were blocked by 5% bovine serum albumin with or without 0.2% Triton X-100 for 1 hour at room temperature. The anti-AChE antibody (1:500; Santa Cruz Biotechnology) was treated to stain AChE protein in cells for 16 hours at 4°C. Subsequently, the corresponding fluorescence-conjugated secondary antibody (Alexa 488–conjugated anti-goat) was treated for 2 hours at room temperature. After three times of washing by PBS, samples were serially dehydrated with ice-cold 50, 75, 95, and 100% ethanol and mounted with DAKO (Carpinteria, CA). Until they were completely dried, samples were observed under a Zeiss laser scanning confocal microscope (Leica SP8, Wetzlar, Germany). Leica confocal software (version 2.61) using a PLAPO 40 0.75 DRY objective was employed to capture the images at excitation 488 nm/emission 500–535 nm.
Apoptosis Detection
An Annexin V–FITC/PI Apoptosis Detection kit (BD Biosciences) was employed to measure the apoptosis of cells by flow cytometry according to the manufacturer’s instructions. In brief, cells were cultured for 24 hours before exposure. After exposure, the floating and adherent cells were gently collected in culture medium. After three times of washing in PBS, cells were incubated in binding buffer [annexin-V/fluorescein isothiocyanate (FITC) and propidium iodide] for 15 minutes at room temperature in dark. To eliminate clumps and debris, cells were passed through the cell strainer. A total of 10,000 events of each sample were acquired by the loader. The data were showed by the quadrants, which were set based on the population of viable unstained cells in the control samples. FACS Aria equipped with the CellQuest Software (BD Biosciences) was used to analyze the results.
Detection of Mitochondrial Membrane Potential
The mitochondrial membrane potential (MMP, Δψ) of cells was measured by JC-1 (Sigma-Aldrich) staining (5 μg/ml) for 30 minutes. The floating and adherent cells were harvested in culture medium. After three times washing by PBS, the cells were stained by JC-1 staining solution and measured by a flow cytometry. Confocal microscopy was used to measure the MMP of live cells in situ. HEK293T cells were cultured on poly-l-lysine–coated coverslips and stained by JC-1. Followed by three times of washing, the coverslips were mounted onto microscope slides. Images were captured by a confocal microscope (Leica SP8). JC-1 generated complexes called J-aggregates with red florescence, which was detected at excitation 561 nm/emission 582 ± 25 nm. The JC-1 monomer exhibited intensive green florescence, which was measured at excitation 488 nm/emission 530 ± 30 nm. FlowJo version 7.6 software was used to analyze the flow cytometry data (Kuete et al., 2015). For the images taken by confocal microscopy, the ratio of red/green fluorescence intensity was analyzed by ImageJ software (Rawak Software Inc., Dresden, Germany).
Transmission Electron Microscopy
HEK293T cells were seeded and grown for 24 hours. Glutaraldehyde (2.5%) in cacodylate buffer (0.1 M, pH 7.4) was used to fix the treated cells. After firming with 1% osmium tetroxide (pH 7.2), samples were dehydrated and embedded in durcupan. A diamond knife was employed to cut samples into 60 nm, which was mounted on Cu-grids. Images were captured by a transmission electron microscope (Philips CM100, Andover, MA).
Hoechst 33342 Staining
Cells were cultured and seeded on poly-l-lysine–coated plates. After exposure and intensive washing with cold PBS, cells were stained by cell-permeable DNA dye Hoechst 33342 (300 μl, 5 μg/ml) at 4°C for 5 minutes. The nuclei were visualized by a confocal microscope (Leica SP8). Cells with chromatin condensation or fragmentation were stained with bright blue and were considered as apoptotic cells. Condensed nuclei were calculated by counting at least 500 cells of three randomly chosen fields from four experiments.
Molecular Docking
The human AChE sequence from UniProtKB P22303 was used. Wild-type and activity-deleted mutant AChE were homology modeled with template 6O4X (Protein Data Bank code). Their structures were prepared by correcting the structure issues, including adding hydrogens, missing loops, calculating partial charges, and break bonds by Amber10: EHT forcefield. The structure of tacrine was built and converted to a three-dimensional structure with minimized energy. The binding site of co-crystallized AChE ligand 9-aminoacridine was used to establish the active pocket. The poses of placement and refinement were set as 180 and 30, which were chosen as triangle matcher and rigid receptor with evaluated scores of London dG and Generalized-Born volume integral/Weighted surface area dG. By using MOE Dock module, the molecular docking simulation between tacrine and residue site in AChE active pocket was carried out.
Other Assays
A Bradford method kit from Bio-Rad was employed to determine the concentration of protein. For statistical analyses, each result represented the mean ± S.D., each with triplicate samples. Statistical significance was determined by paired Student’s t test and one-way or two-way repeated measures of ANOVA with subsequent application of different multiple comparisons methods.
Results
Tacrine Inhibits Trafficking of PRiMA-Linked G4 AChE
Tacrine, a centrally acting AChEI, has been discontinued due to safety concerns: this chemical serves as a representative agent to study the potential threats of AChEIs. The subcellular fractionation was carried out to detect the effects of tacrine on enzyme distribution in cultures having overexpressed AChE catalytic subunit together with PRiMA, i.e., overexpression of the G4 form. The expression level of AChE protein was upregulated by over 30-fold after overexpression of the G4 form of AChE in cultured NG108-15 cells (Supplemental Fig. 1A). The subcellular compartments of cultured NG108-15 cells were separated by linear iodixanol gradients. The protein markers of each fraction were determined by Western blot with specific primary antibodies. At the top of gradient, i.e., fractions 2–4, the plasma membrane (PM) marker (Na+/K+-ATPase) and the Golgi marker (Golgi matrix protein 130) could be identified. At the bottom of gradient, i.e., fractions 8-10, the ER marker (calnexin) was enriched. The last fraction, i.e., fraction 11, was excluded since the aggregated proteins could be contaminated (Fig. 1A). In cells overexpressing G4 AChE at time zero (no tacrine), AChE protein showed a classic transport from ER to Golgi, with most of AChE protein localized in Golgi/PM-enriched fractions (Fig. 1A). The situation was slightly changed with the exposure of tacrine for 8 hours. After exposure of tacrine for 24 hours, AChE localization was almost exclusively found in the ER-enriched fractions, with only very small amount in the Golgi/PM-enriched fractions (Fig. 1A). The quantitative distribution of AChE in different subcellular organelles, after exposure of tacrine for different numbers of hours, is shown in Fig. 1B. To identify the ER-retained AChE, the ER fraction of tacrine-exposed NG108-15 cultures was subjected to sucrose density gradient analysis. Because tacrine inhibited most of the activity of AChE in the ER fractions, Western blot was employed here for the protein recognition in each fraction. With or without tacrine exposure, G1/G2 was the major form of AChE, as compared with G4 (Fig. 1C). This notion was further supported by nonreducing gel as shown in Fig. 1D. The result indicated that tacrine should not affect dimerization or tetramerization of the enzyme in ER.

Tacrine inhibits the trafficking of PRiMA-linked G4 AChE. (A) Cultured NG108-15 cells co-transfected with AChE catalytic subunit (AChET) and PRiMA were treated with tacrine (THA; 20 µM) for different numbers of hours. The cell lysates underwent subcellular fractionation. The distribution of proteins was analyzed by corresponding antibodies. (B) The amount of AChE protein in each fraction was quantified from the blots in (A) by calibrated densitometry. (C) The ER fractions [fractions 8–10 as in (A)] from NG108-15 cells with or without THA exposure were concentrated by ultrafiltration centrifuge tube, then subjected to sucrose density gradient assay. The expression of AChE in different fractions was detected. (D) The ER fractions of cultured NG108-15 cells with or without tacrine exposure were analyzed by nonreducing electrophoresis. Heavy (+PRiMA) and light (−PRiMA) dimers are indicated. (E) Cultured NG108-15 cells were treated with THA (20 µM) for 24 hours. The medium containing THA was removed and intensively washed with cold PBS. Then fresh culture medium was added. Cell lysates were collected after the removal of THA after different numbers of hours for Ellman assay. The THA-treated ER fraction from NG108-15 cells was dialyzed: the dialyzed samples underwent sucrose density gradient analysis (insert). (F) Cell lysates from THA-treated NG108-15 cells with equal amounts of proteins were incubated with Con A, SNA, or agarose only. After centrifuging, the activity of AChE in supernatant was determined by Ellman assay. Percentage of precipitated AChE was calculated as (total input AChE activity − supernatant AChE activity)/total input AChE activity × 100%. The data are normalized to the percentage of control or the amount of agarose-precipitated AChE. Values are in means ± S.D., n = 4, each with triplicate samples. Statistical significance was analyzed by one-way repeated measures ANOVA with subsequent application of Dunnett’s multiple comparisons test. *P < 0.5, **P < 0.01, ***P < 0.001 versus control.
Tacrine is a reversible AChEI, and thus we tested the recovery of enzyme activity after removal of tacrine from the culture. After removal of tacrine in cultured NG108-15 cells and washing with PBS, the activity of AChE was almost completely recovered (Fig. 1E). The activity in ER fraction of tacrine-exposed cultures could be fully recovered after dialysis against buffer (Fig. 1E, insert). Moreover, the sedimentation forms of recovered AChE were not changed. Proper glycosylation of AChE is known to play a role in enzyme trafficking (Chen et al., 2011). Hence, two lectins [concanavalin A (Con A) and Sambucus nigra (SNA)] were used to determine different glycan composition of AChE by specifically precipitating the enzyme. The high-mannose glycan chain, abundant in both precursor and mature AChE, could be specifically precipitated by Con A. Sialic acid, expressed mainly in complex chains of mature AChE, could be bound by SNA (Supplemental Fig. 1B). Similar binding with immobilized Con A and SNA of AChE oligosaccharides in cells treated with or without tacrine was detected, which suggested no change in enzyme glycosylation after exposure to tacrine (Fig. 1F).
The immunofluorescence staining of AChE in cells was employed to visualize the effect of tacrine in AChE protein trafficking. NG108-15 cells having overexpression of G4 AChE were stained with anti-AChE antibody, 4′,6-diamidino-2-phenylindole (DAPI), and phalloidin–iFluor 555 with or without Triton X-100. DAPI and phalloidin were used to stain nucleus and plasma membrane, respectively. Intensive immunoreactivity was detected in both with and without tacrine-exposed cells in presence of Triton X-100, indicating that AChE was expressed at similar level (Fig. 2). Without permeabilization by Triton X-100, the staining of AChE in control cells could be revealed on cell surface; however, the staining of AChE was not detected on cell surface of tacrine-exposed cells (Fig. 2A). The minimal amount of surface AChE in tacrine-exposed culture was further illustrated by quantitation of fluorescence staining (Fig. 2B). Here, we are hypothesizing that tacrine could disrupt the proper membrane targeting of PRiMA-linked G4 AChE to cell surface.

Tacrine blocks the trafficking of AChE to membrane. (A) Cultured NG108-15 cells co-transfected with AChE catalytic subunit (AChET) and PRiMA were treated with or without tacrine (THA; 20 µM) for 24 hours. Cells were immunostained by the anti-AChE antibody, DAPI, and phalloidin–iFluor 555 with or without permeabilization of Triton X-100 (0.2%). Florescence and phase photos were taken. (B) The quantification of florescence intensity of AChE is shown. The values of average florescence intensity per cell are shown as means ± S.D. were analyzed by ImageJ software. n = 4.
Tacrine Induces ER Stress in Neuronal Cells
An induction of BiP expression is an indicative marker of ER stress, as well as a central regulator of the UPR. To examine the effects of tacrine on ER stress, we first exposed the cells with chemical reagent that induced ER stress, namely thapsigargin, serving as a positive control. Treatment of cultured NG108-15 cells with thapsigargin activated the UPR, as indicated by ∼4-fold induction of BiP expression (Fig. 3A). The induction of BiP by applied tacrine in cultured NG108-15 cells was in a dose-dependent manner with the maximal induction by ∼2-fold at ∼50 μM tacrine (Fig. 3B). Unfolded or misfolded protein being accumulated in ER is known to cause stress and cell death. Cleaved caspase 3 (cl-caspase 3), an apoptotic marker, was measured here to reveal the effect of tacrine in inducing cell apoptosis. In a time-dependent manner, tacrine obviously induced expression of cl-caspase 3 in both cultured NG108-15 cells and cortical neurons. After exposure of tacrine for 24 to 48 hours, the induction of cl-caspase 3 reached to 2–3-fold in NG108-15 cells or cortical neurons (Fig. 3C). Tacrine inhibited AChE activity in a dose-dependent manner in cultured NG108-15 cells. Tacrine at ∼50 µM almost completely inhibited the activity of AChE in culture, similar to that of donepezil (Supplemental Fig. 1C). However, the protein expression of AChE was upregulated by over 2-fold after exposure of tacrine in cultured NG108-15 cells or cortical neurons (Fig. 3D). Thus, the expression of AChE protein under ER stress was measured here in cultures. The exposure of thapsigargin induced the expression of AChE in a dose-dependent manner having the maximal induction at ∼3-fold (Fig. 3E, left panel). At the same time, thapsigargin induced the total enzymatic activity of AChE in a dose-dependent manner, reaching to the highest induction of ∼3-fold, as compared with control (Fig. 3E, right panel). Meanwhile, the immunofluorescence staining of AChE-overexpressed cells indicated that thapsigargin caused an undetectable amount of AChE in the cell surface, i.e., no AChE being transported to cell surface (Fig. 3F). The minimal amount of surface AChE in thapsigargin-exposed cells was further analyzed by quantitation of fluorescence staining (Supplemental Fig. 1D).

Tacrine induces ER stress and apoptosis. (A) Cultured NG108-15 cells were exposed with thapsigargin (Tg) for 24 hours. The expression level of BiP was determined by Western blotting and quantified in histograms. (B) Cultured NG108-15 cells were exposed with tacrine (THA) for 24 hours. The expression level of BiP was determined by Western blotting and quantified. (C) Cultured NG108-15 cells and rat cortical neurons were exposed with or without THA at 100 μM for different numbers of hours. The expression level of cl-caspase 3 was determined by Western blot and quantified. (D) Cultured NG108-15 cells and rat cortical neurons were exposed with or without THA from 10 to 100 μM for 24 hours. The expression level of AChE was determined by Western blotting and quantified. (E) Cultured NG108-15 cells were exposed with or without Tg at different concentrations for 24 hours. The expression level of protein was calculated (left panel). AChE activity was determined by Ellman assay (right panel). (F) Cultured NG108-15 cells co-transfected with AChE and PRiMA were exposed with Tg (1 µM) for 24 hours. Cells were immunostained by the anti-AChE antibody with or without permeabilization of Triton X-100 (0.2%). In all cases, α-tubulin served as an internal control. Values are expressed as the fold to basal, percentage of control, or percentage of increase and are in means ± S.D., n = 4, each with triplicate samples. Statistical significance was analyzed by one-way repeated measures of ANOVA with subsequent application of Dunnett’s multiple comparisons test. *P < 0.5, **P < 0.01, ***P < 0.001 versus control.
Tacrine Induces Downstream Signaling of ER Stress
cAMP–cAMP response element-binding protein (CREB) signaling is known to be part of UPR during ER stress (Kikuchi et al., 2016). Here, pCRE-Luc, a report construct containing three copies of cAMP response element (CRE) and tagging upstream of a luciferase gene, was employed to analyze the regulation of cAMP signaling. In pCRE-Luc transfected NG108-15 cells, exposure of tacrine induced transcriptional activity of pCRE-Luc in a dose-dependent manner. The maximum induction of ∼15-fold was revealed at 20 μM tacrine (Fig. 4A, left panel). Besides, thapsigargin induced the activity of pCRE-Luc in a time-dependent manner (Fig. 4A, right panel). Meanwhile, exposure of tacrine, or thapsigargin, increased the CREB phosphorylation in a time-dependent manner (Fig. 4B, left panel). The response time was at least 24 hours because the induction of ER stress had to take time to accumulate the response (Fig. 4B, right panel). The activation of CREB suggested the turn-on of UPR during ER stress in culture.

Tacrine activates downstream signaling of ER stress. (A) Cultured NG108-15 cells were transiently transfected with pCRE-Luc and exposed with tacrine (THA) at different concentrations for 24 hours (left panel) or thapsigargin (Tg) at 1 µM for different numbers of hours (right panel). Luciferase assays were performed. (B) NG108-15 cells were exposed with THA at 100 µM and Tg at 1 µM for different numbers of hours. Total and phosphorylated CREB (p-CREB) (∼42 kDa) were detected by corresponding antibodies (left panel) and quantified in histograms (right panel). (C) Human wild-type ACHE promoter (pAChE-Luc) with CRE-binding site (CAC GTC A) and mutated promoter (pAChEΔCRE-Luc) with unfunctional CRE-binding site (CCC TTA A) are shown. These two cDNAs were transiently transfected in NG108-15 cells, which were then exposed with THA or Tg for 24 hours. Luciferase assays were performed. AP2: activating protein 2; MyoD: myoblast determination protein; NFκB: nuclear factor kappa B; SP1: specificity protein 1. Statistical significance was analyzed by one-way repeated measures of ANOVA with subsequent application of Dunnett’s multiple comparisons test, or two-way repeated measures of ANOVA with subsequent application of Bonferroni’s multiple comparisons test in (C). *P < 0.5, **P < 0.01, ***P < 0.001 versus control. Data are expressed as the fold to basal or percentage of increase and are in means ± S.D., n = 4, each with triplicate samples.
The binding of CRE element on mammalian ACHE gene promoter is critical in cAMP-mediated AChE expression in neuron, which contains only one CRE binding site at ∼2 kb upstream of the ATG start site (Schneider, 2000; Choi et al., 2008). The AChE promoter with or without mutation on the CRE-binding site, named pAChEΔCRE-Luc or pAChE-Luc, was transfected in NG108-15 cells to detect the effect of CRE on transcription of ACHE gene (Fig. 4C, top panel). In pAChE-Luc (intact human promoter) transfected NG108-15 cells, applied tacrine induced the promoter activity in a dose-dependent manner. The tacrine-induced promoter activity was markedly reduced in the mutated promoter, i.e., pAChEΔCRE-Luc, suggesting the role of this CRE site on ACHE gene (Fig. 4C, right panel). In parallel, treatment of thapsigargin induced the activity of pAChE-Luc in a dose-dependent manner, which was markedly reduced in a scenario of pAChEΔCRE-Luc–expressing cells (Fig. 4C, left panel). This CRE site of ACHE promoter could account for the regulation of AChE after ER stress.
Tacrine-Induced ER Stress Depends on Amount of AChE
To find out involvement of AChE in ER stress and apoptosis, cultured NG108-15 cells were transiently transfected with AChEWT catalytic subunit with PRiMA for 24 hours to overexpress the G4 AChE. The transfected pcDNA3 culture served as a negative control, an empty vector for AChE plasmid. This overexpression induced enzymatic activity of AChE at ∼4-fold compared with control culture (Supplemental Fig. 1E), serving as a confirmation of AChE overexpression. After the transfection at 24 hours, cultures were exposed with tacrine from 4 to 48 hours. The protein lysates of cultures were subjected to Western blot assay for BiP and cl-caspase 3 proteins. The exposure of tacrine in control cultures, or G4 AChE overexpressed cells, induced the expressions of BiP and cl-caspase 3 (Fig. 5A, upper panel). In AChE-overexpressed cultures, exposure of tacrine obviously enhanced the expressions of BiP and cl-caspase 3, i.e., the effective curve shifted to the left (Fig. 5A, lower panel), which suggested that overexpression of AChE could accelerate the tacrine-induced ER stress and apoptosis.

Amount of AChE regulates the tacrine-induced ER stress and apoptosis. (A) NG108-15 cells were transiently transfected with pcDNA3 or AChE catalytic subunit (AChET) with PRiMA for 24 hours before application of tacrine (THA; 100 µM) for different numbers of hours. The amounts of cl-caspase 3 and BiP were recognized by Western blot (upper panel) and quantified (lower panel). (B) A schematic diagram of human AChEWT and glycosylation mutant AChE3N→3Q constructs is shown. Human AChEWT contains three N-linked glycosylation sites located at Asn296, Asn381, and Asn495. These sites were site-directed mutated from asparagine to glutamine substitution. The cDNAs encoding AChEWT and glycosylation mutant AChE3N→3Q were subcloned in a pcDNA3 vector under a CMV promoter. HEK293T cells were transiently transfected with AChEWT and AChE3N→3Q with PRiMA for different numbers of hours. The amounts of cl-caspase 3 and BiP were recognized by Western blot (upper panel) and quantified (lower panel). (C) HEK293T cells were transfected with pcDNA3, AChEWT, and AChEMT with PRiMA. After application of THA (100 µM) for different numbers of hours, the cultures were collected for Western blot. (D) Quantification plots from (C) are shown. The expression level was calculated using α-tubulin as an internal control. Statistical significance was analyzed by one-way or two-way repeated measures of ANOVA with subsequent application of Tukey’s or Bonferroni’s multiple comparisons test. *P < 0.5, **P < 0.01, ***P < 0.001 versus control; ##P < 0.01 versus AChEWT + PRiMA group. Values are expressed as the fold to basal or percentage of increase and are in means ± S.D., n = 4, each with triplicate samples.
Three N-linked glycosylation sites (Asn296, Asn381, and Asn495) on ACHE gene were conserved in mammalian AChE, including human, mice, and rat (Velan et al., 1993). A robust decrease of AChE activity and protein stability was identified in the glycosylation-depleted human AChE (Chen et al., 2011). Besides, the depletion of AChE glycosylation led to the newly synthesized protein being stuck in the ER (Chen et al., 2011). Human AChE cDNA and its glycosylation mutant, all-site mutant AChE3N→3Q, were subcloned in a vector under a CMV promoter (Velan et al., 1993; Chen et al., 2011) (Fig. 5B, upper panel). The cDNAs encoding AChEWT, or AChE3N→3Q, with PRiMA cDNA was transfected in cultured HEK293T cells. After transfection for different numbers of hours, the expression of BiP and cl-caspase 3 proteins were measured. As expected, the transfection of AChE3N→3Q with PRiMA, forming unglycosylated G4 AChE, induced the expressions of BiP and cl-caspase 3 proteins after 24 hours, having the maximal induction at 2–3-fold (Fig. 5B). However, the overexpression of wild-type AChE showed no effects on BiP and cl-caspase 3 expression (Fig. 5B). Thus, the ER stress, as induced by glycosylated mutant of AChE in this case, was able to induce subsequently the cell death, similar to that of applied tacrine.
To demonstrate the effects of AChE activity in tacrine-induced ER stress and apoptosis, a mutant of AChE without enzymatic activity was produced. Residues Ser234, Glu365, and His478 of the catalytic triad in AChEWT contributing to AChE enzymatic activity were all mutated to alanine generating AChE (S234A, E365A, H478A), named AChEMT (Supplemental Fig. 1E). This mutated AChEMT construct expressed the AChE protein with no enzymatic activity (Supplemental Fig. 1E). HEK293T cells were employed here to avoid the disturbance of intrinsic AChE since very minimal AChE was found in this cell type (Chen et al., 2011). Cultured HEK293T cells were transiently transfected with pcDNA3, AChEWT, and AChEMT with PRiMA for 24 hours. The same protein size could be detected both in wild-type and mutated AChE protein (Liu et al., 2020). After the transfection at 24 hours, cultures were exposed with tacrine from 2 to 24 hours. The protein lysate of cultures was subjected to Western blot assay for BiP and cl-caspase 3 proteins. Exposure of tacrine in control cultures (pcDNA3-transfected) showed minimal effect on the expressions of BiP and cl-caspase 3 (Fig. 5C). In G4 AChE overexpressed cultures, application of tacrine enhanced the expressions of BiP and cl-caspase 3: this result was consistent with that in NG108-15 cells. However, tacrine also enhanced slightly the expressions of BiP and cl-caspase 3 in AChEMT (no enzymatic activity) overexpressed cultures (Fig. 5C). According to the quantification results, the tacrine-induced BiP and cl-caspase 3 in G4 AChEWT overexpressed cultures were more robust than that in G4 AChEMT overexpressed cultures after 8 hours, or more robust thereafter (Fig. 5D). The pcDNA3-transfected cultures showed no effect on tacrine exposure since the control HEK293T cells contained minimal amount of AChE (Fig. 5, C and D).
The discrepancy of responses to tacrine could be accounted for by loss of tacrine binding to the mutated enzyme, e.g., AChEMT versus AChEWT. By molecular docking simulation, the calculated binding sites of tacrine with wild-type and activity-deleted mutant AChE were analyzed. The binding modes of tacrine with wild-type AChE (Protein Data Bank code, 6O4X; Supplemental Data File 1) and AChEMT (homology modeling with template 6O4X; Supplemental Data File 2) were different. In wild-type AChE, tacrine bound to Trp117 (the acyl pocket active site), Gly152 (oxyanion hole active site), His478 (muted catalytic site), and Asp105 (peripheral anion site) by π-π, π-H, π-H, and H-donor interactions with proposed binding affinities of −8.35, −3.465, −2.93, and −3.8775 kJ/mol, respectively. In AChEMT, tacrine bound to Trp117, Gly152, Ala478 (muted catalytic site), and Asp105 by similar interactions with proposed binding affinities of −4.73, −1.61, −2.48, and −4.005 kJ/mol, respectively (Supplemental Fig. 2). By this docking measurement, we speculated that the binding mode of tacrine with AChEWT should be more conductive in the inhibitory effect than that of AChEMT. Besides, the residues of His478, Trp117, and Gly152 are known to contribute AChE activity. Thus, the proposed binding of tacrine to activity-deleted mutant AChE was weak, which accounted for the negative effect of tacrine in inducing ER stress of AChEMT expressing cells here.
Tacrine Increases Cell Death and Loss of MMP
An extension of ER stress causes cell death. Here, flow cytometry was used to reveal the tacrine-induced cell death and loss of MMP in cultured NG108-15 cells. An annexin V/propidium iodide (PI) apoptosis detection kit was employed to detect cell apoptosis by flow cytometry. Annexin V–conjugated FITC and PI were used to label phosphatidylserine residues on the surface of cells, as well as cellular DNA. A distinctive characteristic of early apoptotic cells is the appearance of phosphatidylserine residues on membrane surface. PI, a non–membrane-permeable dye, could distinguish healthy, apoptotic, and necrotic cells based on the membrane integrity. Hence, the healthy cells (annexin V negative, PI negative), the early apoptotic cells (annexin V positive, PI negative), and the late apoptotic cells (annexin V positive, PI positive) could be seen in the bottom left quadrant, the bottom right quadrant, and the top right quadrant, respectively (Fig. 6A). Certain percentage of cells underwent apoptosis after tacrine exposure in a time-dependent manner (Fig. 6A). H2O2 served as a positive control here (Fig. 6A). In addition, the detection of MMP was performed using JC-1 dye to assess the depolarization of mitochondrial membrane. The MMP loss in culture was increased with the exposure of tacrine in a time-dependent manner (Fig. 6B). Therefore, the alteration of MMP level, induced by tacrine, could result in an increase of cell death.
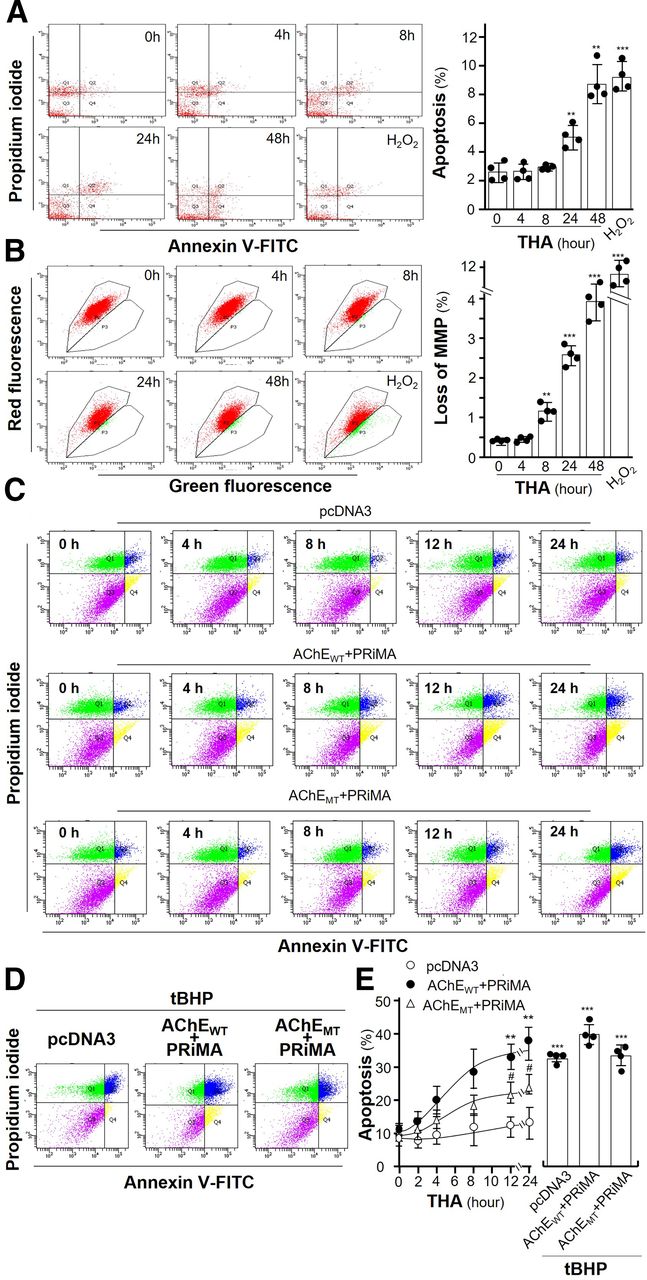
Amount of AChE regulates the tacrine-induced apoptosis. (A) Cultured NG108-15 cells were exposed with or without tacrine (THA; 100 µM) for different numbers of hours. The subsequent disassembled cells were dual labeled by an annexin V/PI apoptosis detection kit and detected by flow cytometry (left panel). Calibration of apoptotic rates is shown (right panel). (B) Cultured NG108-15 cells were treated as in (A) and measured by flow cytometry (left panel). The percentage of MMP loss was quantified (right panel). (C) Cultured HEK293T cells were transiently transfected with pcDNA3, AChEWT, and AChEMT with PRiMA for 24 hours. After application of THA at 100 µM from 2 to 24 hours, apoptotic cells were measured by flow cytometry. (D) HEK293T cells were treated as in (C). After application of tBHP at 100 µM for 6 hours, apoptotic cells were measured by flow cytometry. (E) Calibration of apoptotic rates. Values are in percentage of total cell number or control value and are in means ± S.D., n = 4, each with triplicate samples. Statistical significance was analyzed by one-way or two-way repeated measures of ANOVA with subsequent application of Dunnett’s or Tukey’s multiple comparisons test. **P < 0.01, ***P < 0.001 versus control; #P < 0.05 versus AChEWT + PRiMA group.
To strengthen the observation of cell death, cultured HEK293T cells were transiently transfected with pcDNA3, AChEWT, and AChEMT with PRiMA for 24 hours, followed by exposure of tacrine from 2 to 24 hours, to reveal the cell death under an overexpression system of G4 AChE. Annexin V–FITC and PI-labeled HEK293T cells, transfected with different cDNAs, were subjected to flow cytometry analysis. In pcDNA3-transfected cultures (control), tacrine did not affect the apoptotic cells because HEK293T cells expressed very little AChE (Fig. 6C, upper panel). In G4 AChE expressed cultures, exposure of tacrine starting from 8 hours obviously induced the number of apoptotic cells (Fig. 6C, middle panel). In G4 AChEMT expressed culture, tacrine showed much reduced degree of cell death (Fig. 6C, lower panel). According to the quantification results, tacrine-induced apoptotic cells in G4 AChEWT expressed cultures better than that in G4 AChEMT expressed cultures, i.e., ∼40% versus ∼20% at 24 hours of exposure (Fig. 6, D and E). The findings were in line with the results from apoptotic markers, cl-caspase 3 and BiP. Here, tert-butylhydroperoxide (tBHP), an organic peroxide, served as a positive control to induce statistically significant cell death via apoptosis (Zhao et al., 2005). In all cultures, tBHP induced apoptotic cells by over 35% (Fig. 6, D and E).
The apoptotic cells, under different DNA transfected HEK293T cells, were further illustrated by Hoechst nucleic acid staining. The results of Hoechst staining were consistent with the outcome of flow cytometry. Apoptotic cells were stained blue. In control cultures, tacrine did not affect the percentage of apoptotic cells (Fig. 7A). In G4 AChEWT overexpressed cultures, the exposure of tacrine for 24 hours induced apoptotic cells (Fig. 7A). Tacrine slightly induced apoptotic cells in G4 AChEMT overexpressed culture (Fig. 7A). tBHP served as a positive control here (Fig. 7B). The quantification results showed the tacrine-induced apoptotic cells in AChEWT overexpressed cultures were more robust than those in AChEMT overexpressed cultures, i.e., 70% (AChEWT) versus 25% (AChEMT) (Fig. 7C). In addition, the trafficking of G4 AChE was revealed in HEK293T cells transfected with different DNA constructs. As expected, the membrane transport of G4 AChEWT was highly sensitive to tacrine exposure; however, the trafficking of G4 AChEMT to the membrane was not affected by the present of tacrine (Fig. 7D). This result indicates the specificity of misfolded AChE-tacrine complex in directing ER stress.

The tacrine-induced apoptosis is recognized by Hoechst staining. (A) Cultured HEK293T cells were transiently transfected with pcDNA3, AChEWT, and AChEMT with PRiMA for 24 hours. After exposure of tacrine (THA; 100 µM) for 24 hours, cells were stained by Hoechst 33342 (5 μg/ml). Images were taken by confocal microscope. (B) Cultured HEK293T cells were treated as in (A). After application of tBHP (100 µM) for 6 hours, cells were stained by Hoechst dye. (C) Condensed nuclei were calculated by counting at least 500 cells of three randomly chosen fields. Values are in percentage of total cell number and are in means ± S.D., n = 4, each with triplicate samples. Statistical significance was analyzed by two-way repeated measures of ANOVA with subsequent application of Tukey’s multiple comparisons test. ***P < 0.001 versus pcDNA3 group; ###P < 0.001 versus AChEWT + PRiMA group. (D) Cultured HEK293T cells were treated as in (A). After treatment with or without THA (100 µM) for 48 hours, cells were immunostained by anti-AChE antibody with or without permeabilization of Triton X-100 (0.2%).
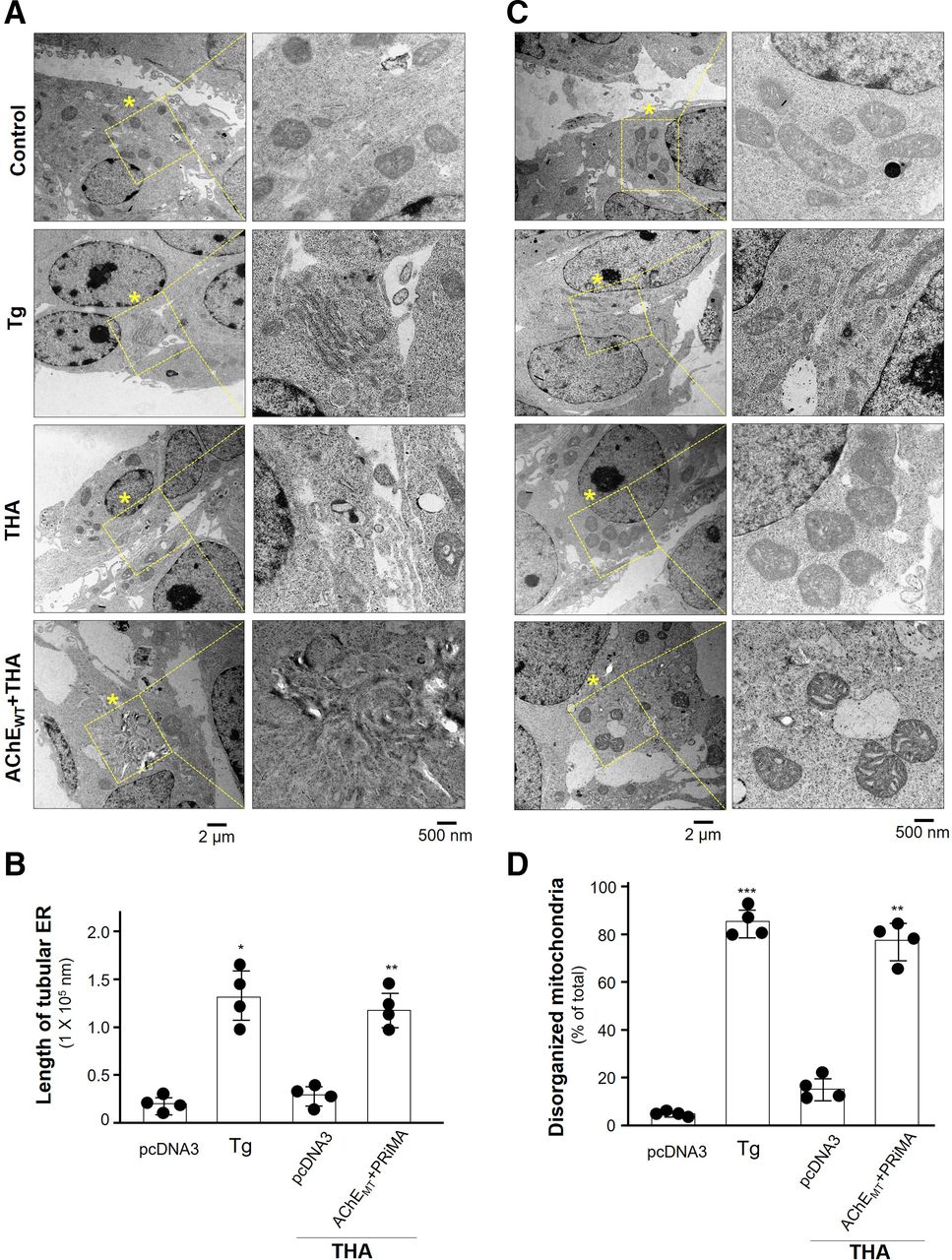
The tacrine-induced ER stress was further illustrated by electron microscope. Normal morphology of ER in cultured HEK293T cells were identified in control and in tacrine-exposed cultures (Fig. 8, A and B). The positive control thapsigargin caused the disrupted ER structure. As expected, this disruption was markedly revealed in the tacrine-exposed cultures having overexpression of G4 AChE. In parallel, the disruption of mitochondrial morphology was identified in tacrine-exposed G4 AChE overexpressed cells, as well as in the case of applied thapsigargin (Fig. 8, C and D).

The ultrastructure of cells showing tacrine-induced ER stress. Cultured HEK293T cells were transiently transfected with AChEWT and PRiMA, or only vector alone as control. After application of PBS (control), thapsigargin (Tg; 1 µM) or tacrine (THA; 100 µM) for 24 hours, the cell ultrastructure was taken by transition electron microscope. (A) Asterisk indicates the magnification structures of ER. One representative result is shown. (B) Quantification of the length of tubular ER (in nanometers) per cell. (C) Drug exposure was as in (A). Asterisk indicates the magnification structures of mitochondria. One representative result is shown. (D) The percentage of disorganized mitochondria in total number of mitochondria was calculated. Twenty cells were counted in each independent experiment, n = 4. Values are in means ± S.D. Statistical significance was analyzed by one-way repeated measures of ANOVA with subsequent application of Dunnett’s multiple comparisons test. *P < 0.05, **P < 0.01, ***P < 0.001.
Mitochondria play critical roles in the process of ER stress-induced cell apoptosis (Lemasters, 2005), and MMP is an important early determinant of mitochondrial apoptotic signaling. The effect of tacrine on change of MMP (ΔΨm) was detected by JC-1 staining. At high MMP cells. JC-1 forms aggregation and yields red fluorescence. At low MMP cells, JC-1 exists as a monomer and exhibits intensive green fluorescence (Martinez-Pastor et al., 2004). Cultured HEK293T cells were transiently transfected with various DNA constructs for 24 hours, followed by exposure of tacrine for another 24 hours. In pcDNA3-transfected culture, tacrine did not affect the change of MMP, i.e., insufficient AChE being expressed in the cells (Fig. 9, A and C). In G4 AChEWT overexpressed cultures, exposure of tacrine for 24 hours induced the loss of MMP, i.e., reduced red/green signal (Fig. 9A, and C). Tacrine induced the minor loss of MMP in G4 AChEMT overexpressed culture (Fig. 9, A and C). Carbonyl cyanide m-chlorophenyl hydrazone served as a positive control here, inducing a low red/green signal in all scenarios (Fig. 9B). The quantification results showed that the applied tacrine induced the loss of MMP in AChEWT overexpressed culture, and this induction was more robust than that in AChEMT overexpressed culture (Fig. 9C).
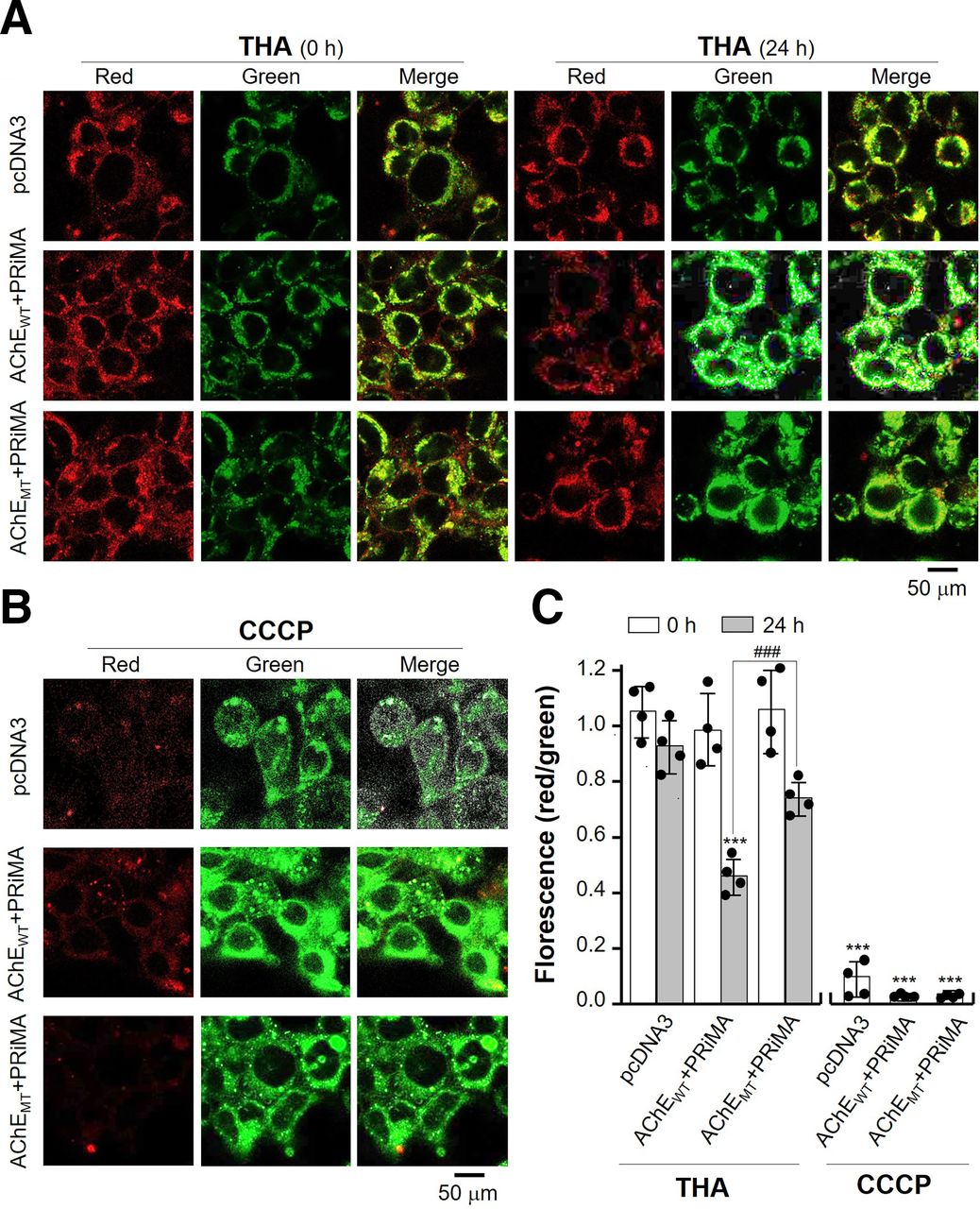
Amount of AChE regulates the tacrine-induced loss of MMP. (A) Cultured HEK293T cells were transiently transfected with pcDNA3, AChEWT, and AChEMT with PRiMA for 24 hours. After exposure of tacrine (THA; 100 µM) for 24 hours, cells were stained by JC-1. Images were taken by confocal microscope. (B) Cultured HEK293T cells were treated as in (A). After application of carbonyl cyanide m-chlorophenyl hydrazone (CCCP; 10 µM; a positive control) for 1 hour, cells were stained by JC-1. (C) The ratio of red/green fluorescence intensity was analyzed by ImageJ software. Values are ratio of red/green florescence and are in means ± S.D., n = 4, each with triplicate samples. Statistical significance was analyzed by two-way repeated measures of ANOVA with subsequent application of Tukey’s multiple comparisons test. ***P < 0.001 versus pcDNA3 group; ###P < 0.001 versus AChEWT + PRiMA group.
AChEIs Inhibit Trafficking of G4 AChE
Besides tacrine, the effects of other AChEIs on trafficking of G4 AChE were analyzed by immunofluorescence staining in AChE overexpressed NG108-15 cells. The staining of AChE could not be detected on the plasma membrane of cells with exposures of rivastigmine, bis(3)-cognitin, bis(7)-cognitin, daurisoline, and dauricine, i.e., these AChEIs causing ER stress (Fig. 10A). In contrast, the staining of AChE was detected on the plasma membrane of cells with exposures of donepezil, galantamine, and huperzine A (Fig. 10A). After application of these AChEIs, AChE possessed same binding affinity with immobilized Con A and SNA, suggesting no change in enzyme glycosylation after binding with AChEIs (Supplemental Fig. 1F). The results are consistent with side effects of AChEIs in clinic being highly variable from drug to drug (Schneider, 2000).

Different AChEIs affect the trafficking of AChE. (A) Cultured NG108-15 cells co-transfected with AChE catalytic subunit (AChET) and PRiMA were exposed with rivastigmine (RI; 10 µM), huperzine A (HupA; 10 µM), galantamine (GAL; 10 µM), donepezil (DPZ; 20 µM), bis(3)-cognitin (B3C; 10 µM), bis(7)-cognitin (B7C; 2 µM), daurisoline (DAS; 10 µM), and dauricine (DAC; 10 µM) for 24 hours. Cells were immunostained with anti-AChE antibody with or without permeabilization of Triton X-100 (0.2%). (B) NG108-15 cells were treated with AChEIs as in (A) plus jatrorrhizine (JAT; 10 µM), lycobetaine (LBT; 10 µM), and thapsigargin (Tg; 1 µM) for 48 hours. Quantitative PCR of BiP and CHOP were determined. Values were normalized with GAPDH (upper panel). The cultures were collected for Western blotting of BiP (lower panel). The expression level of proteins was calculated using α-tubulin as an internal control. (C) HEK293T cells were transiently transfected with pcDNA3, AChEWT with PRiMA for 24 hours. After exposure of DPZ (20 µM) for different numbers of hours, the cultures were collected for Western blotting. (D) Cultured macrophages (RAW 264.7 cells) were exposed with AChEIs, as in (A), and Tg (1 µM) for 48 hours. Values of quantitative PCR were normalized with GAPDH. (E) Cultured RAW 264.7 cells (upper panel), or cultured primary osteoblasts (lower panel), co-transfected with AChE and PRiMA, were exposed with or without tacrine (THA; 100 µM), DPZ (20 µM), RI (10 µM), and Tg (1 µM) for 48 hours. Cells were immunostained by anti-AChE antibody with or without permeabilization of Triton X-100 (0.2%). Data are expressed as the fold of basal value, mean ± S.D., n = 4, each with triplicate samples. Statistical significance was analyzed by paired Student’s t test. *P < 0.05; ***P < 0.001 versus control.
In addition, the mRNA levels of BiP and CHOP, also an indicative marker for ER stress, were investigated after exposures of different AChEIs. Tacrine, rivastigmine, bis(3)-cognitin, daurisoline, and dauricine obviously induced the expressions of BiP and CHOP mRNA (Fig. 10B, upper panel). Donepezil, galantamine, huperzine A, and jatrorrhizine showed no effect on BiP and CHOP mRNA (Fig. 10B, upper panel). Interestingly, lycobetaine increased CHOP mRNA and showed no effects on BiP mRNA. Besides, tacrine, rivastigmine, bis(3)-cognitin, daurisoline, dauricine, and lycobetaine induced the expression of BiP protein (Fig. 10B, lower panel). In contrast, donepezil, galantamine, huperzine A, and jatrorrhizine showed no, or minor, effect on BiP protein expression. The role of donepezil was further demonstrated in G4 AChE overexpressed NG108-15 cells. Despite the enzyme overexpression being expressed in cultures, the cells did not response to challenge of donepezil (Fig. 10C). Thus, there are two classes of AChEIs: ER stress–inducing and non–ER stress–inducing.
The effects of AChEIs on the trafficking of AChE in other cell types expressing AChE were demonstrated here. In RAW 264.7 cells, a macrophage cell line expressing G4 AChE (Liu et al., 2020), tacrine, rivastigmine, bis(3)-cognitin, daurisoline, and dauricine obviously induced the mRNA level of BiP and CHOP (Fig. 10D). Donepezil, galantamine, huperzine A, lycobetaine, and jatrorrhizine showed no effect on changes of BiP and CHOP mRNA (Fig. 10D). In addition, the immunofluorescence staining of macrophage, exposures with tacrine and thapsigargin, caused a minimal amount of AChE in cell surface; however, donepezil showed no effect on the amount of G4 AChE in cell surface (Fig. 10E, upper panel). Similar situation in cultured osteoblasts, tacrine, rivastigmine, and thapsigargin caused a minimal amount of AChE in cell surface (Fig. 10E, lower panel). To reveal the specificity of tacrine on AChE-expressing cells, other membrane protein, e.g., α7 nicotinic acetylcholine receptor (nAChR), was overexpressed in cultures to investigate the effect of tacrine in ER stress. In α7 nAChR overexpressed cultures, the staining of α7 nAChR was fully identified on membrane of cells with the exposure of tacrine (Supplemental Fig. 3). Thus, other membrane proteins might not be involved in tacrine-induced ER stress.
Discussion
AChEIs are the most established therapeutic agents for AD treatment. Other strategies are still at an early stage of clinical development and not in the market yet (Schneider, 2000). AChEIs are likely to accompany us for a long time. Therefore, the possible risk profiles of AChEIs should be considered, and the safest drug can be discovered in targeting the inhibition of AChE activity. Since tacrine was discontinued in 2013 due to hepatotoxicity, the mechanism of hepatotoxicity remains to be fully established. Early alterations of membrane fluidity are likely to play an important role in developing tacrine liver toxicity (Galisteo et al., 2000). Recently, the liver-gut microbiota axis has been reported to modulate the hepatotoxicity of tacrine (Yip et al., 2018). The study of tacrine has never stopped. For the first time, we have identified tacrine, or other AChEIs, in cultured neurons, or cells expressing AChE, caused an accumulation of misfolded AChE being retained in ER: these misfolded proteins were not able transport to Golgi/plasma membrane. The stuck AChE proteins thereafter induced ER stress, as well as the downstream signal cascade of UPR. Once the induced stress is overwhelming, the cooperation of the ER with mitochondria increases the loss of MMP. The cells lost the homeostasis leading to apoptosis. Besides neurons, the inhibitor-induced ER stress could be identified in other AChE expressing cells, e.g., macrophages and osteoblasts.
ER stress in neuronal cells is regarded as a critical aspect in pathogeny of AD (Brewer and Diehl, 2000). The accumulation of misfolded proteins is a fundamental mechanism underlying the induction of ER stress, which subsequently induces neuronal cell death, as well as the cause of neurodegenerative diseases (Kozutsumi et al., 1988). Here, two classes of AChEIs are identified: those inducing ER stress and those not inducing ER stress. The exact distinction between these two classes of AChEIs is not resolved. We are hypothesizing that the distinct binding of AChEI to the enzyme could be a means to account for this discrepancy, i.e., the binding of certain types of AChEIs alters the proper folding of the enzyme during synthesis. For an example, the binding sites of tacrine and rivastigmine, as determined by molecular docking, share the common binding pockets, whereas these binding pockets are in distinction to donepezil and huperzine A. Indeed, the tacrine binding site of human AChE is known to be quite different from that of Torpedo AChE and varies among different AChEIs (Cheung et al., 2012). The reasons behind this discrepancy should be further investigated by comprehensively analyzing the crystal structure of AChE.
The use of AChEIs during AD treatment could be a problem in inducing possible ER stress in neurons, as shown in this study. The are two possible parameters affecting the induction of ER stress: 1) the dosage and time of AChEI treatment and 2) the level of AChE in cells, e.g., neuron at certain brain region. Thus, the AChEI-induced ER stress is believed to be more robust in cholinergic neurons expressing high abundant of AChE. From the clinical reports, a high dose of AChEI consistently has better efficacy than a low dose. This critical paradox, however, is the meaning of greater adverse events (Ali et al., 2015). A low dose of AChEIs has been proven not sufficient to be efficacious for AD treatment (Schneider, 2000). Hence, a higher dose of AChEIs is being considered in treatment. Because action mechanisms of different AChEIs are rather similar, the efficacy outcomes of AChEIs for AD treatment are usually consistent. However, the drug-induced side effects are highly variable from drug to drug, and thus the pharmacological differences of AChEIs must be considered. For example, an elevated level of transaminase was detected in patients with exposure to tacrine (Ali et al., 2015). On the other hand, an increased incidence of anorexia was more likely dose-related with patients treated with donepezil (Schneider, 2000). In protein biosynthesis, the chaperone protein copes with the problem of aggregating protein intermediates whose role is to prevent aggregation and to assist folding. “Chemical or pharmacological chaperons” describes a class of small molecules or drugs that function to alter the folding of protein. These molecules reduce the inherent conformational flexibility of the polypeptide chain via stabilizing a protein in a specific region. However, our results imply the AChEIs function in an opposite way with pharmacological chaperon.
The inclusion of chemical/pharmacological chaperones is generally considered to increase the yield of protein production both in vivo and in vitro (Rajan et al., 2011). Therefore, an emerging strategy of using a reversible enzyme inhibitor as pharmacological chaperone in stabilizing the folded form of a protein in ER is employed to avoid possible protein aggregation (Yu et al., 2007). In the folding of Torpedo AChE, the reversible AChEI and chemical chaperon were able to stabilize the purified enzyme during thermal denaturation, suggesting that enzyme folding could be affected by chaperons even in a test tube (Weiner et al., 2009). In cholinergic molecules, Lester’s group has shown that nicotine could function as a pharmacological chaperone to accelerate ER export of α4β2 nAChR, suppressing ER stress (Srinivasan et al., 2012). In electrophysical measurement, galantamine at low concentration acted as a positive allosteric modulator of assembly of α4β2 nAChRs (Samochocki et al., 2003; Hillmer et al., 2013). These lines of evidence suggest that AChEIs may serve as an allosteric potentiating ligand in affecting the ER stress, as mediated by nAChR.
The current pharmaceutical treatments for AD have either no or very minimal disease modulating effects. The role of immune system in the process of AD development has been proposed (Weiner and Frenkel, 2006; Wisniewski and Goñi, 2015). As the main cause of AD, amyloid-β (Aβ) deposition promotes pathologic innate immune responses, such as astrocyte proliferation and microglial cell activation. The immunotherapies of AD by administration of Aβ-specific antibodies or immunization of Aβ could slow down the memory-robbing progress in mouse models (Weiner and Frenkel, 2006). The US Food and Drug Administration has recently approved the first new AD drug since 2003, which is a Aβ-specific monoclonal antibody. The new drug reduces Aβ levels in the brain and slows the progress of AD. Although doubts about the therapy’s effectiveness and risk are arising due to mixed results in clinical trials, many immune-based therapies of AD are currently either under development or in clinical trials. Moreover, the promising immune-based treatments of AD could be used in combination with AChEIs.
As reported here, tacrine transcriptionally regulates the expression of AChE, which affects the trafficking of G4 AChE from the ER to the Golgi/PM. Eventually, the tacrine-bound misfolded AChE causes ER stress and cell death of neurons. AD is closely associated with protein misfolding, aggregation, and increased ER stress in neurons. The potential threats of tacrine on cultured neurons have been identified in this study, which suggests possible deterioration of AChEI for AD. Although the use of tacrine has been abandoned, rivastigmine, a current AD drug, has been found to inhibit the trafficking of G4 AChE, similar to tacrine. The potential threats of rivastigmine as a treatment of AD therefore must be considered (Nordberg and Svensson, 1998). Tacrine is concentrated in patients’ brains because of the relatively greater lipid solubility, and rivastigmine can easily cross the blood-brain barrier (Pohanka, 2014). Thus, the common point of these two AChEIs suggests a structural cause for blockage of AChE trafficking. Besides, the AChEI-induced ER stress is not restricted to neurons; this occurs in other cell types expressing AChE. Although neurons have the highest expression of AChE, the distribution of this enzyme is found in a broad range of cell types, e.g., muscle, blood cells, bone cells, and skin cells (Freitas Leal et al., 2017; Xu et al., 2018; Liu et al., 2020). The safety guidance for drug design and discovery of AChEIs for different types of medical problems relating to cholinergic deficiency should be considered.
Acknowledgments
We thank Han Yifan for kindly providing bis(7)-cognitin and bis(3)-cognitin.
Authorship Contributions
Participated in research design: Liu, Mak, Tsim.
Conducted experiments: Liu, Mak, Kong, Xia, Kwan, Xu.
Performed data analysis: Liu, Kong.
Wrote or contributed to manuscript writing: Liu, Tsim.
Footnotes
- Received February 24, 2021.
- Accepted August 23, 2021.
This work was supported by the Health and Medical Research Fund, Food and Health Bureau of Hong Kong [Grants HMRF18SC06, 06173886)]; Natural Science Foundation of China [Grant 32001811]; Guangdong Basic and Applied Basic Research Foundation [Grant 2019A1515110603]; Shenzhen Science and Technology Innovation Committee [Grants JCYJ20170413173747440, ZDSYS201707281432317, JCYJ20180306174903174]; China Post-doctoral Science Foundation [Grant 2019M653087]; Hong Kong RGC Theme-based Research Scheme [Grant T13-605/18-W]; and Hong Kong Innovation Technology Fund [Grants UIM/340, UIM/385, ITS/500/18FP, TCPD/17-9, TUYF19SC02].
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- Aβ
- amyloid-β
- AChE
- acetylcholinesterase
- AChEI
- AChE inhibitor
- AChEMT
- mutant AChE
- AChEWT
- wild-type AChE
- AD
- Alzheimer’s disease
- ALP
- alkaline phosphatase
- ATCh
- acetylthiocholine iodide
- BiP
- immunoglobulin heavy chain binding protein
- CHOP
- CAAT/enhancer binding protein homologous protein
- cl-caspase 3
- cl-caspase 3
- CMV
- cytomegalovirus
- Con A
- concanavalin A
- CRE
- cAMP response element
- CREB
- cAMP response element-binding protein
- DAPI
- 4′,6-diamidino-2-phenylindole
- ER
- endoplasmic reticulum
- FITC
- fluorescein isothiocyanate
- GAPDH
- glyceraldehyde 3-phosphate dehydrogenase
- MMP
- mitochondrial membrane potential
- nAChR
- nicotinic acetylcholine receptor
- PCR
- polymerase chain reaction
- PI
- propidium iodide
- PM
- plasma membrane
- PRiMA
- proline-rich membrane anchor
- SNA
- Sambucus nigra
- tBHP
- tert-butylhydroperoxide
- UPR
- unfolded protein response
- Copyright © 2021 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}