


Abstract
The P2X4 receptor is a ligand-gated ion channel activated by extracellular ATP. P2X4 activity is associated with neuropathic pain, vasodilation, and pulmonary secretion and is therefore of therapeutic interest. The structure-activity relationship of P2X4 antagonists is poorly understood. Here we elucidate the structure-activity of 5-(3-bromophenyl)-1,3-dihydro-2H-benzofuro[3,2-e]-1,4-diazepin-2-one (5-BDBD) at human P2X4 by combining pharmacology, electrophysiology, molecular modeling, and medicinal chemistry. 5-BDBD antagonized P2X4 in a noncompetitive manner but lacked effect at human P2X2. Molecular modeling and site-directed mutagenesis suggested an allosteric binding site for 5-BDBD located between two subunits in the body region of P2X4, with M109, F178, Y300, and I312 on one subunit and R301 on the neighboring subunit as key residues involved in antagonist binding. The bromine group of 5-BDBD was redundant for the antagonist activity of 5-BDBD, although an interaction between the carbonyl group of 5-BDBD and R301 in P2X4 was associated with 5-BDBD activity. 5-BDBD could inhibit the closed channel but poorly inhibited the channel in the open/desensitizing state. We hypothesize that this is due to constriction of the allosteric site after transition from closed to open channel state. We propose that M109, F178, Y300, R301, and I312 are key residues for 5-BDBD binding; provide a structural explanation of how they contribute to 5-BDBD antagonism; and highlight that the limited action of 5-BDBD on open versus closed channels is due to a conformational change in the allosteric site.
SIGNIFICANCE STATEMENT Activity of P2X4 receptor is associated with neuropathic pain, inflammation, and vasodilatation. Molecular information regarding small-molecule interaction with P2X4 is very limited. Here, this study provides a structural explanation for the action of the small-molecule antagonist 5-BDBD at the human P2X4 receptor.
Introduction
P2X receptors (P2X1–7) mediate the fast biologic effects of extracellular ATP. They are a family of ligand-gated cation channels whose activation results in elevated cytoplasmic Ca2+ and membrane depolarization. P2X receptors are attractive drug targets, and recently, clinically efficacious antagonists have been developed for some subtypes (Richards et al., 2019; Smith et al., 2020). P2X4 is a moderately desensitizing ion channel (Fountain and North, 2006) that is activated by nanomolar extracellular ATP. Its activity has been implicated in flow-dependent vasodilation and remodeling of arteries (Yamamoto et al., 2006), cardiac contractility (Yang et al., 2014), production of inflammatory mediators (Layhadi et al., 2018), calcium signaling in human leukocytes (Layhadi and Fountain, 2019), surfactant secretion by type II alveolar epithelial cells (Miklavc et al., 2011), and neuropathic pain (Tsuda et al., 2003). As such, P2X4 is a promising therapeutic target with some recent advancement in selective antibody antagonists (Williams et al., 2019) and small-molecule antagonists (Stokes et al., 2017; Illes et al., 2021), including PSB-12062 (Hernandez-Olmos et al., 2012), BX-430 (Ase et al., 2015), NP-1815-PX (Matsumura et al., 2016), and 5-(3-bromophenyl)-1,3-dihydro-2H-benzofuro[3,2-e]-1,4-diazepin-2-one (5-BDBD) (Fischer et al., 2004). Despite an increase in small molecules that antagonize P2X4, there remains limited information regarding the structure-activity relationship between antagonistic ligands and the P2X4 receptor. Information regarding the inhibitory mechanism of the benzodiazepine derivative 5-BDBD is contrasting and contradictory. Studies have suggested that 5-BDBD competes with ATP at the orthosteric binding site (Balazs et al., 2013; Coddou et al., 2019). In such studies, 5-BDBD causes a rightward shift in the ATP concentration-response curve with no reduction in the response maxima and with a potency similar to the competitive antagonist TNP-ATP et al., 2013). In contradiction to these studies, Abdelrahman et al. (2017) demonstrated that TNP-ATP is capable of displacing [35S] ATPγS from P2X4, but 5-BDBD could not, suggesting 5-BDBD has a negative allosteric rather than competitive mode of action. To this end, we have combined pharmacology, electrophysiology, in silico predictions, molecular dynamics, and medicinal chemistry to understand the structure-activity relationship of 5-BDBD action at the human P2X4 receptor.
Materials and Methods
Cells and Cell Culture
The parental 1321N1 human astrocytoma cell line (RRID:CVCL_0110) and 1321N1 cells stably expressing either human P2X4 or human P2X2 receptors were maintained at 37°C and 5% CO2 in DMEM (Lonza) supplemented with l-glutamine (4.1 mM), FBS (10% v/v; HyClone), penicillin/streptomycin (50 U/ml and 50 µg/ml, respectively; Gibco), and glucose (4.5 g/l). Cells were passaged twice weekly upon reaching 70%–80% confluence and discarded upon reaching the 20th passage.
Drugs and Salts
NaCl, KCl, d-glucose, HEPES, MgCl2, CaCl2, and ATP were purchased from Sigma. 5-BDBD and BX-430 were purchased from Tocris Biosciences. 5-BDBD analogs with the following modifications were purchased from Vitas-M laboratories (R2-H, R3-Br; R2-H; R2-H, R3-CH3; R2-H, R3-F; R1-Br, R2-H; R2-H, R1-Br; R2-H, R3-Cl; R1-Cl, R2-H).
Site-Directed Mutagenesis and Transient
Transfection Human P2X4 cloned in pcDNA3.1 was mutated by polymerase chain reaction using the Q5 Site-Directed mutagenesis kit (New England Biolabs), and mutagenic primers were designed using NEBaseChanger software (New England Biolabs). Cycling conditions for mutagenesis were as follows: Initial denaturation occurred at 98°C for 30 seconds, and this was followed by 25 cycles of 98°C for 10 seconds, 50–60°C for 20 seconds, and 72°C for 5 minutes. A final extension was performed at 72°C for 2 minutes. Mutations were confirmed by sequencing, and plasmid DNA was prepared using an ENZA Plasmid kit (Omega Bio-tek). For transient transfection of parental 1321N1 cells with pcDNA3.1 containing wild-type or mutated human P2X4 receptor, 1.25 × 104 cells/well were seeded in 96-well plates and cultured overnight at 37°C and 5% CO2. Cells were transfected with a mixture of Lipofectamine 2000 (0.5 μl/well) and 300 ng/well plasmid DNA in Opti-MEM. Transiently transfected cells were used for experiments 72 hours after transfection.
Production of Lentiviral Particles and Stable Transfection
In brief, 9 × 106 human embryonic kidney (HEK) 293/17 cells were incubated overnight in 150-mm culture dishes at 37°C and 5% CO2. HEK293/17 were cotransfected with three plasmids; a lentiviral vector coding the viral genome with the transgene, psPAX2 containing a CMV enhancer, chicken beta-actin promoter, rabbit beta-globin acceptor site (CAG) promoter, and pMD2.G, which encodes the vesicular stomatitis virus envelope glycoprotein. The plasmids (24 µg DNA) and Lipofectamine 2000 were incubated together at room temperature for 20 minutes in Opti-MEM. The medium was replaced in the culture dishes with DMEM containing 1% (v/v) FBS, and the plasmid mix was added in a drop-wise manner. Between days 2 and 4, the media were collected daily, passed through a 0.45-μm polyethersulfone filter (Whatman), and stored at 4°C. The virus was concentrated with Lenti-X Concentrator (Clontech Laboratories) at 4°C for 72 hours and then centrifuged at 1500 g for 45 minutes at 4°C. The supernatant was aspirated, pellet was resuspended in DMEM, and suspension was stored at −80°C. Viral titers were quantified using a QuickTiter Lentivirus Titer Kit (Cell Biolabs Inc) according to manufacturer’s instructions. Absorbance was measured at 450 nm using a Flexstation 3 plate reader (Molecular Devices). To stably transfect 1321N1 astrocytoma cells, 2 × 105 cells were added to a 15-ml Falcon tube containing DMEM, polybrene (8 μg/ml; Sigma), and the viral media described above prior to centrifugation at 1400 g for 1 hour at 37°C. Cells exhibiting mCherry fluorescence were sorted using a BD FACSAria III Sorter (BD Biosciences) with excitation at 587 nm and emission at 610 nm.
Intracellular Ca2± Measurements
1321N1 cells stably expressing human P2X2 or human P2X4 were seeded at 2.5 × 103 cells/well in poly-d-lysine–coated 96-well plates 24 hours prior to loading with Fura-2 AM calcium indicator dye (Abcam). 1321N1 cells transiently transfected with wild-type and mutant P2X4 receptors were loaded with Fura-2 AM 72 hours after transfection. Loading with Fura-2 AM was achieved by incubating cells for 1 hour at 37°C in loading solution containing 130 mM NaCl, 5 mM KCl, 8 mM d-glucose, 10 mM HEPES, 1.2 mM MgCl2, 1.5 mM CaCl2, 2 μM Fura-2 AM, and 0.01% (w/v) pluronic-F127, pH 7.4. After loading, the buffer was replaced with salt-buffered saline (loading solution without Fura-2 AM and pluronic-F127). Cells were warmed for 10 minutes prior to calcium measurements. Fura-2 was measured at excitation wavelengths 340 nm and 380 nm with emission wavelength 520 nm using a Flexstation 3 plate reader. Sampling interval was 3.5 seconds with three reads/well. Fura-2 ratio was calculated using Softmax Pro v5.4, and responses were quantified using peak measurements.
Electrophysiology
Cells were resuspended at 1 × 106 cells/ml in a solution containing 140 mM NaCl, 4 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 5 mM d-glucose, and 10 mM HEPES, pH 7.4. Whole-cell patch-clamp electrophysiology was performed using a Nanion Port-a-Patch system fitted with an eight-valve gravity-fed perfusion panel. Total solution exchange occurred in <150 milliseconds. Internal solution contained 140 mM CsF, 10 mM NaCl, 2 mM EGTA, and 10 mM HEPES, pH 7.2. All recordings were performed in external solution containing 140 mM NaCl, 2 mM KCl, 2 mM CaCl2, 1 mM MgCl2, and HEPES, pH 7.4. In preincubation studies, 5-BDBD was perfused across the cells for 3 minutes prior to stimulation with ATP. In all other studies, ATP was perfused onto the cells for 2 seconds, which was followed by immediate perfusion of ATP ± 5-BDBD, BX-430, or the DMSO vehicle control for 5 seconds. ATP, 5-BDBD, and BX-430 were used at concentrations of 30 µM, 20 µM, and 10 µM for all experiments, respectively. The DMSO vehicle control was at a final concentration of 0.1% (v/v). Acquisition protocols were run using HEKA PatchMaster software (RRID:SCR_000034). Data were sampled at 1 kHz and filtered at 10 kHz. Cells were held at −80 mV in all experiments, which were performed at room temperature (21–23°C). The values for τ were calculated by fitting a single exponential decay curve to the desensitization phase of the current traces using OriginPro (v2019, Originlabs).
Homology Modeling and Ligand Docking
One hundred homology models for the human P2X4 receptor in the closed state were generated in Modeler 9v19 (Sali and Blundell, 1993) using the structure of the zebrafish P2X4 ortholog (62% pairwise sequence identity; Protein Data Bank identifier: 4DW0) as a template following a standard protocol (Stavrou et al., 2020). The 10 top-ranking models were prepared as a receptor ensemble for Rosetta ligand docking. 5-BDBD was prepared in Marvin (v21.11; ChemAxon, https://www.chemaxon.com), and multiple conformers were generated using the frog2 server (Miteva et al., 2010). Following the docking strategy outlined in Lemmon and Meiler (2012), two series of Rosetta ligand docking (Davis and Baker, 2009) were run, sampling broadly the orthosteric site for ATP binding and a region equivalent to the previously identified allosteric binding site in P2X7 (Allsopp et al., 2017) generating 10,000 docking poses for each combination of site and ensemble structure. From each site, the 1000 docked complexes with the lowest total Rosetta energy were clustered (Roe and Cheatham, 2013) to identify groups of alternative binding modes. Representative poses with the lowest Rosetta interface δ scores from the largest cluster and best scoring poses were visualized in PyMol and served as starting structures for molecular dynamics simulations for further analysis. The 5-BDBD analog three-dimensional models were generated using CACTVS (National Cancer Institute) and were prepared for docking using LigPrep software (www.schrodinger.com). Induced-fit docking of the allosteric site on the homology model above was performed using the automated extended sampling protocol within the Schrödinger Maestro suite (www.schrodinger.com) using an OPLS3e force field. The top 1000 docking poses were retained from 10,000 docking poses. Structures within 30 kcal mol−1 of the lowest energy structure were retained, and representative docks were visualized using Chimera.
Molecular Dynamics
Molecular dynamics simulations were run in AMBER18 using the ff14SB and GAFF force fields (Case et al., 2018). 5-BDBD parameters were generated in antechamber using atomic population charges with additive bond charge corrections (AM1-BCC) charges. The extracellular domain of the P2X4 receptor models including docked 5-BDBD, was surrounded with a cubic 12-Å TIP3P water box and charge-neutralized at 0.15 M Na+ and Cl- (Machado and Pantano, 2020). System equilibration started with three rounds of steepest descent energy minimization followed by conjugate gradient minimization with restraints on nonhydrogen atoms in the first round, restraints on nonsolvent molecules in the second round, and no restraints in round three. The system was then heated at constant volume from 0–300 K within 50 ps with position restraints on protein and ligand (2.0 kcal mol−1 Å−2). This was followed by a 100-ps equilibration step and 500-ns production runs. All simulations steps used periodic boundary conditions, the SHAKE algorithm for bonds involving hydrogens, a Langevin thermostat for temperature control, and a Monte Carlo barostat for the equilibration step and production runs. Replicates for the allosteric site simulations were set up by placing 5-BDBD in symmetrically equivalent positions between different subunits. Trajectories and ligand binding were analyzed in cpptraj (Roe and Cheatham, 2013).
General Chemistry
All reagents and solvents were purchased from Merck, Fisher Scientific, and Fluorochem and used as received. NMR solvents were purchased from Goss Scientific. NMR data were recorded with a Bruker instrument operating at the specified frequency and with the described solvent. Shifts are recorded in ppm and referenced to the solvent residual peak. High-resolution mass spectrometry was provided by the Engineering and Physical Sciences Research Council (EPSRC) National Mass Spectrometry Facility at Swansea. Melting points were recorded on a Stuart Scientific Melting Point Apparatus SMP3.
Chemical Synthesis of 5-BDBD Analogs
Analogs of 5-BDBD with the following modifications were synthesized with the general method below: R1-H, R2-Cl, R3-H; R1-H, R2-F, R3-H; R1-H, R2-CF3, R3-H; R1-Cl, R2-H, R3-H; R1-F, R2-H, R3-H; R1-CF3, R2-H, R3-H; R1-Br, R2-Br, R3-H; R1-Cl, R2-Cl, R3-H; R1-F, R2-F, R3-H; R1-CF3, R2-CF3, R3-H.
Synthesis of the compounds follows a reported procedure (Fischer et al., 2004) with minor modifications. Intermediate purification is not necessary and was only performed for the final product. A mixture of 5-substituted-2-hydroxybenzonitrile (a, 250 mg, 1 Eq), 3-substituted-phenacyl bromide (b, 1 Eq), and triethylamine (1.1 Eq) in 2.5 ml of dimethylformamide was stirred at 70°C until completion (2–6 hours) was confirmed by TLC. The reaction mixture was cooled down to room temperature, diluted with 100 ml of ethyl acetate, and washed with water (3 × 30 ml) and brine (1 × 30 ml). The organic layer was dried over sodium sulfate and evaporated under reduced pressure. The residue (crude c) was dissolved in 10 ml of ethanol, and 1.1 equivalents of sodium ethoxide were added thereto. The mixture was stirred at 80°C in a sealed tube for 2–12 hours until completion was confirmed by TLC. The reaction mixture was cooled down to room temperature, diluted with 100 ml of ethyl acetate, and washed with water (3 × 30 ml) and brine (1 × 30 ml). The organic layer was dried over sodium sulfate and evaporated under reduced pressure. A stirring mixture of the residue (crude d) and sodium hydrogen carbonate (4 Eq) in 30 ml of chloroform was cooled down to 0°C. Bromoacetyl bromide (1.1 Eq) was slowly added, and the mixture was then stirred for 1–2 hours, until completion was confirmed by TLC. The mixture was diluted with 100 ml of ethyl acetate and washed with a saturated solution of sodium hydrogen carbonate (1 × 30 ml) and with brine (1 × 30 ml). The organic layer was dried over sodium sulfate and evaporated under reduced pressure. A mixture of the residue (crude e), 7 ml of diethyl ether, NH3-dioxane 0.5 M (10 Eq), and 1.5 g of anhydrous sodium sulfate was stirred in a sealed tube for 3 days. The mixture was diluted with 100 ml of ethyl acetate and washed with water (3 × 50 ml) and with brine (1 × 50 ml). The organic layer was dried over sodium sulfate and evaporated under reduced pressure. The product was purified by flash chromatography and then triturated with diethyl ether to give the pure product (f). When necessary, an additional purification by preparative high-performance liquid chromatography (HPLC) was performed. Purity and characterization were assessed according to the attached methods. Supporting information regarding synthesized compounds, including high-resolution mass spectrometry and NMR spectroscopy, is given in Supplemental Material.
Flash Chromatography
All silica employed was Davisil Silica 60A, particle size 40–63 micron, purchased from Fisher Scientific. Dichloromethane (solvent A) and methanol (solvent B) were purchased either from Fisher Scientific or Merck. Ten-gram SNAP columns purchased from Biotage were refilled with the silica described above using vacuum packing. Crude samples were adsorbed on 1 g of silica and loaded on a pre-equilibrated column. The sample was eluted with one column volume of solvent A and a linear gradient to 10% solvent B over ten column volumes, and then the concentration was maintained constant for 2 CVersus Collection parameters were set to monitor at 254 and 280 nm, with a collection threshold of 30 mAU.
Preparative HPLC
All preparative HPLC work was performed on an Agilent Technologies 1200 Infinity series equipped with an Agilent Eclipse XDB-C18, 5 μM, 21.2 × 150mm. Samples were detected by UV absorbance at 210 nm. All solvents were HPLC-grade and purchased either from Merck or Fisher Scientific. The samples were eluted with water/acetonitrile 95/5 + 0.05% trifluoroacetic acid (solvent A) and acetonitrile/water 95/5 + 0.05% trifluoroacetic acid (solvent B) using the gradient method reported below. It is important to note that some samples produce split peaks in the presence of trifluoroacetic acid (TFA), likely because of the equilibrium between the open and closed form of the diazepinone ring; removing the acid from solutions A and B prevents the issue. The samples were dissolved in solvent B and filtered on 0.45 µM polytetrafluoroethylene (PTFE) filters before injection. The fractions containing the product were collected, and the acetonitrile was removed by evaporation under reduced pressure. The remaining water was freeze-dried to yield the dry product. Gradient was 100% A until minute 3, then linear gradient to 100% B in 7 minutes followed by 5 minutes at 100% B. In 5 minutes, the eluent is brought back to 100% A before the following analysis.
Analytical HPLC
All analytical HPLC measurements were performed on an Agilent Technologies 1200 series equipped with an Agilent Eclipse XDB-C18, 5 μM, 4.6 × 150mm. Samples were detected by UV absorbance at 210 nm. The column compartment was maintained at 40°C. All solvents were HPLC-grade and purchased either from Merck or Fisher Scientific. The samples were eluted with water + 0.05% trifluoroacetic acid (solvent A) and acetonitrile + 0.05% trifluoroacetic acid (solvent B) using the gradients in the methods below. The samples were dissolved in solvent B and filtered on 0.45-µM polytetrafluoroethylene (PTFE) filters before injection. Solvent B was also used as a blank. Data analysis was performed with OpenLAB CDS ChemStation Edition by Agilent Technologies. Purity was determined by peak integration. The blank was subtracted from the signal of the sample prior to integration. It is important to note that some samples produce split peaks in the presence of trifluoroacetic acid (TFA), likely due to the equilibrium between the open and closed form of the diazepinone ring; removing the acid from solutions A and B prevents the issue. This will be specified with the data reported for each compound. Method 1 was 100% A until minute 3, then linear gradient to 95% B in 7 minutes followed by 5 minutes at 95% B. In 5 minutes, the eluent is brought back to 100% A before the following analysis. Method 2 was 100% A until minute 2, then linear gradient to 95% B in 3 minutes followed by 5 minutes at 95% B. In 5 minutes, the eluent is brought back to 100% A before the following analysis.
Experimental Design and Statistical Analysis
Experimental design, analysis, and disclosure follow the guidance outlined by Curtis et al., (2018). Studies were designed to groups of equal size using randomization and blinded analysis when technically or practically feasible. Data are expressed as mean ± S.D., and N is the number of technical replicates. Statistical analysis was performed using independent values from technical or biologic repeats. Statistical analysis was only performed on dataset in which N ≥ 5 using OriginPro software (v2019; OriginLab). A threshold for statistical analysis of p < 0.05 was applied throughout. Data were first tested with a Shapiro-Wilk test for normality and Levene's test for equality of variance. Population means were compared with an ANOVA for parametric analysis with Dunnett’s post hoc test and Mann-Whitney test for nonparametric analysis. No outliers were excluded from any analysis or presentation of data. Data normalization to control values has been applied to control for interexperimental variation when applicable. For transformed dataset (e.g., data expressed as % control), test data points are normalized to control values within the same technical repeat. When effects are represented as % or fold change, the mean value is given proceeded 95% confidence interval values in squared brackets.
For antagonist IC50 data sets, the normalized response was plotted against Log10 antagonist concentration and fitted with a modified Hill equation as below:

wherein k = Michaelis constant, and n = slope of the curve. Data points were equally weighted for nonlinear fitting of concentration-response relationships.
Pairwise comparison of EC50 and IC50 values generated in curve shift experiments was conducted using a paired sample t test. τ was estimated by fitting a single exponential decay curve to the desensitization phase of current traces:

wherein Y0 = offset, A = amplitude, and t = time constant.
Nomenclature of Targets and Ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the International Union of Basic and Clinical Pharmacology/British Pharmacological Society (IUPHAR/BPS) Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
Results
5-BDBD Activity at Human P2X4, Human P2X2, and Chimeric Receptors
ATP could evoke concentration-dependent intracellular Ca2+ responses in 1321N1 astrocytoma cells stably expressing either human P2X4 or human P2X2 receptors (Fig. 1A) with EC50 values 458 ± 102 nM (N = 6) and 891 ± 137 nM (N = 6), respectively. ATP-evoked responses were not observed in untransfected cells. 5-BDBD antagonized human P2X4 with an IC50 780 ± 68 nM (N = 8) (Fig. 1B) but had a negligible effect on human P2X2 across the concentration range tested (1–30 µM) (Fig. 1B). 5-BDBD caused a decrease in the human P2X4 response maxima (p < 0.05; N = 6) and small but statistically significant increase in ATP EC50 (control vs. ≥10 μM 5-BDBD; p < 0.05; N = 6), indicating a negative allosteric mechanism of action (Fig. 1C). At 30 μM 5-BDBD, the maximum concentration tested, the ATP response maximum was reduced by 68% [61%–74%]. Next we took advantage of the human P2X2 receptor insensitivity to 5-BDBD to make a series of receptor chimeras, substituting sequential 10 amino-acid sections in the ectodomain of the human P2X4 receptor with the equivalent residues of P2X2. The majority of chimeras were functional and mediated ATP-evoked Ca2+ responses when expressed in 1321N1, with the exception of substitutions at amino-acid positions 71–80, 101–110, 131–140, 201–210, 221–220, 261–270, 281–290, and 291–300, which were nonresponsive to ATP (≤100 μM; N = 5). 5-BDBD sensitivity was retained in all P2X4-P2X2 chimeric receptors (N = 5; p > 0.05) except for the chimera baring substitutions at amino acid positions 81–90, which resulted in a 59% [53%–65%] (N = 5; p < 0.05) loss in 5-BDBD activity (Fig. 1D). These amino acids were located toward the top of the body region of the receptor and extended toward the orthosteric site for ATP binding (Fig. 1E).

5-BDBD displays an allosteric mode of action (A) ATP-evoked Ca2+ responses from 1321N1 astrocytoma cells stably expressing human P2X2 (open circles) or P2X4 (closed circles; N = 6). (B) 5-BDBD inhibition (30-minute incubation) of P2X4 (closed circles) but not P2X2 receptors (open circles; N = 8). Both receptors activated with 1 μM ATP. (C) Effect of varying 5-BDBD concentrations (30-minute incubation) on ATP concentration-response relationship in 1321N1 astrocytoma cells stably expressing P2X4 (N = 6). (D) Sequential mutation of 10-amino-acid stretches in the ectodomain of P2X4 to the equivalent residues in P2X2 identified a region between residues 81 and 90 important for 5-BDBD activity. Substitutions between 71–80, 101–110, 131–140, 201–210, 221–220, 261–270, 281–290, and 291–300 produced nonfunctional chimeras (N = 5; * p < 0.05 ANOVA with Dunnett’s post hoc test). (E) Homology model of P2X4 in the ATP-bound (beige) open state based on zebrafish P2X4 (Protein Data Bank:4DW1). Residues 81–90 can be located toward the top of the body region (red) and extended toward the orthosteric site (blue).
In Silico Identification and Interrogation of an Allosteric Binding Site for 5-BDBD
To explore putative binding poses of 5-BDBD to the P2X4 receptor, the orthosteric ATP binding site and a pocket equivalent to the allosteric site identified in P2X7 (Bin Dayel et al., 2019) were sampled using Rosetta ligand docking. Docking was performed using a homology model of human P2X4 in the closed conformation. Comparing the top docking scores for both sites clearly pointed toward an allosteric binding mode with best scores of −17.5 and −15.0 for allosteric and orthosteric poses, respectively. In the predicted allosteric binding pose 5-BDBD sat between two P2X4 subunits with residues of three β strands from the β sheet located in the “body” of the first P2X4 subunit (V105, L107, M109; F296, K298, Y300; T310, I312) forming one side of the cavity (Fig. 2A; Supplemental Fig. 1). The other side of the cavity comprised three regions, first F178 and L179 adjacent to the aforementioned β sheet, second residues L79, R82, W84, and D88 and third, contributions from residues W164, Y299, R301, and E307 of the second subunit (Fig. 2A). Although the highest-ranked predicted allosteric docking poses of the largest cluster shared common features, there were also differences. For instance, it was not conclusive how much R301 and R82 were involved in hydrogen bonding to the 5-BDBD carbonyl group or whether E307 or Y300 might be involved in coordinating the 5-BDBD amide-NH.

Molecular modeling identified an allosteric binding site for 5-BDBD. (A) Representative 5-BDBD docking pose from molecular dynamics simulations. Relevant residues are shown as sticks, and potential hydrogen bonding is indicated by dashes. (B) Carbonyl-guanidine distances between 5-BDBD and R301 (blue) and R82 (red), respectively, for a representative molecular dynamics simulation. (C) Amide-carboxylate oxygen distances between 5-BDBD and E307. Blue and red represent the two carboxylate oxygens. (D) Distance between the Y300 hydroxyl group oxygen and the amide-NH (red) and carbonyl oxygen (blue). (E) Presence of hydrophobic interactions between 5-BDBD and relevant residues. For comparison residues are color-coded as in Fig. 3B.

Site-directed mutagenesis of the allosteric site identified F81, M109, F178, Y300, R301, and I312 as important residues for 5-BDBD inhibition (A) Alignment of amino acids predicted to interact with 5-BDBD in the binding site between P2X1-7 (P2X4 numbering). Blue and orange depict which subunit the amino acid residues are located on. (B) Inhibitory activity of 5-BDBD against mutant P2X4 receptors (N = 5; * p < 0.05 ANOVA with Dunnett’s post hoc test). (C) A representative image of 5-BDBD binding to the wild-type P2X4 receptor. Residues implicated in 5-BDBD (purple) binding are highlighted in red. (D) Representative images of 5-BDBD binding to the mutant receptors M109H, F178L, Y300A, R301K, and I312G. The mutated residue is labeled, and the distances between the amino acids and potential interactions with 5-BDBD are shown by dashes. Images were generated using Chimera.
Predicted binding modes identified via Rosetta ligand docking were further explored by four 500-ns molecular dynamics (MD) simulations of the extracellular domain of the human P2X4 receptor. The three simulations for the allosteric binding mode and the control simulation for an orthosteric binding mode remained stable in all simulations with Cα-root-mean-square deviation (RMSD) values of less than 3.5 Å (Supplemental Fig. 2, A–D). In the simulation of the orthosteric mode the ligand did not remain in its starting position but moved ∼7 Å out of the orthosteric site (Supplemental Fig. 2H), supporting the notion that orthosteric binding was less favorable. For the MD simulations with 5-BDBD located in the allosteric binding site, such movement out of the binding pocket was not observed (Supplemental Fig. 2, E–G), and these simulations were further analyzed for the presence of specific interactions. Although not fully conclusive, the simulations favored R301 over R82 as an interaction partner for the 5-BDBD carbonyl, with the guanidinium-carbonyl interaction forming stably in two replicates (Fig. 2B). From ligand docking, the 5-BDBD amide-NH could potentially interact with Y300 or E307. In all three replicates the distance between E307 and 5-BDBD NH quickly increased to values unfavorable for hydrogen bonding (Fig. 2C). For Y300, the hydroxyl group moved at least partially into a position where a hydrogen bond to 5-BDBD-NH was feasible (Fig. 2D). Calculating hydrophobic contacts between 5-BDBD and the receptor suggested contributions from L79, W84, V105, L107, M109, F178, Y299, and I312 to antagonist binding (Fig. 2E). From the analysis of the molecular dynamics simulations, a picture emerged wherein R301 and Y300 from different subunits were predicted to be key residues for coordinating the carbonyl and amide group, and a series of hydrophobic residues in the binding pocket were contributing to 5-BDBD binding.
Interrogation of the 5-BDBD Binding Pose by Receptor Mutagenesis and Medicinal Chemistry
We further tested the fidelity of the in silico docking pose by generating a series of point mutations in human P2X4, substituting single residues for their equivalent in human P2X2 (Fig. 3A). Docking and MD simulations suggested that W84, L107, M109, F178, and I312 might contribute favorably to 5-BDBD binding. We investigated the key residues predicted to interact with 5-BDBD and those that form the hydrophobic lining of the pocket. W84A, D88A, L107A, G305T, and E307T substitutions had no effect of the activity of 5-BDBD (N = 5, p > 0.05; Fig. 3B). In support of MD stimulations, R301K substitution resulted in a 71% [54%–86%] reduction in 5-BDBD activity (N = 5, p < 0.05; Fig. 3B), and E307T substitution caused no statistically significant reduction in 5-BDBD activity (N = 5, p > 0.05) (Fig. 3B). Modeling of the R301K mutation suggested that the shorter chain length of lysine (K) versus arginine (R) creates a distance unfavorable (>5Å) for hydrogen bond formation between the amine-carbonyl groups of the residue and ligand (Fig. 3, C and D). In addition, R82K or R82A substitutions had no effect on 5-BDBD activity (N = 6, p > 0.05; Fig. 3B). These data are in agreement with MD simulations that favor R301 over R82 in interacting with 5-BDBD (Fig. 2C). A 38% (23%–52%) loss of 5-BDBD activity at the Y300A mutant receptor (N = 5, p < 0.05; Fig. 3B) supported a role for Y300 in 5-BDBD interactions (Fig. 2C), and the unfavorability of E307 identified in MD stimulations was supported by intact 5-BDBD activity at the E307T mutant (N = 5, p > 0.05; Fig. 3C).
Hydrophobicity of residue 107 is conserved in human P2X2 (Fig. 3A), and consequently, the L107A mutation conserved 5-BDBD activity (Fig. 3B). However, hydrophobicity is not conserved at residues 81 and 109 in P2X2 (Fig. 3A), and mutation to the basic residue histidine equivalent to these positions in P2X2 resulted in a reduction in 5-BDBD activity of 43% [35%–51%] and 39% [24%–54%] (N = 5, p < 0.05; Fig. 3B), respectively. Mutation of F178 to L178, which is in close proximity to the 5-BDBD Br-phenyl moiety in the preferred docking pose, was not tolerated and resulted in loss of 5-BDBD activity by 57% [36%–76%] (N = 5, p < 0.05; Fig. 3B). Finally, when I312 was mutated, which showed a fairly consistent interaction with the 5-BDBD benzofurane moiety, the activity of 5-BDBD was reduced (N = 5, p < 0.05; Fig. 3B). Although there was a 14% [8%–19%] loss of 5-BDBD activity in the W84A mutant, the MD and modeling predicted a larger reduction in activity, and this result was unexpected (Fig. 3B). Agreement between modeling and experimental data supported the role of F178, Y300, R301, and I312G as four key anchoring points for the 5-BDBD ligand in addition to M109 in the hydrophobic lining of the binding pocket.
To further probe the proposed binding pocket, we generated or purchased several analogs of 5-BDBD that would allow the ligand chemical space in relation to the pocket to be explored. The 5-BDBD analogs had modifications at position R1, R2, and R3 (Fig. 4A). Halogen substitutions (Cl and F) of Br at position R2 were well tolerated with similar activity and predicted docking poses to 5-BDBD (N = 5; Fig. 4, B and C; Supplemental Fig. 3), although substitution with a bulkier trifluoromethyl (CF3) group caused a 42% [35%–48%] reduction in activity (N = 5; Fig. 4, B and C). Interestingly, substitution of Br with H led to a retention in antagonist potency and slight enhancement of inhibitory activity (N = 5; Fig. 4, B and C), revealing the dispensability of Br at position R2. Strikingly, movement to Br from R2 to R3 was not tolerated, leading to complete loss of antagonist activity (N = 6; Fig. 4, B and C). Likewise, substitutions of other chemical groups at position R3 were also not tolerated (Fig. 4, B and C). Docking analysis of ligands with R3 substitutions revealed an inability to fully enter the pocket. This was likely due to the Y300 and R301 side chains anchoring the amide of 5-BDBD, which would lead to a steric clash between the β-sheet in region A72-W84 and R3 when replacing the H with larger substitutions (Supplemental Fig. 4). For R2, docking and molecular dynamics simulations predicted that the Br either pointed toward the solvent or, by rotation of the phenyl ring, into the hydrophobic binding pocket. For both scenarios, chemically similar substitutions are likely to be tolerated. Docking predicted that the R1 position pointed deeper toward the hydrophobic pocket lining, and thus we tested the effect of new chemical groups at this position by paired substitutions at R1 and R2 positions. In these experiments, substitution of H with Br at R1 caused complete loss of antagonist activity (N = 5; Fig. 4, B and C). Complete loss of activity was also observed for the R1:R2 F and R1:R2 CF3 substitutions (Fig. 4, B and C), although the R1:R2 Cl substitution was tolerated and resulted in a 13% [7%–18%] enhancement antagonist activity (N = 5; Fig. 4, B and C). Docking analysis revealed a similar pose for R1:R2 Cl as 5-BDBD (Supplemental Fig. 5), although other R1:R2 substituents were unable to penetrate into the binding site (Supplemental Fig. 5). As the R2 Br was not required for 5-BDBD, we further investigated R1 substitutions in the absence of R2 Br. Surprisingly, now R1 substitutions were fairly well tolerated with zero or modest changes in antagonist activity (Fig. 4, B and C). Docking of these ligands revealed an almost mirror image of the docking pose observed for 5-BDBD and R2 analogs, with the R1 group now protruding from the binding site and R2 position facing into the pocket (Supplemental Fig. 6). Further in silico measurement of the docking poses was made to fully understand the effect of chemical substitution on antagonist activity. Quantification of the distance between the carbonyl group of 5-BDBD and analogs and R301 indicated a correlation between distance and antagonist activity (Fig. 4D). Analysis revealed that if the carbonyl group of 5-BDBD analogs were within 4 Å of the guanidinium group of R301, then antagonist activity was retained, although distances >8 Å resulted in loss of activity (Fig. 4D). The activity of 5-BDBD was also related to the depth of ligand penetration into the binding pocket (Fig. 4E).
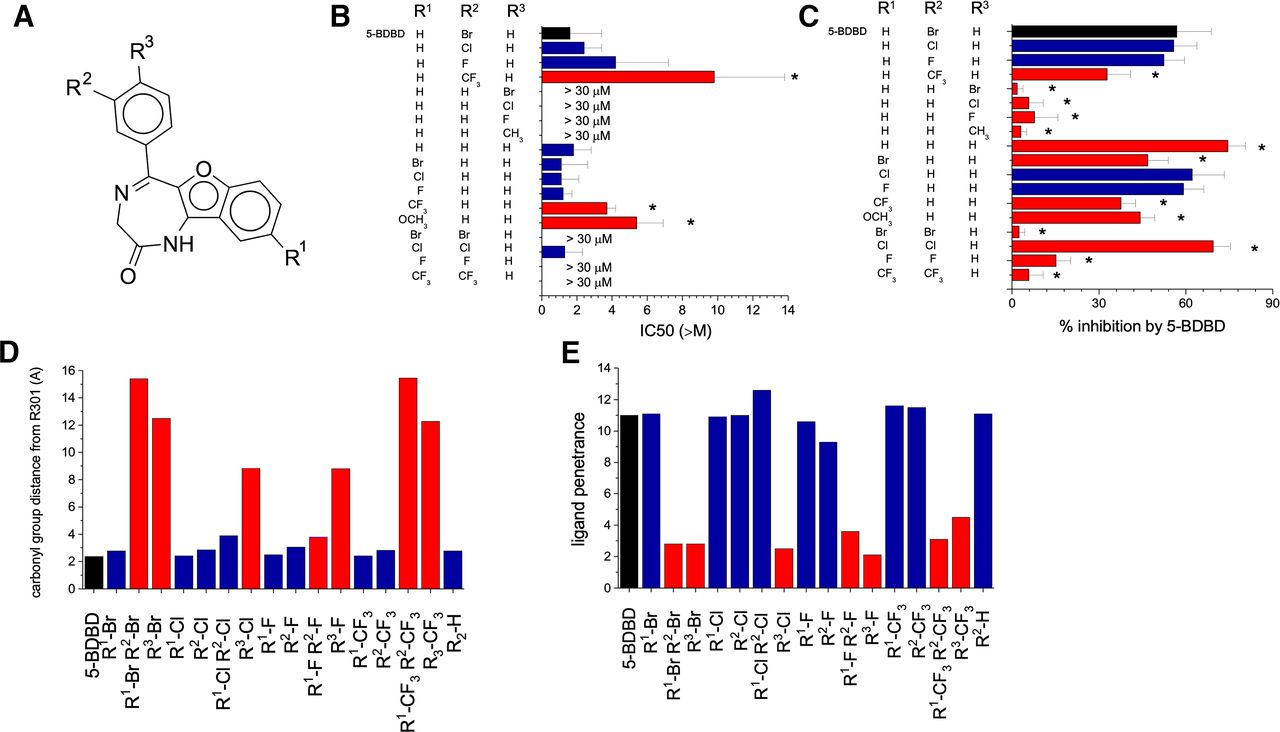
5-BDBD analogs identified key structural interactions between 5-BDBD and P2X4. (A) The structure of 5-BDBD and the positions of the halogen substitutions at R1, R2 and R3. Unmodified 5-BDBD has a bromine at position R2. (B and C) The IC50 values (B) and inhibitory activity (C) of the 5-BDBD analogs toward P2X4 stably expressed in 1321N1 astrocytoma cells (600 µM ATP and 30 µM 5-BDBD analog were used to quantify inhibitory activity; N = 3; * p < 0.05 ANOVA with Dunnett’s post hoc test). (D) The distance between the carbonyl group of 5-BDBD/5-BDBD analogs and R301 in the binding pocket as determined through docking studies. (E) The penetration distance of 5-BDBD/5-BDBD analogs into the predicted binding site relative to the opening of the pocket as determined by docking studies. Images were generated using Chimera.
Receptor State Dependency of 5-BDBD Activity Is Associated with Conformational Changes in the Binding Pocket
Analysis of the identified binding pocket in homology models of the P2X4 receptor in the closed versus the open state revealed a conformational change (Fig. 5A) that could possibly result in a state-dependent action of 5-BDBD. In silico measurement of the residues lining the entrance to the pocket revealed a constriction of the opening relative to L107 when the channel moved from a closed to open state (Fig. 5B; Supplemental Table 1). Consistently, docking analysis failed to dock 5-BDBD within the allosteric binding pocket of the open channel. This analysis predicted an ability of 5-BDBD to bind better and effectively antagonize human P2X4 in the closed state but have more limited activity in the open channel conformation. To test this hypothesis, we compared the ability of 5-BDBD to block human P2X4 currents after preincubation with 5-BDBD (incubation with closed state) compared with application of 5-BDBD when the channel was bound with ATP. This was achieved using whole-cell patch clamp electrophysiology and rapid solution switching. In these experiments, incubation with 5-BDBD prior to ATP challenge effectively inhibited channel activity (Fig. 5C), although 5-BDBD exhibited very limited activity when coapplied with ATP (Fig. 5D). In contrast, BX-430, which is predicted to share some common residues for binding with those identified in this study (Ase et al., 2019), efficiently antagonized P2X4 upon coapplication with ATP (Fig. 5E). The differences between 5-BDBD and BX-430 were apparent when comparing the current decay constants in response to antagonist application (Fig. 5F), wherein 5-BDBD and BX-430 caused a 1.6 [1.3–2.0]- and 14 [12.1–16.1]-fold decrease in the current decay time constant, respectively.

Constriction of the P2X4 allosteric binding site in the open state may prevent the activity of 5-BDBD. (A) Representative image depicting the predicted constriction of the 5-BDBD binding pocket and conformational changes between P2X4 in the closed (blue) and open (red) states. (B) The relative distance between amino acids T76, S77, L79, R82, and W84 on one subunit with the atoms CD1 or CD2 of L107 on the other subunit in the closed and open channel. (C) Representative electrophysiology trace of a cell stimulated with 30 µM ATP in the absence of 5-BDBD (black) or after a 3-minute preincubation with 20 µM 5-BDBD (red). (D) Representative electrophysiology trace of cells stimulated with 30 µM ATP for 2 seconds prior to the addition of 30 µM ATP and 20 µM 5-BDBD (red) or ATP and DMSO (black) during the desensitization stage for 5 seconds. (E) Representative electrophysiology trace of cells stimulated with 30 µM ATP for 2 seconds prior to the addition of 30 µM ATP and 10 µM BX-430 (red), or ATP and DMSO (black), during the desensitization stage for 5 seconds. (F) The time constant values for current decay (τ) for the desensitization stage in the presence of DMSO, 5-BDBD, or BX-430. Traces and data are representative of 17, 7, and 5 cells for the vehicle control, 5-BDBD, and BX-430, respectively. *p < 0.05 vs. vehicle, **p < 0.05 vs. 5-BDBD. Images were generated using Chimera.
Discussion
The complementary approaches of molecular modeling, receptor mutagenesis, and ligand medicinal chemistry supported a negative allosteric action of 5-BDBD by binding to an intersubunit pocket located in the body region of human P2X4. We propose that M109, F178, Y300, R301, and I312 are key anchor points for 5-BDBD binding, provide a structural explanation of how they contribute to 5-BDBD antagonism, and highlight that the limited action of 5-BDBD on open versus closed channels is due to a conformational change in the proposed binding pocket.
The current mechanism of action for 5-BDBD remains controversial. Initial studies demonstrated that 5-BDBD competed with ATP for the orthosteric site (Balazs et al., 2013; Coddou et al., 2019). Conversely, recent studies by Abdelrahman et al., (2017) used a radioactively labeled derivative of the ATP analog ATPγS ([35S]ATPγS) to dispute the competitive nature of 5-BDBD. The authors found that TNP-ATP could displace [35S]ATPγS, but neither 5-BDBD or the allosteric antagonist paroxetine competed with [35S]ATPγS for the orthosteric site. Our data agree with the latter observations, and there is growing evidence to support an allosteric mode of action for other P2X4 antagonists, including PSB-12054, PSB-12062, and BX-430 (Hernandez-Olmos et al., 2012; Ase et al., 2015). Notably, the studies reporting the PSB compounds, the study reporting 5-BDBD as an allosteric antagonist, and our own study use 1321N1 cells, whereas those showing 5-BDBD as a competitive inhibitor use HEK293 cells. The key difference between these two cell types is the presence or absence of endogenous P2 receptors. The 1321N1 cell line does not express endogenous P2 receptors, whereas HEK293 expresses both P2Y1 and P2Y2 (Schachter et al., 1997). There is some evidence that P2Y2 can influence P2X7 responses in macrophages (Thorstenberg et al., 2018). However, whether endogenous P2Y receptors can influence P2X4 responses is something that is yet to be determined, and the choice of cell line must be carefully considered.
In-depth analysis of the predicted allosteric site identified several potentially interacting residues on the α-helices and β-subunits of P2X4, including F178 (α2), R82, W84 (β3), V105, L107, M109 (β4), Y300 (β13), and I312 (β14) on one subunit and R301 (β13), G305, and E307 (β14) on the neighboring subunit. Site-directed mutagenesis identified that M109, F178, Y300, I312, and R301 as residues contributing most to 5-BDBD activity, whereas W84, L107, and G305 were only marginally implicated. Indeed, Y300 and I312 were found to be critical for the inhibitory activity of BX-430 toward P2X4 (Ase et al., 2019). Although 5-BDBD and BX-430 are chemically distinct (albeit both contain bromines), it is feasible that both could bind to a similar site in P2X4 given the chemical promiscuity of the equivalent site in P2X7. Interestingly, I312 may be intrinsically linked with channel opening in P2X4, as mutation of this residue to leucine and glycine could disrupt BX-430 inhibition, whereas mutation to tyrosine and tryptophan could convert BX-430 into a positive allosteric modulator (Ase et al., 2019).
The R301 residue in P2X4 was found to be implicated in 5-BDBD activity. Here, the guanidinium group was predicted to form hydrogen bonds with the carbonyl on the diazepinone ring of 5-BDBD. When R301 was mutated to lysine, which shares similar chemical properties, in silico predictions suggested that the R301K mutation increased the interaction distance between the lysine and the carbonyl group. Further in silico modeling of 5-BDBD analogs reinforced the link between R301 to carbonyl distance and inhibitory activity. It is also noteworthy that R301 (with the exception of P2X5) is not conserved in other human P2X receptor subtypes. These findings could contribute to the generation of more potent and selective antagonists.
To date, R301 has not previously been directly implicated in the activity of antagonists at P2X4. However, previous studies in P2X7 showed that mutating the D92 residue could destabilize the β-sheet containing Y291-E301. This indirectly reduced the participation of K300 (equivalent to R301 in P2X4) in the binding pocket from 85% to 23%, and antagonist activity was subsequently weakened. Notably, in P2X7, the movements of β13 and β14 strands in the upper body domain also seem to be tightly coupled to channel opening (Karasawa and Kawate, 2016). It is plausible that this is also likely for P2X4, and interactions between 5-BDBD with R301 and I312 on the β13 and β14 strands, respectively, may stabilize the closed conformation and inhibit the necessary movements of these β-strands to initiate channel opening upon ATP binding (Karasawa and Kawate, 2016). Furthermore, it could also be hypothesized that mutation of F81 may limit 5-BDBD binding in a similar manner to D92 in P2X7. In the predicted binding pose, F81 is located on the periphery of the β3-strand and is not within proximity of 5-BDBD in the proposed binding site. Therefore, it is unlikely that F81 directly interacts with 5-BDBD but instead induces a conformational change along the β3-sheet, thereby affecting the involvement of potentially interacting residues lining the pocket. Mutation of P2X7 residues 81–94 (77–86 in P2X4) and 112–118 (109–115 in P2X4) prevented inhibition of P2X7 by AZ10606120 (Allsopp et al., 2017). This region has since been implicated in the activity of numerous chemically distinct antagonists of P2X7, including A438079, A740003, A804598, GW791343, JNJ47965567, AZ11645373, Brilliant Blue G, KN-62, calmidazolium, and ZINC58368839 (Bin Dayel et al., 2019).
We had initially hypothesized that the bromine would be critical to the inhibitory activity of 5-BDBD. However, substitution of the bromine with other halogens at position R2 or moving the bromine to position R1 was tolerable and did not significantly alter the activity of 5-BDBD. These observations support those made previously, whereby an R2-F analog of 5-BDBD was shown to have comparable biologic activity to the native molecule (Wang et al., 2017). Moreover, the redundancy of the halogen was verified when an analog lacking halogens had greater inhibitory capacity. All these analogs could likely form stable hydrogen bonds with R301, which we hypothesized to be a critical interaction for 5-BDBD inhibition. Interestingly, in the study mentioned above by Wang and colleagues (2017), analogs wherein the NH group of 5-BDBD was replaced with an N-CH3 had reduced affinity for P2X4, potentially supporting our proposed interactions between 5-BDBD-NH with Y300. Conversely, halogen substitutions to R3 were not tolerated. As aforementioned, this was most likely due to a steric clash with residues lining the binding pocket, preventing the molecule from entering the site.
To support our pharmacological observations, whole-cell patch-clamp electrophysiological readings were conducted. We found that pretreatment with 5-BDBD was able to inhibit P2X4 channel opening, but application of 5-BDBD to desensitizing P2X4 could poorly inhibit. Investigations into the respective allosteric site in P2X7 (Karasawa and Kawate, 2016) showed that the allosteric cavity shrinks upon receptor activation, allowing the lower body region to widen for full channel opening. Indeed, comparisons between the homology models of P2X4 in the open and closed state highlighted that the allosteric site in P2X4 in the open state was also smaller and less accessible. In the open state, the β3 (containing T76, S77, L79, R82, and W84) and β4 sheets (containing L107) came much closer together, and the residues lining these β sheets appeared to occupy and occlude the binding site. Therefore, upon channel opening, it is plausible that the entry of 5-BDBD to this site is limited, resulting in a weakened/delayed ability to inhibit P2X4. During the desensitization phase, the conformation of P2X4 begins to relax, potentially increasing the availability of the binding pocket. Given that 5-BDBD and BX-430 are a similar molecular weight and occupy a similar volume, why BX-430 can inhibit the desensitizing channel much faster than 5-BDBD is unknown. Regarding the chemical properties of 5-BDBD and BX-430, 5-BDBD has one rotatable bond, whereas BX-430 has three. In general, molecules with fewer rotatable bonds are preferable as ligands because they have a smaller conformational entropy change upon binding (Gao et al., 2010). However, in this particular case it could be possible that the enhanced molecular flexibility of BX-430 allows it to adapt to the conformational changes of P2X4 during desensitization and adopt the orientation necessary for binding much quicker than 5-BDBD.
Taken together, we show that 5-BDBD acts as a negative allosteric modulator of human P2X4 by making intersubunit interactions in the body region. The observations made here provide key insights into the comparable but contrasting activities of 5-BDBD and BX-430 toward human P2X4. Furthermore, this study enhances our understanding of the structural interactions underlying P2X4 inhibition and provides critical information to aid in the development of novel, more potent antagonists to treat P2X4-driven pathologies (Braganca and Correia-de-Sa, 2020).
Acknowledgments
Molecular graphics and analyses were performed with UCSF Chimera, which was developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco with support from National Institutes of Health (NIH) P41-GM103311. The authors thank the Engineering and Physical Sciences Research Council (EPSRC) National Mass Spectrometry Facility, University of Swansea, for providing high-resolution mass spectrometry data. This research used the ALICE High Performance Computing Facility (University of Leicester).
Authorship Contributions
Participated in research design: Bidula, Searcey, Schmid, Fountain.
Conducted experiments: Bidula, Bin Nadzirin, Cominetti, Hickey, Cullum.
Performed data analysis: Bidula, Bin Nadzirin, Cominetti, Hickey, Cullum, Schmid.
Wrote or contributed to the writing of the manuscript: Bidula, Cominetti, Schmid, Fountain.
Footnotes
- Received August 27, 2021.
- Accepted October 17, 2021.
This work was funded by a British Heart Foundation project grant [PG/16/69/32194] awarded to Fountain and a Malaysian Government Ph.D. studentship award to Bin Nadzirin.
The authors declare that they have no conflicts of interest with the contents of this article.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- AM
- acetoxymethyl
- 5-BDBD
- 5-(3-bromophenyl)-1,3-dihydro-2H-benzofuro[3,2-e]-1,4-diazepin-2-one
- DMEM
- Dulbecco’s modified Eagle’s medium
- HEK
- human embryonic kidney
- HPLC
- high-performance liquid chromatography
- MD
- molecular dynamics
- TLC
- thin-layer chromatography
- TNP-ATP
- 2',3′-O-trinitrophenyl-ATP
- Copyright © 2021 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}