


Visual Overview
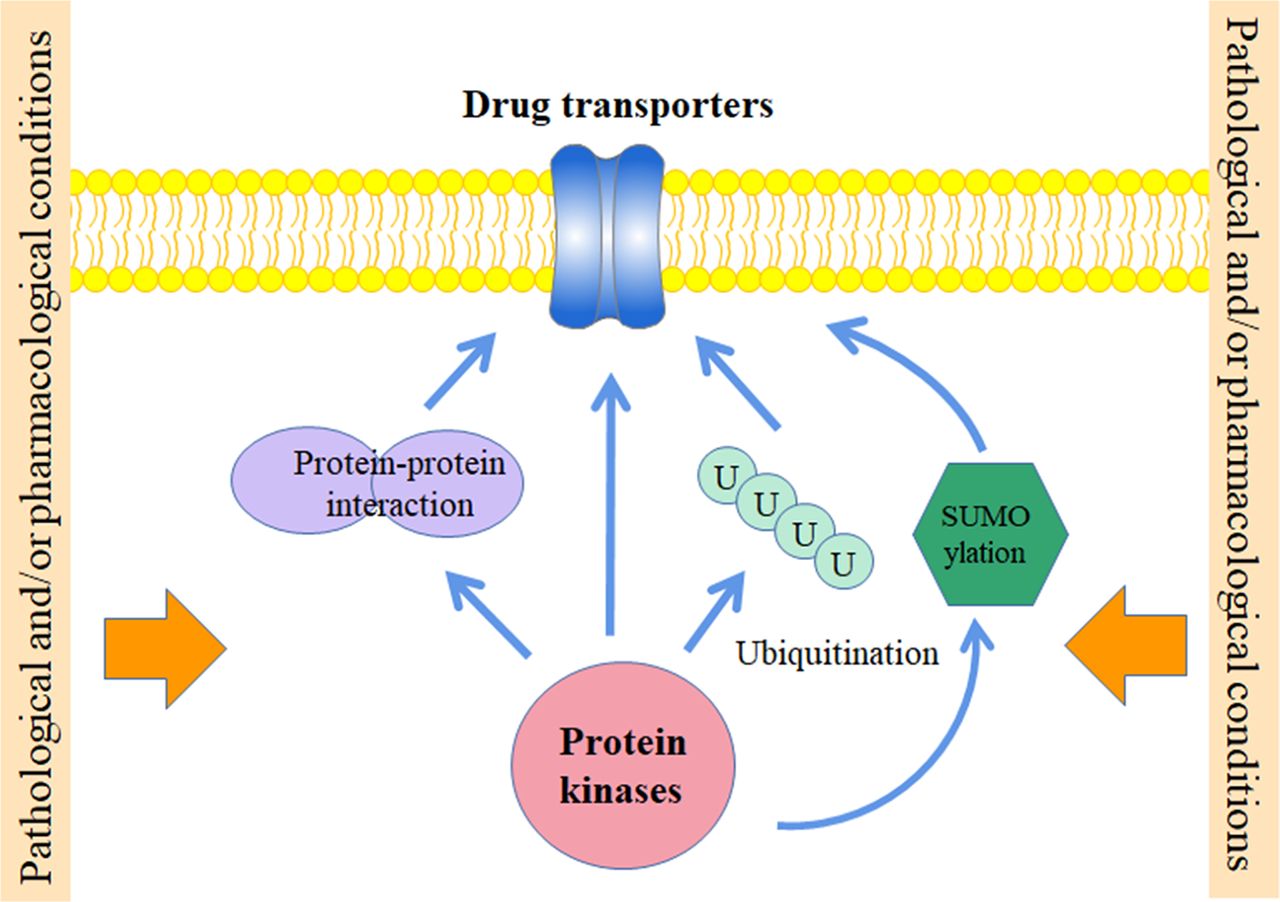
Abstract
Drug transporters are modulators for drug absorption, distribution, and excretion. Key drug transporters including P-glycoprotein and breast cancer resistance protein of the ABC superfamily; organic anion transporting polypeptide 1B1 and 1B3, organic anion transporter 1 and 3, and organic cation transporter 2, as well as multidrug and toxin extrusion 1 and 2 of the SLC superfamily have been recommended by regulatory agencies to be investigated and evaluated in drug-drug interaction (DDI) studies due to their important roles in determining the efficacy, toxicity and DDI of various drugs. Drug transporters are subjected to multiple levels of control and post-translational modifications (PTMs) provide rapid and versatile ways of regulation. Under pathologic and/or pharmacological conditions, PTMs may be altered in the cellular system, leading to functional changes of transporter proteins. Phosphorylation is by far the most actively investigated form of PTMs in the regulation of transporters. Further, studies in recent years also found that protein kinases coordinate with other PTMs for the dynamic control of these membrane proteins. Here we summarized the regulation of major drug transporters by protein kinases and their cross-talking with other PTMs that may generate a complex regulatory network for fine-tuning the function of these important drug processing modulators.
SIGNIFICANCE STATEMENT Kinases regulate drug transporters in versatile manners; Kinase regulation cross-talks with other PTMs, forming a complex network for transporter regulation; Pathological and/or pharmacological conditions may alter PTMs and affect transporter function with different molecular mechanisms.
Introduction
Drug transporters are key determinants for the accumulation of drugs within cells, hence often directly related to therapeutic efficacy, toxicity, and drug-drug interaction (DDI) of various medicines. Drug transporters are also well recognized as important drug targets and essential factors that are involved in inter-individual differences in response to drugs (Hong, 2017). In US Food and Drug Administration (FDA) and European Medicines Agency (EMA) guidelines for drug interaction studies, selected members of the ATP-binding cassette (ABC) superfamily (including P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP)) and solute carrier (SLC) superfamily (including organic anion transporting polypeptide 1B1 and 1B3 (OATP1B1 and 1B3), organic anion transporter 1 and 3 (OAT1 and 3), and organic cation transporter 2 (OCT2), multidrug and toxin extrusion 1 and 2 (MATE1 and 2)) are recommended to be investigated and evaluated in DDI studies (Center for Drug Evaluation and Research, 2012; 2017; Committee for Human Medicinal Products, 2012), suggesting the crucial roles of these transporters in absorption, distribution, and excretion of drugs. Studies so far have been mainly focused on transcriptional regulation of these proteins and DDI studies usually concerned competitive substrates or inhibitors that directly affect transport function. However, drug transporters are transmembrane proteins that also need to be tightly regulated post-translationally and different kinds of post-translational modifications (PTMs) may affect their trafficking, internalization, stability as well as conformation, which in turn lead to altered transport function (Xu and You et al., 2017). Phosphorylation is by far the most actively studied and versatile form of PTM, it not only affects proteins in different aspects, but also cross-talks with other kinds of PTMs and works in concert for the regulation of protein functions under different physiologic and pathologic conditions (Crawford et al., 2018). Here we summarized regulation of the above-mentioned drug transporters by protein kinases and also discussed how other kinds of PTMs interact with phosphorylation during the regulatory process.
Overview of major drug transporters
ABC family transporters
The ABC superfamily transporters are active transmembrane proteins that can be found ubiquitously in both prokaryotes and eukaryotes. A diverse array of substrates, including lipids, amino acids, sugars, bile salts, peptides, steroids, endogenous metabolites, ions, drugs, and other xenobiotics have been demonstrated to be substrates of ABC transporters (Deng et al., 2014). In eukaryotes, ABC family members function as efflux transporters and excrete substrates across the cellular membrane against a concentration gradient by using the energy released from adenosine-triphosphate (ATP) hydrolysis. P-gp and BCRP can be found in different organs and tissues and are also robustly expressed in cancer cells. They are major concerns in the overall efficacy of a wide variety of pharmacological agents and often the main contributing factors in regard to multidrug resistance (MDR) (Czuba et al., 2018).
1. P-gp
P-gp (also known as ABCB1 or MDR1) is a glycoprotein with a molecular weight of 160-170 kDa and consists of two transmembrane domains (TMD) and two nucleotide binding domains (NBD) (Mollazadeh et al., 2018). The first X-ray structure of mouse P-gp was reported in 2009 (Aller et al., 2009) and its refined structure was published a few years later (Li et al., 2014). More recently, cryo-electron microscopy (cryo-EM) structures of human P-gp in complex with taxol (3.6Å resolution) or zosuquidar (3.9Å resolution) were reported. It was found in the higher resolution structure that a single taxol molecule is located in the central cavity formed by the closing of a gate region compose of transmembrane helix 4 (TM4) and TM10, with a concomitant closure of the inter-NBD gap. When the taxol-bound and zosuquidar-bound P-gp structures were superimposed, a number of small but significant structural differences localized primarily in TMD2 and NBD2 were found. These minor structural differences in the protein-drug interaction sites are amplified to NBDs, controlling NBD movement and ATPase activity of P-gp (Alam et al., 2019).
P-gp is localized at the apical membrane of the gastrointestinal tract, liver, kidney, and capillary endothelial cells of the brain and testis (Raub et al., 2006), mediating the efflux of endogenous and exogenous compounds from cells into the urine and bile, protecting the body against cellular toxicants and xenobiotics (Anreddy et al., 2014). Pharmaceutic agents transported by P-gp include anti-neoplastic drugs, antibiotics, calcium channel blockers, HIV protease inhibitors, antiepileptic drugs, and antihypertensive agents (Liu, 2019). Overexpression of P-gp has been demonstrated in various cancers including acute myeloid leukemia, childhood tumors, breast cancers, hematologic malignancies, and solid tumors (Kvackajová-Kisucká et al., 2001). Since many compounds used in cancer chemotherapy are P-gp substrates, overexpression of the transporter confers significant resistance to a wide range of anti-tumor drugs (Thomas and Coley, 2003).
2. BCRP
BCRP (also known as ABCG2) contains 655 amino acid residues and six transmembrane helices. Hydropathy profile analysis predicts that the transporter protein only contains one TMD and one NBD, hence BCRP is considered as a half transporter and needs to form oligomers for its proper function (Ni et al., 2010). Human ABCG2 structure in complex with two antigen-binding fragments of 5D3 (5D3-Fab) was determined by cryo-EM in 2017. The ABCG2-5D3(Fab) complex showed a molecular mass of ∼250 kDa and maintained twofold symmetry. ABCG2 transmembrane helices and intracellular loops are both smaller than B-subfamily ABC transporters, leading to a shorter distance between the NBDs and the membrane. Structural analysis of ABCG2-5D3(Fab) revealed an inward-open conformation with a deep, slit-like cavity (cavity 1) that is primarily lined by TM2 and TM5a from the two opposing ABCG2 monomers. The geometry of cavity 1 makes it suitable to accommodate substrates with flat, polycyclic, and hydrophobic features. A smaller cavity (cavity 2) located below the EL3 external loops was also observed but owing to its less pronounced hydrophobic surface, it may only have a lower affinity for substrate (Taylor et al., 2017).
BCRP is widely expressed in placental tissue, small intestine, brain, colon, liver, ovary, and kidney. It is involved in limiting the oral bioavailability and transport across the blood-brain barrier, blood-testis barrier and maternal-fetal barrier of selected substrates, playing an important role in protecting the fetus and adult against toxins and xenobiotics (Anreddy et al., 2014). BCRP was found to transport anti-tumor drugs, tyrosine kinase inhibitors, anthracyclines, camptothecin analogs, and photosensitizers (Mao and Unadkat, 2015). Human BCRP is essential for both innate and acquired multidrug resistance, drug bioavailability regulation, prognosis prediction of both hematopoietic and solid tumors, and the protection of cancer stem cells (Ni et al., 2010). Although BCRP has a minor role in uric acid transport, dysfunction of the transporter is linked to disease states associated with hyperuricemia, which include gout, kidney disease, and hypertension (Ishikawa et al., 2013).
SLC family transporters
Transporters that belong to the SLC superfamily are considered as gatekeepers of the cellular milieu, responding to different metabolic states in a dynamic way. As altered metabolism is one of the hallmarks of cancer, SLC proteins may serve important roles in the development and progression of cancer (Pizzagalli et al., 2021). Most SLC transporters are responsible for the uptake of small molecules (including nutrients and xenobiotics), but transporters that show efflux or bidirectional function are also found in the superfamily. Substrates of SLCs are usually compounds that possess unfavorable physicochemical properties for lipid bilayer diffusion, and different SLC family members facilitate the permeation of a wide variety of structurally diverse agents across the cell membranes (César-Razquin et al., 2015) ). SLCs act coordinately with ABC transporters, serving important roles in drug disposition and significantly affecting the clinical efficacy of many drugs.
1. OATP1B1 and 1B3 that belong to the SLCO family
Generally, members of the OATP (or SLCO) family contain 643-722 amino acid residues and are predicted to consist of 12 TM segments with both amino and carboxyl termini located intracellularly (Stieger and Hagenbuch, 2014). Multiple predicted and/or confirmed N-glycosylation sites are located at the second and fifth extracellular loops, and several conserved cysteine residues are found in the large fifth extracellular loops. OATPs also have a large third intracellular loop which contains possible phosphorylation sites and other important regulatory motifs (Hagenbuch and Gui, 2008).
OATP1B1 (also known as SLCO1B1) and OATP1B3 (also known as SLCO1B3) are key uptake transporters that are expressed on the sinusoidal membrane of hepatocytes and play important roles in hepatic drug transport. Substrates of these OATPs include endogenous compounds such as bile acids, hormones, steroid sulfates, glucuronide conjugates, and peptides as well as xenobiotics and numerous drugs including statins, antivirals, antibiotics, and anticancer drugs (Roth et al., 2012). OATP1B1 and 1B3 were also found to be expressed in different types of cancers. However, data relating to the use of these transporters as biomarkers have been conflicting (Thakkar et al., 2015) and further investigation is needed for their potential impact on the efficacy of chemotherapeutics (Schulte and Ho, 2019).
2. OAT1, OAT3 and OCT2 that belong to the SLC22 family
SLC22 family members are predicted to have similar structural features, consisting of 12 transmembrane helices, a large glycosylated extracellular loop between TM1 and 2, and a large intracellular loop between TM6 and 7. Consensus sequences for phosphorylation were proposed to be localized at the large intracellular loop (Koepsell, 2013).
OAT1 (also known as SLC22A6), OAT3 (also known as SLC22A8), and OCT2 (also known as SLC22A2) are renal transporters that are predominantly expressed on the basolateral side of proximal tubular cells. Compared with OATPs, OATs transport smaller and more hydrophilic organic anions. Both OAT1 and 3 are anion-exchanging antiporters that serve important roles in maintaining systemic levels of endogenous substrates such as uric acid and facilitating active renal secretion of drugs into the urine. Drug substrates of OAT1 and 3 include antibiotics, antivirals, histamine H2 receptor antagonists, diuretics, non-steroidal anti-inflammatory drugs (NSAIDs), statins, uricosurics, and toxins (Rizwan and Burckhardt, 2007). OAT1 and 3 show a large degree of overlapping substrate specificity, but according to machine learning analysis, OAT3 may transport drugs with slightly more cationic characteristics (Nigam et al., 2020). On the other hand, OCT2 is responsible for the disposition and renal clearance of mostly cationic drugs and endogenous compounds. Substrates of OCTs include a broad range of structurally unrelated small organic cations such as steroids, hormones, monoamine neurotransmitters, numerous drugs, and other xenobiotics. OCTs are bi-directional facilitative transporters that mediate the passive facilitated diffusion of organic cations down the electrochemical gradient. However, OCT2 typically acts as an uptake transporter in vivo, transporting drug substrates from the blood into the proximal tubular cell as the first step for renal elimination (Wagner et al., 2016).
3. MATE1 and MATE2 that belong to the SLC47 family
The mammalian MATE transporters have thirteen predicted transmembrane helices, with the carboxyl terminus located extracellularly. However, mutation of TM13 only showed little impact on transporter ligand binding, suggesting only the core 12 transmembrane helices are involved in transport function (Zhang et al., 2012). It was proposed that TM13 may play a role in stabilizing MATEs in the membrane bilayer or assisting their interaction with other partner proteins (Kusakizako et al., 2020).
SLC47 family members are involved in the excretion of endogenous and exogenous toxic electrolytes from the human body through urine and bile. MATE1 (also known as SLC47A1) is highly expressed on the canalicular membrane (bile side) of hepatocytes and the brush-border membrane (urine side) of proximal tubule cells (Dresser et al., 2001). A high level of MATE1 was also reported in skeletal muscle; while expression of the transporter is relatively low in the adrenal gland, testes, and heart (Otsuka et al., 2005). On the other hand, MATE2 (also known as SLC47A2) and its splice variant MATE2-K are exclusively expressed in the apical membrane of kidney proximal tubular cells. However, MATE2 is not functional in classic in vitro transporter systems, and cell lines expressing MATE2-K are the preferred model for studies of MATE2. Both MATEs are electroneutral, Na+-independent, pH-dependent proton antiporters and were shown to be responsible for renal transepithelial transport of a wide range of structurally diverse, low molecular weight organic cations. Major cationic drugs excreted by MATEs include the antiviral drug acyclovir, histamine H2 receptor antagonist cimetidine, and antidiabetic metformin (Nigam, 2015). The substrate spectrum of MATEs was found to overlap with that of OCTs and many clinical inhibitors of OCTs are also potent MATEs inhibitors, hence modulating the function of transporters from both families (Winter et al., 2011). Some anionic compounds including estrone-3-sulfate, acyclovir, ganciclovir, and zwitterionic drugs such as cephalexin and cephradine are also substrates of MATEs (Tanihara et al., 2007).
Regulation of drug transporters by protein kinases
Phosphorylation is by far the most diverse and well-studied kind of PTMs. It is typically defined as the reversible addition of a negatively charged phosphate group to residues such as serine, threonine, or tyrosine (or to a much less extent lysine and arginine). The presence of this heavily charged group is essential for changing the hydrophobicity and charge property of the corresponding protein region, resulting in changes in protein conformation, cell localization, or interactions with other proteins (Barford, 1996). The phosphorylation state of the target protein is dynamically controlled by different protein kinases and phosphoprotein phosphatases, and the opposite process of phosphorylation/dephosphorylation is commonly used in signaling events, acting as functional on/off switches, and interconnecting with other types of post-translational modifications (Mayati et al., 2017). Although in most cases the evidence for direct phosphorylation of drug transporters is still lacking, quite a few kinases have been demonstrated to regulate the expression and function of these important ADME modulators.
ABC family transporters
ABC family transporters have been demonstrated to be regulated by different kinases. Protein kinase C (PKC) was shown to regulate P-gp in several early reports (Crawford et al., 2018). In multidrug-resistant (MDR) human KB-V1 cells, phosphorylation and activity of P-gp were demonstrated to be induced upon the treatment by phorbol 12-myristate 13-acetate (PMA), a PKC activator (Chamber et al., 1992). Within the linker region of P-gp sequence, Ser 661 and Ser 671, and one or more of Ser 667, 675, and 683, were identified to be PKC phosphorylation sites using membrane vesicles of KB-V1 cells incubated with purified protein kinase C (Chamber et al., 1993). Evidence of direct interaction between PKC and P-gp has been reported in cancer cells and PMA was found to enhance the association between the kinase and the transporter (Yang et al., 1996). However, whether the direct phosphorylation of P-gp by PKC is crucial for its proper function remains controversial (Katayama et al., 2014). Because simultaneously mutating all five serine residues exhibited no effect on protein targeting and multidrug resistance of P-gp (Germann et al., 1996). The oncogenic serine/threonine kinase Pim-1 was also shown to directly interact with P-gp in drug-resistant HL60/VCR leukemia cells, 8226/Dox6 myeloma cells, and OVCAR-8-Pgp ovarian carcinoma cells. P-gp is likely phosphorylated at Ser683 because the serine residue is located within a Pim-1 phosphorylation consensus sequence (QDRKLS). Inhibition of Pim-1 reduced glycosylated P-gp on the cell surface and suppressed the transporter function, sensitizing cells over-expressing P-gp to doxorubicin (Xie et al., 2010). P-gp was also found to be regulated by the MEK-ERK-RSK pathway in the human colorectal cancer cells, HCT-15 and SW620-14, as well as in the MDR1-transduced human breast cancer cells, MCF-7/MDR and MDA-MB-231/MDR. Treatment of U0126, a mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) kinase inhibitor, suppressed P-gp function. Further analysis of MDA-MB-231/MDR cells with pulse-chase experiments revealed that U0126 promoted P-gp degradation but showed no effect on the biosynthesis of the protein (Katayama et al., 2007).
In addition to serine/threonine kinases, tyrosine kinases were also found to regulate P-gp. Flk-1, a receptor for vascular endothelial growth factor (VEGF), was found to be involved in the regulation of P-gp. VEGF was demonstrated to acutely and reversibly reduced P-gp function without altering protein level of the transporter in isolated capillaries. The suppressive effect was blocked by the inhibitor of Flk-1. Additionally, VEGF increased Tyr-14 phosphorylation of caveolin-1, which was inhibited by Src inhibitor PP2, suggesting that Flk-1 and cytoplasmic tyrosine kinase Src are involved in the regulation of P-gp in the blood-brain-barrier (BBB), possibly through the phosphorylation of caveolin-1 (Hawkins et al., 2010). Keratinocyte growth factor receptor (KGFR, also known as FGFR2 IIIb), a receptor tyrosine kinase that is primarily localized on epithelial cells, was demonstrated to be involved in P-gp regulation. Short-term treatment (10 ng/ml, 1 hour) of Caco-2 cells with keratinocyte growth factor-2 (KGF2) resulted in increased cell surface expression and activity of P-gp; while long-term treatment of the growth factor (10 ng/ml, 24 hour) increased mRNA and protein level as well as function of the transporter. These effects were blocked by selective FGFR antagonist PD-161570 and PD-98058, a potent inhibitor of Erk1/2, or knock-down of Erk1/2 by small interfering RNA, suggesting that regulation of P-gp is mediated by FGFR and MAPK-dependent (Saksena et al., 2013). Receptor tyrosine kinase epidermal growth factor receptor (EGFR) is likely related to P-gp regulation as well. Epidermal growth factor (EGF) was found to increase the phosphorylation of P-gp by 20–50% in human MDR breast cancer cell line MCF-7/AdrR, and such an effect was accompanied by stimulation of phospholipase C (PLC) activity (Yang et al., 1997). Cetuximab, an EGFR recombinant antibody, was shown to change the expression and function of different drug transporters through EGFR signaling in proximal tubule cells. Among them, membrane level and transport activity of P-gp was increased by cetuximab, likely through PI3K/Akt and MAPK/ERK pathways, for inhibitors of these pathways resulted in a similar effect on P-gp as cetuximab (Caetano-Pino et al., 2017). Taken together, it seems that multiple signaling pathways are involved in the regulation of P-gp (Table 1).
Effects of kinases on P-gp and BCRP
BCRP is regulated by Pim-1L, the 44 kD Pim-1 isoform that is located primarily on the plasma membrane. The kinase has been experimentally shown to co-immunoprecipitate with BCRP when exogenously expressed in HEK293T cells or endogenously expressed in the prostate cancer cell line CWR-R1, and directly phosphorylated BCRP at Thr362 (Xie et al., 2008). The phosphorylation of BCRP may affect its cell surface expression. SGI-1776, a potent inhibitor for Pim kinases, was shown to reduce BCRP level at the cell membrane and increased uptake of substrate drugs in cells expressing high levels of the transporter (Natarajan et al., 2013). Pim-1 may affect BCRP total protein level as well. Darby and co-workers demonstrated that by treating breast cancer cells with two imidazo-[1,2-b]-pyridazine-based Pim-1 inhibitors, the potency of flavopiridol, mitoxantrone, topotecan, and doxorubicin on BCRP-expressed cells was significantly increased. This was due to a significant time-dependent reduction of BCRP level induced by the inhibitors (Darby et al., 2015).
Cell surface level of BCRP was increased while no change was observed in total protein level after EGF treatment on side population cells of mice bone marrow (Goodell et al., 1996; Mogi et al., 2003), LLC-PK1 (Takada et al., 2005) and MCF-7 cells (To and Tomlinson, 2013). However, treating cells with PI3K inhibitor LY294002 or PPARγ agonists that activate PTEN reduced cell surface expression of BCRP, suggesting that EGF may stabilize BCRP at the plasma membrane through the PI3K/Akt pathway (Crawford et al., 2018). Interestingly, while cetuximab up-regulated P-gp, it suppressed the membrane expression and activity of BCRP induced by EGF. The effect was demonstrated to be mediated by PI3K/Akt and MAPK/ERK pathways as well (Caetano-Pinto et al., 2017). In a recent study on BCRP regulation in obesity, it was found that the transporter is regulated by non-receptor tyrosine kinase Janus kinase 3 (JAK3). JAK3 directly associates with and phosphorylates BCRP and promotes membrane localization of the transporter. When JAK3 was knockout in mice, intestinal BCRP expression was significantly reduced, resulting in compromised colonic drug efflux and barrier functions. Three tyrosine residues within BCRP sequence, i.e., Tyr123, Tyr273, and Tyr336, exhibit strong NetPhos prediction score and was proposed to be potential substrate phosphorylation sites for JAK3 (Mishra et al., 2019). Regulation of BCRP by different kinases was also summarized in Table 1.
SLC family transporters
Quite a few reports regarding kinase regulation of SLC family members are related to the promiscuous role of PKC in modulating the endocytosis and trafficking of these transporters (Czuba et al., 2018). In COS-7 cells over-expressing OAT1, PKC was demonstrated to down-regulate uptake function of the transporter. The trafficking of OAT1 was altered by PMA, resulting in accelerated OAT1 internalization, which occurs partly through a dynamin- and clathrin-dependent pathway (Zhang et al., 2008). Activation of PKC was also found to increase endocytosis of OAT3 in COS-7 cells over-expressing the transporter, resulting in reduced cell surface level and activity (Duan et al., 2010). Membrane level and uptake function of OATP1B1 were also suppressed by PMA. Activation of PKC affected both internalization and recycling of OATP1B1 in HEK-293 cells over-expressing the transporter, reducing its cell surface level and transport activity (Hong et al., 2015). In human hepatocytes, though activation of PKC also reduced the function of OATP1B3, PMA treatment exhibited no effect on surface or total protein level of the transporter. Analysis with anti-phosphor-Ser/Thr/Tyr antibody revealed that PKC activation increased phosphorylation of OATP1B3, suggesting a novel inhibitory mechanism is involved (Powell et al., 2014). Interestingly, the effect of PKC on drug transporters may be isoform-specific. Although the effect of PKC on drug transporter function is mostly inhibitory, it was also reported that PKC zeta (PKCζ), an atypical type of PKC, directly interacts with OAT3 in rat kidney. When PKCζ was activated by insulin or EGF, uptake function of the transporter was induced. PKCζ may also induce OAT1 function because activation of the kinase also increased uptake of para-aminohippurate (PAH), a dual substrate of OAT1 and 3, in OAT3-null mice. Moreover, uptake of adefovir, an OAT1-specific substrate, was enhanced in rat renal slices after insulin stimulation (Barros et al., 2009). SLC family transporters were found to be regulated by other kinases as well. The effect of insulin-like growth factor 1 (IGF-1) on OAT3 was investigated in COS-7 cells stably over-expressing the transporter. It was found that IGF-1 stimulated both function and protein level of OAT3 through protein kinase A (PKA) signaling pathway. H89 abrogated the effect of IGF-1 on OAT3. Regulation by PKA seems to involve direct phosphorylation of OAT3 because H89 reduced phosphorylation of the transporter after IGF-1 treatment, and PKA activator Bt2-cAMP increased phosphorylation of OAT3 (Zhang et al., 2020). Human OAT1 was found to be regulated by serum- and glucocorticoid-inducible kinase 2 (SGK2) in OAT-1 expressing COS-7 cells. Co-immunoprecipitation analysis revealed that SGK2 was associated with OAT1 and enhanced protein stability of the transporter, hence increasing its expression level and uptake function (Xu et al., 2016a). Studies on HEK-293 cells stably expressing OCT2 showed that PKA, phosphatidylinositol 3-kinase (PI3K), as well as calmodulin (CaM)-dependent protein kinases may be involved in regulation of the transporter. PKA activator forskolin suppressed uptake of 4-[4-(dimethylamino)styryl]-N- methylpyridinium (ASP+) by OCT2; while PI3K inhibitor wortmannin significantly stimulated transport activity. On the other hand, inhibitors of Ca2+/CaM-dependent protein kinase II (CaMKII) and myosin light chain kinase (MLCK) both resulted in reduced uptake of ASP+, suggesting a CaM-dependent signaling pathway may be involved in OCT2 regulation (Cetinkaya et al., 2003). Protein kinases involved in several pathways were analyzed for the regulation of MATE1 and 2 in HEK-293 cells over-expressing these transporters. It was found that p56lck tyrosine kinase inhibitor aminogenistein reduced MATE1-mediated ASP+ uptake; while selective CK2 inhibitor tetrabromobenzimidazole (TBBz) stimulated the uptake function of MATE1. On the other hand, both aminogenistein and TBBz induced the uptake activity of MATE2. A potential CK2 phosphorylation site was detected at position Ser402 of MATE1 and putative phosphorylation sites in MATE2 include Tyr104 for p56lck and Ser3, Ser508, and Ser519 for CK2. When the efflux function of MATE was analyzed, it was found that CK2 inhibitor reduced function of MATE1 and the effect of TBBz on MATE2 disappeared, which is in strong contrast to its dramatic up-regulation effect on the uptake function of the transporters, and it was proposed that TBBz may stabilize MATEs in an uptake configuration. Additionally, activators of PKA (forskolin) and PKC (1,2-dioctanoyl-sn-glycerol, DOG) significantly reduced MATE2K efflux function, implicating a regulatory role of these kinases in the process (Kantauskaitė et al., 2020).
SLC transporters are also subjected to regulation by tyrosine kinases. In OCT2-expressing HEK293 cells, it was found that dasatinib, an oral Bcr-Abl and Src-family kinase inhibitor for the treatment of leukemia, is a potent inhibitor of the transporter. Immnuoprecipitation analysis demonstrated that OCT2 is phosphorylated and further investigation revealed that Yes1, a Src family member, is responsible for the tyrosine phosphorylation of OCT2. Purified Yes1 was found to directly phosphorylate OCT2 and several tyrosine residues, including Tyr241, Tyr362, and Tyr377, were identified to be essential for Yes1-mediated phosphorylation. Tyr362 was proposed to be the major phosphotyrosine site with greater functional relevance because it is localized close to the substrate-binding domain of OCT2 and may provide the negative charge that leads to the increased binding ability for positively charged organic cations (Sprowl et al., 2016). In a recent study that used HEK-293 cells over-expressing OATP1B1 to assess the effect of TKIs on the transporter, it was found that among the 46 FDA-approved TKIs tested, 29 of them significantly inhibited the uptake function of OATP1B1. Nilotinib was shown to be the most potent inhibitor and suppressed OATP1B1 function in a non-competitive manner. Further investigation identified Lyn, another Src family member, to be the protein kinase that phosphorylates OATP1B1. It was proposed that Tyr645, which localizes at the border of predicted TM12 and carboxyl- terminus of the transporter, is an important site of phosphorylation and is likely involved in OATP1B1 response to nilotinib (Hayden et al., 2021). Kinase regulation of SLC family transporters is summarized in Table 2.
Effects of kinases on SLC transporters
It should be noted that though kinase-related effects summarized here mostly occur at the post-translational level, i.e., treatment of kinase modulators affect cellular localization, trafficking, stability, and/or conformation of the transporters, kinases may change the transcription level of these proteins as well. For example, treatment of EGF increased mRNA and protein levels of BCRP in cytotrophoblasts, BeWo, and MCF-7 cells. EGFR inhibitor 4-(3-chloroanillino)-6,7-dimethoxyquinazoline (AG1478) or mitogen-activated protein kinase (MAPK) cascade inhibitor 2'-amino-3′methoxy-flavone (PD 98059) abrogated the effect of EGF on BCRP, implicating that EGF up-regulated BCRP through the activation of MAPK pathway (Meyer zu Schwabedissen et al., 2006). Such kind of effect can also be considered as a PTM effect by kinases, for it may occur through a post-translational modification of certain proteins such as transcription factors, which in turn affect gene expression of the drug transporter. In a study that aimed to evaluate the effect of TKIs on drug resistance, it was found that induction of P-gp expression in colon adenocarcinoma LS180 cells by TKIs such as erlotinib, gefitinib, nilotinib, sorafenib, and vandetanib was mediated by pregnane X receptor (PXR) (Harmsen et al., 2013). Quite a few studies have shown that PXR exists as a phosphoprotein and is modified by kinases such as PKC (Ding and Staudinger, 2005), PKA (Lichti-Kaiser et al., 2009), and Cyclin-dependent kinase 2 (Cdk2) (Qin et al., 2022). Therefore, the effect of the above-mentioned TKIs on P-gp may be mediated by the phosphorylation of PXR by corresponding kinases. However, information on such kind of regulation is quite limited and future investigation is warranted.
Cross-talking between kinase regulation and other post-translational modifications
In addition to the regulation of phosphorylation, other post-translational modifications such as oligomerization (protein-protein interaction), glycosylation, ubiquitination, and SUMOlyation have been demonstrated to be important regulatory mechanisms for the function of drug transporters. Interestingly, many of them were found to cross-talk with effects caused by kinases and work coordinately to modulate the expression level, targeting, re-location, and/or conformation of these membrane proteins. The cross-talking between PTMs suggests a complex regulatory network for fine-tuning the function of these transporter proteins. It also implicates that the regulatory role of protein kinases may not actually involve direct phosphorylation of the transporters, but rather exert through other PTMs, especially when considering the fact that only few direct phosphorylation sites have been well defined in the above-mentioned drug transporters so far. Disruption of other PTMs may therefore also result in different responses of transporters toward kinase modulators.
Cross-talk with protein-protein interactions
As mentioned in the previous section, the transport activity of P-gp is regulated by VEGF receptor flk-1 and Src. The regulatory role of these kinases did not relied on direct phosphorylation of P-gp, but rather on the phosphorylation of caveolin-1 (Cav-1) that is associated with the transporter. However, neither protein level of the transporter nor the association of Cav-1 and P-gp was altered in the study (Hawkins et al., 2010), which differed from a previous report that indicated Tyr14 phosphorylation of Cav-1 affected its interaction with P-gp and reduced efflux activity of the transporter (Barakat et al., 2007). Reasons for the discrepancy included that protein expression and function may change in brain capillary endothelial cells, which are sensitive to cultural conditions; and the induced association of Cav-1 and P-gp may be too unstable to survive tissue lysis and protein extraction or phosphorylation of caveolin-1 may not be involved in VEGF signaling to P-glycoprotein (Hawkins et al., 2010). A more recent study with breast cancer cells demonstrated that in addition to Cav-1 and Src, receptor for activated C kinase 1 (Rack1) also interacts with P-gp. Rack1 acts as a scaffold protein and mediates the interaction between Src and the transporter. Moreover, the phosphorylation of Cav-1 by Src is Rack1 dependent, knock-down of Rack1 or Src suppressed phosphorylation of Cav-1 and increased its association with P-gp, which in turn led to reduced P-gp transport activity. Elevated expression of Rack-1 and Src has been reported to associate with chemoresistance of multiple cancer. Therefore, Rack-1 may serve as a recruiting hub and facilitates the phosphorylation of Cav-1, releasing its suppressive effect on P-gp and increasing the efflux activity of the transporter (Fan et al., 2019). On the other hand, when brain microvascular endothelial hCMEC/D3 cells were challenged with acute oxidative stress, both Abl and Src kinases are involved in the phosphorylation of Cav-1. Increased Tyr14 phosphorylation of Cav-1 induced dynamin-dependent internalization of Cav1 together with P-gp, which led to reduced P-gp level in the membrane fraction and loss of P-gp function (Hoshi et al., 2020). These results implicated that though phosphorylation of Cav-1 serves a regulatory role for P-gp function, the effect of phosphorylation posed on the transporter may be cell-type specific and differ upon different treatments.
Knock-down of Pim-1L in drug-resistant prostate cancer cells with siRNA abolished dimerization/oligomerization of endogenous BCRP. Additionally, the association between tagged-BCRP was disrupted in T362A mutant, suggesting that phosphorylation at Thr362 is important for BCRP oligomerization, which is essential for the transport function (Xie et al., 2008). JAK3-mediated phosphorylation of BCRP was demonstrated to promote membrane localization of the transporter and enhanced its transport activity. The tyrosine phosphorylation of BCRP is essential for its association with β-catenin. Further, knockdown of β-catenin using specific shRNA resulted in significantly reduced BCRP surface localization and efflux function (Mishra et al., 2019). Cross-talking between kinase regulation and protein-protein interaction of P-gp and BCRP is summarized in Fig. 1.

Regulation of P-gp and BCRP by the cross-talking of PTMs. The phosphorylation of Cav-1 by Src is Rack1 dependent in breast cancer cells, and Cav-1 phosphorylation reduces its association with P-gp, leading to increased transporter function. On the other hand, increased Cav-1 phosphorylation by Src may increase its internalization together with P-gp in hCMEC/D3 cells. UBE2R1 phosphorylation by RSK1 induced self-ubiquitination and protected P-gp from degradation. Pim-1 may affect ubiquitination and degradation of P-gp; while the isoform Pim-1L phosphorylates BCRP and may promote oligomerization of the transporter. JAK3 affects BCRP function by regulating its interaction with β-catenin. Whether ubiquitination or SUMOlyation modulates kinases involved in transporter function regulation is possible but unknown, hence shown as red dotted arrows. BCRP needs to form oligomers for its function on the plasma membrane and is indicated as such. It should be noted that in most cases, a specific kind of PTM cross-talking is investigated with a specific cellular system or even an over-expressing cell line, so whether the phenomena observed are cell-type specific or generalizable remain unclear and further investigation is needed.
Although homo- and hetero-oligomerization of SLC transporters have been reported, whether protein-protein interaction of these drug transporters cross-talk with protein kinase regulation remains largely unknown. The few cases reported so far mainly concern the effect of PKC on the interaction of OAT family members with E3 ubiquitin-protein ligase neural precursor cell expressed developmentally down-regulated protein 4 (Nedd4), which determines the ubiquitination status of the transporter and will be discussed further in the following section.
Cross-talk with ubiquitination
As mentioned previously, Pim-1 regulates P-gp function by altering the level of the mature, 170 kD glycoprotein. Such an effect was achieved by changing the stability of P-gp protein. Ubiquitinated 150 kD P-gp level was increased after knock-down of Pim-1 in HL60/VCR cells, suggesting that Pim-1 protected P-gp from proteasomal degradation and enabled the transporter further glycosylated to the 170 kD mature form and targeted to the cell surface for its proper function (Xie et al., 2010). The inhibition of MAPK pathway kinases MAPK/ERK kinases (MEKs), extracellular signal-regulated kinases (ERKs), and p90 ribosomal S6 kinases (RSKs) with inhibitors or specific siRNAs has been shown to promote the degradation of P-gp and suppress its transport function (Katayama et al., 2007). It was later demonstrated in colorectal cancer cells that RSK1 is associated with UBE2R1 (CDC34), a member of the ubiquitin-conjugating enzyme (E2) family, and phosphorylated the E2 enzyme at Thr162. UBE2R1 phosphorylation by RSK1 induced self-ubiquitination and rapid degradation of the enzyme, protecting the ubiquitin-proteasomal degradation of P-gp (Katayama et al., 2016). Effects of kinase regulation of P-gp mediated by ubiquitination is also summarized in Fig. 1.
A series of studies using cell lines over-expressing OAT1 or OAT3 demonstrated that these OATs are ubiquitinated by Nedd4-2, a member of the HECT E3 ubiquitin ligase family. Ubiquitination of OATs leads to reduced function of the transporter and PKC regulation is involved in the process. By altering phosphorylation of Nedd4-2, PKC accelerated endocytosis and reduced cell surface level of the transporter in short-term activation (Xu et al., 2017) or targeted the transporter to proteolytic system for degradation in long-term activation (Xu et al., 2016b). Nedd4-2 is regulated by other protein kinases as well and may affect OAT function in different manners. In the case of OAT3, the phosphorylation of Nedd4-2 by SGK1 weaken the interaction between OAT3 and Nedd4-2, which reduced ubiquitination of the transporter and increased its level on the plasma membrane (Wang and You, 2017). SGK2, an isoform of SGK1, was found to directly associate with OAT1. Overexpression of SGK2 impaired interaction between Nedd4-2 and OAT1, enhancing cell surface expression and uptake function of the transporter (Xu et al., 2016a). AG490, a specific inhibitor of the Janus tyrosine kinase 2 (JAK2), was demonstrated to induce a time- and concentration-dependent inhibition of OAT3 transport activity. Such an effect is also related to Nedd4-2 phosphorylation. AG490 treatment led to reduced Nedd4-2 phosphorylation, which strengthened the association of Nedd4-2 with OAT3 and enhanced ubiquitination and degradation of the transporter (Zhang et al., 2018). The cross-talking between kinase regulation and ubiquitination in OATs is shown in Fig. 2.

Regulation of drug transporters of the SLC family by the cross-talking of PTMs. The effect of PKC or SGK2 on OAT1 is likely mediated by ubiquitination of the transporter, at which Nedd4-2 is involved. Nedd4-2 was also found to be phosphorylated by SGK1 or JAK2 and in turn, regulates OAT3 protein level and activity. The effect of PKA on OAT3 may be mediated by SUMOlyation. It should be noted that in most cases, a specific kind of PTM cross-talking is investigated with a specific cellular system or even an over-expressing cell line, so whether the phenomena observed are cell-type specific or generalizable remain unclear and further investigation is needed.
Although interplay studies mainly investigated how protein kinases affect drug transporters through other PTMs, it is likely that other PTMs may affect protein kinases and in turn regulate the function and/or protein level of drug transporters as well. For example, it has been demonstrated that PI3K/Akt pathway is involved in the regulation of P-gp and BCRP (Caetano-Pinto et al., 2017), hence activity change of the related kinases may affect the function of these transporters. It was found that K63-linked ubiquitination is essential for Thr308 phosphorylation of Akt (Wang et al., 2012); while the K48-linked ubiquitination acts as a turn-off switch for the kinase (Wu et al., 2011). Differentiated regulation of Akt by ubiquitination hence may result in opposite responses of the transporters.
Cross-talk with SUMOylation
SUMOylation may also interact with protein kinases in transporter regulation. It was found that activation of PKA with Bt2-cAMP accelerated recycling of OAT3 from intracellular compartments to the cell surface and decelerated degradation of the transporter. Immunoprecipitation analysis revealed that OAT3 was SUMOylated by SUMO-2 and SUMO-3 but not by SUMO-1. Moreover, increased SUMOylation of OAT3 by SUMO-2 was observed with Bt2-cAMP treatment and the enhanced SUMOylation was abrogated in the presence of PKA-specific inhibitor H-89. Therefore, cross-talk may exist between PKA and SUMOylation and coordinately regulate the function of OAT3 (Wang et al., 2019) (Fig. 2).
There is a possibility that SUMOlyation may also affect protein kinase and in turn regulate transporter function. For example, Pim-1 has a constitutively active conformation and no additional phosphorylation is required for its activation. Therefore, enzymatic activity of the kinase is almost exclusively determined by its protein level. Pim-1 was shown to be modified in vitro and in cultured cells through SUMOylation within a consensus SUMOylation motif (IK169DE171). SUMOylation induced ubiquitin-mediated degradation of Pim-1 by recruiting Really Interesting New Gene (RING) Finger Protein 4 (RNF4), a SUMO-targeted ubiquitin ligase. Interestingly, though SUMOylation promoted the degradation of Pim-1, it also enhanced the activity of the kinase in vitro (Iyer et al., 2017). Since Pim-1 has been shown to regulate drug transporters such as P-gp (Xie et al., 2010) and BCRP (Xie et al., 2008), changes in SUMOlyationit may likely affect function of these transporters through Pim-1.
Cross-talk with glycosylation
Glycosylation has been shown to affect the targeting and/or stability of drug transporters such as P-gp (Schinkel et al., 1993), BCRP (Nakagawa et al., 2009), OAT1 (Tanaka et al., 2004), and OATP1B1(Yao et al., 2012). Although report regarding the interaction between phosphorylation and glycosylation is lacking, cross-talking may exist in these PTMs as well. Phosphorylation is a long-known regulatory mechanism for the activity of essential glycosylation enzymes such as glycosyltransferse (McLawhon et al., 1981) and altered kinase activity may thus modulate the glycosylation process. On the other hand, N-glycosylation has been demonstrated to be essential for membrane interactions and structural arrangement of kinases such as EGFR (Kaszuba et al., 2015), which was shown to regulate the function of drug transporters including P-gp (Yang et al., 1997) and BCRP (Crawford et al., 2018). Therefore, the interplay between glycosylation and kinase regulation is well worth exploring in future studies.
Discussion
Although their important roles in the absorption, distribution, and excretion of drugs are unquestionable, our knowledge of post-transcriptional regulation of drug transporters is still quite limited. Altered kinase activity is commonly found in pathologic and/or pharmacological conditions, hence may lead to functional change of drug transporters through different molecular mechanisms. For example, compromised JAK3 expression in the intestine during obesity may lead to suppressed tyrosine phosphorylation of BCRP, resulting in reduced activity of the transporter (Mishra et al., 2019). In addition, protein kinases are the second largest group of drug targets after G protein-coupled receptors and more than sixty tyrosine kinase inhibitors (TKIs) are in clinical use so far. A recent study showed that 97.1% of patients treated with TKIs also concomitantly taking at least one other kind of drug. The median number of additional drugs was 4, and 47.4% of patients experienced at least once the DDI that was potentially mediated by TKIs (Ergen et al., 2019), suggesting that altered kinase activity leading to the functional change of drug transporters is highly likely in clinical applications. Moreover, protein kinases can modulate transporters through other PTMs. Phosphorylation may provide a proper conformation for the occurrence of other kinds of PTMs, or modulate important components involved in these PTMs, precisely regulating protein level, localization, stability, and function of transporters in response to various kinds of environmental cues. Information regarding drug transporter post-translational regulation is accumulating in recent years. However, the extent to which PTMs regulate transporters is far from being fully appreciated, especially for the interconnecting network among different PTMs. Pathologic and pharmacological conditions may affect other PTMs in addition to kinase-related phosphorylation. For example, N-glycosylation status of drug transporters such as MRP2, NTCP, OATP1B1, 1B3, and 2B1 has been shown to decrease in human non-alcoholic steatohepatitis (NASH) livers (Clarke et al., 2017) . As mentioned above, N-glycosylation is important for targeting and/or stability of different drug transporter, such a change in PTM may affect the function of these proteins during the pathologic condition. Also, the dysregulation of deubiquitinating enzymes (DUBs) has been shown in neurodegenerative diseases and cancers (Mcdonell et al., 2013; Murtaza et al., 2015), and several ubiquitination-related drugs are in preclinical investigation recently (Harrigan et al., 2018). Changes in glycosylation and/or ubiquitination status may interconnect with alteration of kinase expression and function, and work in concert to regulate the activity of different drug transporters. Therefore, a fully understanding of the coordinative relationship is essential to get insight of the regulatory mechanisms of drug transporters. More in-depth investigations are warranted for a comprehensive view of the highly orchestrated regulatory network, which will provide invaluable information for the evaluation of pharmacodynamic and pharmacokinetic responses of therapeutic agents and prediction of possible drug-drug interaction.
Finally, it should be pointed out that many of the PTM studies summarized in this review used a single cell line or even an over-expressing cellular system for the analysis, so whether the phenomena observed are generalizable remain unclear. Systematic studies using different cell lines and in vivo systems are needed to get a more complete picture of the regulatory mechanism(s) of drug transporters. Additionally, studies of kinase effects mostly relied on kinase inhibitors, which are small molecules that can directly interact with drug transporters. In such cases, altered transporter activity by these modulators is likely not related to their inhibitory role on kinase activity. Therefore, proper experimental design is required and caution should be taken in interpreting such kind of data.
Authorship Contributions
Participated in research design: Hong
Performed data analysis: Wang, Hong
Wrote or contributed to the writing of the manuscript: Hong
Footnotes
- Received August 5, 2022.
- Accepted October 3, 2022.
This work was supported by Natural Science Foundation of Guangdong Province [grant number 2022A1515010552] and National Natural Science Foundation of China [grant number U1832101 and 81373473] to MH. No author has an actual or perceived conflict of interest with the contents of this article.
Abbreviations
- BCRP
- breast cancer resistance protein
- MATE
- Multidrug and toxin extrusion
- P-gp
- P-glycoprotein
- OAT
- organic anion transporter
- OATP
- organic anion transporting polypeptide
- OCT
- organic cation transporter
- PTM
- post-translational modification
- Copyright © 2022 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}