


Abstract
N-Methyl-d-aspartate (NMDA) receptors are modulated by extracellular spermine and protons and are blocked in a voltage-dependent manner by spermine and polyamine derivatives such as N1-dansyl-spermine (N1-DnsSpm). The effects of mutations in the first and third transmembrane domains (M1 and M3) and the pore-forming loop (M2) of NMDA receptor subunits were studied. Surprisingly, some mutations in M2 and M3 of the NR1 subunit, including mutations at W608 and N616 in M2, reduced spermine stimulation and proton inhibition. These mutations may have long-range allosteric effects or may change spermine- and pH-dependent gating processes rather than directly affecting the binding sites for these modulators because spermine stimulation and proton inhibition are not voltage dependent and are thought to involve binding sites outside the pore-forming regions of the receptor. A number of mutations in M1–M3, including mutations at tryptophan and tyrosine residues near the extracellular sides of M1 and M3, reduced block by spermine and N1-DnsSpm. The effects of these mutants on channel block were characterized in detail by using N1-DnsSpm, which produces block but not stimulation of NMDA receptors. Block by N1-DnsSpm was studied by using voltage ramps analyzed with the Woodhull model of channel block. Mutations at W563 (in M1) and E621 (immediately after M2) in the NR1a subunit and at Y646 (in M3) and N616 (in the M2 loop) in the NR2B subunit reduced the affinity for N1-DnsSpm without affecting the voltage dependence of block. These residues may form part of a binding site for N1-DnsSpm. Mutation of a tryptophan residue at position W607 in the M2 region of NR2B greatly reduced block by N1-DnsSpm, and N1-DnsSpm could easily permeate channels containing this mutation. The results suggest that at least parts of the M1 and M3 segments contribute to the pore or vestibule of the NMDA channel and that a tryptophan in M2 (W607 in NR2B) may contribute to the narrow constriction of the pore.
A number of modulators and noncompetitive antagonists, including spermine and protons, affect the activation of NMDA receptors (1, 2), and there are complex interactions between some of these modulators (Fig.1A). Spermine has several macroscopic effects on NMDA receptors, including stimulation (Fig. 1A, 1 and 2) and voltage-dependent block (Fig. 1A, 3). Protons inhibit NMDA receptors (Fig. 1A, 5), with a tonic inhibition of ≈50% at physiological pH (3-5). The glycine-independent form of spermine stimulation (Fig. 1A, 1), seen with saturating concentrations of glycine, may involve relief of proton inhibition (Fig. 1A, 6), processes that are also influenced by the alternatively spliced insert encoded by exon 5 in the NR1 subunit (Fig. 1A, 9). Voltage-dependent inhibition by spermine is due to an open-channel block, and spermine can also permeate NMDA channels (Fig. 1A, 11). Spermine is a weak NMDA channel blocker, being active at high micromolar concentrations, but some polyamine derivatives, such as N1-DnsSpm, are potent channel blockers, being active at nanomolar to micromolar concentrations (1, 6). N1-DnsSpm is a useful new tool with which to study the structure and properties of glutamate receptor channels (6).

Modulation and mutagenesis of NMDA receptors. A, Diagram of NMDA receptor showing effects of spermine, N1-DnsSpm, pH, and ifenprodil. Numbers, various macroscopic effects and interactions of these modulators. B, Relative positions of M1–M4 segments in NR1 and NR2 subunits. NR1 and NR2 subunits have different-sized amino- and carboxyl-terminal domains.sp, signal peptide. Inset, proposed topology of glutamate receptor subunits, with an extracellular amino-terminal domain, an intracellular carboxyl-terminal domain, three membrane-spanning domains (M1, M3, and M4), and a reentrant loop (M2) that contributes to the ion-permeation pathway (10, 42, 43).Below schematic, amino acid sequences of NR1a (residues 561–650), NR2A (residues 556–648), and NR2B (residues 557–649) between the beginning of M1 and the end of M3.Vertical lines, amino acids are numbered at intervals of 10 residues. Horizontal lines above each sequence, positions of M1–M3. Gaps, introduced at three positions in NR1a to maximize homology with NR2A and NR2B.Dots above amino acid, positions at which mutations were studied; the particular mutations (W-to-L, D-to-N, E-to-Q, and so on) are shown below each amino acid. Residues are numbered from the initiator methionine in each subunit (20, 27).
NMDA receptors are hetero-oligomers composed of combinations of NR1 and NR2 subunits in as-yet undefined combinations and stoichiometries (7-9). The topology of NMDA receptor subunits may be similar to that proposed for α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and kainate receptor subunits and kainate binding proteins, with an extracellular amino-terminal domain, three membrane-spanning domains (M1, M3, and M4), and a reentrant loop (M2) that contributes to the ion channel pore (10, 11) (Fig. 1B). Mutations at asparagine residues, such as NR1a(N616Q), in the M2 regions of NR1 and NR2 subunits influence Ca2+ permeability and block by Mg2+, and these residues may form the narrowest constriction of the channel pore (Fig. 1A, 10) (12-17).
In this study, we initially set out to determine the role of residues in and around the M1–M3 regions of NMDA receptor subunits in voltage-dependent block by spermine and N1-DnsSpm. The focus of the experiments was on mutations at aspartate, glutamate, tryptophan, and tyrosine residues because these amino acids are involved in binding of spermidine to PotD, a bacterial polyamine binding protein (18, 19). Because spermine has multiple effects on NMDA receptors, we also examined stimulation by spermine and found, surprisingly, that some mutations in the M2 region reduced spermine stimulation. These mutations also reduced inhibition by protons, an effect that may be largely responsible for the loss of spermine stimulation seen at pH 7.5. A number of mutations in the M1–M3 regions of NR1 and NR2B were found to affect block by spermine and N1-DnsSpm. Residues near the extracellular side of the M1 and M3 regions, as well as residues in M2, form part of a binding site for N1-DnsSpm, suggesting that at least part of the M1 and M3 segments contributes to the pore or vestibule of the ion channel.
Experimental Procedures
cDNA clones and site-directed mutagenesis.
The wild-type NR1a and NR1b clones (20, 21) and the NR1a mutants E594Q, E595Q, E596Q, E597Q, E598Q, D599N, T602G, S604A, S605A, A606S, W608L, F609L, S610A, N616Q, and E621Q (15) were gifts from Dr. S. Nakanishi (Institute for Immunology, Kyoto University Faculty of Medicine, Kyoto, Japan). The NR1a(N616R) and NR1a(S617N) mutants (14) were gifts from Dr. R. J. Dingledine (Department of Pharmacology, Emory University School of Medicine, Atlanta, GA). The wild-type NR2A and NR2B clones (22) were gifts from Dr. P. H. Seeburg (Center for Molecular Biology, University of Heidelberg, Germany).
The NR1b(N637Q) mutant was prepared by inserting a 1.6-kbBglII fragment of NR1a(N616Q) into the corresponding sites of NR1b (which contains the 21-amino acid insert encoded by exon 5). The preparation of NR1a(N616G), NR2A(N614Q), NR2A(N615Q), NR2A(N615G), NR2B(N615Q), and NR2B(N616Q) has been previously described (6). Other NR1 mutants were prepared by using a 2.6-kbSphI/SalI fragment of plasmid pN60 (20) inserted into the same sites of M13mp18 (23). Similarly, the NR2 mutants were prepared using a 2.2-kb BamHI/XmaI fragment of pBSNR2A and a 2.1-kb BamHI/SphI fragment of NR2B inserted into the same sites of M13mp18 and M13mp19, respectively. Mutagenesis was carried out according to the method of Kunkel et al. (24) or Sayers et al. (25) with the Sculptorin vitro mutagenesis system (Amersham International, Buckinghamshire, UK). The oligonucleotides (antisense strand) for preparation of NR1a mutants were 5′-CTA CTA GCA ACA ACA GTG TGC TC-3′ for W563L, 5′-CGG TCC AGC AGGAGC AGC ATC ACA GC-3′ for Y578L, 5′-CTG AAG CGG TTC AGC AGG TAC-3′ for D581N, 5′-GCA GGA CGC CCA AGG AAA ACC AC-3′ for W611L, 5′-CGA AAC CAG CCA ACA CCA TGC CT-3′ for W636L, and 5′-AAG TTG GCA GTGAGG GAA GCC ACT AT-3′ for Y647L. The oligonucleotides (sense strand) for preparation of NR2 mutants were 5′-AAA AGC TAT ATT GCT CCT CTG GG-3′ for NR2A(W606L), 5′-CGC TGA CGT GTT GGT GAT GAT GTT-3′ for NR2B(W559L), 5′-CAA AGC AAT TTT GTT ACT CTG GG-3′ for NR2B(W607L), 5′-TTG GTT ACT CTT GGG TCT GGT GT-3′ for NR2B(W610L), and 5′-TCC TGG CCA GCT TGA CTG CCA ACT TAG-3′ for NR2B(Y646L). Mutated DNA fragments were isolated from the replicative form of M13 and religated into the corresponding sites of pN60, pBSNR2A, and pBSNR2B. Mutations were confirmed by DNA sequencing (26). Amino acids are numbered from the initiator methionine in NR1 and NR2 clones (20, 27) (Fig. 1B).
Expression in oocytes and voltage-clamp recording.
The preparation of cRNAs and the preparation, injection, and maintenance of oocytes were carried out as previously described (28-30). Oocytes were injected with NR1 plus NR2 cRNAs in a ratio of 1:5 (0.2–4 ng of NR1 plus 1–20 ng of NR2). Macroscopic currents were recorded with a two-electrode voltage-clamp using a GeneClamp 500 amplifier (Axon Instruments, Foster City, CA) or an OC-725 amplifier (Warner Instruments, Hamden, CT) as previously described (28, 30). Oocytes were continuously superfused with a saline solution consisting of 96 mm NaCl, 2 mm KCl, 1.8 mmBaCl2, and 10 mm HEPES, pH 7.5, and in most experiments, oocytes were injected with K+-1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (100 nl of 40 mm, pH 7.0–7.4) on the day of recording (28). To study the pH sensitivity of NMDA receptors, glutamate was applied in buffer at a particular pH with a 20–30-sec wash at the same pH before and after application of glutamate. I-V relationships were measured by using linear voltage ramps from −150 to +30 mV over 6 sec as previously described (6).
Data analysis.
Data analysis and curve fitting were carried out using Axograph (Axon Instruments) or SigmaPlot (Jandel Scientific, San Rafael, CA) on Macintosh computers. To obtain IC50 and Hill slope (nH
) values of antagonists, concentration-inhibition curves were fit to eq. 1: Equation 2where Iglu is the agonist-induced current, and Imax is the maximum response. For analysis of the voltage dependence of block by N1-DnsSpm, data were analyzed using the model of Woodhull (31) by fitting the data to eq. 3:
Equation 2where Iglu is the agonist-induced current, and Imax is the maximum response. For analysis of the voltage dependence of block by N1-DnsSpm, data were analyzed using the model of Woodhull (31) by fitting the data to eq. 3:
Materials.
Glutamate and glycine were purchased from Sigma Chemical (St. Louis, MO). Spermine tetrahydrochloride was purchased from Calbiochem (San Diego, CA). N1-DnsSpm was provided by Drs. J. Renault and N. Seiler (University of Rennes, France). Ifenprodil was from Synthélabo Recherche (Bagneux, France). Other reagents were from Sigma Chemical or Fisher Scientific (Pittsburgh, PA).
Results
Spermine stimulation and pH sensitivity.
Spermine has four macroscopic effects on NMDA receptors (Fig. 1A, 1–4) that can be studied in relative isolation by the use of particular subunit combinations and by manipulating the concentrations of glutamate and glycine and the holding potential (1, 32, 33). The glycine-independent form of spermine stimulation (Fig. 1A, 1) was studied at NR1a/NR2B receptors activated by saturating concentrations of glutamate and glycine in oocytes voltage-clamped at −20 mV (Figs.2A and 3A). A number of mutations in the M1–M3 regions of NR1a, including W563L, S610A, and W636L, produced a small increase in spermine stimulation. However, a surprising finding was that mutations at W608, W611, and N616 in M2 and at Y647 in M3 of NR1areduced or abolished spermine stimulation (Figs. 2A and 3A). In the case of N616, any of three mutations (N-to-Q, N-to-R, or N-to-G) reduced spermine stimulation. Mutations at the equivalent positions in the M2 and M3 regions of NR2B (W607L, W610L, N615Q, Y646L) had no effect or increased spermine stimulation (Fig. 3, A and B). Thus, the decrease in spermine stimulation seen with mutations in M2 and M3 is largely selective for the NR1a subunit.
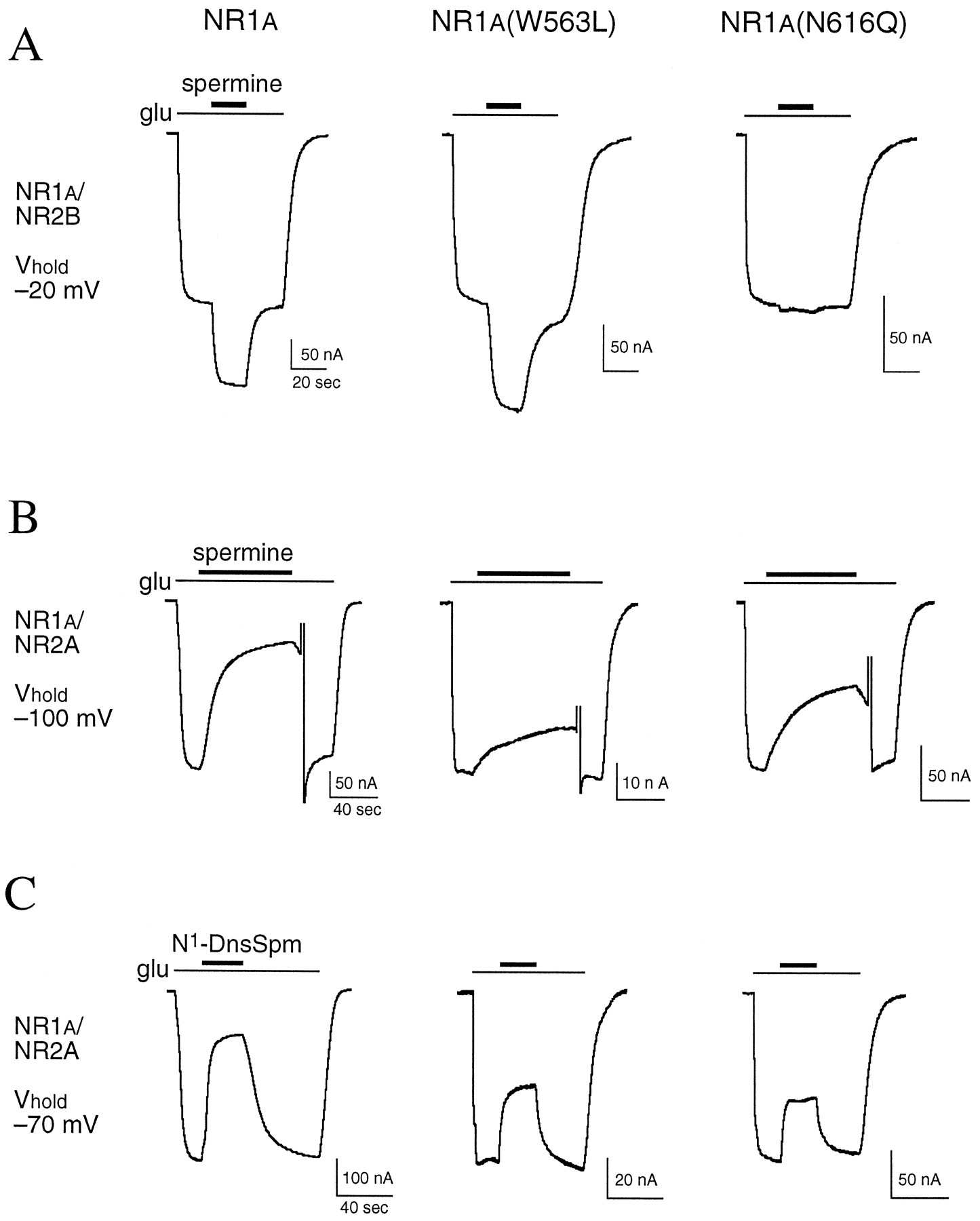
Screening mutant NMDA receptors. A, The effects of spermine (100 μm) on responses to glutamate (glu; 10 μm, with 10 μmglycine) were studied on oocytes expressing NR1a/NR2B receptors with wild-type or mutant NR1a subunits and voltage-clamped at −20 mV. B, The effects of spermine (10 μm) were studied on oocytes expressing NR1a/NR2A receptors with wild-type or mutant NR1a subunits and voltage-clamped at −100 mV. During recovery from block by spermine, the oocytes were briefly depolarized to +40 mV to speed recovery. C, The effects of N1-DnsSpm (1 μm) were determined in the same oocytes as shown in B, but oocytes were voltage-clamped at −70 mV. Horizontal calibration bars, 20 sec in A, 40 sec in B and C.

Screening mutant NMDA receptors. A, The effects of 100 μm spermine were determined at NR1a/NR2B receptors containing wild-type and mutant subunits in oocytes voltage-clamped at −20 mV. The effects of 10 μm spermine (B) and 1 μm N1-DnsSpm (C) were determined at NR1a/NR2A receptors containing wild-type and mutant subunits in oocytes voltage-clamped at −100 mV (spermine) or −70 mV (N1-DnsSpm). For experiments with NR1a mutants, the mutants were coexpressed with wild-type NR2B (A) or wild-type NR2A (B and C). Similarly, for experiments with NR2B mutants (A) and NR2A mutants (B and C), the mutants were coexpressed with wild-type NR1a. In all paradigms, NMDA receptors were activated by glutamate (10 μm; with 10 μm glycine), and currents measured in the presence of spermine and N1-DnsSpm are expressed as a percentage of the control response to glutamate in each oocyte. Solid horizontal line, control response to glutamate (100%). Dashed horizontal line, mean effect of spermine or N1-DnsSpm at wild-type NR1a/NR2 receptors. Values are mean ± standard error from 4–14 oocytes for each mutant and from 20–66 oocytes for wild-type receptors, which were measured in all batches of oocytes. †, p < 0.05, ∗, p < 0.01 compared with wild-type NR1a/NR2 receptors (one-way analysis of variance withpost hoc Dunnett’s test).
Because spermine can alter the affinity of NMDA receptors for glutamate and glycine (Fig. 1A, 2 and 4), a decrease in spermine stimulation would arise if the affinity for glutamate were reduced and an increase in stimulation would arise if the affinity for glycine were reduced (30). However, mutations in the M2 and M3 regions of NR1a that reduced spermine stimulation had no effect or produced a small increase in sensitivity to glutamate and glycine (Table 1). Thus, the effects of these mutants on sensitivity to glycine-independent spermine stimulation are not due to changes in the affinity for glutamate and glycine. Spermine produces another form of stimulation at NMDA receptors, “glycine-dependent stimulation,” which involves an increase in the affinity of the receptor for glycine (Fig. 1A, 2). Thus, a larger fold stimulation by spermine is seen in the presence of subsaturating concentrations of glycine. Experiments were carried out to determine whether glycine-dependent stimulation occurs at NR1a(N616Q)/NR2B receptors (at which glycine-independent stimulation is lost). The effects of spermine (100 μm) were determined using a saturating concentration of glycine (10 μm) and concentrations (0.1 μm for wild-type, 0.01 μm for N616Q) below the EC50 value for glycine (see Table 1). At wild-type NR1a/NR2B receptors, spermine potentiated glutamate currents by 35 ± 2% (10 μm glycine) and 108 ± 11% (0.1 μm glycine; six oocytes), and at NR1a(N616Q)/NR2B receptors, spermine potentiated glutamate currents by 2 ± 1% (10 μm glycine) and 100 ± 7% (0.01 μm glycine; six oocytes). Thus, glycine-dependent spermine stimulation is not reduced by the NR1a(N616Q) mutation.
Sensitivity of mutant NMDA receptors to glutamate and glycine
Stimulation by spermine is dependent on extracellular pH (5). NMDA receptors are inhibited by protons, and at pH 7.5 (the conditions used for the experiments represented in Figs. 2 and 3), spermine stimulation may involve relief of proton inhibition (Fig. 1A, 6). If mutations in NR1 changed pH sensitivity of the NMDA receptor (affecting mechanism 5, Fig. 1A), this could indirectly alter the effects of spermine (Fig. 1A, 6). We therefore carried out experiments to determine whether the mutants alter pH sensitivity (Fig.4). Mutations at W608, N616, and Y647 in NR1a significantly reduced proton inhibition, with the largest effect seen with the NR1a(N616Q) mutation (Fig. 4). Mutations at the equivalent positions in NR2B (W607L, N615Q, and Y646L), at nearby positions in NR2B (e.g., W610L, N616Q), or at a number of other residues in M1–M3 of NR1a (e.g., W563L, S617N, W636L) had no effect on pH sensitivity (Fig. 4C). We also tested the NR1a(N616Q) mutant coexpressed with NR2A (rather than NR2B). Similar to effects at NR1a/NR2B receptors, the NR1a(N616Q) mutation reduced pH sensitivity of NR1a/NR2A receptors (Fig. 4C). Thus, the effects of mutations at NR1a(N616Q) are independent of the type of NR2 subunit coexpressed with NR1a. In the NR2A subunit, mutation NR2A(N614Q) but not mutations at NR2A(N615) also produced a small change in pH sensitivity (Fig. 4C).

Proton inhibition of mutant NMDA receptors. A, Inward currents induced by glutamate (10 μm; with 10 μm glycine) at pH 8.5, 7.5, 7.0, and 6.5 in oocytes expressing NR1a/NR2B and NR1a(N616Q)/NR2B receptors and voltage-clamped at −70 mV. B, Inhibition by protons was determined using the experimental protocols shown in A. Values are mean ± standard error from seven to nine oocytes for each subunit combination. Responses to glutamate at a given pH (Iglu) are expressed as a fraction of the response to glutamate at pH 8.5 [Iglu(8.5)]. C, IC50 values for proton inhibition were determined by curve fitting of data from pH inhibition curves, similar to those shown in B, for various NR1/NR2 subunit combinations. For these analyses, the raw data (glutamate current versus pH) were fit to the equation Iglu = Imax/[1 + (pH/IC50)n], in which Iglu is the glutamate current at a given pH, Imax is the maximum glutamate-induced current, andn is the slope factor. Dashed line, mean IC50 value at the respective wild-type receptors (hatched bars). Values are mean ± standard error from 4–12 oocytes for each mutant and from 41 (NR1a/NR2B), 4 (NR1b/NR2B), and 21 (NR1a/NR2A) oocytes for wild-type receptors. ∗,p < 0.01 compared with respective wild-type receptor (one-way analysis of variance with post hocDunnett’s test, or unpaired t test).
Proton sensitivity of NMDA receptors is influenced by the 21-amino acid insert encoded by exon 5 in the NR1 subunit (Fig. 1A, 9). Splice variants of NR1 that contain the insert, such as NR1b, are less sensitive to proton inhibition than variants that lack the insert, such as NR1a (5). To determine whether the effects of mutations at N616 were influenced by the presence of the exon 5 insert, we measured the pH sensitivity of receptors containing an N-to-Q mutation at N637 in NR1b(which contains the exon 5 insert), a position equivalent to the N616Q mutation in NR1a (which lacks the insert). The pH sensitivity of wild-type NR1b/NR2B receptors was reduced compared with wild-type NR1a/NR2B receptors (5), and mutation NR1b(N637Q) produced a large decrease in pH sensitivity of NR1b/NR2B receptors, similar to the effects of the NR1a(N616Q) mutation at NR1a/NR2B receptors (Fig. 4C). Thus, the effects of mutations at N616 are independent of the exon 5 insert.
Experiments were carried out to determine whether a change in pH sensitivity could be responsible for the change in spermine stimulation seen with some mutations in NR1a. If spermine stimulation involves a relief of proton inhibition (Fig. 1A, 6) and the change in spermine stimulation is secondary to a change in pH sensitivity in mutant receptors, then spermine stimulation should be restored at mutants such NR1a(N616Q) by decreasing the extracellular pH. To test this hypothesis, we measured the effects of spermine at different pH in wild-type and mutant channels (Fig.5). At receptors containing the W608L, W611L, N616Q, and Y647L mutations in NR1a, spermine stimulation was partially restored at acidic pH (Fig. 5). The smallest effects of spermine were seen with the NR1a(N616Q) mutation, which has the largest effect on proton inhibition (Figs. 4and 5). Thus, interactions between pH and spermine remain intact at these mutants. The results suggest that spermine and protons may share a common binding site or common determinant of their coupling to channel gating.

Interactions of spermine and protons. A, Responses to glutamate (glu, 10 μm; with 10 μm glycine) were measured in the absence and presence of 100 μm spermine at different extracellular pH in oocytes voltage-clamped at −20 mV. Data are from the same oocyte for each subunit combination. Right, traces normalized to the glutamate response immediately before application of spermine at each pH. B, Effects of spermine were measured at different extracellular pH using the protocols shown in A at NR1a/NR2B receptors containing wild-type and mutant NR1a subunits. Responses measured in the presence of spermine are expressed as a percentage of the control response to glutamate at each pH. Values are mean ± standard error from four to six oocytes for each subunit combination.
Inhibition by the noncompetitive antagonist ifenprodil (28) was also studied at NR1a/NR2B receptors containing each of the NR1a mutants listed in Fig. 3. Mutations at NR1a(W608), NR1a(N616), and NR1a(Y647) produced small decreases in block by ifenprodil (data not shown). The effect on ifenprodil inhibition may be secondary to the changes in pH sensitivity seen with these NR1amutants because block by ifenprodil is dependent on extracellular pH (Fig. 1A, 7 and 8), and a decrease in proton inhibition would be predicted to reduce ifenprodil inhibition (34). Other NR1a mutations had no effect on inhibition by ifenprodil (data not shown).
Voltage-dependent block of NMDA receptors by extracellular Mg2+ is influenced by residue N616 in NR1a (13-15). Inhibition of native NMDA receptors by protons is not voltage dependent, and protons do not seem to act as classic channel blockers at NMDA receptors (3, 4). Because of the effects of NR1a(N616) mutations on proton inhibition, experiments were carried out to determine whether protons produce a voltage-dependent block of recombinant NMDA receptors. In one set of experiments, glutamate-induced currents were measured at different pH in oocytes voltage-clamped at −20, −70, and −100 mV, and in other experiments, I-V curves were constructed by using voltage ramps (−100 to +40 mV) at different extracellular pH (6.5–8.5). Using these paradigms, pH inhibition was not voltage dependent at NR1a/NR2B or NR1a(N616Q)/NR2B receptors (data not shown). To determine whether there is an interaction between protons and Mg2+ at NMDA channels, we measured block by 100 μm Mg2+ at different extracellular pH (7.0–8.5) in oocytes voltage-clamped at −20 to −70 mV and by using voltage ramps (Fig. 6). Block of NR1a/NR2B receptors by Mg2+was not affected by changes in pH when studied in oocytes voltage-clamped at different holding potentials (data not shown) or by voltage ramps (Fig. 6). Thus, over a pH range of 7.0–8.5 and over a voltage range of −100 to +40 mV, there is no apparent interaction between Mg2+ block and proton inhibition (Fig.6).

Block of NMDA channels by extracellular Mg2+ is not pH sensitive. A, I-V curves were constructed by voltage ramps (−100 to +40 mV over 4 sec) in an oocyte expressing NR1a/NR2B receptors activated by glutamate (glu, 10 μm; with 10 μmglycine) in the absence and presence of 100 μmMg2+ at pH 7.0, 7.5, and 8.5. Leak currents have been subtracted. (Note the different scales on the ordinate at each pH.) B, Glutamate currents measured in the presence of Mg2+ are expressed as a fraction of the control current at each pH. Data around the reversal potentials have been masked for presentation.
Block by spermine and N1-DnsSpm.
We initially screened block by spermine and N1-DnsSpm at NR1a mutants coexpressed with NR2A (Fig. 2, B and C, and Fig. 3, B and C). For these experiments, NR1a mutants were coexpressed with NR2A to avoid the complication of spermine stimulation, which occurs at NR1a/NR2B but not NR1a/NR2A receptors. Those analyses showed that mutations W563L, D581N, N616Q, N616R, and E621Q in NR1a reduced block by spermine and/or N1-DnsSpm (Fig. 3, B and C). In contrast, mutations N616G and Y647L in NR1a increased block by N1-DnsSpm.
To characterize in more detail the effects of some mutations on polyamine block, we measured I-V relationships for block by N1-DnsSpm at NR1a/NR2B receptors containing wild-type and mutant subunits (Fig.7; Table2). For these experiments, we used NR2B rather than NR2A to allow a more direct comparison of the effects of the mutants on block by N1-DnsSpm with their effects on spermine stimulation and pH sensitivity. N1-DnsSpm blocks but does not stimulate NR1a/NR2B receptors (6). For some of the NR1amutants, the effects of N1-DnsSpm were also determined when the mutants were coexpressed with NR2A rather than NR2B. In the case of NR1a mutants that had pronounced effects, we studied mutations at the equivalent positions in NR2B (Table 2).

Voltage-dependence of block by N1-DnsSpm. A, I-V relationships for responses to glutamate (10 μm; with 10 μm glycine) in the absence (control) and presence of 1 μm N1-DnsSpm were constructed by using voltage ramps (−150 to +30 mV over 6 sec;inset) with oocytes expressing NR1a/NR2B and NR1a(W563L)/NR2B receptors. Leak currents have been subtracted. B, Currents in the presence of N1-DnsSpm are expressed as a fraction of the glutamate-induced current (Iglu), over the range −16 to −110 mV. Values ofK d(0) and zδ were derived by fitting the data to text eq. 3. Smooth lines, fits.
Voltage-dependent block by N1-DnsSpm at mutant NMDA receptors
Examples of I-V curves measured by voltage ramps at NR1a/NR2B at NR1a(W563L)/NR2B receptors are shown in Fig. 7A. The data from these experiments were analyzed with the Woodhull model of voltage-dependent channel block (31) by fitting the fractional response measured in the presence of N1-DnsSpm to eq. 3 to obtain values for theKd(0) and zδ of N1-DnsSpm (Fig. 7B). As previously reported (6), block by N1-DnsSpm was steeply voltage dependent with a zδ value of 2.6–2.7 and aKd(0) value of 0.3–0.7 mm at wild-type NR1/NR2 channels (Fig. 7; Table2). Mutations W563L and E621Q in NR1a, which reduce block by N1-DnsSpm, increased theKd(0) value by 3–7 fold without affecting zδ, suggesting that these mutations reduce the affinity of binding of N1-DnsSpm. A similar effect was seen with mutations NR2B(N616Q) and NR2B(Y646L) (Table 2), again indicating a selective effect of these mutants on the affinity of binding of N1-DnsSpm. In the NR2A subunit, the residue at the position equivalent to NR2B(N616) is NR2A(N615). Similar to the NR2B(N616Q) mutation, an NR2A(N615Q) mutation increased theKd(0) value of N1-DnsSpm by ∼10-fold without changing zδ. Mutations at the positions equivalent or adjacent to NR2B(N616Q) and NR2B(Y646L) in NR1a (i.e., N616Q, S617N, and Y647L) had a different profile. The NR1a(N616Q) mutation reduced zδ (see also Ref. 6) and may interfere with the accessibility of one of the charged groups on N1-DnsSpm, whereas NR1a(Y647L) increased block by N1-DnsSpm due to a decrease inKd(0) (i.e., an increase in affinity) (Table 2). Mutations at NR2B(W559L) and NR2B(N615Q) also decreased zδ, similar to the NR1a(N616Q) mutation (Table2).
For some NR1a mutants that altered theKd(0) value of N1-DnsSpm, we also determined the IC50 value of N1-DnsSpm by measuring concentration-inhibition curves for steady state responses at −70 mV. The shift in the IC50 value seen in these experiments was similar to the change inKd(0) value determined using voltage ramps (Table 2). Thus, the IC50 value at NR1a(W563L)/NR2B (1.2 ± 0.2 μm; six oocytes) was increased by 6-fold and that at NR1a(Y647L)/NR2B (0.07 ± 0.01 μm; six oocytes) was decreased by 3-fold compared with the IC50 value at wild-type NR1a/NR2B receptors (0.20 ± 0.02 μm; eight oocytes). We have previously shown that the NR2B(N616Q) mutation increases the IC50value of N1-DnsSpm by 8-fold (6), similar to its effect on Kd(0) (Table 2). Thus, a change in the potency of N1-DnsSpm at these mutants can be accounted for entirely by a change inKd(0) value.
Some mutations in M1–M3 that altered block by spermine and N1-DnsSpm also increased the potencies of glutamate and glycine. These mutations, which include NR1a(W563L), NR1a(N616Q), and NR1a(Y647L), could alter the gating of NMDA channels or the coupling of agonist binding to channel gating, thereby increasing the apparent affinities for glutamate and glycine (Table 1). Thus, some residues that interact with channel blockers such as N1-DnsSpm may also participate in gating of the channels.
Block by 0.1 μm or 1 μmN1-DnsSpm was measured by voltage ramps at all of the subunit combinations listed in Table 2, and all of the mutants, with the exception of NR2B(W607L), showed a steeply voltage-dependent block that was well fit by eq. 3. At wild-type and mutant NR1/NR2B receptors, with the exception of NR1a/NR2B(W607L) and NR1a(N616G)/NR2B, there was little or no relief from block at extreme hyperpolarized potentials down to −150 mV. Examples are shown in Fig.8A for wild-type NR1a/NR2B and NR1a(W608L)/NR2B channels. The NR1a(W608L) mutation is in a position equivalent to the NR2B(W607L) mutation. However, at NR1a/NR2B(W607L) receptors, N1-DnsSpm produced a very shallow voltage-dependent block that was incomplete, with some recovery of the glutamate current at extreme negative membrane potentials (Fig. 8A). The recovery from block by N1-DnsSpm at NR1a/NR2B(W607L) receptors is shown quantitatively in Fig.8B, in which the fractional block at −50, −100, and −145 mV is shown for wild-type and mutant receptors. At NR1a/NR2B and NR1a(W608L)/NR2B receptors, block by N1-DnsSpm was almost complete at −100 mV and showed no recovery at −145 mV. At NR1a/NR2B(W607L) receptors, block by 1 μm N1-DnsSpm at −50 mV was similar to that at wild-type NR1a/NR2B receptors, but there was only a modest increase in block at −100 mV and some recovery of the glutamate-induced current at −145 mV (Fig.8B). A similar effect was seen with 10 μm (rather than 1 μm) N1-DnsSpm (data not shown). The shallow slope conductance and partial recovery from block at NR1a/NR2B(W607L) receptors are reminiscent of the effects of spermine at wild-type receptors (35, 36) and of N1-DnsSpm at receptors containing NR1a(N616G) (Fig. 8) or NR2A(N615G), at which N1-DnsSpm seems to permeate the NMDA channel (6). At receptors containing NR1a(N616G), there is a clear region of negative slope conductance for the block by N1-DnsSpm (Fig. 8). However, with NR2B(W607L), the slope conductance was very shallow, and the data were not well fit by the Woodhull model using eq. 3 because recovery from block developed very easily, presumably due to permeation of N1-DnsSpm.
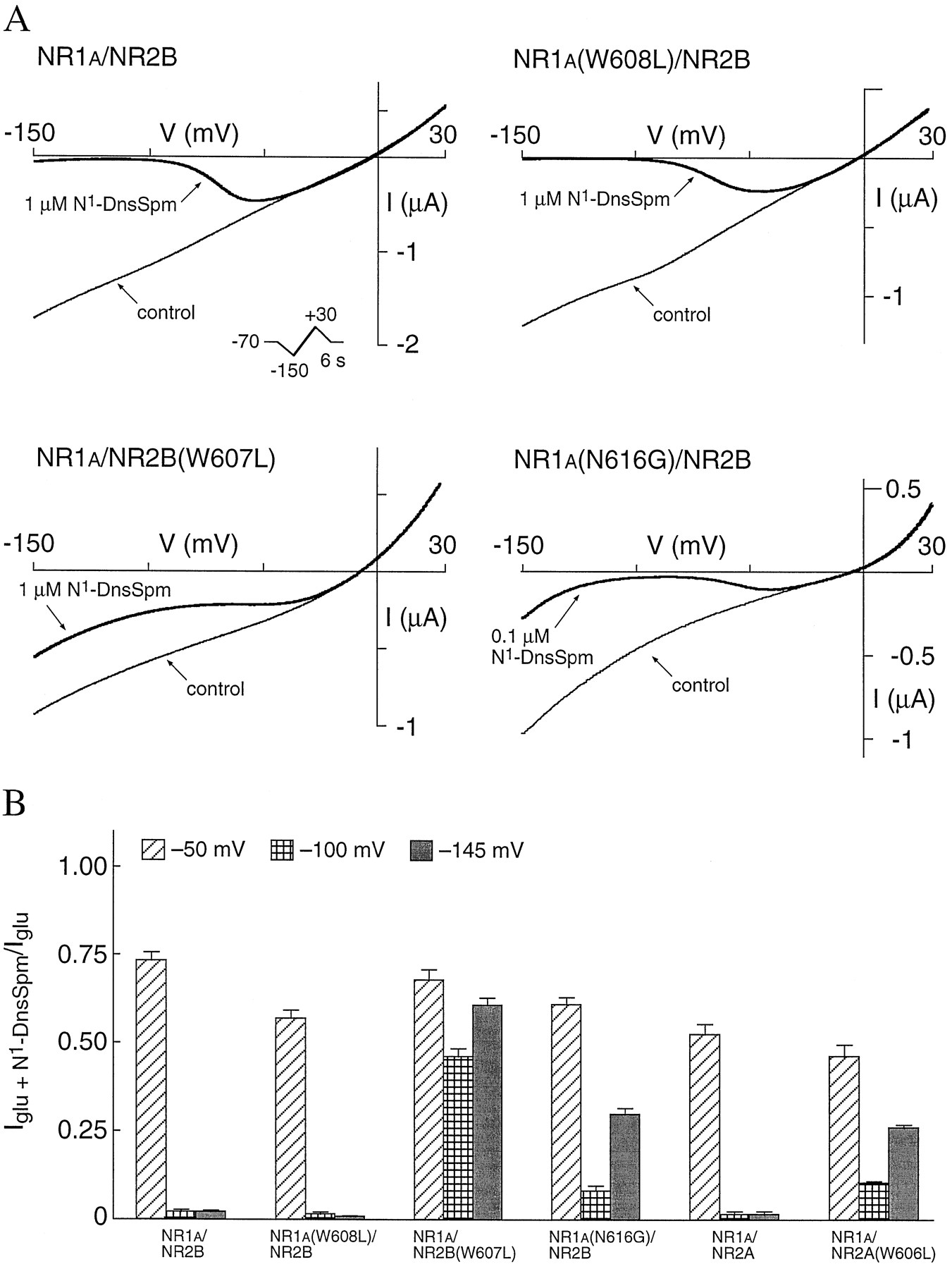
N1-DnsSpm permeates NR1a/NR2B(W607L) and NR1a/NR2A(W606L) channels. A, I-V relationships for responses to glutamate (10 μm; with 10 μm glycine) in the absence (control) and presence of 1 μm or 0.1 μm N1-DnsSpm were constructed by using voltage ramps (−150 to +30 mV over 6 sec;inset) on oocytes expressing wild-type and mutant receptors. Leak currents have been subtracted. B, Currents in the presence of N1-DnsSpm are expressed as a fraction of the glutamate-induced current (Iglu) at −50, −100, and −145 mV at each receptor type. Values are mean ± standard error from 7–15 oocytes for each subunit combination. Note the incomplete block and very shallow slope conductance with NR1a/NR2B(W607L) in A and the partial recovery from block at −145 mV with NR1a/NR2B(W607L), NR1a(N616G)/NR2B, and NR1a/NR2A(W606L) in B.
Because of the properties seen with NR2B(W607L), we also studied a W-to-L mutation at the equivalent position in NR2A, NR2A(W606L). Block and permeation of N1-DnsSpm were studied by using voltage ramps at NR1a/NR2A and NR1a/NR2A(W606L) channels (Fig. 8B). There was little or no permeation of N1-DnsSpm at wild-type NR1a/NR2A channels, but, similar to the effects seen with the NR2B(W607L) mutation, there was recovery from block (presumably reflecting permeation of N1-DnsSpm) at receptors containing NR2A(W606L) (Fig. 8B). Block by N1-DnsSpm at NR1a/NR2A(W606L) channels was more pronounced than the block at NR1a/NR2B(W607L) channels, and the portion of the I-V curve that showed a steep voltage-dependence could be fit to the Woodhull model (eq. 3). With this analysis, the NR2A(W606L) mutation had no significant effect on the values of eitherKd(0) or zδ of block by N1-DnsSpm (Table 2).
Discussion
The major effects of the mutations that we studied are summarized in Fig. 9. Mutations at some residues in M2, in particular at the critical asparagine residues, including NR1a(N616) and NR2B(N615), have previously been shown to affect permeation and block by inorganic divalent cations (13-17). Here, we extended characterization of the pore-forming M2 region by determining the effects of mutations in M2 on modulation and block by polyamines and protons. In addition, residues in the M1 and M3 regions that affect sensitivity to channel blockers and modulators have been identified.

Summary of the effects of mutations in M1–M3. A, The effects of mutations at residues in NR1a and, in some cases, at the corresponding position in NR2B are summarized.Upward arrows, an increase. Downward arrows, a decrease. Dashes, no effect of the mutation on a particular experimental paradigm. Thickness of the arrows, relative magnitude of the effects. In the case of N1-DnsSpm, the effects of mutations on the values ofK d(0) and zδ are included to facilitate comparisons of the mechanisms that may influence block. Thus, a decrease in block by N1-DnsSpm at receptors containing NR1a(W563L) is due entirely to an increase in K d(0), whereas a decrease in block at receptors containing NR1a(N616Q) is due to a decrease in voltage-dependence (zδ). nd, not determined. ∗, Mutation NR2B(W607L) greatly increased the apparent permeation of N1-DnsSpm, and block at NR1a/NR2B(W607L) channels was very shallow. The equivalent mutation in NR2A, NR2A(W606L), also increased permeation of N1-DnsSpm, and the mutation had no significant effect on the values of K d(0) and zδ (Table 2). B, Putative transmembrane topology of NR1 and NR2B subunits and the relative positions of the key residues described in this study (•). ○, Positions of residues E342 and D669 in NR1, which influence spermine stimulation and proton inhibition (32, 33). The binding sites for glutamate (Glu) and glycine (Gly) are formed by regions in the distal part of the amino-terminal domain and the M3–M4 loop (37, 38). Mutations at residues such as NR1a(N616) and NR1a(Y647) may alter spermine stimulation, proton inhibition, and sensitivity to glutamate and glycine by disrupting channel gating and/or by long-range allosteric effects that alter the binding and coupling of these agents. Because of their effects on the affinity [i.e.,K d(0)] of block by N1-DnsSpm, residues NR1a(W563), NR2B(N616), and NR2B(Y646) are proposed to form part of a binding site for N1-DnsSpm and to contribute directly to the pore or vestibule of the ion channel. Other residues, including NR2B(W559), NR1a(N616), and NR1a(Y647), may influence or interfere with binding of N1-DnsSpm. Residue W607 in NR2B may contribute to a narrow constriction in the pore and/or form part of the binding site for N1-DnsSpm because a NR2B(W607L) mutation greatly increases permeation of N1-DnsSpm.
Block by spermine is voltage dependent and presumably involves a spermine binding site within the channel pore. In contrast, spermine stimulation and proton inhibition are not voltage dependent, and these effects are unlikely to involve direct interactions of spermine and protons with residues in the pore-forming regions of the channel. Thus, a surprising finding was that mutations at W608, W611, and N616 in M2 and at Y647 in M3 of NR1a altered spermine stimulation and proton inhibition. In the case of mutations at W608, N616, and Y647 in NR1a, a decrease in spermine stimulation was correlated with a decrease in proton inhibition. These effects are similar to those of mutations at residues E342 and D669, which are located in the extracellular domains of NR1a (32, 33) (Fig. 9B). The change in sensitivity to spermine may be secondary to a change in pH sensitivity if spermine functions to relieve proton inhibition (mechanism 6, Fig. 1A) (5). Alternatively, the mutations may disrupt a common determinant that couples the spermine and proton binding sites to channel gating. In this case, the effects of spermine and protons would not be functionally interdependent, and spermine stimulation would not involve a relief of proton inhibition (i.e., mechanism 6, Fig. 1A, would not exist). Rather, the separate and opposing effects of spermine and protons may be mediated through a structural motif that involves residues W608, N616, and Y647 in NR1a or at least through a motif that is disrupted by mutations at these residues.
With the NR1a(N616) mutants, the largest effect on pH sensitivity was seen with an N-to-Q mutation, whereas N-to-R or N-to-G mutations at this position produced smaller changes in pH sensitivity. This profile is different from the effects of N616 mutations on Mg2+ block, in which substitution of the positively charged R residue produces the largest reduction in Mg2+ block, and on cation permeability, in which the N-to-G mutation produces the largest increase in pore size (12-15). Proton inhibition was reduced in receptors containing an N-to-Q mutation at N637 in the NR1b variant of NR1, similar to the reduction seen in receptors with an N-to-Q mutation at the equivalent position, N616, in NR1a. The NR1bvariant contains the exon 5 insert, which itself reduces proton inhibition, possibly by interacting with a proton sensor on the receptor. These observations, together with the lack of voltage-dependence of proton inhibition and the lack of interaction of protons and Mg2+, suggest that NR1a(N616) does not contribute directly to a proton sensor. Mutations at N616, and at other residues in M2 and M3, may indirectly alter sensitivity to spermine and pH through subtle disruptions of the subunit protein structure or disruptions to gating processes. Changes in gating produced by mutations at residues in the M1–M3 region could also account for the increase in sensitivity to glutamate and glycine seen with these mutants. The glutamate and glycine binding sites are thought to be formed by residues in the extracellular amino-terminal and M3–M4 loop regions (37, 38) (Fig. 9B), and residues in M1–M3 are unlikely to directly affect agonist binding. Indeed, the NR1a(N616Q) mutation was recently shown to affect the coupling of cation permeation to channel gating (39), which may also account for some of the effects of this mutant seen in the current study.
The effects of mutations in M2 and M3 on pH sensitivity were selective for the NR1 subunit. Mutations at W608, N616, and Y647 in NR1a reduced pH sensitivity, whereas mutations at the equivalent or adjacent positions in NR2B did not (Fig. 9). Thus, mutations in NR1 may affect a proton sensor and a spermine binding site located on the NR1 subunit. It is notable that mutations at NR1a(N616) produce receptors with a complex pharmacological phenotype. Thus, in addition to effects on block and permeation of divalent cations, mutations at NR1a(N616) produce changes in pH sensitivity and agonist potency, which should be kept in mind when evaluating the macroscopic effects of mutations at this position.
Block by spermine and N1-DnsSpm was influenced by mutations in M1–M3. Using the Woodhull model of voltage-dependent channel block (31), we determined the effects of the mutations on theKd(0) and zδ of N1-DnsSpm block. In this analysis, z is the valence of N1-DnsSpm, which is +3 at pH 7.5 (6), and δ is the fraction of the membrane electrical field sensed by the blocker. As previously shown (6), block by N1-DnsSpm was strongly voltage dependent with a zδ of 2.6–2.7 at wild-type channels. This suggests that the binding site is very deep within the channel, although there are a number of potential limitations to use of this model to analyze block by N1-DnsSpm. For example, it is not known if only one molecule of N1-DnsSpm enters the channel at one time or if the entire polyamine tail on the molecule enters the membrane field (6). Nevertheless, the data derived from voltage ramps were well-fit by eq. 3, and these analyses provide some information about amino acid residues that seem to form part of a polyamine binding site and residues that may interfere with block by polyamines.
The proposed topology of the M1–M3 regions of NR1 and NR2 subunits (40-43) and the proposed positions of residues in the M1–M3 regions are shown in Fig. 9B. Thus, M2 is proposed to be a hair-pin loop with the critical asparagine residues that form the narrowest part of the pore (N616 in NR1 and NR2B) near the top of the loop and NR1a(E621), immediately after M2, at the cytoplasmic side of the pore. Residues W563 and Y647 in NR1a are close to the extracellular ends of M1 and M3, respectively, as are the corresponding residues, W559 and Y646, in NR2B. Residues at which mutations selectively increase theKd(0) value (i.e., decrease the affinity) of N1-DnsSpm may contribute directly to a binding site for N1-DnsSpm; these include W563 and E621 in NR1a, and N616 and Y646 in NR2B. Interestingly, the corresponding residues in the other subunit have different effects. For example, mutation NR1a(W563L) increased theKd(0) value, whereas mutation NR2B(W559L) did not. A Y-to-L mutation at NR1a(Y647L), equivalent to NR2B(Y646L), actually increased N1-DnsSpm block by increasing the affinity of binding (Fig. 9). This mutation, however, did not affect block by spermine. Thus, residue NR1a(Y647) may normally interfere with binding of the head group of N1-DnsSpm at wild-type channels, possibly through steric hindrance because the Y-to-L mutation reduces the size of the side chain at this position. Residues N616 in NR1a and N615 and W607 in NR2B may also normally interfere with binding of N1-DnsSpm because mutations at those positions alter the zδ and/or apparent permeation of N1-DnsSpm. Some residues in the pore-forming region that are involved in binding channel blockers may also play a role in gating processes because mutations at W563, N616, and Y647 in NR1 increased sensitivity to glutamate and glycine (Fig. 9A).
We previously found that an N-to-G mutation at NR1a(N616G) or NR2A(N615G) increased the potency of N1-DnsSpm and that N1-DnsSpm could easily permeate the channel of receptors containing these mutants. This is presumably because the N-to-G mutation increases pore size, allowing passage of the bulky naphthalene head group of N1-DnsSpm (6). These results are consistent with the finding, based on the permeability of organic cations, that the asparagine residues seem to form the narrowest part of the channel (12). An effect of the second (N616) rather than the first (N615) asparagine residue in the M2 region of NR2B on block by N1-DnsSpm (Fig. 9A) is consistent with a staggered position of the M2 regions in NR1 and NR2 subunits, with the second asparagine in NR2 having a function similar to that of N616 in NR1a (12) (Fig. 9B). The W-to-L mutation at NR2B(W607L) produced channels with a very shallow and incomplete block by N1-DnsSpm, suggesting that N1-DnsSpm can easily permeate these channels. Thus, NR2B(W607) may also form part of the narrowest constriction of the channel, and the W-to-L mutation may increase the size of the pore, allowing passage of N1-DnsSpm. Importantly, a W-to-L mutation at the equivalent position (W608) in NR1adid not affect block by N1-DnsSpm, whereas a mutation at the equivalent position in NR2A, NR2A(W606L), did increase permeation of N1-DnsSpm. This suggests that only in NR2 subunits is the tryptophan residue at this position critical for permeation of N1-DnsSpm. The NR2B(W607L) mutation is presumably sufficient to make the pore large enough to allow passage of the bulky-head group of N1-DnsSpm.
In a model of the structure of M2 based on cysteine-accessibility mutagenesis, the tryptophan residues in the NR1 and NR2C subunits at positions equivalent to NR2B(W607) were proposed to face the lumen of the channel (40); this is consistent with a direct interaction of NR2B(W607) with N1-DnsSpm in the channel pore. However, in the model proposed by Kuner et al. (40), the narrow constriction of the channel is formed by the critical asparagine residues, and the residues at positions equivalent to NR2B(W607) lie some distance below the narrow constriction. Indeed, the tryptophan residue in NR2C was accessible to cytoplasmic but not to extracellular thiol reagents after cysteine mutagenesis (40). Thus, mutations at the tryptophan residue (W607 in NR2B, W606 in NR2A) would not be expected to alter permeation of blockers, such as N1-DnsSpm, entering the channel from the extracellular side.
Although it is difficult to reconcile our data with the data of Kuneret al. (12, 40), there are a number of possibilities that could account for the effects of the NR2B(W607L) mutant. First, residue NR2B(W607) may contribute to the narrow constriction of the channel together with the asparagines in NR1 and NR2 subunits. This would require that W607 lies in the same plane as the asparagines rather than below them, as in the model by Kuner et al. In this case, all or part of the M2 loop may exist parallel to the plane of the membrane rather than perpendicular to it, as was proposed for the structure of the loop region in a cyclic nucleotide-gated channel (44). Second, the effects of W607L may reflect changes in the binding of N1-DnsSpm rather than changes in pore size. In this case, the relief from block at extreme negative potentials would reflect some process other than permeation of N1-DnsSpm. This seems unlikely because N-to-G mutations such as NR1a(N616G), which increase pore size (12), also increase permeation of N1-DnsSpm as determined by the relief of block at extreme negative potentials (6). Furthermore, the NR2A(W606L) mutation, which increased the permeation of N1-DnsSpm, did not affect the values of eitherKd(0) or zδ for block by N1-DnsSpm. This suggests that the binding and unbinding of N1-DnsSpm within NR1a/NR2A(W606L) channels remain intact and that the NR2A(W606L) mutation alters the ability of N1-DnsSpm to pass through the channel. Finally, we cannot exclude the possibility that the NR2B(W607L) and NR2A(W606L) mutations disrupt the asparagine residues further along the M2 loop or cause a general disruption of the secondary structure or positioning of the M2 loop, which could affect block and permeation of N1-DnsSpm. However, this seems unlikely given the lack of effect of a corresponding mutation in the NR1 subunit, NR1a(W608L).
Acknowledgments
We are grateful to Drs. S. Nakanishi and P. H. Seeburg for providing the wild-type NR1 and NR2 clones, to Drs. S. Nakanishi and R. J. Dingledine for providing some of the NR1 mutants, to James Chao for technical assistance with some experiments, and to Drs. J. Renault and N. Seiler for providing N1-DnsSpm.
Footnotes
- Received April 7, 1997.
- Accepted June 19, 1997.
-
Send reprint requests to: Dr. Keith Williams, Department of Pharmacology, University of Pennsylvania School of Medicine, Philadelphia, PA 19104-6084.
-
This work was supported by United States Public Health Service Grant NS35047 from the National Institute of Neurological Disorders and Stroke, a Grant-in-Aid from the American Heart Association, a Grant-in-Aid from the Tokyo Biochemical Research Foundation, and a grant from the Japan Health Science Foundation.
Abbreviations
- NMDA
- N-methyl-d-aspartate
- N1-DnsSpm
- N1-dansyl-spermine
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- I-V
- current-voltage
- The American Society for Pharmacology and Experimental Therapeutics
References
MolPharm articles become freely available 12 months after publication, and remain freely available for 5 years.Non-open access articles that fall outside this five year window are available only to institutional subscribers and current ASPET members, or through the article purchase feature at the bottom of the page.
|





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}