


Abstract
Zn2+ is abundant in the brain, where it plays a role in the function of a number of enzymes, structural proteins, and transcription factors. Zn2+ is also found in synaptic vesicles and is released into synapses achieving concentrations in the range of 100 to 300 μM [Proc Natl Acad Sci USA1997;94:13386–13387; Mol Pharmacol1997;51:1015–1023]. Therefore, Zn2+may play a physiological role in regulating the function of postsynaptic channels and receptors. We characterized the effect of Zn2+ on the functional properties of the β2-adrenergic receptor (β2AR). We found that physiological concentrations of Zn2+ increased agonist affinity and enhanced cAMP accumulation stimulated by submaximal concentrations of the βAR agonist isoproterenol. These results provide evidence that Zn2+ released at nerve terminals may modulate signals generated by the β2AR in vivo.
Several groups have used Zn2+ together with engineered metal ion binding sites as a method to study G protein-coupled receptor (GPCR) structure (Sheikh et al., 1996, 1999; Thirstrup et al., 1996;Elling et al., 1999). In these studies, Zn2+ is used as a ligand for a binding pocket introduced into a receptor by one or more histidine substitutions. This method has provided insight into the proximity and relative orientation of transmembrane domains (Thirstrup et al., 1996), as well as the mechanism of receptor activation (Sheikh et al., 1996, 1999; Elling et al., 1999). However, there is considerable evidence that Zn2+ may be a physiological regulator of receptor function. Zn2+ is an abundant ion in the central nervous system (Schetz et al., 1999) and may be present at high enough concentrations in specific synapses to have a physiological role in regulating GPCR function in vivo. The β2-adrenergic receptor (β2AR) mediates adrenergic responses in both the central nervous system and the sympathetic nervous system. We therefore examined the effects of Zn2+ on β2AR function. At low concentrations (1–20 μM), Zn2+ increases agonist affinity and enhances cAMP accumulation in response to submaximal concentrations of a β-agonist. At high concentrations (>500 μM), Zn2+ alters both K Dand B max values for the antagonist dihydroalprenolol (DHA). These results demonstrate that Zn2+ is a positive allosteric modulator of agonist binding for the β2AR and suggest that Zn2+ may be a physiologically relevant regulator of β2AR function in vivo.
Experimental Procedures
Materials.
[3H]DHA (111.8 Ci/mmol) and guanosine-5′-O-(3-[35S]thio)triphosphate (GTPγS; 1250 Ci/mmol) were purchased from PerkinElmer Life Sciences (Boston, MA). Unlabeled GTPγS was purchased from Roche Molecular Biochemicals (Indianapolis, IN). GDP, zinc chloride, isoproterenol (ISO), and alprenolol were purchased from Sigma (St. Louis, MO). Nickel sulfate was obtained from Aldrich Chemical (Milwaukee, WI). Cobalt chloride was from Mallinckrodt (Chesterfield, MO). EDTA (disodium salt) was purchased from Fisher Scientific (Fair Lawn, NJ). Baculovirus expression vector pVL1392 and BaculoGold transfection kit were obtained from BD PharMingen (San Diego, CA). SF 900 II medium was obtained from Invitrogen (Carlsbad, CA). Fetal calf serum was obtained from Gemini (Calabases, CA) and gentamicin was obtained from Roche Molecular Biochemicals (Mannheim, Germany). Glass fiber filters (GF/C filters) and nitrocellulose filters were purchased from Schleicher & Schuell (Keene, NH).
Membrane Preparation.
For infection, Sf9 cells were sedimented for 2 h at 1g and suspended in fresh medium. Cells were seeded at 3.0 × 106 cells/ml, infected with recombinant baculovirus for the β2AR and/or membrane-tethered Gsα (tetGsα), and cultured for 48 h. All the membrane preparation steps were done at 4°C, as described elsewhere (Lee et al., 1999). Cells were harvested by centrifugation (10 min at 10,000g), washed once with phosphate-buffered saline and recentrifuged, and then resuspended in lysis buffer (10 mM Tris-HCl, pH 7.4, with 1 mM EDTA) and lysed using 25 strokes of a Dounce homogenizer. Nuclei and unbroken cells were removed by centrifugation (5 min at 500g). The supernatant was removed and centrifuged (30 min at 40,000g). The resulting pellet was resuspended in 20 ml of lysis buffer (10 mM Tris-HCl, pH 7.4, alone) and recentrifuged. Membranes were resuspended at 0.5 to 1.5 mg of protein/ml in binding buffer (75 mM Tris-HCl, pH 7.4) and stored at −80°C until use.
Urea Treatment of Membranes.
Membranes were extracted with 7 M urea to remove G protein subunits (Lim and Neubig, 2001). The urea solution was prepared by dissolution of urea crystals in a buffer containing 50 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 1 mM EDTA with protease inhibitors (25 μg/ml leupeptin and 16 μg/ml benzamidine) at room temperature and then kept on ice until use. Sf9 membranes expressing the β2AR were pelleted (40,000g for 10 min) and resuspended to ∼1 mg/ml in freshly prepared 7 M urea solution. The membranes were homogenized 10 times and incubated for 30 min with stirring. The membranes were centrifuged at 40,000g for 30 min then washed with 75 mM Tris-HCl, pH 7.4, containing protease inhibitors. The final pellet was resuspended to a final concentration of ∼1 mg/ml and stored in aliquots at −80°C.
Expression and Purification of β2AR Receptor.
The human β2AR, epitope tagged at the amino terminus with the FLAG eptiope (Sigma) and tagged at the carboxy terminus with six histidines, was expressed in Sf9 cells and purified as described previously (Kobilka, 1995).
Membrane Binding Assays.
Antagonist binding assays were done with membranes expressing β2AR. Membranes (25 μg of protein) were suspended in 500 μl of binding buffer incubated with different concentrations of divalent ions in the presence of 1 nM [3H]DHA. Saturation binding experiments were done on the β2AR expressed in Sf9 membranes. Membranes (50 μg of protein) were suspended in 500 μl of binding buffer supplemented with 100 pM to 10 nM [3H]dihydroalprenolol and 0.2% bovine serum albumin. The binding buffer contained only 75 mM Tris-HCl, pH 7.5. Nonspecific binding was assessed with 10 μM alprenolol. Incubations were performed for 1 h at room temperature with shaking at 230 rpm. Competition binding experiments were carried out with 1 nM [3H]dihydroalprenolol in the presence of increasing concentrations of (−)-isoproterenol and different concentrations of divalent ions.
Solubilized Binding Assays.
Binding assays on purified, detergent-solubilized receptor were carried out in 100-μl volumes in high-salt buffer (20 mM Tris-HCl, pH 7.5, 500 mM NaCl, and 0.1% n-dodecyl maltoside). The binding assays were stopped and free [3H]DHA separated from bound by desalting on 2-ml Sephadex G50 column (4 × 0.5 cm) by using ice-cold high-salt buffer. Nonspecific binding was determined in the presence of 10 μM alprenolol.
Dissociation Rate Kinetic Assay.
The effect of zinc on the rate of antagonist dissociation from β2AR was examined by measuring the k off in the absence and presence of 1 mM Zn2+. Membranes were suspended in 75 mM Tris-HCl, pH 7.5, with 1 nM [3H]DHA for 30 min at room temperature (shaking at 230 rpm). At time zero, total binding was determined and a saturating amount of cold alprenolol (final 10−5M) or cold alprenolol (final 10−5 M) and ZnCl2 (final concentration, 1 mM) was added to tubes containing membranes and [3H]DHA. Bound [3H]DHA was measured at 5-min intervals.
[35S]GTPγS Binding.
β2AR with tetGsα were coexpressed in Sf9 cells and membranes were prepared as described above. Membranes were pelleted by a 15-min centrifugation at 4°C at 15,000g and resuspended in buffer containing 75 mM Tris-HCl, pH 7.5. Sf9 membranes (10 μg of protein/tube) were suspended in 500 μl of magnesium binding buffer (75 mM Tris-HCl, pH 7.5, and 1 mM MgCl2) supplemented with 0.05% (w/v) bovine serum albumin, 1 nM [35S]GTPγS (0.25 μCi/tube), 1 μM GDP with or without 10 μM isoproterenol in presence or absence of Zn2+. Incubations were performed at 25°C and shaking at 250 rpm for 1 h. Nonspecific binding was determined in the presence of 10 μM GTPγS and was less than 0.2% of total binding. Bound [35S]GTPγS was separated from free [35S]GTPγS by filtration through GF/C filters, followed by three washes with 3 ml of cold magnesium binding buffer. Filter-bound radioactivity was determined by liquid scintillation counting.
cAMP Accumulation.
The production of cAMP was determined by adenylyl cyclase activation FlashPlate assay (PerkinElmer Life Sciences), in which 96-well plates are coated with solid scintillant to which anti-cAMP antibody has been bound. Briefly, HEK293 cells expressing human β2AR were detached and washed four times in 1× phosphate-buffered saline without Ca2+/Mg2+, and then resuspended to a density of approximately 2 × 106 cells/ml in stimulation buffer (1× phosphate-buffered saline, without calcium/magnesium, with 700 μM 3-isobutyl-1-methylxanthine, 0.1% protease-free bovine serum albumin, and 0.09% chloroacetamide) from PerkinElmer Life Sciences. Ligands (25 μl each) were diluted in Milli-Q water with various concentrations and dispensed to the flashplate. Resuspended whole cells (50 μl) were added to the ligand-loaded plate and stimulated at 37°C for 10 min before lysing cells with 100 μl of Detection Buffer (Invitrogen) containing 125I-cAMP, permeabilizer, and 0.09% sodium azide as provided by the manufacturer. After 2 h of incubation at room temperature, radioactivity was counted. To determine the concentrations of cAMP in the sample, cAMP standards were run in the same plate and expressed as picomoles per well.
Miscellaneous.
Protein was determined using the DC protein assay kit (Bio-Rad, Hercules, CA). Data were analyzed by nonlinear regression analysis with Prism program (GraphPad Software, San Diego, CA).
Results
Effect of Zn2+ on Antagonist Binding.
To examine the effect of Zn2+ on antagonist binding, we performed radioligand binding studies by using [3H]DHA, a neutral antagonist. As shown in Fig.1A, a significant effect of Zn2+ on DHA binding was observed only at concentrations of Zn2+ >500 μM, where DHA binding was reduced by 40 ± 2% (Fig. 1A). Saturation binding studies in the presence or absence of 1 mM Zn2+caused an increase of nearly 3-fold in apparentK D value for DHA (1.4 ± 0.2 nM control, 4.9 ± 1.2 nM with Zn2+) with a 40% decrease in B max (Fig. 1B). These results suggest that Zn2+ may function as a noncompetitive blocker of antagonist binding; however, we found that 1 mM Zn2+ slowed the rate of DHA dissociation (Fig.1C). Moreover, the effect of Zn2+ on antagonist binding is not fully reversible in membrane-bound β2AR (Fig. 2A). Note that Zn2+ also inhibits binding of antagonist to purified, detergent-solubilized β2AR; however, this inhibition is almost completely reversible after chelation of Zn2+ by EDTA (Fig. 2B). These results suggest that some of the effects of Zn2+ on antagonist binding may be caused by nonspecific effects of Zn2+ on phospholipids.
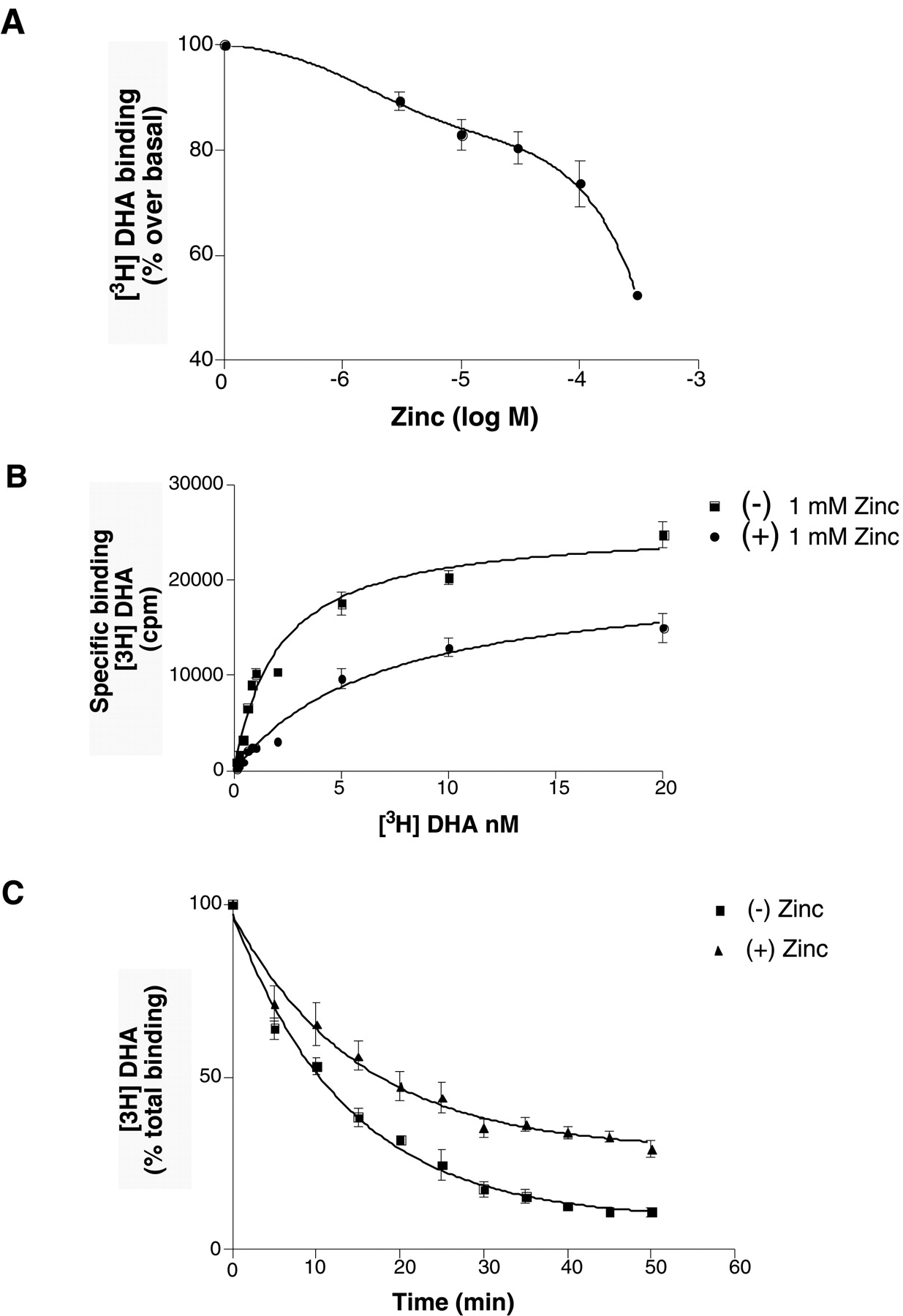
Effect of zinc on antagonist binding in membranes expressing β2AR. A, inhibition of [3H]DHA binding to β2AR by zinc. Sf9 membranes expressing β2AR (15.2 pmol/mg) were incubated with different concentrations of zinc in the presence of 1 nM [3H]DHA. The DHA binding in the absence of zinc is referred to as basal binding and was set to 100%. Maximum inhibition of antagonist binding was around 1 mM Zn2+. B, saturation binding of [3H]DHA (100 pM–20 nM) on membranes expressing β2AR (8.7 pmol/mg) in the presence and absence of 1 mM Zn2+. Zn2+ caused an increase of nearly 3-fold in apparent K D value for DHA (1.489 ± 0.23 nM control, 4.877 ± 1.178 nM with Zn2+) and ∼40% decrease in B max value. Data were best fit to monophasic saturation hyperbolae. These data are representative of three different experiments done in triplicate. C, effect of zinc on the rate of antagonist dissociation from β2AR. Dissociation rate kinetics measured for [3H]DHA binding to membranes expressing β2AR (7 pmol/mg) was determined in the presence (triangles) or absence (squares) of 1 mM Zn2+ as described under Experimental Procedures. Data shown are the mean ± S.D. of three independent experiments performed in triplicate.

Reversibility of the Zn2+ effect on antagonist binding. Membrane-bound (A) or purified β2AR (B) was preincubated with either 1 mM ZnCl2, 5 mM EDTA, or nothing for 30 min on ice. After the pretreatment period, radioligand (2 nM [3H]DHA) and either 5 mM EDTA or nothing was added and incubated for 1 h at room temperature. The membranes were then harvested by rapid filtration and soluble receptor was harvested by gel filtration as described under Experimental Procedures. Data shown are the mean ± S.D. of three independent experiments performed either in duplicate for purified protein or triplicate for membranes.
Effect of Zn2+ on High- and Low-Affinity Agonist Binding.
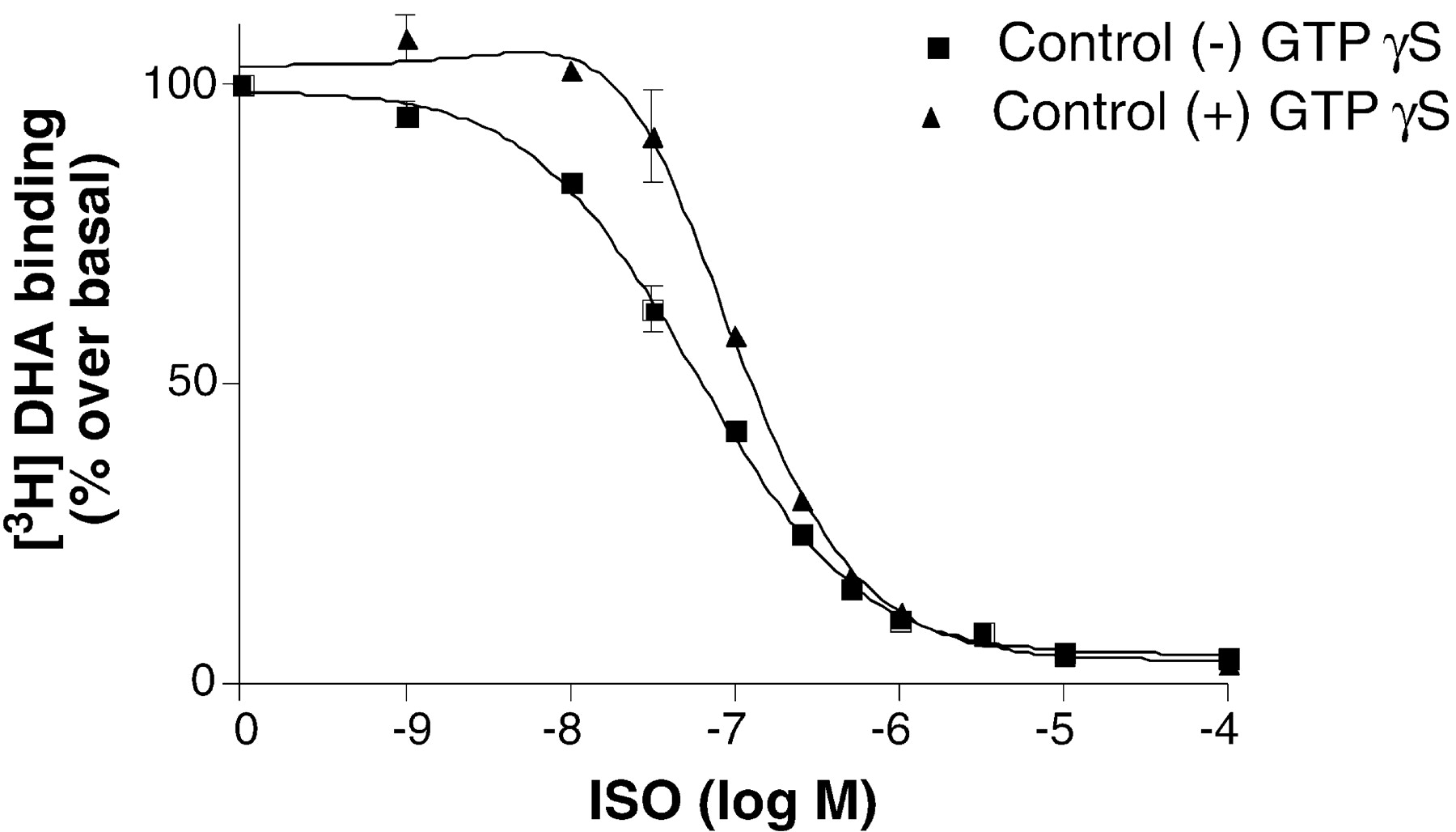
We examined the effect of Zn2+ on agonist binding in membranes expressing the β2AR with and without Gsα. For these experiments, we used tetGsα. We have shown previously that tetGsα couples more efficiently to β2AR than does wild-type Gsα (Lee et al., 1999). In membranes expressing the β2AR with tetGsα, we observed a biphasic curve with a high-affinity, GTPγS-sensitive component (Fig. 3). To study the effect of Zn2+ on agonist binding affinity, isoproterenol competition binding studies were done using 1 nM antagonist [3H]DHA in the presence of varying concentrations of Zn2+ (Fig.4, A–C). As shown in Fig. 4A, as little as 20 μM Zn2+ resulted in a change of the apparent biphasic nature of the competition curve with an increase inK H. We also observed a decrease inK L, which was even greater at 100 μM Zn2+ (Fig. 4B), a concentration at which we observed no effects on antagonist binding. A smaller decrease inK L was observed at 1 mM Zn2+ (Fig. 4C), a concentration that also reduces antagonist affinity. We observed similar effects on agonist binding affinity in membranes expressing the β2AR without tetGsα, where only a small amount of high-affinity binding is detected (Fig.5A), as well as in membranes treated with 7 M urea to remove heterotrimeric G proteins (Fig. 5B). Thus, at relatively low concentrations, Zn2+ induces an increase in agonist affinity for the uncoupled receptor.

Effect of GTPγS on isoproterenol competition of DHA binding to Sf9 membranes expressing β2AR together with tetGsα. Isoproterenol competition in presence and absence of unlabeled GTPγS (10 μM) was performed as described underExperimental Procedures. Reaction mixtures contained 10 μg of protein/tube. The membranes expressing the protein were 15.7 pmol/mg. Data shown are the mean ± S.D. of three independent experiments performed in triplicate. Binding data were analyzed for best fit to two affinity states.

Modulation of agonist binding by Zn2+. A to C, isoproterenol competition binding was performed on membranes expressing β2AR (12.7 pmol/mg) together with tetGsα in the presence of 20 μM Zn2+ (A), 100 μM Zn2+ (B), and 1 mM Zn2+ (C). The antagonist binding observed in the absence of any competitor was set to 100%. In A to C, binding in the absence of Zn2+ is shown for reference. In the presence of 20 to 100 μM Zn2+, GTPγS-sensitive high-affinity binding is lost, yet the affinity of the low-affinity binding is increased. Reaction mixtures contained 1 nM [3H]DHA and isoproterenol at the indicated concentrations. Binding data were analyzed for best fit to two affinity states. Data represent the mean of three independent experiments. Each experiment was performed in triplicate.

Effect of Zn2+ on agonist binding does not depend on Gs. A, effect of 20 μM Zn2+ on agonist binding affinity in Sf9 membranes expressing β2AR without tetGsα. Isoproterenol competition binding experiments were performed as described under Experimental Procedures. The competition binding was performed with 1 nM [3H]DHA in the presence of different concentrations of isoproterenol with and without 20 μM Zn2+. B, competition binding experiments performed on membranes treated with 7 M urea to remove G proteins. Urea treatment of membranes is described underExperimental Procedures. Experiments shown in A and B above were done on membranes expressing the β2AR with a carboxyl-terminal hexahistidine sequence. C, effect of 20 μM Zn2+ on agonist binding affinity in membranes expressing β2AR without a carboxy-terminal His tag. Data represent the mean of two independent experiments. Each experiment was done in triplicate.
The receptor used in the studies described thus far contains a hexahistidine tail on the carboxyl terminus to facilitate receptor purification. However, we observed identical effects of Zn2+ on antagonist binding (data not shown) and on agonist binding in membranes expressing a β2AR without a carboxyl-terminal hexahistidine sequence (Fig. 5C). Therefore, binding of Zn2+ to this carboxyl-terminal hexahistidine sequence is not responsible for the observed effects of Zn2+ on receptor function.
To further investigate the effect of Zn2+ on the GTPγS-insensitive agonist binding affinity, we performed a modified competition binding experiment in which 100 nM isoproterenol competes for binding sites with 1 nM [3H]DHA in the presence of 10 μM GTPγS and varying concentrations of Zn2+ (Fig. 6A). Also shown is the effect of Zn2+ on [3H]DHA binding in the absence of isoproterenol. In the presence of increasing concentrations of Zn2+, 100 nM isoproterenol becomes more effective at displacing [3H]DHA. The maximal effect of Zn2+ on isoproterenol affinity occurs at ∼30 μM with an IC50 of 3.0 μM. Similar results were obtained with membranes that had been extracted with 7 M urea to remove G proteins (Fig. 6B).

Effect of zinc on the agonist affinity. A, effect of Zn2+ on [3H]DHA binding in presence and absence of 100 nM isoproterenol was determined using Sf9 membranes expressing β2AR (7 pmol/mg). Assay mixtures contained ∼25 μg of membrane protein, 1 nM [3H]DHA, and 10 μM GTPγS. Nonspecific binding was less than 10% of total binding. Data represent the mean ± S.D. of three independent experiments. Each of these experiments was performed in triplicate. B, effect of Zn2+ on agonist affinity in urea-treated membranes expressing β2AR (12.6 pmol/mg). The reaction mixtures contained ∼20 μg of membrane protein and 2 nM [3H]DHA. Data represent the mean ± S.D. of two independent experiments. Each of these experiments was performed in triplicate. C, effect of Zn2+ on agonist affinity in purified, detergent-solubilized β2AR. Soluble binding was performed as described under Experimental Procedures. The assay mixture contained purified receptor, with or without 1 μM isoproterenol, and 2 nM [3H]DHA, in the absence or presence of the indicated concentrations of zinc. Data obtained were from three independent experiments done in duplicate. All the binding data were analyzed for best fit to two affinity states.
To confirm that the Zn2+-mediated increase in agonist affinity was caused by a direct interaction between Zn2+ and the β2AR, we examined the influence of Zn2+ on purified, detergent-solubilized β2AR. As shown in Fig.6C, Zn2+ enhanced the ability of 1 μM isoproterenol to displace [3H]DHA in a soluble binding assay.
Effects of Ni2+ and Co2+ on Agonist and Antagonist Binding.
To determine the specificity of the functional effects of Zn2+ on receptor function we compared the effect of Ni2+ and Co2+on agonist and antagonist binding (Fig.7). Co2+ had an effect on agonist affinity similar to Zn2+, but no significant effect on antagonist affinity (Fig. 7A). Ni2+ had no significant effect on either agonist or antagonist affinity (Fig. 7B).

Effect of divalent ions on the agonist affinity. The effect of Co2+ (A) and Ni2+ (B) on [3H]DHA binding in presence and absence of 100 nM isoproterenol was determined using Sf9 membranes expressing β2AR (7 pmol/mg). Assay mixtures contained ∼25 μg of membrane protein, 1 nM [3H]DHA, and 10 μM GTPγS. Nonspecific binding was less than 10% of total binding. Data represent the mean ± S.D. of three independent experiments. Each of these experiments was performed in triplicate. Binding data were analyzed for best fit to two affinity states.
Inhibition of Gs Function by Zn2+.
Our competition binding studies (Fig. 4) suggest that Zn2+uncouples the receptor from Gs. To further examine the effect of Zn2+ on the interaction of the β2AR and Gsα, receptor-mediated GTPγS binding was performed in Sf9 membranes coexpressing β2AR and tetGsα. Consistent with the results of Sheikh et al. (1999), we found that Zn2+ inhibited stimulation of GTPγS binding by the agonist isoproterenol with an IC50 of 7.2 μM (Fig.8). However, we also found that Zn2+ inhibited basal GTPγS binding to purified Gsα (data not shown), suggesting that the uncoupling of β2AR and Gs is caused by a direct effect of Zn2+ on Gsα, possibly by competing for the Mg2+ binding site.

Effect of Zn2+ on [35S]GTPγS binding in Sf9 membranes coexpressing β2AR (12.7 pmol/mg) and tetGsα. GTPγS binding was done on Sf9 membranes in presence of different concentrations of zinc. (−)-Isoproterenol (10 μM) was used as agonist. Zn2+ inhibits receptor-stimulated GTPγS binding to tetGsα (IC50 of 7.2 μM) (left). The effect of Zn2+ on GTPγS binding to membranes expressing tetGsα alone (right).
Effect of Zn2+ on Isoproterenol-Stimulated cAMP Accumulation.
The studies described above were done on membrane fragments or purified receptor in which Zn2+ has equal access to both cytoplasmic and extracellular domains of the β2AR. Therefore, they do not provide information about the location of the Zn2+binding site(s) responsible for the functional effects of Zn2+ on agonist and antagonist binding. In vivo, Zn2+ released from synaptic vesicles would have access to extracellular domains of the receptor. Diffusion or transport of Zn2+ across the plasma membrane would limit access to intracellular domains. We therefore examined the effect of Zn2+ on isoproterenol-stimulated cAMP accumulation in intact HEK293 cells expressing the wild-type (non–histidine-tagged) β2AR. Isoproterenol dose-response studies revealed that at Zn2+concentrations 100 μM or greater the maximal cAMP accumulation was decreased (Fig. 9A). When these data were plotted as the percentage of the maximal isoproterenol-stimulated cAMP, we observed a small decrease in EC50 in the presence of 1 μM Zn2+ (Fig. 9B). To further examine the effect of Zn2+ on isoproterenol-stimulated cAMP accumulation, cells were exposed to increasing concentrations of Zn2+ in the presence of 0 to 1 nM isoproterenol. Maximal cAMP accumulation was observed at 1 μM Zn2+ in the presence of isoproterenol, but Zn2+ had no effect in the absence of isoproterenol (Fig. 9C). Moreover, Zn2+ did not augment cAMP accumulation stimulated by either 10 or 100 μM forskolin (Fig. 9D).

Effect of zinc on isoproterenol-stimulated cAMP accumulation in HEK293 cells. A, ISO dose-response studies were done in HEK293 cells expressing nonhistidine-tagged β2AR in the presence of 0, 1, and 1000 μM Zn2+. The cAMP assay was performed as described under Experimental Procedures. B, data in A replotted as the percentage of the maximal isoproterenol-stimulated cAMP to facilitate comparison of the effects of Zn2+ on the EC50 for isoproterenol. C, Zn2+ dose-response studies done in the presence of 0, 0.1, 0.3, 0.6, and 1 nM ISO. D, Zn2+ dose-response studies done in the presence of 10 and 100 μM forskolin. Data represented are from three independent experiments performed in triplicate.
Discussion
Our results demonstrate that at micromolar concentrations, Zn2+ is a positive allosteric modulator of agonist binding; at higher concentrations, Zn2+has complex effects on antagonist binding. Allosteric modulation of ligand binding has been observed for several GPCRs. These modulators include physiologically relevant ions as well as small organic molecules. A large number of compounds allosterically increase binding affinity for muscarinic receptor antagonists. Allosteric regulation can be demonstrated in all five subtypes of muscarinic receptors, but the m2 receptor seems to be the most sensitive (Tucek and Proska, 1995). Although most allosteric modulators for the muscarinic receptors primarily affect antagonist binding, allosteric modulation of agonist binding has also been described previously (Jakubik et al., 1997).
In addition to the muscarinic receptor, allosteric compounds have been identified for the A1 adenosine receptor. The 2-amino-3-benzoylthiophene PD 81,723 has been shown to enhance agonist binding and G protein coupling in a Mg2+-dependent manner (Bhattacharya and Linden, 1995; Musser et al., 1999).
The function of several Gi-coupled receptors has been shown to be modulated by amiloride analogs and by Na+. This effect has been particularly well characterized for the α2-adrenergic receptor (Motulsky and Insel, 1983; Limbird, 1984; Horstman et al., 1990) and for the dopamine D2 (Neve, 1991) and D4 subtypes (Schetz and Sibley, 2001). Na+ both uncouples the receptor from Gi and reduces agonist affinity. The Na+-sensitive site has been identified in the α2-adrenergic receptor (Horstman et al., 1990) and the D4 dopamine receptor (Schetz and Sibley, 2001) as a conserved Asp residue within the cytoplasmic side of transmembrane 2. This Na+ sensitivity may be physiologically relevant, because relatively high local concentrations may be achieved near the cytoplasmic side of the receptor after membrane depolarization.
Recently, the function of the calcium receptor has been shown to be positively modulated by certain l-amino acids (Conigrave et al., 2000a,b). This may be physiologically relevant because nutrient and Ca2+ homeostasis may be regulated in a coordinate manner.
Zn2+ is an abundant divalent cation in the central nervous system and is released from some synaptic vesicles (Schetz et al., 1999; Weiss et al., 2000). Zn2+has been shown to be a noncompetitive blocker of dopamine uptake by the dopamine transporter (Norregaard et al., 1998) and modulates the function of several ligand-gated ion channels. Glycine-induced currents in freshly dissociated rat dorsal motor nucleus neurons are potentiated at low concentrations of Zn2+ (<3 μM) but inhibited at higher concentrations (>10 μM) (Doi et al., 1999). Zn2+ inhibits γ-aminobutyratergic currents by slowing the transition rate from closed to open and by accelerating the deactivation kinetics (Barberis et al., 2000). Of particular interest is the observation that relatively low concentrations of Zn2+ (∼10 μM) potentiate both agonist binding and peak current in the 5-hydroxytryptamine 3 receptor (Hubbard and Lummis, 2000).
Less is known about the regulation of G protein-coupled receptor function by Zn2+. At relatively high concentrations (>100 μM), Zn2+ has been shown to inhibit antagonist binding to the dopamine receptor (D1, D2, and D4 subtypes) (Schetz and Sibley, 1997, 2001). However, the effect of Zn2+ on agonist binding or G protein activation was not examined. In the case of the D4 dopamine receptor, the Zn2+ binding site that alters antagonist binding is different from the Na+ binding site (Schetz and Sibley, 2001). Zn2+ at high concentrations (>1 mM) also inhibits binding to the M1 muscarinic receptor (Lu and Hulme, 2000) and the μ-opioid receptor (Thirstrup et al., 1996). However, to our knowledge, there has been no report of Zn2+ increasing agonist affinity at these or other GPCRs.
Our findings with the human β2AR constitute the first report of Zn2+ as a positive allosteric modulator of agonist binding for a GPCR. The effect of Zn2+ on agonist binding is caused by a direct effect of Zn2+ on the β2AR, because it was observed in membranes treated with 7 M urea, a concentration shown previously to strip membranes of both α and βγ G protein subunits (Lim and Neubig, 2001) (Fig. 5B). Moreover, we observed that Zn2+enhanced agonist affinity in purified β2AR (Fig. 6B).
While enhancing agonist affinity, Zn2+ seems to uncouple the receptor from Gs, probably due to a direct effect of Zn2+ on Gs, because Zn2+inhibited basal GTPγS binding to purified Gs (data not shown). GTP binds to Gαs as a GTP-Mg2+ complex (Birnbaumer and Birnbaumer, 1995). Zn2+ may form a complex with GTP or interact directly with the Mg2+binding site on Gαs. However, in intact cells, we observed a small increase in isoproterenol-stimulated cAMP accumulation (Fig. 9B). This stimulatory effect of Zn2+ on cAMP accumulation is most probably caused by a direct effect of Zn2+ on the β2AR, rather than on downstream signaling components, for several reasons. The plasma membrane limits access of Zn2+ to intracellular signaling components. We observe no effect of Zn2+ in the absence of the β-agonist. As discussed above, the effect of Zn2+ on Gs is inhibitory and occurs at Zn2+ concentrations greater than 1 μM. This inhibitory effect may explain the inhibition of cAMP accumulation that we observe at higher Zn2+ concentrations (Fig. 9). Moreover, Zn2+ has been reported to be an inhibitor of adenylyl cyclase (Tesmer et al., 1999). Finally, the cAMP accumulation experiments were done in the presence of the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine, and Zn2+did not augment cAMP-stimulated by forskolin (Fig. 9D). Therefore, the effect of Zn2+ cannot be explained by inhibition of cAMP hydrolysis. Therefore, we would predict that the Zn2+ enhances cAMP accumulation by an interaction with an extracellular domain of the β2AR, possibly the same domain responsible for the positive allosteric effect on agonist binding.
The effect of Zn2+ on antagonist binding seems to be biphasic (Figs. 1A and 5A). A small (<20%) decrease in antagonist binding occurs with an IC50 comparable with that for the Zn2+ effect on agonist affinity (∼5 μM; Fig. 5A); however, a larger effect is observed at concentrations >100 μM. At 1 mM Zn2+, bothB max and K Dvalues for [3H]DHA binding are altered, consistent with the idea that Zn2+ is a noncompetitive blocker of this antagonist. However, Zn2+ also slows the rate of [3H]DHA dissociation (Fig. 1C), and the effects of 1 mM Zn2+ on antagonist binding are not fully reversible in membrane-bound receptor (Fig. 2A). The fact that binding is almost completely reversible in purified, detergent-soluble receptor suggests that some of the effects of Zn2+ on antagonist binding may be caused by interactions between Zn2+ and phospholipids. Previous studies showed that Zn2+ can alter the properties of phospholipids by interactions with the polar head groups (Binder et al., 2001). Thus, the effects of Zn2+ on antagonist binding are complex and cannot be explained by noncompetitive inhibition. However, because these effects occur only at relatively high concentrations of Zn2+ (>100 μM), they are not likely to be physiologically important.
Our results are consistent with the existence of at least two Zn2+ binding sites in the β2AR, one primarily affecting agonist binding and one primarily affecting antagonist binding. This is further supported by the differential effects of other divalent cations on agonist and antagonist binding. Like Zn2+, Co2+ enhances agonist binding but has no significant effect on antagonist binding (Fig. 7A). Ni2+ has only a small effect on antagonist binding, but no significant effect on agonist binding (Fig. 7B). Thus, the divalent cation binding site influencing agonist binding can accommodate Zn2+ and Co2+, but not Ni2+, whereas the binding site influencing antagonist binding accommodates Zn2+and to a much lesser extent Ni2+, but not Co2+.
In conclusion, Zn2+ has complex effects on the functional properties of the β2AR. Zn2+ binding to a high affinity site (IC50 of ∼5 μM) enhances agonist affinity and agonist-stimulated cAMP accumulation. Zn2+binding to a low-affinity site (IC50 of >500 μM) inhibits antagonist binding, yet slows antagonist dissociation. The effects of Zn2+ on agonist binding and cAMP accumulation occur at concentrations of Zn2+ that may be achieved within a synapse. Thus, Zn2+ may be a physiological modulator of β2AR function.
Footnotes
- Received April 12, 2001.
- Accepted September 26, 2001.
-
This work was supported in part by National Institutes of Health grant 5-RO1-NS28471 and the Mathers Charitable Foundation.
Abbreviations
- β2AR
- β2-adrenergic receptor
- DHA
- [3H]dihydroalprenolol
- GTPγS
- guanosine-5′-O-(3-thio)triphosphate
- ISO
- (−)-isoproterenol
- GPCR
- G protein-coupled receptor
- NDM
- n-dodecyl maltoside
- HEK
- human embryonic kidney
- PD 81,723
- (2-amino-4,5-dimethyl-3-thienyl)-[3-(trifluoromethyl)phenyl]methanone
- The American Society for Pharmacology and Experimental Therapeutics
References
MolPharm articles become freely available 12 months after publication, and remain freely available for 5 years.Non-open access articles that fall outside this five year window are available only to institutional subscribers and current ASPET members, or through the article purchase feature at the bottom of the page.
|





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}