


Abstract
In this study, the patch-clamp technique was used to determine the effects of galantamine, a cholinesterase inhibitor and a nicotinic allosteric potentiating ligand (APL) used for treatment of Alzheimer's disease, on synaptic transmission in brain slices. In rat hippocampal and human cerebral cortical slices, 1 μM galantamine, acting as a nicotinic APL, increased γ-aminobutyric acid (GABA) release triggered by 10 μM acetylcholine (ACh). Likewise, 1 μM galantamine, acting as an APL on presynaptically located nicotinic receptors (nAChRs) that are tonically active, potentiated glutamatergic or GABA-ergic transmission between Schaffer collaterals and CA1 neurons in rat hippocampal slices. The cholinesterase inhibitors rivastigmine, donepezil, and metrifonate, which are devoid of nicotinic APL action, did not affect synaptic transmission. Exogenous application of ACh indicated that high and low levels of nAChR activation in the Schaffer collaterals inhibit and facilitate, respectively, glutamate release onto CA1 neurons. The finding then that the nAChR antagonists methyllycaconitine and dihydro-β-erythroidine facilitated glutamatergic transmission between Schaffer collaterals and CA1 neurons indicated that in a single hippocampal slice, the inhibitory action of strongly, tonically activated nAChRs in some glutamatergic fibers prevails over the facilitatory action of weakly, tonically activated nAChRs in other glutamatergic fibers synapsing onto a given neuron. Galantamine is known to sensitize nAChRs to activation by low, but not high agonist concentrations. Therefore, at 1 μM, galantamine is likely to increase facilitation of synaptic transmission by weakly, tonically activated nAChRs just enough to override inhibition by strongly, tonically activated nAChRs. In conclusion, the nicotinic APL action can be an important determinant of the therapeutic effectiveness of galantamine.
Alzheimer's disease (AD) is a neurodegenerative disorder that afflicts millions worldwide. It is characterized by a progressive decline of intellectual abilities, which eventually becomes severe enough to interfere with social or occupational individual functioning and ultimately leads to death (Chung and Cummings, 2000). Although the cause of the disorder remains unknown, there is a strong correlation between brain cholinergic dysfunction and the severity of AD symptoms (Coyle et al., 1983; Nordberg, 1999). Post-mortem biopsy studies have shown that as the condition worsens, progressive loss of basal forebrain cholinergic neurons, which innervate the entire cortical mantle (Cullen et al., 1997), is accompanied by incremental loss of nAChRs in cerebral cortical neurons (Nordberg, 1999; Perry et al., 2000). Therefore, increasing brain nicotinic functions to a level sufficient to improve synaptic plasticity and neuronal survival emerges as a promising therapeutic approach for treatment of AD patients.
Two nAChR subtypes are found in abundance in the mammalian central nervous system (CNS). One binds nicotine with high affinity and is composed of α4- and β2-subunits; the other binds α-bungarotoxin and is most probably a homomeric α7-nAChR (Lindstrom, 1997). These receptors are located postsynaptically, where they mediate fast synaptic transmission (Albuquerque et al., 2000a), and presynaptically, where they modulate synaptic transmission mediated by numerous neurotransmitters, including glutamate, GABA, serotonin, ACh, and noradrenaline (Albuquerque et al., 2000b).
At the neuromuscular junction, cholinergic nicotinic function can be enhanced by cholinesterase inhibitors and nicotinic agonists (Taylor, 1982). In the nervous systems, however, the effects of these agents are more complex. First, neuronal nAChRs, particularly α7-nAChRs, are much more prone to agonist-induced desensitization than muscle nAChRs. Thus, nicotinic agonists only transiently increase nicotinic function in CNS neurons (Alkondon et al., 2000b). Furthermore, unlike muscle nAChRs, some neuronal nAChRs, including α7-nAChRs, recognize both ACh and its metabolite choline as full agonists; the EC50 values for ACh and choline as α7-nAChR agonists are approximately 140 μM and 1.6 mM, respectively (Albuquerque et al., 2000b). Therefore, cholinesterase inhibition may not necessarily enhance functions mediated by these nAChRs. In fact, cholinesterase inhibitors do not affect α7-nAChR-mediated synaptic transmission evoked by low-frequency stimulation of cholinergic fibers in chick ciliary ganglia (Zhang et al., 1996).
An alternative means to increase nicotinic functions in the brain is to “sensitize” the nAChRs to activation by the endogenous agonist(s). In the middle 1980s, the anticholinesterase physostigmine was shown to activate frog muscle nAChRs (Shaw et al., 1985). Subsequent studies not only confirmed the nicotinic agonistic activity of physostigmine, but also demonstrated that such an effect is insensitive to blockade by classical nicotinic antagonists (Okonjo et al., 1991; Pereira et al., 1993). The agonistic activity of physostigmine-like compounds, initially referred to as “noncompetitive agonists” (Storch et al., 1995), is the result of their binding to a site close to, but distinct from, the ACh-binding site on nAChR α-subunits (Schrattenholz et al., 1993). Noncompetitive agonists are weak agonists; by themselves, they cannot induce macroscopic nicotinic responses (Pereira et al., 1994;Storch et al., 1995). They can, however, potentiate the nAChR activity induced by classical nAChR agonists, and are, therefore, also referred to as nicotinic APLs (Maelicke and Albuquerque, 1996; Schrattenholz et al., 1996).
Galantamine, an alkaloid originally obtained from bulbs of snowdrops, is a weak cholinesterase inhibitor and a powerful nicotinic APL that seems to be more effective and less toxic than most cholinesterase inhibitors currently used to treat AD patients (Woodruff-Pak et al., 2001). However, the effects of galantamine on neuronal functions in the CNS and the relevance of its nicotinic APL action remain elusive. Thus, the present study was designed to investigate whether galantamine affects synaptic transmission and, if so, by what mechanism. Evidence is provided that galantamine, acting as an APL on presynaptically located nAChRs that are weakly, tonically activated, induces long-lasting facilitation of glutamatergic or GABA-ergic transmission between the Schaffer collaterals and CA1 neurons in rat hippocampal slices. Such an effect is also observed when hippocampal slices are exposed to the anticholinesterase and nicotinic APL methyl-galantamine, but is not observed when the slices are exposed to cholinesterase inhibitors devoid of nicotinic APL action, including methamidophos, donepezil, rivastigmine, and metrifonate (Fig.1). Furthermore, via its nicotinic APL action, galantamine potentiates GABA release triggered by low ACh concentrations exogenously applied to rat hippocampal and human cerebral cortical slices. Taken together these results indicate that the nicotinic APL action can contribute to the cognitive improvement observed in AD patients treated with galantamine.

Chemical structures of tested cholinesterase inhibitors. In addition to inhibiting cholinesterase, galantamine and methyl-galantamine act as APLs at nAChRs. All the other cholinesterase inhibitors are devoid of the nicotinic APL action.
Materials and Methods
Rat Hippocampal Slices.
Slices of 250-μm thickness were obtained from the hippocampi of 15- to 25-day-old Sprague-Dawley rats according to the procedure described previously (Alkondon et al., 1999). The slices were stored in a holding chamber containing artificial cerebrospinal fluid (ACSF) bubbled with 95% O2 and 5% CO2 and were maintained at room temperature. Each slice, as needed, was transferred to a recording chamber (capacity of 2.0 ml) and held submerged by two nylon fibers. The recording chamber was continuously perfused with bubbled ACSF, which had the following composition: 125 mM NaCl, 25 mM NaHCO3, 2.5 mM KCl, 1.25 mM NaH2PO4, 2 mM CaCl2, 1 mM MgCl2, and 25 mM glucose (osmolarity ∼ 340 mOsM).
Cultured Hippocampal Neurons.
Primary cultures were prepared from the hippocampi of 16- to 18-day-old fetal Sprague-Dawley rats according to the procedure described elsewhere (Pereira et al., 1993).
Human Cerebral Cortical Slices.
Slices of 250-μm thickness were prepared from specimens of the human lateral neocortex obtained from the temporal or frontal cortical lobe of three male and three female patients according to the procedure described by Alkondon et al. (2000b).
Electrophysiological Recordings.
By means of the whole-cell mode of the patch-clamp technique, postsynaptic currents (PSCs) were recorded from neurons of the CA1 pyramidal layer of rat hippocampal slices in response to afferent stimulation. Test solutions were applied to the slices through a set of coplanar-parallel glass tubes (400 μm i.d.) glued together and assembled on a motor driven system (Newport Corporation, Irvine, CA) controlled by microcomputer. The tubes were placed at a distance of approximately 100 to 150 μm from the slice, and the gravity-driven flow rate was adjusted to 1.0 ml/min. Each tube was connected to a different reservoir filled with test solution. Evoked PSCs were recorded after application of a supramaximal 20- to 60-μs electrical stimulus via a bipolar electrode made of thin platinum wires (50- to 100-μm diameter). The stimulus was delivered by an isolated stimulator unit (Digitimer Ltd., Garden City, England) connected to a digital-to-analog interface (TL-1 DMA; Axon Instruments, Union City, CA). The platinum electrode was positioned in the stratum radiatum approximately 250 to 300 μm away from the cell body of the neurons in the CA1 pyramidal layer of the hippocampal slices. Possible changes in series resistance were detected by applying online a hyperpolarizing pulse (5 mV) before the stimulus pulse. Excitatory and inhibitory postsynaptic currents (EPSCs and IPSCs, respectively) evoked by field stimulation were pharmacologically identified and isolated by the application of antagonists of the excitatory α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) andN-methyl-d-aspartate (NMDA) receptors, 20 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and 50 μM 2-amino-5-phosphonovaleric acid (APV), respectively, or the antagonist of the inhibitory GABAA receptor, 100 μM picrotoxin.
ACh-induced GABA-ergic PSCs were recorded from CA1 stratum radiatum interneurons in rat hippocampal slices or from neurons in the human cerebral cortical slices. Whole-cell currents were recorded from hippocampal neurons in culture. U-tubes were used to deliver short pulses (2- to 30-s duration) of ACh and agonists of various ligand-gated channels to hippocampal neurons in culture and slices. Membrane potential and spontaneously occurring action potentials were recorded from current-clamped neurons in the CA1 pyramidal layer of rat hippocampal slices.
Electrophysiological signals were recorded by means of an Axopatch 200A (Axon Instruments) or an LM-EPC-7 patch-clamp system (List Electronics, Heidelberg, Germany), filtered at 2 kHz, and either stored on VCR tapes or directly sampled by a microcomputer using the pClamp6 software (Axon Instruments). Low resistance (2–5 MΩ) electrodes were pulled from borosilicate capillary glass (World Precision Instruments, New Haven, CT) and filled with internal solution. The composition of the internal solution used for voltage-clamp recordings from neurons in the CA1 pyramidal layer was: 80 mM CsCl, 80 mM CsF, 10 mM EGTA, 22.5 mM CsOH, 10 mM HEPES, and 5 mM QX-314 (pH adjusted to 7.3 with CsOH; 340 mOsM). The composition of the internal solution used for recording from CA1 interneurons and from human cerebral cortical neurons was: 130 mM Cs-methane sulfonate, 10 mM CsCl, 2 mM MgCl2, 5 mM QX-314, 10 mM EGTA, and 10 mM HEPES (pH adjusted to 7.3 with CsOH; 340 mOsM). The internal solution for current-clamp recordings from neurons in the CA1 pyramidal layer of rat hippocampal slices had the following composition: 130 mM K-gluconate, 20 mM KCl, 10 mM EGTA, and 10 mM HEPES (pH adjusted to 7.3 with KOH; 340 mOsM). In most experiments, biocytin (0.5%) was included in the internal solution for later identification of the neuron type. All experiments were performed in the presence of the muscarinic receptor antagonist atropine (1 μM) and at room temperature (20–22°C).
Data Analysis.
Peak amplitude, 10 to 90% rise time and decay-time constant of field stimulation-evoked PSCs and of spontaneously occurring action potentials were determined using the pClamp6 software. Spontaneously occurring and ACh-triggered IPSCs were analyzed using the Continuous Data Recording software (Dempster, 1989). Results are presented as means ± S.E.M. and were compared for their statistical significance using the Student's t test or one-way ANOVA followed by Dunnett's test.
Drugs.
Galantamine HBr and methyl-galantamine Br were provided by Boehringer Ingelheim GmbH (Ingelheim, Germany). A 100 mM stock solution of methamidophos (99.8%; Bayer AG, Leverkussen, Germany) was prepared and kept at −20°C, and working dilutions were made daily just before the experiments. Donepezil HCl and rivastigmine hydrogen tartrate were kindly provided by Prof. Madeleine M. Joullie and Michael S. Leonard (Department of Chemistry, University of Pennsylvania, Philadelphia). Janssen Research Foundation also provided a pure sample of Donepezil HCl. Methyllycaconitine (MLA) citrate was a gift from Prof. M. H. Benn (Department of Chemistry, University of Calgary, Alberta, Canada). Dihydro-β-erythroidine (DHβE) hydrobromide was a gift from Merck (Rahway, NJ). All other chemicals were purchased from Sigma (St. Louis, MO). A 250 mM stock solution of picrotoxin was made in dimethyl sulfoxide, and dilutions were made in the ACSF. NaOH was used to dissolve CNQX and APV (the 10 mM stock solution of CNQX had 12.5 mM NaOH and the 50 mM stock solution of APV had 0.5 M of NaOH). Donepezil and rivastigmine were dissolved in DMSO and diluted further with ACSF.
Results
Effects of Galantamine on Glutamatergic Transmission in Rat Hippocampal Slices: Time and Concentration Dependence.
In the continuous presence of the GABAA receptor antagonist picrotoxin (100 μM), inward PSCs were recorded at −60 mV from neurons in the CA1 pyramidal layer of rat hippocampal slices in response to field stimulation of the Schaffer collaterals. These currents were glutamatergic in nature, because they were reversibly inhibited by exposure of the slices to 20 μM CNQX and 50 μM APV (Fig. 2A), and they are, herein, referred to as EPSCs. After recording stable responses for at least 5 min under control conditions, EPSCs were recorded during a subsequent 5-min perfusion of the slices with ACSF containing 1 μM galantamine. At the end of the 5-min exposure of the slices to galantamine, there was an increase in the amplitude of EPSCs (Fig. 2A). The potentiating effect of galantamine was reversible upon wash.

Galantamine potentiates glutamatergic synaptic transmission in rat hippocampal slices. A, samples of EPSCs evoked by field stimulation of the Schaffer collaterals and recorded from a CA1 pyramidal neuron continuously perfused with 100 μM picrotoxin and 1 μM atropine under various experimental conditions. B, plot of normalized EPSC amplitudes versus recording time in the absence and in the presence of 1 μM galantamine. EPSCs were evoked every 5 s. The average amplitude of the first set of six consecutive events recorded from a neuron was taken as 1 and was used to normalize the average amplitude of every consecutive set of six events recorded at any given time thereafter. Each point and error bar represent mean and S.E.M., respectively, of results obtained from six neurons. C, plot of the normalized EPSC amplitudes recorded in the presence of various concentrations of galantamine. Each concentration was tested on a slice that had not been previously exposed to galantamine. The amplitudes of events evoked at a frequency of 0.2/s for 5 min were averaged. The averaged amplitude of 60 events recorded in the absence of galantamine was taken as 1 and was used to normalize the averaged amplitude of events recorded at the same frequency for 5 min in the presence of the drug. Holding potential, −60 mV. Graph and error bars represent mean and S.E.M., respectively, of results obtained from three to seven experiments. ∗∗, p < 0.01; according to the unpaired Student's t test.
To examine the time course of the effect of galantamine on the amplitude of evoked EPSCs, currents were triggered by electrical pulses delivered every 5 s for 5 min under control conditions, for 5 min during perfusion with ACSF containing 1 μM galantamine, and for 5 min during washing of the preparations with galantamine-free ACSF. After a 30-s exposure of the neurons to galantamine, there was an increase in the peak amplitude of the EPSCs (Fig. 2B). The effect was sustained during the time the neurons were exposed to galantamine and was reversed within 1 min of perfusion of the slices with galantamine-free ACSF (Fig. 2B).
To analyze the concentration-response relationship for galantamine-induced potentiation of EPSCs, the average of the amplitudes of EPSCs recorded for 5 min under control conditions from a given neuron was taken as 1 and was used to normalize the average of the amplitudes of EPSCs recorded during the 5-min exposure of that neuron to galantamine. The plot of normalized EPSC amplitudes versus concentrations of galantamine revealed that the concentration-response relationship was bell-shaped and that the maximal effect occurred at 1 μM galantamine (Fig. 2C; Table 1). Galantamine had no significant effect on the kinetics of EPSCs. The decay-time constants of the currents were 124.8 ± 11.3 ms under control conditions and 125.6 ± 11.0 ms in the presence of 1 μM galantamine (n = 6 neurons).
Cholinesterase inhibition and potentiation of glutamatergic transmission by structurally unrelated cholinesterase inhibitors
The use of pharmacological agents identified two components in the EPSCs recorded from CA1 neurons in rat hippocampal slices. EPSCs recorded when the slices were continuously perfused with ACSF containing 50 μM APV in addition to 100 μM picrotoxin had approximately the same amplitude as those recorded in the absence of APV but had a faster decay phase. These currents were mediated by AMPA/kainate receptors because they could be blocked by CNQX (Fig.3). On the other hand, EPSCs recorded when the slices were continuously perfused with ACSF containing 20 μM CNQX in addition to 100 μM picrotoxin had approximately the same decay-time constant as those recorded in the absence of CNQX but had a smaller amplitude. These currents were mediated by NMDA receptors because they could be blocked by APV (Fig. 3).

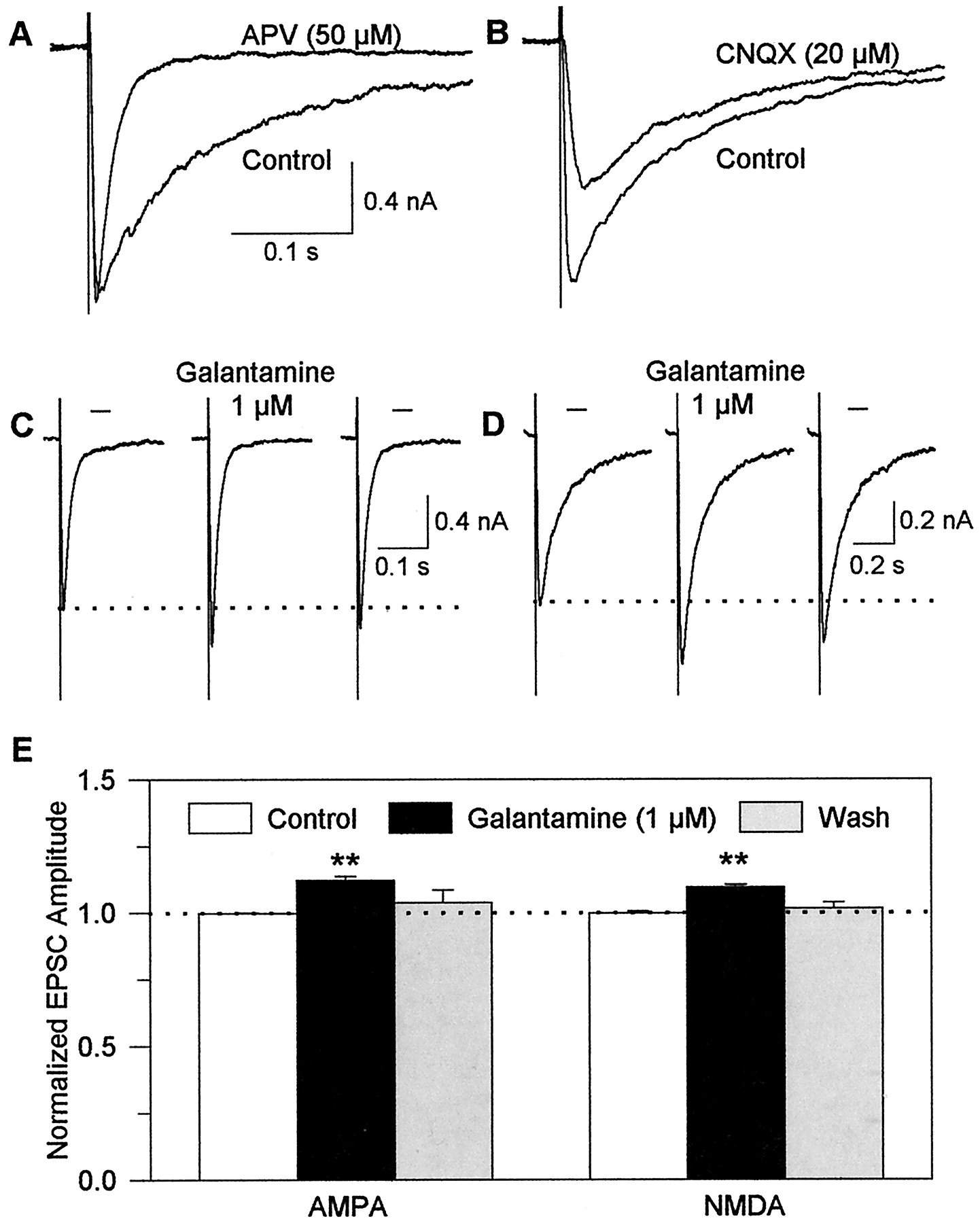
Galantamine potentiates evoked glutamatergic transmission mediated by both AMPA and NMDA receptors in rat hippocampal slices. A, samples of EPSCs evoked by field stimulation of the Schaffer collaterals and recorded from a CA1 pyramidal neuron in the absence and in the presence of the NMDA receptor blocker APV (50 μM). B, sample recordings of EPSCs evoked in the absence and in the presence of the AMPA receptor blocker CNQX (20 μM). C, sample recordings of AMPA EPSCs obtained from a CA1 pyramidal neuron under control conditions, in the presence of 1 μM galantamine after the neuron had been exposed for 5 min to the drug and at 10 min after wash with ACSF. D, sample recordings of NMDA EPSCs obtained from another neuron before, at 5 min after beginning of exposure to 1 μM galantamine, and at 10 min after wash. E, plot of amplitudes of AMPA or NMDA EPSCs under the different experimental conditions. The amplitudes of events evoked at a frequency of 0.2/s for 5 min were averaged. The averaged amplitude of events recorded in the absence of galantamine was taken as 1 and was used to normalize the averaged amplitude of events recorded at the same frequency for 5 min in the presence of the drug. Graph and error bars represent mean and S.E.M., respectively, of results obtained from five experiments. ∗∗, p< 0.01 according to the unpaired Student's t test.
To analyze the effects of galantamine on each component of the field stimulation-evoked EPSCs, currents were recorded in the presence of 100 μM picrotoxin plus 50 μM APV (Fig. 3C) or 20 μM CNQX (Fig.3D). Galantamine (1 μM) increased the amplitude (Fig. 3, C and E) and had no effect on the decay phase of the pharmacologically isolated AMPA/kainate EPSCs evoked by field stimulation of the Schaffer collaterals. In the absence and in the presence of 1 μM galantamine, the decay-time constants of these currents were 12.4 ± 0.46 ms and 12.7 ± 0.23 ms, respectively (n = 5 neurons). Galantamine (1 μM) also increased the amplitude and had no effect on the decay phase of field stimulation-evoked NMDA EPSCs (Fig. 3, D and E). The decay-time constants of these currents were 175.7 ± 7.74 ms and 185.4 ± 6.24 ms in the absence and in the presence of 1 μM galantamine, respectively (n = 4 neurons).
The sum of the magnitude of the effects of galantamine on the amplitudes of pharmacologically isolated AMPA/kainate and NMDA EPSCs was approximately equal to the magnitude of the drug's effect on the amplitudes of EPSCs recorded in the absence of the AMPA receptor blocker CNQX and the NMDA receptor antagonist APV (see Fig. 2). This finding indicates that a common mechanism of action underlies galantamine-induced potentiation of EPSCs mediated by AMPA/kainate and NMDA receptors.
Galantamine Does Not Alter the Activity of Postsynaptic Glutamate Receptors.
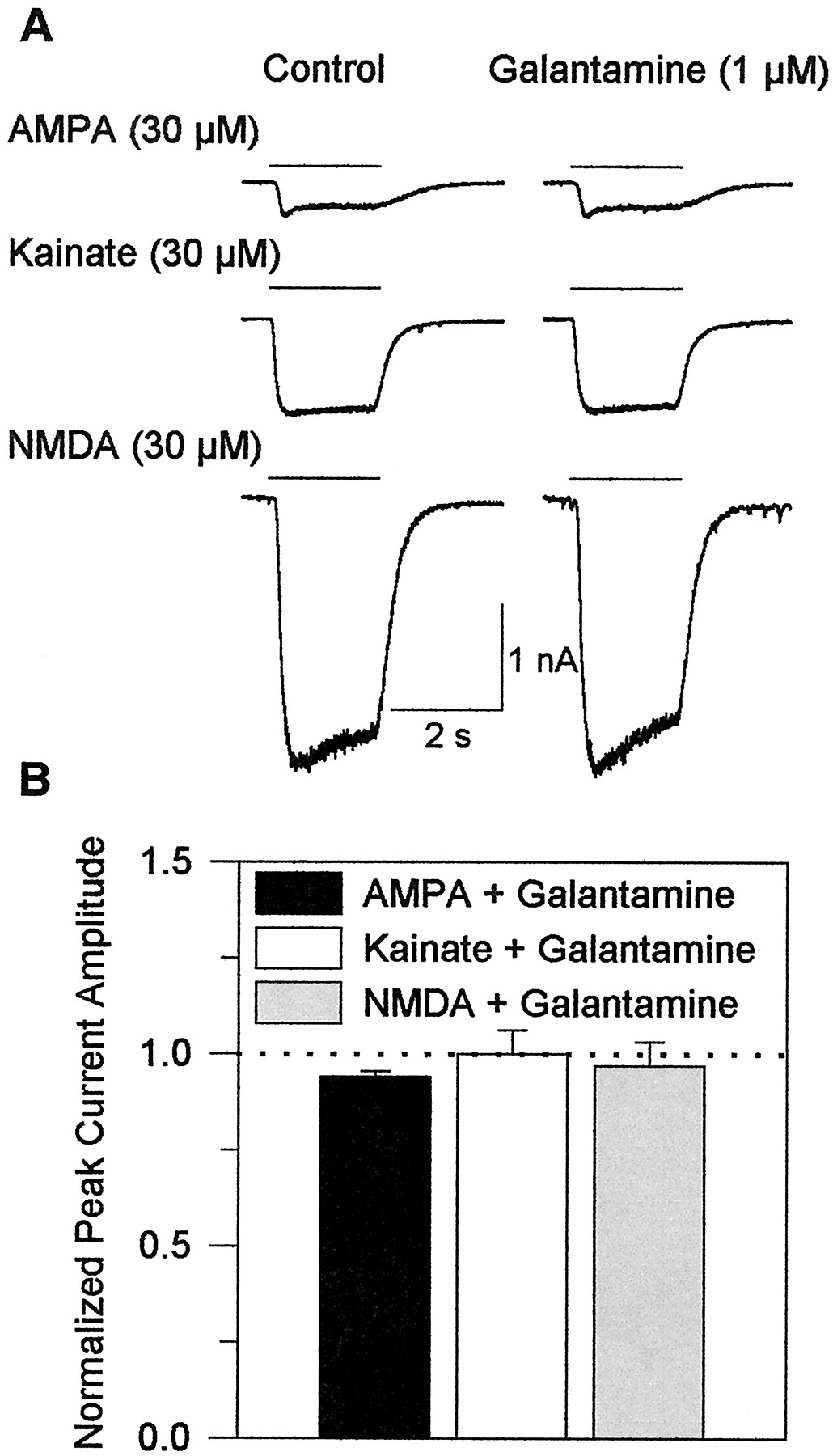
The above results suggest that galantamine facilitates glutamate transmission via a presynaptic mechanism of action. They do not rule out, however, the possibility that galantamine also alters the sensitivity of the postsynaptic glutamatergic receptors to glutamate. To address this possibility, whole-cell currents evoked by 2-s pulses of AMPA, kainate, or NMDA (each at 30 μM, a subsaturating concentration) were recorded from cultured hippocampal neurons before and during their perfusion with external solution containing 1 μM galantamine. Galantamine was present in both the background perfusion and the agonist solution. After a 10-min exposure of the neurons to 1 μM galantamine, there were no apparent changes in the amplitude or kinetics of currents evoked by each agonist (Fig.4, A and B).

Galantamine has no effect on glutamate receptors in cultured hippocampal neurons. A, sample recordings of whole-cell currents evoked by application of AMPA, kainate, or NMDA together with 10 μM glycine to neurons in the absence and in the presence of galantamine after the neurons had been exposed for 8 min to the drug. B, peak amplitudes of agonist-evoked currents in the absence of galantamine were taken as 1 and were used to normalize the peak amplitudes of currents recorded in the presence of the drug. Each graph and error bar represent mean and S. E M., respectively, of results obtained from five to six experiments. Holding potential, −50 mV.
Galantamine on Membrane and Action Potentials in Hippocampal Neurons.
Alterations in membrane and action potentials could also account for the effects of galantamine on synaptic transmission; however, 5- to 10-min exposures of rat hippocampal slices to 1 or 10 μM galantamine had no significant effect on the membrane potential or the peak amplitude, rate of rise, duration, and frequency of spontaneously occurring action potentials recorded from CA1 pyramidal neurons (Table 2). Furthermore, galantamine caused no change in the input resistance of the neurons (Table 2).
Effect of galantamine on membrane potential and action potential (AP) recorded from CA1 pyramidal neurons of rat hippocampal slices
Effects of Cholinesterase Inhibition and nAChR Activation on Field Stimulation-Evoked EPSCs in Rat Hippocampal Slices.
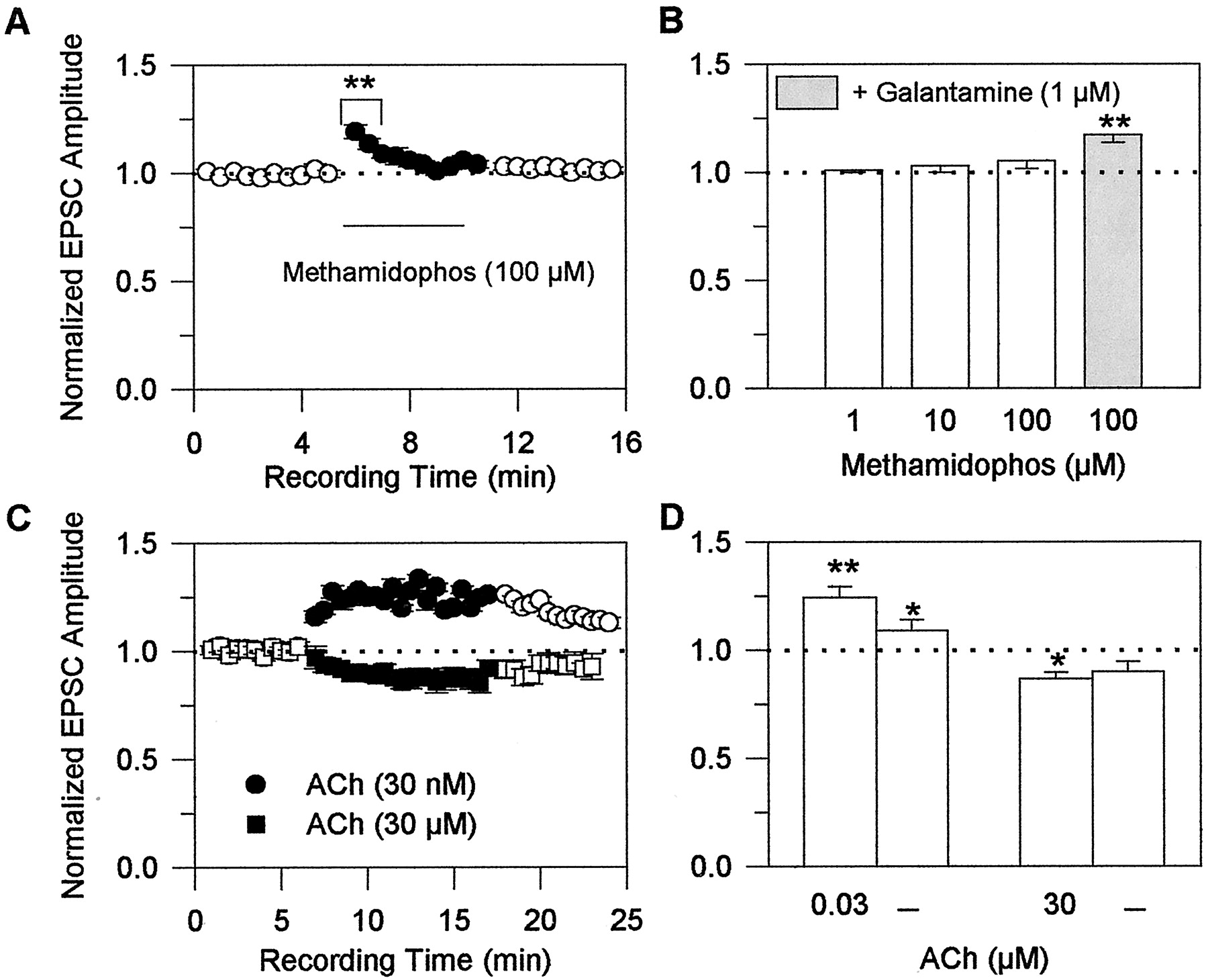
To determine the effect of cholinesterase inhibition on glutamatergic transmission, field stimulation-evoked EPSCs were recorded from CA1 neurons before, during, and after their exposure to methamidophos, a cholinesterase inhibitor devoid of the nicotinic APL action (Camara et al., 1997; Fig.1). Methamidophos was tested at concentrations that inhibit cholinesterase activity in the hippocampus by approximately 10, 30, and 100% (i.e., 1, 10, and 100 μM) (Camara et al., 1997). No significant changes in EPSC amplitudes were observed when the slices were exposed to 1 and 10 μM methamidophos. An increase in the EPSC amplitude was observed 30 s after exposure of the slices to 100 μM methamidophos; however, this effect decreased with time, becoming negligible after a 2-min exposure of the preparations to 100 μM methamidophos (Fig. 5A). Because of the transient nature of the effect of cholinesterase inhibition on glutamatergic transmission, the net result of a 5-min exposure of the slices to 1 to 100 μM methamidophos was no apparent change in the amplitude of EPSCs (Fig. 5B; Table 1).
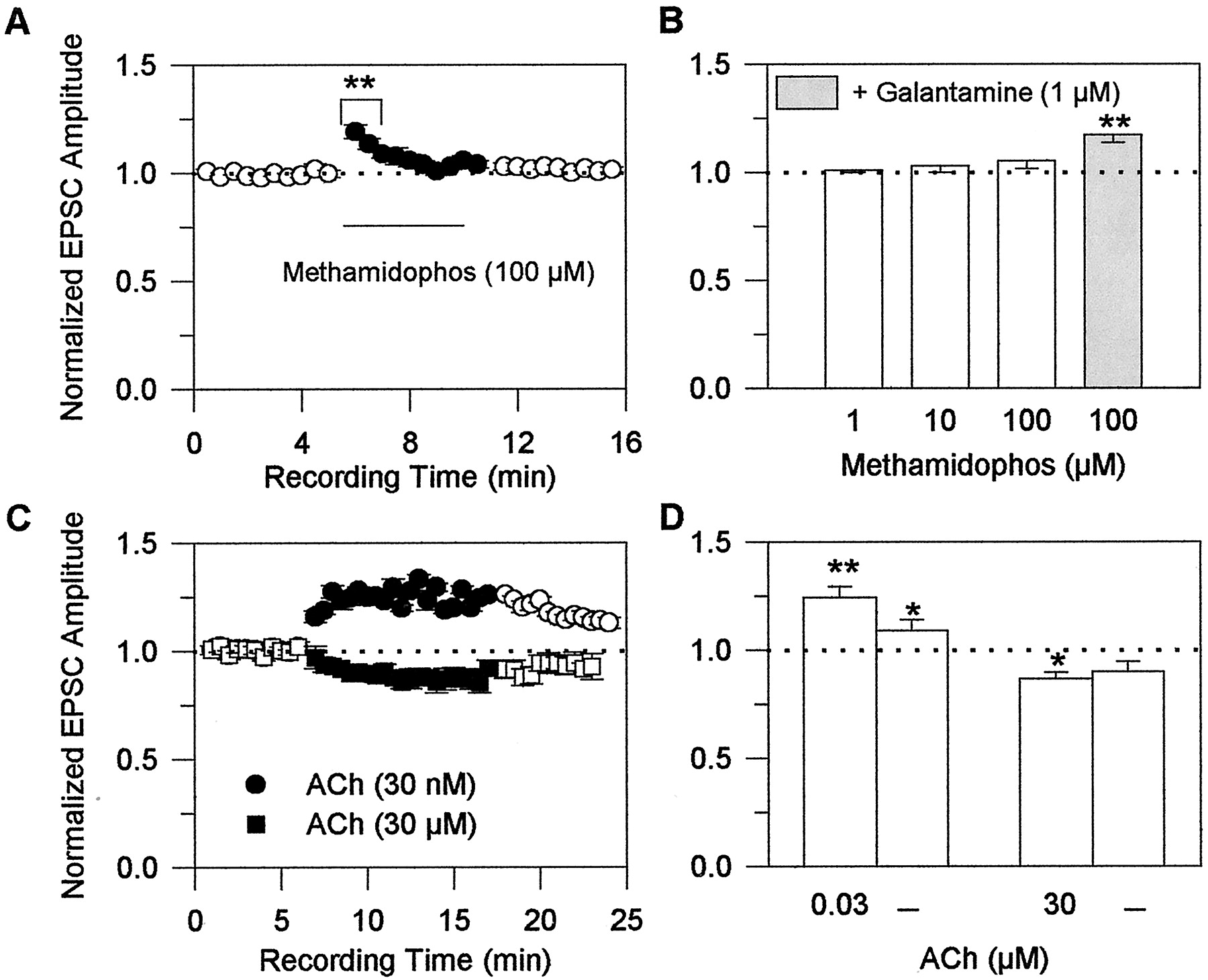
Effects of the acetylcholinesterase inhibitor methamidophos and of exogenously applied ACh on glutamatergic transmission in rat hippocampal slices. A, normalized amplitudes of EPSCs evoked by field stimulation of Schaffer collaterals and recorded from CA1 pyramidal neurons before, during, and after perfusion of the slices with 100 μM methamidophos were plotted against recording time. EPSCs were evoked every 5 s. The average amplitude of the first set of six consecutive events recorded from a neuron was taken as 1 and was used to normalize the average amplitude of every consecutive set of six events recorded thereafter. Each point and bar represent mean and S.E.M., respectively, of results obtained from five experiments. B, plot of normalized EPSC amplitudes recorded from CA1 pyramidal neurons in the presence of various concentrations of methamidophos alone or in admixture with 1 μM galantamine. Each concentration was tested on a neuron that had not been previously exposed to methamidophos. The amplitudes of events evoked at a frequency of 0.2/s for 5 min were averaged. The averaged amplitude of events recorded in the absence of methamidophos was taken as 1 and was used to normalize the averaged amplitude of events recorded at the same frequency for 5 min in the presence of methamidophos or methamidophos-plus-galantamine. Graph and error bars represent mean and S.E.M., respectively, of results obtained from five to seven experiments. C, normalized amplitudes of evoked EPSCs recorded from CA1 pyramidal neurons before, during, and after perfusion of the slices with 0.03 or 30 μM ACh. Amplitudes were normalized according to the protocol described in A. Each point and bar represent mean and S.E.M., respectively, of results obtained from six experiments. D, plot of the normalized EPSC amplitudes recorded from CA1 pyramidal neurons in the presence of the different concentrations of ACh and after a 10-min wash of the neurons with ACSF. Values were normalized according to the protocol described in B. Each graph and error bar represent the mean and S.E.M., respectively, of results obtained from six to seven experiments. Holding potential, −60 mV. ∗, p < 0.05 and ∗∗, p < 0.01 according to the unpaired Student's t test.
After a 10-min perfusion of the slices with ACSF containing 100 μM methamidophos, the magnitude of the effect of 1 μM galantamine on the amplitude of evoked EPSCs was the same as that seen in preparations that had not been previously exposed to methamidophos (Fig. 5B). In the absence (see Fig. 2B) and in the presence (Fig. 5B) of methamidophos, a 5-min exposure of the slices to 1 μM galantamine increased the amplitude of field stimulation-evoked EPSCs to 120 ± 2.9% and 117 ± 1.9% of control, respectively. Thus, the effect of galantamine on glutamatergic transmission is unrelated to its anticholinesterase activity.
Structurally unrelated cholinesterase inhibitors with or without the nicotinic APL action were also tested for their ability to alter glutamatergic transmission in rat hippocampal slices. The net effect of a 5-min exposure of hippocampal slices to the cholinesterase inhibitor and nicotinic APL methyl-galantamine was similar to that observed when the slices were exposed to galantamine; methyl-galantamine caused a 20% increase in the EPSC amplitudes (see Table 1). On the other hand, the net effect of a 5-min perfusion of hippocampal slices to three other cholinesterase inhibitors devoid of nicotinic APL action (i.e., metrifonate, donepezil, and rivastigmine) resembled that observed when the slices were exposed to methamidophos; metrifonate, donepezil, and rivastigmine caused no significant changes in the amplitudes of EPSCs recorded from CA1 pyramidal neurons in response to field stimulation of the Schaffer collaterals (see Table 1).
To examine the effects of nicotinic agonists on glutamatergic transmission, EPSCs were evoked by field stimulation of the Schaffer collaterals every 5 s for several min before, during, and after exposure of the slices to 30 nM or 30 μM ACh in the continuous presence of atropine (Fig. 5C). At 30 s after exposure of the slices to 30 nM ACh, there was an enhancement in the EPSC amplitudes (Fig. 5C). This effect reached a plateau in 3 min and was maintained at that level for 10 min (Fig. 5C). The potentiating effect of ACh on glutamatergic transmission was not promptly reversed. It took longer than 5 min of washing for the effect to subside (Fig. 5C). In contrast, at 30 s after perfusion of the same slices with ACSF containing 30 μM ACh, there was a reduction of the amplitude of field stimulation-evoked EPSCs; this effect was maintained during a 10-min exposure to the agonist (Fig. 5C). The net effects of a 5-min exposure of the slices to 30 nM and 30 μM ACh were an increase and a decrease, respectively, of the amplitude of field stimulation-evoked EPSCs (Fig.5D). These results suggest that low levels of activation of nAChRs in Schaffer collaterals synapsing onto the neurons from which recordings are obtained facilitate glutamate release, whereas high levels of nAChR activation in Schaffer collaterals synapsing onto the neurons under study reduce glutamate release (Fig. 6).

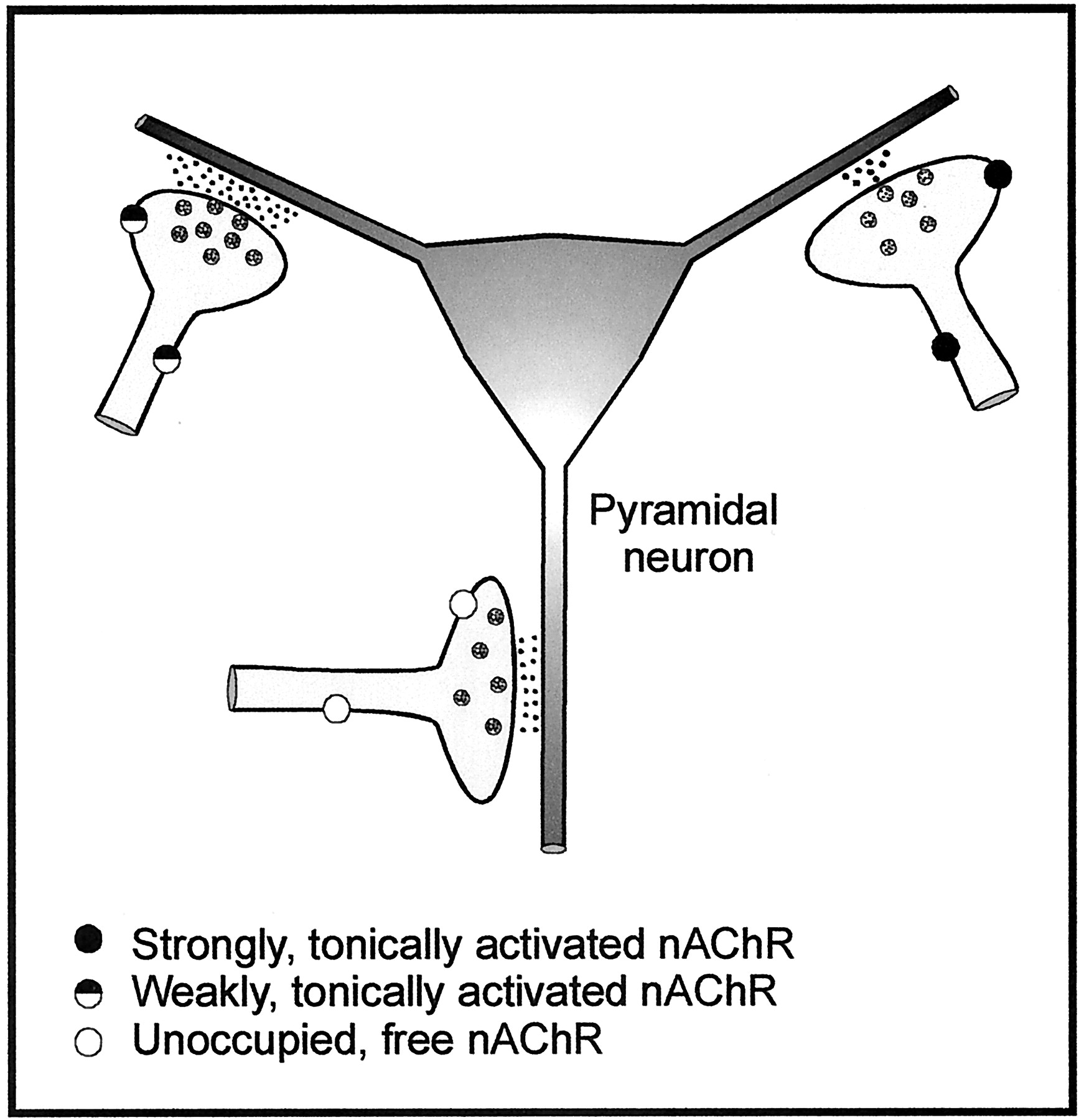
Schematic representation of a neuronal circuitry depicting possible mechanisms of nicotinic cholinergic regulation of glutamatergic synaptic transmission between the Schaffer collaterals and CA1 neurons in the hippocampus. This simplified schematic model shows a neuronal network comprised of glutamatergic fibers synapsing onto a pyramidal neuron. According to this model, strongly, tonically activated nAChRs in glutamatergic fibers reduce glutamate release onto a pyramidal neuron. On the other hand, moderately, tonically activated nAChRs in glutamatergic fibers increase glutamate release onto a pyramidal neuron. This model lends support to the concept that block of nAChRs by MLA and DHβE relieves the prevailing inhibitory tonic nicotinic activity and thereby facilitates overall synaptic transmission between Schaffer collaterals and CA1 neurons. Because galantamine is known to sensitize nAChRs to activation by low, but not high, agonist concentrations, this model also supports the contention that galantamine increases facilitation of synaptic transmission by weakly, tonically activated nAChRs just enough to override inhibition by strongly, tonically activated nAChRs. Agonist-free nAChRs in the resting state may be activated to different levels by elevated ambient concentrations of endogenous agonists (ACh or choline) and/or by exposure to exogenous nicotinic agonists.
Involvement of α7-nAChRs in Galantamine-Induced Potentiation of Glutamatergic Transmission in Rat Hippocampal Slices.
To determine the contribution of the different nAChR subtypes to regulation of glutamatergic transmission between the Schaffer collaterals and neurons in the CA1 pyramidal layer of the hippocampus, field stimulation-evoked EPSCs were recorded from CA1 neurons before, during, and after perfusion of hippocampal slices with ACSF containing the α7-nAChR antagonist MLA (10 nM) and/or DHβE, a nicotinic antagonist that at 10 μM reduces by ∼40 and 90% the activity of α7- and α4β2-nAChRs, respectively (Alkondon et al., 1999). A 10-min exposure of the slices to MLA increased by ∼20% the amplitude of the evoked EPSCs (Fig. 7A). Approximately 10% enhancement of the amplitude of evoked EPSCs was observed when the slices were exposed for 10 min to DHβE (Fig. 7A). Potentiation of glutamatergic transmission by DHβE was fully reversed after 10 min of washing of the slices with DHβE-free ACSF; the average amplitudes of EPSC recorded after washing the preparations were 104 ± 2% of those recorded under control conditions (n = 5 neurons). The effect of MLA was only partially reversed after 10 min of washing the preparations with MLA-free ACSF; the average EPSC amplitudes recorded after 10 min of washing the preparations were 108 ± 1% of those recorded under control conditions (n = 5 neurons). The effects of DHβE and MLA on evoked EPSCs were not additive. In fact, the total effect of both antagonists together was ∼5% smaller than that of MLA alone and ∼5% higher than that of DHβE alone (Fig. 7A), suggesting a competitive interaction between the two antagonists with the same receptor (i.e., the α7-nAChR). Considering that neither MLA nor DHβE affects the postsynaptic glutamatergic receptors (Alkondon et al., 1999), the present findings suggest that in a single hippocampal slice the net effect of α7-nAChRs tonically activated to different levels on Schaffer collaterals is a reduction of glutamate release onto CA1 neurons.

Nicotinic receptor antagonists block galantamine-induced potentiation and ACh-induced changes in glutamatergic transmission in rat hippocampal slices. A, effects of MLA and/or DHβE on glutamatergic transmission. Plot of normalized EPSC amplitudes recorded from neurons in the CA1 pyramidal layer of rat hippocampal slices under different conditions. Events were recorded from neurons in the absence of drugs and during perfusion of the neurons with ACSF containing nAChR antagonists. Each perfusion lasted 5 min. The average of EPSC amplitudes recorded at a frequency of 0.2/s for 5 min before exposure of the neurons to any drug were taken as 1 and were used to normalize the averaged amplitudes of EPSCs recorded at a frequency of 0.2/s for 5 min from neurons at the different experimental conditions. B, effects of ACh and galantamine on glutamatergic transmission in the presence of MLA and DHβE. Events were recorded from neurons continuously perfused with ACSF containing MLA and DHβE before and during their exposure to 30 nM or 30 μM ACh or 1 μM galantamine. Each perfusion lasted 5 min. Results were normalized according to the same procedure as that described in A. In each plot, graph and error bars represent mean and S.E.M., respectively, of results obtained from five experiments. All EPSCs were recorded at −60 mV in the presence of 1 μM atropine, 100 μM picrotoxin, and 100 μM methamidophos. ∗∗, p< 0.01 according to one-way ANOVA followed by Dunnett's test.
In the presence of 10 nM MLA alone or in admixture with 10 μM DHβE (see Fig. 7B), 1 μM galantamine did not alter glutamatergic transmission between the Schaffer collaterals and CA1 neurons in the hippocampal slices. After 10 min of perfusion of the slices with ACSF containing MLA or the admixture of MLA and DHβE, the average EPSC amplitudes were 121 ± 1% and 117 ± 5% of those recorded under control conditions (Fig. 7A). After an additional 5-min perfusion of the slices with ACSF containing 1 μM galantamine in addition to MLA or MLA-plus-DHβE, the average EPSC amplitudes were 122 ± 1% and 115 ± 7%, respectively, of those recorded under control conditions (n = 5 neurons). However, when the α7-nAChRs were only partially blocked by 10 μM DHβE, the potentiating effect of galantamine on glutamatergic transmission added up to that of DHβE. After a 10-min perfusion of hippocampal slices with ACSF containing DHβE, the average EPSC amplitudes were 110 ± 1% of those recorded under control conditions (n = 5 neurons). After a subsequent 5-min perfusion of the slices containing galantamine in addition to DHβE, the average EPSC amplitudes were 115 ± 1% of those recorded under control conditions (n = 5 neurons); the additional 5% increase in the EPSC amplitudes observed in the presence of galantamine was statistically significant (p < 0.05 according to the paired Student's t test).
Potentiation and inhibition of evoked EPSCs by 30 nM and 30 μM ACh, respectively, could not be observed in slices that had been pre-exposed for 10 min to the admixture of MLA and DHβE (Fig. 7B). The fact that the potentiating effect of galantamine and 30 nM ACh on evoked EPSCs did not add up to that of the nicotinic antagonists could not be explained by saturation of the system, because a 5-min exposure of the preparations to the K+-channel blocker 4-aminopyridine (100 μM) increased the amplitudes of evoked EPSCs to 229 ± 14% of control (n = 3 neurons). These results support the concept that changes induced in the EPSCs by exogenously applied galantamine and ACh were the result of the interaction of these compounds with nAChRs.
One could argue that the potentiating effects of 30 nM ACh on evoked EPSCs were the result of nAChR desensitization rather than activation. However, MLA and DHβE suppressed 30 nM ACh-induced enhancement of EPSC amplitude when they were applied to the slices after the onset of the potentiating effect of ACh. For example, the mean peak EPSC amplitude recorded from six CA1 pyramidal neurons after a 5-min exposure of the hippocampal slices to 30 nM ACh was 124.5 ± 5.1% of that recorded under control conditions. During subsequent exposure of the preparations to ACSF containing ACh and the antagonists, the mean peak EPSC amplitude decreased significantly to 107.5 ± 4.1% of control (p < 0.05 according to the one-way ANOVA) returning to levels that were not statistically different from those observed in the presence of the antagonists alone (i.e., 117 ± 5% of control; see Fig. 7A). Taken together these results indicate that the effect of 30 nM ACh on evoked glutamatergic transmission is the result of ACh-induced nAChR activation.
Effects of the Monoclonal Antibody FK-1 on Galantamine-Induced Potentiation of Glutamatergic Transmission in Rat Hippocampal Slices.
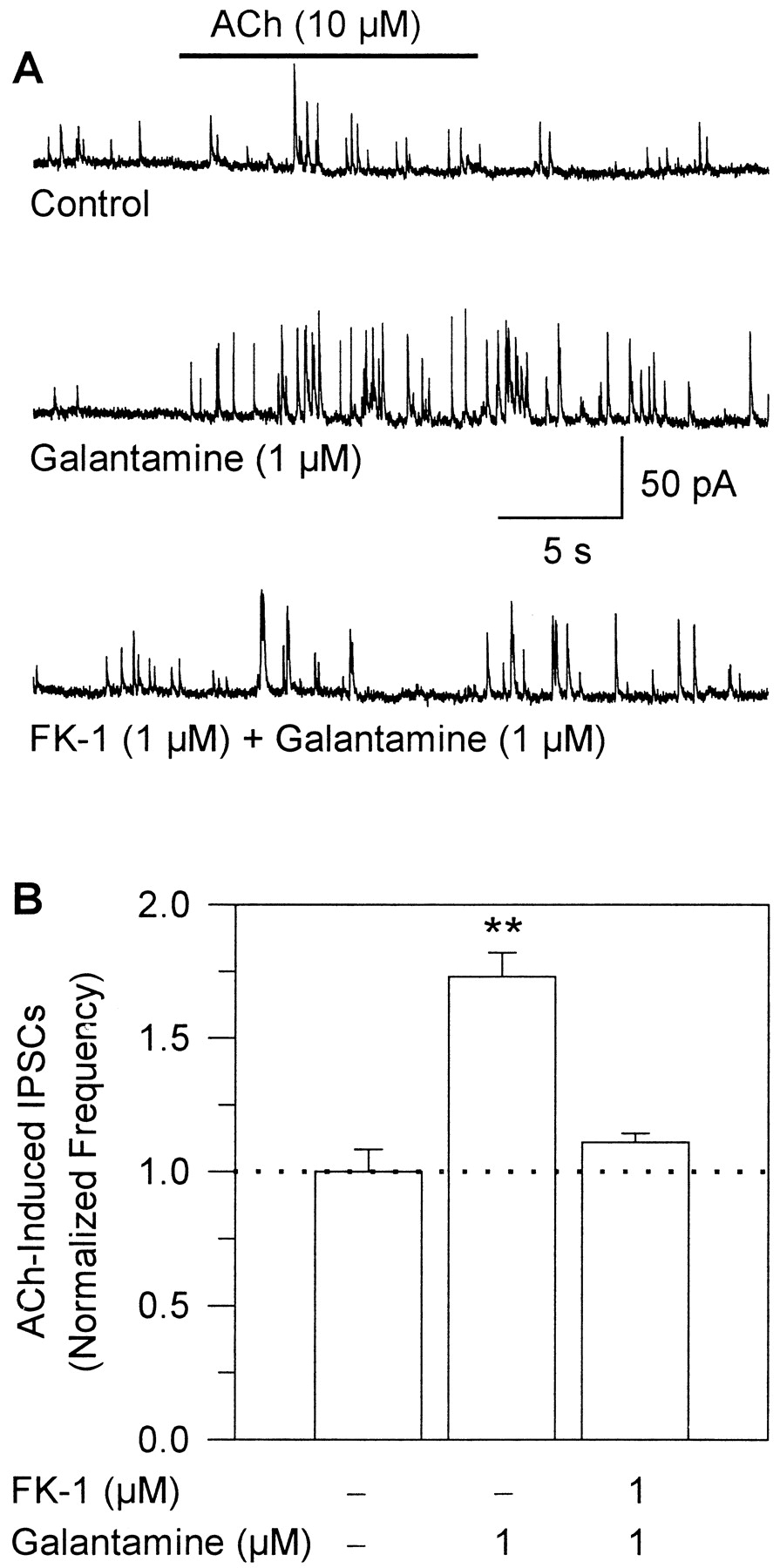
To investigate the mechanism underlying the effects of galantamine on glutamatergic synaptic transmission, the effects of 1 μM galantamine on field stimulation-evoked EPSCs were analyzed before and 30 min after perfusion of the hippocampal slices with ACSF containing the monoclonal antibody FK-1 (1 μM). This antibody specifically recognizes the binding region of APLs on the α-subunit of the nAChRs (Schrattenholz et al., 1993) and is a functional inhibitor of the noncompetitive agonistic (Pereira et al., 1993) and allosteric potentiating actions (Schrattenholz et al., 1996) of nicotinic APLs.
As mentioned previously, 1 μM galantamine increased the amplitude of field stimulation-evoked EPSCs, such an effect being fully reversible upon washing of the preparations with galantamine-free ACSF (Fig.8, A and B). A subsequent 30-min perfusion of the slices with ACSF containing 1 μM FK-1 caused no significant change in the amplitude of the evoked EPSCs (Fig. 8, A and B). Thereafter, in the continued presence of FK-1, 1 μM galantamine failed to increase the amplitude of the evoked EPSCs (Fig. 8, A and B). These findings indicate that galantamine potentiates glutamatergic transmission in the hippocampus by interacting with the APL-binding region on nAChRs.

Galantamine-induced potentiation of glutamatergic transmission in rat hippocampal slices is mediated via a nicotinic APL action. A, recording samples of field stimulation-evoked EPSCs recorded from a CA1 pyramidal neuron under various experimental conditions. Recordings were obtained in the following sequence: before exposure of the neuron to drugs; in the presence of 1 μM galantamine after the neuron had been perfused for 5 min with ACSF containing the drug; after a 10-min wash with ACSF; in the presence of 1 μM FK-1 after the neuron had been exposed for 25 min with ACSF containing the antibody; in the presence of FK-1-plus-galantamine after the neuron had been exposed for an additional 5 min to the admixture of FK-1 and galantamine; and after a 10-min wash with ACSF. B, plot of normalized EPSC peak amplitudes under the different experimental conditions. The average of EPSC amplitudes recorded at a frequency of 0.2/s for 5 min before exposure of the neurons to any drug were taken as 1 and were used to normalize the averaged amplitudes of EPSCs recorded at the same frequency for 5 min under the different experimental conditions. All EPSCs were recorded at −60 mV in the presence of 1 μM atropine, 100 μM picrotoxin, and 100 μM methamidophos. Graph and error bars represent mean and S.E.M., respectively, of results obtained from five experiments. ∗∗, p < 0.01 according to one-way ANOVA followed by Dunnett's test.
Effects of Galantamine on GABA-ergic Transmission in the Rat Hippocampus.
When 20 μM CNQX and 50 μM APV were both present in the ACSF, inward PSCs were recorded at −60 mV from neurons in the CA1 pyramidal layer of the hippocampal slices in response to field stimulation of the Schaffer collaterals (Fig.9A). These currents, which are herein referred to as IPSCs, were GABA-ergic in nature because they were inhibited by 100 μM picrotoxin (Fig. 9A).

Galantamine potentiates evoked GABA-ergic transmission in rat hippocampal slices. A, samples of IPSCs evoked by field stimulation of the Schaffer collaterals and recorded from a CA1 pyramidal neuron continuously perfused with ACSF containing 20 μM CNQX, 50 μM APV, 1 μM atropine, and 100 μM methamidophos. B, plot of normalized IPSC amplitudes versus recording time. IPSCs were evoked every 5 s. The average amplitude of the first set of six consecutive events recorded from a neuron was taken as 1 and was used to normalize the average amplitude of every consecutive set of six events recorded at any given time thereafter. Each point and error bar represent mean and S.E.M., respectively, of results obtained from five experiments. C, plot of normalized IPSC amplitudes recorded before, during, and after exposure of the neurons to 1 μM galantamine. The amplitudes of events evoked under control conditions at a frequency of 0.2/s for 5 min were averaged, taken as 1, and used to normalize the averaged amplitude of events recorded at the same frequency for 5 min in the presence of the drug and after a 5-min wash with ACSF. Each graph and error bar represent mean and S.E.M., respectively, of results obtained from five experiments. Holding potential, −60 mV. ∗∗, p < 0.01 according to the unpaired Student's t test.
After a 5-min perfusion of the slices with ACSF containing 1 μM galantamine, there was an increase in the amplitude of evoked IPSCs (Fig. 9A). The effect, observed within 30 s of exposure of the slices to galantamine, was sustained for as long as the drug was present (Fig. 9B) and was fully reversible upon washing of the slices with galantamine-free ACSF (Fig. 9, A–C). Galantamine had no significant effect on the kinetics of field stimulation-evoked IPSCs. In the absence and in the presence of 1 μM galantamine, the decay-time constants of IPSCs were 76.8 ± 17.7 ms and 78.5 ± 18.5 ms, respectively (n = 5 neurons).
Galantamine (1 μM) did not affect the amplitude or frequency of miniature IPSCs (mIPSCs) recorded from CA1 pyramidal neurons in the presence of 300 nM TTX. In the absence of galantamine, the mean frequency and amplitude of mIPSCs recorded from pyramidal neurons voltage clamped at −60 mV were 1.2 ± 0.25 Hz and 24.3 ± 2.9 pA, respectively. After a 10-min exposure to 1 μM galantamine, the mean frequency and amplitude of mIPSCs were 1.3 ± 0.34 Hz and 21.7 ± 4.2 pA, respectively. Also, the amplitudes of whole-cell currents evoked by 2-s pulses of 30 μM GABA in cultured hippocampal neurons exposed for 5 to 10 min to 1 μM galantamine were 97.5 ±3.8% of those of GABA-evoked whole-cell currents recorded before exposure of the neurons to the drug (n = 3 neurons). These findings indicate that galantamine does not alter the activity of postsynaptic GABAA receptors and suggest that galantamine-induced potentiation of evoked IPSCs is the result of a presynaptic action.
Effects of Galantamine on ACh-Triggered GABA-ergic IPSCs in Rat Hippocampal Slices.
A different protocol was used to verify whether the effects of galantamine on GABA-ergic transmission are mediated via its interaction with nAChRs present on GABA-ergic neurons synapsing onto the neurons from which recordings were obtained. In this protocol, a methanesulfonate-based internal solution was used and IPSCs were selectively recorded from CA1 stratum radiatum interneurons voltage-clamped at 0 mV. In the presence of muscarinic receptor antagonist atropine, U-tube application of 10 μM ACh to the slices enhanced the frequency of spontaneous IPSCs recorded from the interneurons (Fig. 10A). Previous studies have demonstrated that this effect of ACh is mediated by activation of α7- and α4β2-nAChRs on GABA-ergic neurons synapsing onto neurons from which recordings are obtained (Alkondon et al., 1999).
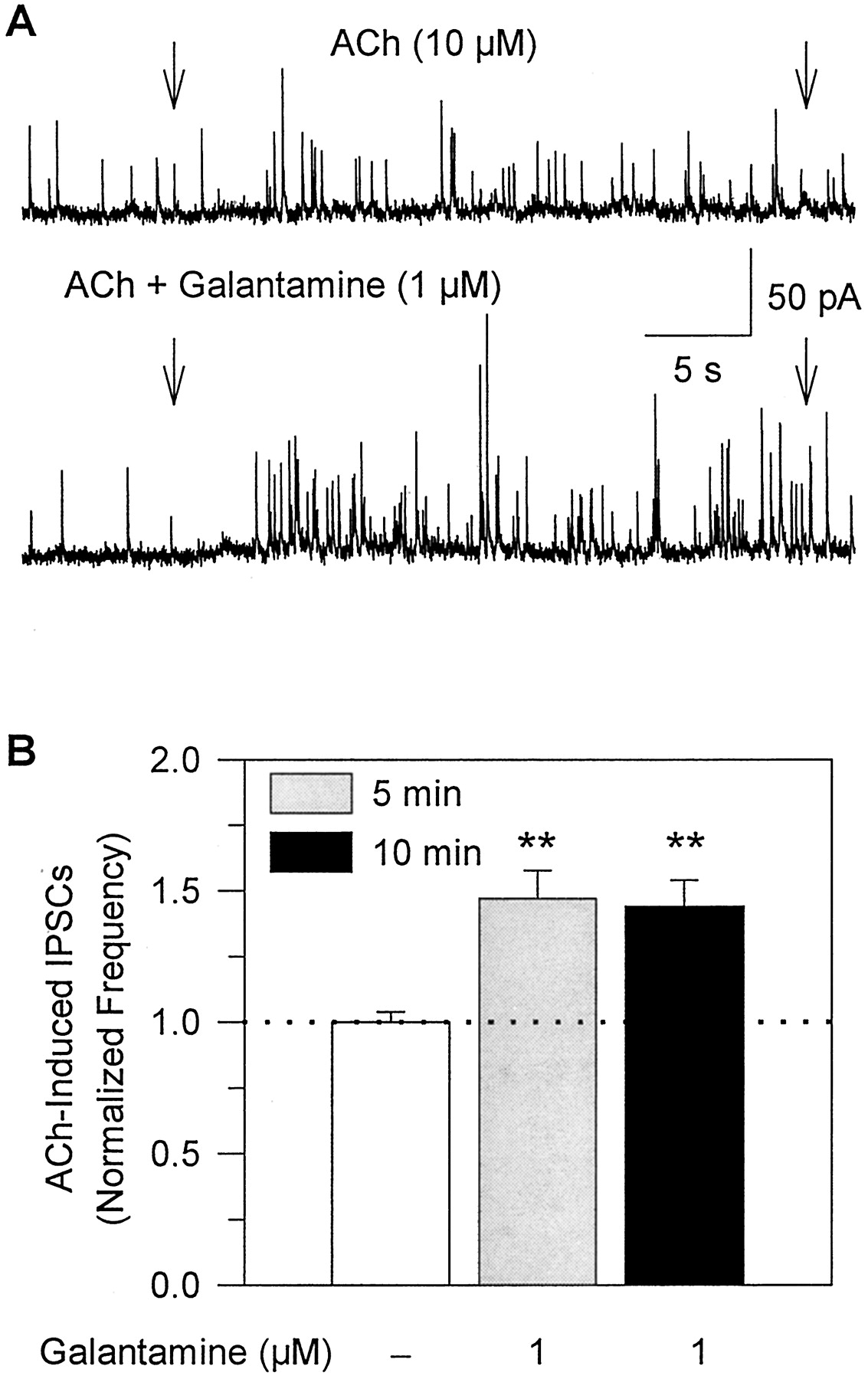
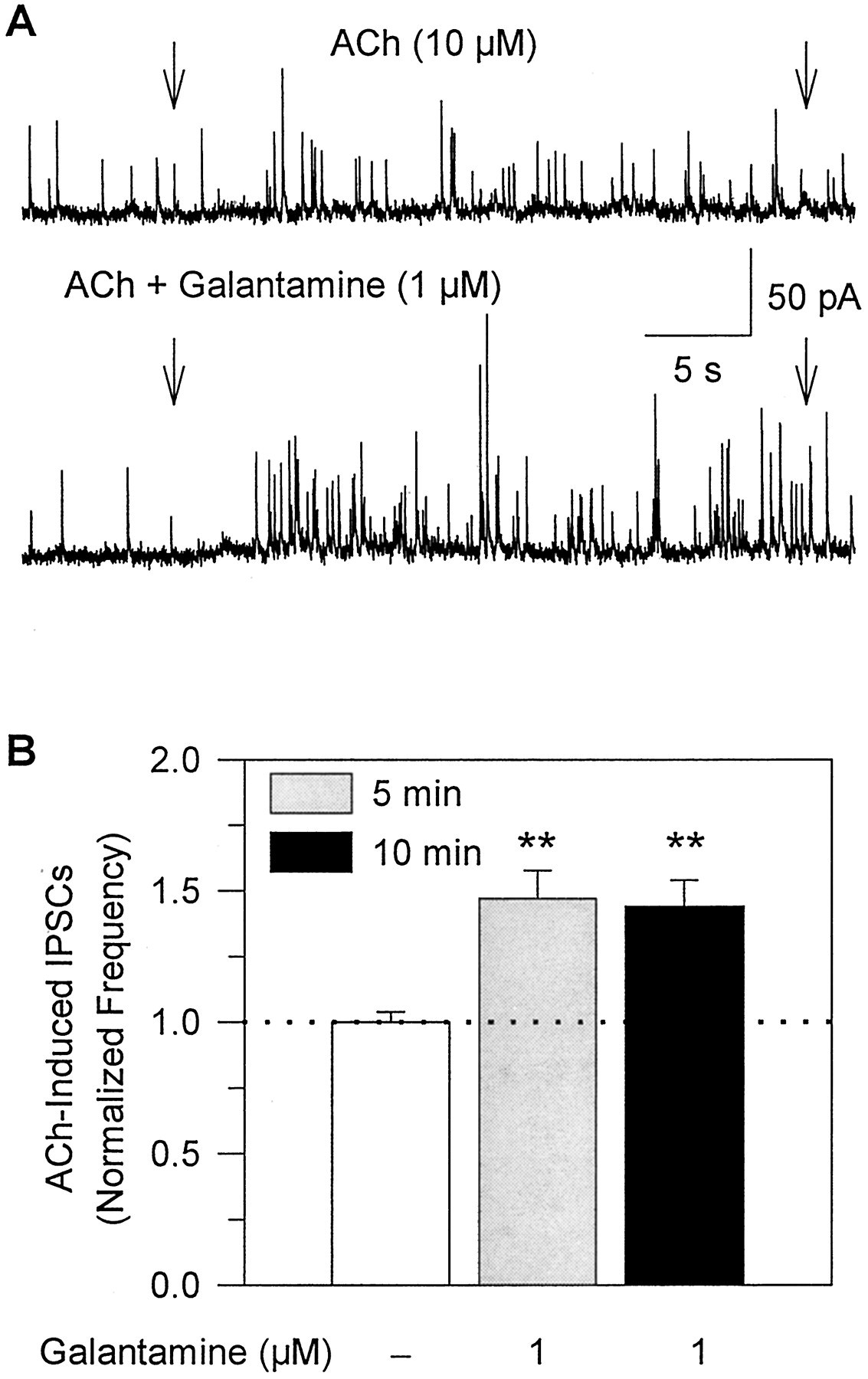
Galantamine potentiates the effect of ACh on GABA-ergic transmission in rat hippocampal slices. A, recording samples of GABA-ergic PSCs obtained from a CA1 stratum radiatum interneuron at 0 mV. Arrows indicate duration of pulse application of 10 μM ACh alone or in admixture with galantamine (1 μM). The admixture of galantamine-plus-ACh was applied to the neuron after the slice had been perfused for 5 min with ACSF containing 1 μM galantamine. B, plot of the frequency of ACh-triggered IPSCs in the absence and in the presence of galantamine after the neurons had been exposed to galantamine for 5 or 10 min. Number of IPSCs recorded for 24 s after beginning the agonist pulse was counted. The frequency of ACh-induced IPSCs in the absence of galantamine was taken as 1 and was used to normalize the frequency of ACh-triggered IPSCs in the presence of galantamine. Graph and error bars represent mean and S.E.M., respectively, of results obtained from five interneurons in the CA1 stratum radiatum. ∗∗,p <0.01 according to the paired Student'st test.
A 5- to 10-min perfusion of hippocampal slices with galantamine-containing ACSF did not result in changes in amplitude or frequency of spontaneously occurring IPSCs recorded from CA1 interneurons. In interneurons voltage clamped at 0 mV, the mean amplitudes of spontaneously occurring GABA-ergic IPSCs recorded in the absence and in the presence of 1 μM galantamine were 16.8 ± 0.60 pA and 16.5 ± 0.72 pA, respectively (n = 5 neurons). In addition, the frequency of spontaneous IPSCs recorded in the absence and in the presence of galantamine was 0.68 ± 0.06 Hz and 0.69 ± 0.05 Hz, respectively. In contrast, the frequency of ACh-evoked IPSCs was significantly increased by galantamine. The frequency of ACh-triggered IPSCs recorded when 10 μM ACh was coapplied with 1 μM galantamine after a 5-min perfusion of the slices with 1 μM galantamine-containing ACSF was ∼50% higher than that recorded before exposure of the slices to galantamine (Fig. 10, A and B). The effect of galantamine was sustained for as long the drug was present (Fig. 10B), and was reversed after washout of the drug. Thus, galantamine-induced potentiation of GABA-ergic transmission is mediated by the interaction of the drug with nAChRs located on GABA-ergic neurons synapsing onto the neurons under study.
Effects of Galantamine on ACh-Triggered IPSCs in Human Cerebral Cortical Slices.
To determine whether the effects of galantamine on synaptic transmission observed in the rat preparation can be extended to humans, experiments were performed in neurons of human cerebral cortical slices according to techniques described previously (Alkondon et al., 2000b). In the continuous presence of the muscarinic receptor antagonist atropine (1 μM), U-tube application of 10 μM ACh to neurons in human cerebral cortical slices triggered IPSCs (Fig.11). This effect of ACh is the result of its interaction with α4β2-nAChRs present on GABA-ergic neurons synapsing onto the neurons from which recordings are obtained (Alkondon et al., 2000b).

Galantamine potentiates the effect of ACh on GABA-ergic transmission in human cerebral cortical slices. A, samples of IPSCs recorded from a human cerebral cortical interneuron at 0 mV. The top trace represents the control ACh response; the middle trace shows the ACh response recorded after a 5-min exposure of the neurons to 1 μM galantamine, and the bottom trace shows the ACh response recorded in the presence of the FK-1 and galantamine after the neurons had been exposed for 10 min to FK-1 and for an additional 5 min to the admixture of galantamine-plus-FK-1. B, plot of the frequency of ACh-triggered IPSCs recorded from human cerebral cortical interneurons under the different experimental conditions. The number of IPSCs recorded for 24 s after beginning the agonist pulse was counted. The frequency of ACh-induced IPSCs in the absence of galantamine was taken as 1 and was used to normalize the frequency of ACh-triggered IPSCs in the presence of galantamine or galantamine-plus-FK-1. Graph and error bars represent mean and S.E.M., respectively, of results obtained from three interneurons in human cerebral cortical slices. ∗∗, p <0.01 according to the paired Student'st test.
Galantamine (1 μM) did not alter the frequency (0.44 ± 0.10 Hz in control and 0.46 ± 0.11 Hz in presence of drug) or amplitude (21.7 ± 2.0 pA in control and 21.5 ± 1.3 pA in the presence of drug) of spontaneously occurring IPSCs recorded from human cerebral cortical neurons. However, it increased substantially the frequency of ACh-triggered IPSCs (Fig. 11A). The frequency of ACh-triggered IPSCs recorded when 10 μM ACh was coapplied with 1 μM galantamine after a 5-min perfusion of the slices with 1 μM galantamine-containing ACSF was ∼80% higher than that recorded before exposure of the slices to galantamine (Fig. 11B). Galantamine-induced potentiation of ACh-triggered IPSCs was blocked after perfusion of the slices with ACSF containing the monoclonal antibody FK-1 (1 μM; Fig. 11, A and B). These results demonstrate that in the human brain galantamine, by acting as a nicotinic APL, can potentiate ACh-induced facilitation of GABA-ergic transmission.
Discussion
The present study demonstrates that galantamine facilitates both excitatory and inhibitory transmissions in the rat and human brains and that these effects are the result of the nicotinic APL action of galantamine. The concentrations at which galantamine facilitates synaptic transmission are very similar to those achieved in the brain of rats treated with memory-enhancing doses of the drug (Bores et al., 1996). Thus, facilitation of synaptic transmission by galantamine is likely to underlie its therapeutic effectiveness in AD.
Galantamine Potentiates Glutamatergic Transmission by Increasing Glutamate Release via a Cholinesterase-Unrelated Mechanism.
Galantamine (0.5–3 μM) enhanced the amplitude of EPSCs recorded from CA1 neurons in response to field stimulation of the Schaffer collaterals in rat hippocampal slices. The rapid onset and reversibility of the effect suggested that galantamine interacts with an extracellular target. Several lines of evidence indicated a presynaptic site of action for galantamine: 1) the drug had no effect on postsynaptic glutamatergic receptors; 2) the amplitudes of both AMPA/kainate and NMDA components of evoked EPSCs were increased by galantamine; and 3) the sum of the magnitude of the effects of galantamine on both components was equal to the magnitude of the drug's effect on compounded EPSCs. Although galantamine-induced potentiation of EPSCs was the result of a presynaptic action, it could not be explained by changes in Na+ and K+ conductances, because the drug caused no changes in the membrane properties of CA1 pyramidal neurons.
Even though the concentrations at which galantamine facilitates glutamatergic transmission are well within the range (0.4–4.0 μM) reported to inhibit by 50% brain cholinesterase activity (Sweeney et al., 1989; Bickel et al., 1991; Bores et al., 1996), the anticholinesterase activity per se cannot explain the effect of galantamine on glutamatergic transmission. First, long-lasting facilitation of glutamate release by prolonged exposure (≥5 min) of hippocampal slices to galantamine could not be observed when the slices were exposed to cholinesterase inhibitors devoid of nicotinic APL activity, including methamidophos, metrifonate, donepezil, and rivastigmine, each tested at nearly saturating concentrations for cholinesterase inhibition. Second, the concentration dependence and dynamics of the effects of methamidophos on evoked EPSCs differed drastically from those of galantamine. Whereas the effects of methamidophos on evoked EPSCs were only observed at concentrations that inhibit by 100% cholinesterase activity (Camara et al., 1997), those of galantamine were observed at concentrations that inhibit cholinesterase activity by ∼50%. In addition, galantamine-induced facilitation of glutamatergic transmission lasted for as long as the neurons were exposed to galantamine, whereas methamidophos-induced potentiation of evoked EPSCs was transient, lasting no longer than 1 min. Third, the magnitude of galantamine's effect on evoked EPSCs in hippocampal slices in which cholinesterase was blocked by methamidophos was the same as that observed in control slices.
Galantamine-Induced Facilitation of Synaptic Transmission in the Rat Hippocampus and the Human Cerebral Cortex Is Mediated by Its Action as a Nicotinic APL.
In addition to increasing the amplitude of evoked EPSCs, galantamine, via a presynaptic action, also increased the amplitude of evoked IPSCs in the rat hippocampus and the frequency of IPSCs triggered by low concentrations of ACh in the rat hippocampus and human cerebral cortex. As reported in previous studies, GABA-ergic transmission in the rat hippocampus and human cerebral cortex is modulated by presynaptically located α7- and/or α4β2-nAChRs (Alkondon et al., 1999; 2000b). In this study, exogenous application of nAChR antagonists (MLA and DHβE) and different concentrations of ACh to hippocampal slices led to the conclusions that α7-nAChRs are present and tonically active in the Schaffer collaterals and that, depending on the degree of receptor activity, glutamate release onto CA1 neurons can be facilitated or inhibited (see Fig. 6).
Exogenous application of ACh to hippocampal slices had a bi-modal effect on evoked glutamatergic transmission between Schaffer collaterals and CA1 neurons; transmission was facilitated by 30 nM ACh and inhibited by 30 μM ACh. Facilitation of glutamatergic transmission by low level of α7-nAChR activation was probably the result of increased intracellular Ca2+concentrations (Radcliffe et al., 1999; Albuquerque et al., 2000b). In contrast, inhibition of synaptic transmission was probably the result of α7-nAChR activation to a degree that causes enough depolarization to inactivate Na+ channels and to dampen the active propagation of action potentials (Alkondon et al., 2000a). The finding that MLA and DHβE potentiate glutamatergic transmission between Schaffer collaterals and CA1 neurons indicated that, overall, this transmission is preset by the inhibitory action of strongly, tonically activated nAChRs in some glutamatergic fibers prevailing over the potential facilitatory action of weakly, tonically activated nAChRs in other glutamatergic fibers synapsing onto a CA1 neuron. Tonically activated nAChRs are also known to modulate synaptic transmission in other areas of the CNS (Cordero-Erausquin and Changeux, 2001).
The potentiating effect of galantamine on evoked glutamatergic transmission in hippocampal slices is mediated by its interaction as an APL with presynaptically located α7-nAChRs, because the effect is observed in the presence of α7-nAChR antagonists or FK1, which functionally antagonizes nicotinic APL actions (Pereira et al., 1993;Schrattenholz et al., 1993, 1996). Considering that galantamine sensitizes nAChRs to activation by low, but not high, agonist concentrations (Schrattenholz et al., 1996; Samochocki et al., 2000), it can be concluded that at 1 μM galantamine increases facilitation of synaptic transmission by weakly, tonically activated nAChRs just enough to override inhibition by strongly, tonically activated nAChRs. Unoccupied, resting nAChRs may also exist in the Schaffer collaterals (see Fig. 6). However, it is unlikely that galantamine-induced potentiation of glutamatergic transmission is the result of direct activation of these free receptors, because galantamine is a very weak nAChR agonist (Pereira et al., 1994; Storch et al., 1995).
The bell-shaped concentration-response relationship for galantamine-induced potentiation of glutamatergic transmission in hippocampal slices can be explained by the fact that up to 1 μM galantamine increases agonist-induced nAChR activation and reduces/prevents agonist-induced nAChR desensitization, whereas at higher concentrations, it inhibits agonist-induced nAChR activation (Pereira et al., 1993; Schrattenholz et al., 1996; Samochocki et al., 2000).
The effects of galantamine on field stimulation-evoked GABA-ergic transmission in rat hippocampal slices and on ACh-triggered GABA release in rat hippocampal and human cerebral cortical slices could also be accounted for by its interaction with nAChRs located on GABA-ergic neurons/axons synapsing onto the neurons from which recordings were obtained. The finding that galantamine alone did not alter the frequency of spontaneously occurring IPSCs in rat hippocampal slices ruled out a direct agonist action of the drug on presynaptically located nAChRs that are in the resting, agonist-free state. Finally, block by FK1 of galantamine-induced potentiation of ACh-triggered GABA release in rat hippocampal and human cerebral cortical slices demonstrated that the effect was the result of the nicotinic APL action of galantamine.
Clinical Relevance of Galantamine Actions as a Nicotinic APL on Synaptic Transmission in the Brain.
In the CNS, glutamatergic, GABA-ergic, and cholinergic activities have been associated with cognitive processing (Menschik and Finkel, 1998) and other forms of synaptic plasticity, including dendritic spine motility and shaping (Papa and Segal, 1996; Fischer et al., 2000; Shoop et al., 2001). Cholinergic, glutamatergic, and GABA-ergic malfunctions have also been associated with cognitive impairment in AD patients (Mohr et al., 1986;Farber et al., 1998). Significant progress has been made in understanding the role of nAChRs in the pathology of AD (Maelicke and Albuquerque, 1996; Nordberg, 1999) and their relation to cognitive function (Rezvani and Levin, 2001). Modulation of GABA-ergic and glutamatergic transmissions by nAChRs seems to underlie the ability of nicotinic agonists to improve learning and memory in animal models and humans (Rezvani and Levin, 2001), and can be a major determinant of the therapeutic effectiveness of galantamine in AD patients (Tariot et al., 2000; Coyle and Kershaw, 2001).
Galantamine has been shown to improve the performance of AD patients in memory tests (Bickel et al., 1991; Tariot et al., 2000; Coyle and Kershaw, 2001) and of rats in the step-down passive test (Bores et al., 1996). There is a striking similarity between the dose dependence of galantamine-induced enhancement of step-down passive avoidance in rats (Bores et al., 1996) and the concentration dependence of galantamine-induced facilitation of transmitter release (Fig. 2). In both conditions, there is a bell-shaped relationship between dose/concentration and effect. In addition, the brain concentration of galantamine achieved after treatment of rats with the most behaviorally active dose (2.5 mg/kg, p.o.) is ∼0.8 μM (Bores et al., 1996). This concentration is about the same as that at which galantamine exerted its highest effect on synaptic transmission. This remarkable similarity further underscores the relevance of modulation of transmitter release to the reported memory-enhancing effects of galantamine in AD patients.
In conclusion, the present study provides evidence that at concentrations that are behaviorally effective in enhancing cognitive tasks in animal models, galantamine, acting primarily as an APL at presynaptically located nAChRs, facilitates glutamatergic and GABA-ergic transmissions in both rat and human brains. The development of drugs with nicotinic APL action represents a novel and effective approach to drug therapy for AD and other diseases linked to impaired nicotinic functions in the CNS.
Acknowledgments
We thank Mabel Zelle, Barbara Marrow, and Bhagavathy Alkondon for technical support.
Footnotes
- Received June 25, 2001.
- Accepted February 4, 2002.
-
This work was supported by a grant from the Janssen Pharmaceutical Research Foundation, by United States Army Medical and Research Development Command Contract DAMD-17-95-C-5063, and by United States Public Health Service Grant NS25296 (to E.X.A.). A preliminary account of this study was presented at the 2000 Annual Meeting of the Society for Neurosciences (Soc Neurosci Abstr 26:1914, abstr. 716.5).
Abbreviations
- ACh
- acetylcholine
- ACSF
- artificial cerebrospinal fluid
- AD
- Alzheimer's disease
- AMPA
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- ANOVA
- analysis of variance
- APL
- allosteric potentiating ligand
- APV
- 2-amino-5-phosphonovaleric acid
- CNQX
- 6-cyano-7-nitroquinoxaline-2,3-dione
- CNS
- central nervous system
- DHβE
- dihydro-β-erythroidine
- EPSC
- excitatory postsynaptic current
- GABA
- γ-aminobutyric acid
- IPSC
- inhibitory postsynaptic current
- mIPSC
- miniature inhibitory postsynaptic current
- MLA
- methyllycaconitine
- nAChR
- nicotinic acetylcholine receptor
- NMDA
- N-methyl-d-aspartate
- PSC
- postsynaptic current
- The American Society for Pharmacology and Experimental Therapeutics
References
MolPharm articles become freely available 12 months after publication, and remain freely available for 5 years.Non-open access articles that fall outside this five year window are available only to institutional subscribers and current ASPET members, or through the article purchase feature at the bottom of the page.
|





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}