


Abstract
Although neurosteroids have rapid effects on GABAA receptors, study of steroid actions at GABA receptors has been hampered by a lack of pharmacological antagonists. In this study, we report the synthesis and characterization of a steroid analog, (3α,5α)-17-phenylandrost-16-en-3-ol (17PA), that selectively antagonized neurosteroid potentiation of GABA responses. We examined 17PA using the α1β2γ2 subunit combination expressed in Xenopus laevis oocytes. 17PA had little or no effect on baseline GABA responses but antagonized both the response augmentation and the direct gating of GABA receptors by 5α-reduced potentiating steroids. The effect was selective for 5α-reduced potentiating steroids; 5β-reduced potentiators were only weakly affected. Likewise, 17PA did not affect barbiturate and benzodiazepine potentiation. 17PA acted primarily by shifting the concentration response for steroid potentiation to the right, suggesting the possibility of a competitive component to the antagonism. 17PA also antagonized 5α-reduced steroid potentiation and gating in hippocampal neurons and inhibited anesthetic actions in X. laevis tadpoles. Analogous to benzodiazepine site antagonists, the development of neurosteroid antagonists may help clarify the role of GABA-potentiating neurosteroids in health and disease.
GABAA receptors are important participants in central nervous system inhibition and are the target of many clinically important compounds, including benzodiazepines, barbiturates, neuroactive steroids, and other general anesthetics (Macdonald and Olsen, 1994; Belelli et al., 1999). By allosterically potentiating GABAA receptor-mediated responses, these compounds generally increase inhibition and decrease overall activity of neuronal circuits (Macdonald and Olsen, 1994). Neuroactive steroids are particularly interesting because they may be present endogenously at concentrations that modulate GABAA receptor function (Robel and Baulieu, 1994).
Many important questions remain unanswered regarding steroid actions at GABAA receptors. Despite concerted efforts, a steroid binding site on the GABAA receptor has not been identified. GABA-potentiating steroids have two distinct structural classes: 5α-reduced and 5β-reduced (Covey et al., 2001). Both classes potentiate GABAA responses with similar potency and efficacy; whether the two classes work at the same site or by the same mechanisms is unclear. In addition, the role of endogenous neurosteroids in modulating GABAA receptors is obscure. New pharmacological tools for exploring neuroactive steroids, including neurosteroid antagonists, would advance efforts to understand the interaction of steroids with GABAA receptors.
Several earlier studies explored potential steroid antagonists. [3H]Flunitrazepam binding assays suggest that 3β-hydroxysteroids competitively antagonize the actions of the potentiating steroids (3α,5α)-3-hydroxypregnan-20-one (3α5αP) and (3α,5β)-3-hydroxypregnan-20-one (3α5βP), two standard representatives of the 5α-reduced and 5β-reduced classes of potentiating steroids (Prince and Simmonds, 1992, 1993). In electrophysiological studies, however, 3β-hydroxypregnane steroids noncompetitively antagonize potentiating steroids through activation-dependent block of GABAA receptors (Wang et al., 2002). The block by 3β-hydroxysteroids is not specific; barbiturate potentiation is reduced, as are responses to GABA itself at high GABA concentrations (Wang et al., 2002). Therefore, 3β-hydroxysteroids, although interesting functional antagonists of steroid potentiation, lack the specificity desired of a tool for exploring steroid potentiation of GABAA receptors.
In the present study, we examined the actions of a synthetic neuroactive steroid analog that was inactive as a potentiator at GABAA receptors in our functional screens, (3α,5α)-17-phenylandrost-16-en-3-ol (17PA). 17PA represents the first described selective antagonist of neurosteroid potentiation at GABAA receptors. It is noteworthy that 17PA antagonized the physiological and anesthetic actions of 5α-reduced steroids but had little effect on 5β-reduced steroids. Furthermore, 17PA inhibited direct gating of GABAA receptor channels by 5α-reduced but not 5β-reduced steroids. Inhibition of 5α-reduced steroid potentiation decreased with increasing potentiating steroid concentration, suggesting the possibility of a competitive interaction between the antagonist and potentiating steroids. 17PA did not antagonize barbiturate or benzodiazepine potentiation. Our results suggest that 5α-reduced and 5β-reduced steroids are likely to possess different sites or mechanisms of potentiation and support the hypothesis that GABAA receptors are critical participants in the behavioral anesthesia induced by neuroactive steroids.
Materials and Methods
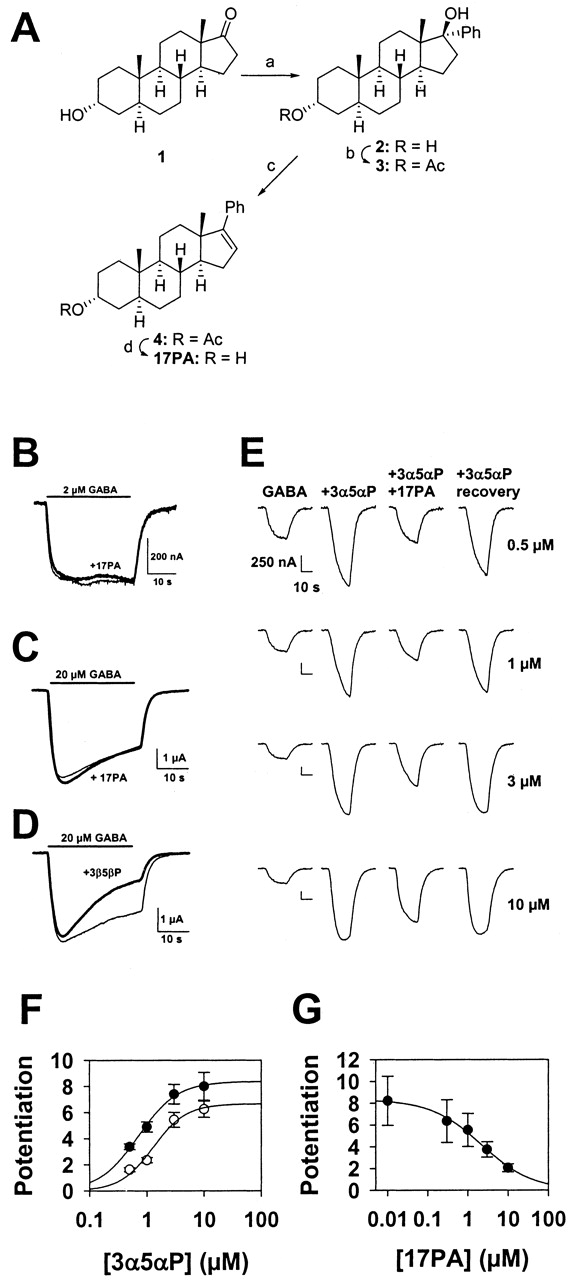
17PA Synthesis.Fig. 1A shows the reaction scheme for the synthesis of 17PA.

17PA effects on 5α-reduced neurosteroid potentiation of GABAA receptors. A, synthetic scheme for 17PA. Reagents and conditions: a, PhLi, tetrahydrofuran, 0°C to 25°C, 5 h, 44%; b, Ac2O, pyridine, 4-(N,N-dimethylamino)pyridine, 25°C, 5 min, 95%; c, MsCl, Et3N, CH2Cl2, 0°C, 40 min, 99%; and d, NaOH, H2O, MeOH, 65°C, 1 h, 95%. B to D, 17PA effect on GABA responses. B, 17PA (10 μM; thick trace) had no effect on responses to 2 μM GABA in an X. laevis oocyte expressing the α1β2γ2 GABAA receptor subunit combination. C, oocyte responses to high GABA concentrations were unaffected by 10 μM 17PA. D, in contrast, the GABA response from the same oocyte depicted in C was markedly depressed by 3β5βP (10 μM). For B to D, thin traces represent responses to GABA alone; thick traces represent steroid co-application with GABA. E, effects of 10 μM 17PA on increasing potentiating steroid concentrations. Note that the 17PA effect became weaker as potentiating steroid concentration was increased. F, summary of experiments like that shown in E (n = 11 oocytes). Solid symbols represent 3α5αP effects in the absence of 17PA; open symbols represent 3α5αP effects in the presence of 10 μM 17PA. The solid lines are fits of the Hill equation with the minimum level of potentiation constrained to zero but with maximum potentiation unconstrained. Parameters for the fits in the absence of 17PA were 0.7 μM for the EC50, 8.4 for maximum potentiation, and 1.4 for the Hill coefficient. Parameters for fits in the presence of 17PA were 1.3 μM for the EC50, 6.7 for maximum potentiation, and 1.5 for the Hill coefficient. G, summary of the effects of varied concentrations of 17PA against a fixed 3α5αP concentration (500 nM). The IC50 for 17PA estimated from a fit to the Hill equation was 2.2 μM. Data were averaged from three oocytes.
(3α,5α,17β)-17-Phenylandrostane-3,17-diol (Compound 2). Phenyllithium (1.8 M in cyclohexane-ether, 2.77 ml) was added to compound 1 (290 mg, 1.0 mmol) in anhydrous tetrahydrofuran (25 ml) at 0°C. The resultant solution was stirred at ambient temperature for 5 h. Saturated NH4Cl (5 ml) was added, and the product was extracted with EtOAc. The combined extracts were washed with NH4Cl and brine and dried over Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (silica gel; hexanes/EtOAc, 8:1) to give compound 2 (162 mg, 44%).
Compound 2 was obtained as white crystals: mp. 193°C to 196°C (EtOAc-hexanes). 1H NMR δ 0.75 (s, 3H), 1.04 (s, 3H), 2.08 (m, 1H), 2.37 (m, 1H), 3.97 (m, 1H), 7.23 to 7.41 (m, 5H); 13C NMR δ 11.1, 14.9, 20.3, 24.3, 28.4, 28.9, 31.6, 32.0, 33.6, 35.8, 36.0, 36.2, 38.6, 39.1, 46.7, 49.1, 53.7, 66.4, 86.0, 126.7, 127.2 (2 × C), 127.3 (2 × C), 146.1; IR νmax 3403, 2926, 1446, 1002, 908 cm-1. Anal. Calcd for C25H36O2: C, 81.47; H, 9.85; found: C, 81.60; H, 9.89.
(3α,5α,17β)-17-Phenylandrostane-3,17-diol, 3-acetate (Compound 3). 4-(N,N-Dimethylamino)pyridine (16 mg) was added to the solution of compound 2 (160 mg, 0.43 mmol) in Ac2O (0.5 ml) and pyridine (1.5 ml). The mixture was stirred at room temperature for 5 min and poured into 10% NaHCO3 (20 ml). After the mixture was stirred for 10 min, the product was extracted with EtOAc. The combined extracts were washed with 5% HCl and water and dried over Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (silica gel; hexanes/EtOAc, 10:1) to give compound 3 (169 mg, 95%).
Compound 3 was obtained as white crystals: mp. 196°C to 198°C. 1H NMR δ 0.77 (s, 3H), 1.05 (s, 3H), 1.98 (s, 3H), 2.09 (m, 1H), 2.38 (m, 1H), 4.96 (m, 1H), 7.23 to 7.42 (m, 5H); 13C NMR δ 11.3, 14.9, 20.3, 21.5, 24.3, 26.0, 28.3, 31.6, 32.7, 32.8, 33.6, 35.7, 36.2, 38.6, 40.0, 46.7, 49.1, 53.6, 70.1, 86.0, 126.7, 127.2 (2 × C), 127.4 (2 × C), 146.1, 170.6; IR νmax 3511, 2927, 1719, 1445, 1269, 1024 cm-1. Anal. Calcd for C27H38O3: C, 78.98; H, 9.33; found: C, 79.04; H, 9.22.
(3α,5α)-17-Phenylandrost-16-en-3-ol Acetate (Compound 4). Et3N (0.60 ml, 4.31 mmol) and MsCl (0.17 ml, 2.20 mmol) were added to compound 3 (160 mg, 0.39 mmol) in CH2Cl2 (15 ml) at 0°C and stirred for 40 min. After CH2Cl2 was removed under reduced pressure, the residue was purified by column chromatography (silica gel; hexanes/EtOAc, 15:1) to give compound 4 (151 mg, 99%).
Compound 4 was obtained as white crystals: mp. 152°C to 153°C (hexanes). 1H NMR δ 0.85 (s, 3H), 1.02 (s, 3H), 2.05 (s, 3H), 2.10 (m, 1H), 5.02 (m, 1H), 5.90 (m, 1H), 7.19 to 7.39 (m, 5H); 13C NMR δ 11.3, 16.7, 20.8, 21.5, 26.1, 28.3, 31.5, 31.8, 32.7, 32.9, 34.0, 35.5, 36.0, 40.3, 47.4, 54.5, 57.6, 70.1, 126.7 (3 × C), 127.2, 128.0 (2 × C), 137.4, 154.9, 170.7; IR νmax 2948, 1724, 1494, 1446, 1374, 1246 cm-1. Anal. Calcd for C27H36O2: C, 82.61; H, 9.24; found: C, 82.48; H, 9.02.
17PA. 5N NaOH (1 ml) was added to compound 4 (145 mg, 0.37 mmol) in MeOH (15 ml). The mixture was refluxed for 1 h and cooled to room temperature. Water was added, and the product was extracted with CH2Cl2. The combined extracts were washed with water to neutral and dried over Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (silica gel; hexanes/EtOAc, 10:1) to give compound 5 (123 mg, 95%).
Compound 17PA was obtained as white crystals: mp. 174.5°C to 175.5°C (EtOAc-hexanes). 1H NMR δ 0.83 (s, 3H), 1.02 (s, 3H), 2.02 (m, 2H), 2.20 (m, 1H), 4.04 (m, 1H), 5.89 (m, 1H), 7.18 to 7.42 (m, 5H); 13C NMR δ 11.2, 16.7, 20.8, 28.5, 29.0, 31.5, 31.8, 32.0, 34.0, 35.5, 35.9, 36.3, 39.3, 47.4, 54.6, 57.6, 66.5, 126.6, 126.7 (2 × C), 127.2, 128.0 (2 × C), 137.4, 154.9; IR νmax 3305, 2928, 1494, 1444, 1002 cm-1. Anal. Calcd for C25H34O: C, 85.66; H, 9.78; found: C, 85.74; H, 9.95.
(3β,5α)-3-hydroxypregnan-20-one sulfate was obtained from Steraloids (Newport, RI), and all other drugs were obtained from Sigma/RBI (Natick, MA). Steroids were dissolved in DMSO, with final working concentrations of DMSO typically ≤0.1%. DMSO concentration was matched across all working solutions to account for any actions of solvent alone. Pentobarbital was dissolved in 0.1 N NaOH.
GABA Receptor Expression in Xenopus laevis Oocytes. Stage V to VI oocytes were harvested from mature female X. laevis (Xenopus I, Ann Arbor, MI) and were defolliculated and injected with cRNA encoding rat GABAA receptor α1, β2, and γ2L subunits. Capped cRNA was transcribed using the mMESSAGE mMachine kit (Ambion, Austin, TX). Oocytes were incubated up to 5 days at 18°C in ND96 medium containing: 96 mM NaCl, 1 mM KCl, 1 mM MgCl2, 2 mM CaCl2, and 5 mM HEPES at pH 7.4, supplemented with pyruvate (5 mM), penicillin (100 U/ml), streptomycin (100 μg/ml), and gentamycin (50 μg/ml). Two-electrode voltage-clamp experiments were performed 2 to 5 days after RNA injection. Extracellular recording solution was ND96 medium, and 1-MΩ intracellular recording pipettes were filled with 3 M KCl. Cells were clamped at -70 mV for all experiments, and current at the end of 20- to 60-s drug applications was measured for quantification of current amplitudes. Steroids and other modulators were co-applied with GABA by gravity perfusion.
Hippocampal Culture Recordings. Primary cultures of hippocampal cells were prepared from 1- to 3-day old postnatal Sprague-Dawley rats and plated on microdroplets of collagen using established methods (Mennerick et al., 1995). In brief, minced hippocampal slices were dissociated with 1 mg/ml papain in oxygenated Leibovitz L-15 medium and triturated in modified Eagle's medium containing 5% horse serum, 5% fetal calf serum, 17 mM d-glucose, 400 μM glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin. Dissociated cells were plated at 75 cells/mm2 onto 35-mm plastic culture dishes previously sprayed with collagen microdroplets (Mennerick et al., 1995). Cultures were treated with cytosine arabinoside (5-10 μM) after 3 days in vitro to halt glial proliferation.
Electrophysiological recordings were done 8 to 21 days after plating. Whole-cell recordings at room temperature were performed using an Axopatch 1D amplifier (Axon Instruments Inc., Union City, CA) interfaced to a Pentium III-based computer via a Digidata 1200 acquisition board (Axon Instruments Inc.). Access resistance was typically <12 MΩ and was not compensated. The extracellular recording solution consisted of: 138 mM NaCl, 4 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose, 10 mM HEPES, 0.025 mM d-2-aminophosphonovalerate, 0.001 mM 1,2,3,4-tetrahydro-6-nitro-2,3-dioxobenzo[f]quinoxaline-7-sulfonamide, and 0.0002 mM tetrodotoxin, pH 7.25. Solutions were exchanged via a local multibarrel perfusion pipette with a common delivery port placed 0.5 mm from the cell under study. The pipette solution contained: 140 mM CsCl, 4 mM NaCl, 0.5 mM CaCl2, 5 mM EGTA, and 10 mM HEPES, pH 7.25. In some experiments (Fig. 4, C and D), CsCl was replaced with Cs methanesulfonate, and neurons were clamped at -20 mV.
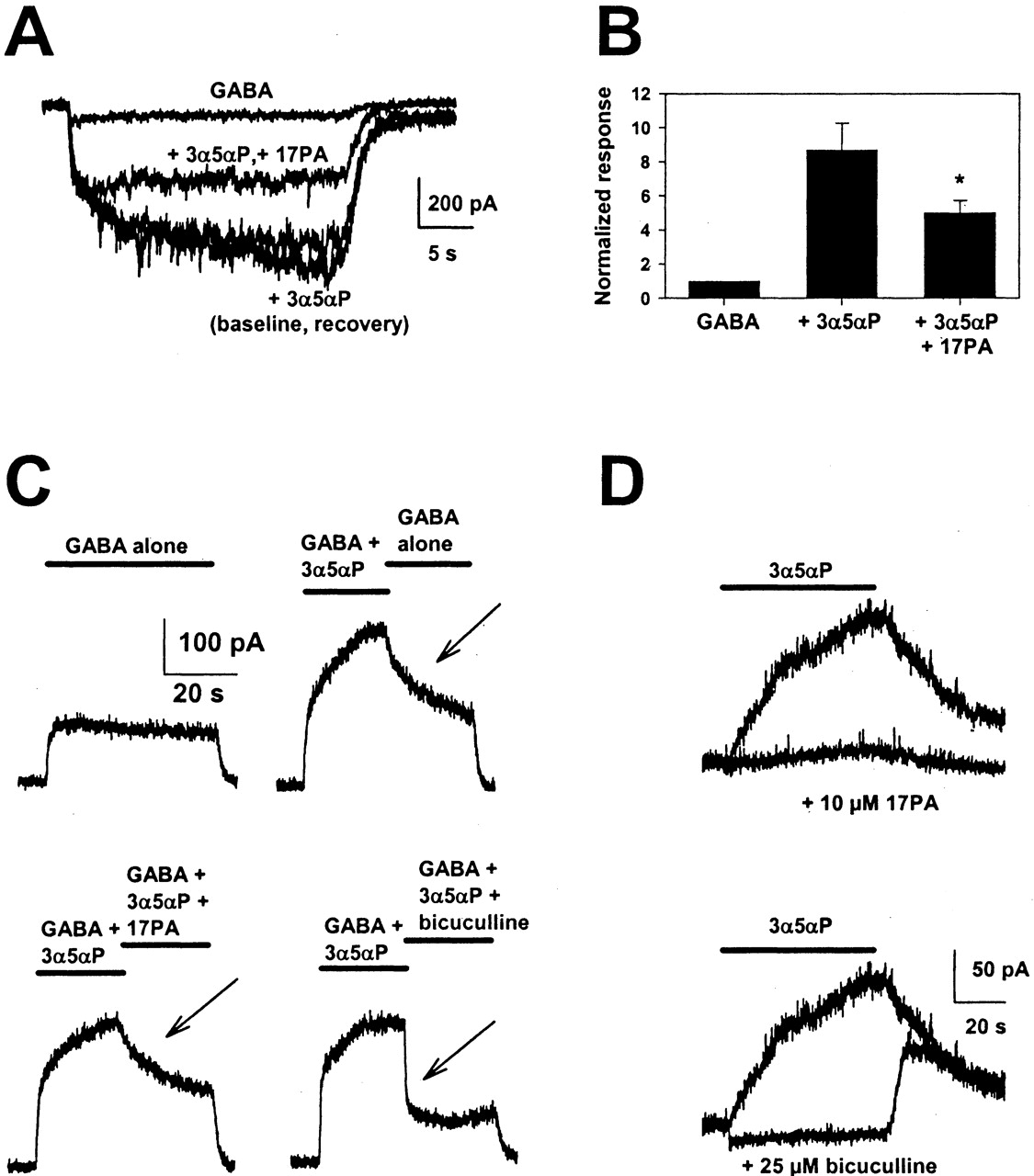
17PA effects in cultured hippocampal neurons. A, 17PA effect on GABA currents potentiated by 3α5αP in hippocampal neurons. Drug concentrations were 0.5 μM GABA, 0.1 μM 3α5αP, and 10 μM 17PA. B, summary of results from the experiment depicted in C over five neurons. Amplitudes of potentiated currents were depressed 40 ± 3% by 17PA (p < 0.05). C, block kinetics tracked with potentiator dissociation. The indicated protocols were performed on a hippocampal neuron using 0.5 μM GABA, 0.1 μM 3α5αP, and 10 μM 17PA. See text for details. Arrows denote the decay of currents under various conditions. D, 17PA and bicuculline effects on direct gating in hippocampal neurons. Top, 3α5αP (200 nM) gated a slowly activating and deactivating current in a hippocampal neuron that was nearly abolished by 10 μM 17PA. Bottom, in the same cell, the current gated by 3α5αP is superimposed on a trace in which 25 μM bicuculline was co-applied with 3α5αP. Note the tail current upon bicuculline washout.
Tadpole Anesthesia. Groups of 10 early prelimb bud X. laevis tadpoles (Nasco, Fort Atkinson, WI) were placed in 100 ml of oxygenated Ringer's solution containing various concentrations of compound (Wittmer et al., 1996). Compounds were added from a 10 mM DMSO stock (final DMSO concentration ≤ 0.2%). After equilibrating at room temperature for 3 h, tadpoles were evaluated for loss of righting reflex (LRR) or loss of swimming reflex (LSR) behavioral endpoints. LRR was defined as failure of the tadpole to right itself within 5 s after being flipped by a smooth glass rod. LSR was defined by a failure of purposeful tail movement within 10 s of gently sliding the tadpole around the beaker with a glass rod for 5 s. Control beakers containing up to 0.6% DMSO produced no LRR in tadpoles.
Data Analysis. Electrophysiology data acquisition and analysis were performed with pCLAMP (Axon Instruments Inc.). Data plotting and curve fitting were done with Sigma Plot (SPSS Inc., Chicago, IL). Data are presented as mean ± S.E. Statistical differences were determined using a two-tailed Student's t test. Fitting of concentration-response data for electrophysiology and behavioral assays was performed using the Hill equation: r = Rmax/[1 + (EC50/[conc])nH], where Rmax is the maximum effect, [conc] is the steroid concentration, EC50 is the half-maximal effective concentration, and nH is the Hill coefficient.
Results
Synthesis of the Neuroactive Steroid Antagonist 17PA. As noted above, because modifications of 3β-hydroxysteroids result in noncompetitive, activation-dependent GABA receptor antagonists rather than specific antagonists of neuroactive steroids (Wang et al., 2002), we have recently shifted our focus toward altering substituents at or near C17, another region of neuroactive steroids known to be critical for interactions at GABAA receptors (Phillipps, 1975; Covey et al., 2001). The steroid 17PA was prepared as part of this effort (Fig. 1A).
Effects of 17PA on Potentiating Steroids.Fig. 1B shows the lack of effect of 17PA on GABA responses in X. laevis oocytes expressing the α1β2γ2 subunit combination. GABA was used at 2 μM in this experiment, representing a response amplitude approximately 5-fold below the EC50 (Wang et al., 2002). At 20 μM GABA, responses were still unaffected (Fig. 1C). Overall, 17PA altered responses to 2 μM GABA by -5 ± 4% (n = 8) and responses to 20 μM GABA by +2 ± 1% (n = 3). Previous results have suggested that 3β-hydroxysteroids might be selective antagonists of neuroactive steroids (Prince and Simmonds, 1992, 1993). However, our previous results suggested that 3β-hydroxysteroids may be activation-dependent noncompetitive GABA receptor antagonists (Wang et al., 2002). In agreement with these results, the putative steroid antagonist 3β5βP directly inhibited responses to 20 μM GABA (Fig. 1D). Thus, 17PA, unlike 3β-hydroxysteroids, is not a direct antagonist of GABA responses.
Although it did not affect GABA responses, 10 μM 17PA suppressed the potentiation of GABA responses evoked by the endogenous potentiating neurosteroid 3α5αP (Fig. 1, E-G). Increasing the concentration of potentiator largely overcame the 17PA effect, suggesting that a competitive mechanism might contribute to the interaction (Fig. 1, E and F). However, analysis of concentration-response curves predicted a maximum response that was smaller than the maximum in the absence of potentiator (Fig. 1F). In addition, when directly tested against 20 μM 3α5αP, 17PA still significantly reduced the potentiated response (data not shown). Steroid solubility and receptor desensitization precluded study of potentiator concentrations above 20 μM. These results suggest that the mechanism of 17PA block is not simple competition, although potential explanations for the decreased maximum response are discussed below. When 3α5αP concentration was fixed at 500 nM, an IC50 of approximately 2 μM was observed (Fig. 1G).
Specificity of 17PA. We also assessed the ability of 17PA to antagonize the potentiation of GABA responses by two other classes of commonly used GABAA receptor modulators, benzodiazepines and barbiturates. 17PA had no effect on potentiation evoked by either pentobarbital or lorazepam (Fig. 2), suggesting specificity of the antagonist for potentiating steroids.

Potentiation by barbiturates and benzodiazepines was unaffected by 17PA. A, potentiation by 20 μM pentobarbital (Ptb) was unaffected by 10 μM 17PA. B, lorazepam (Lzpm; 0.1 μM) potentiation was also unaffected. GABA was used at 2 μM in both experiments. C, summary of the pentobarbital experiments (n = 12). D, summary of the lorazepam experiments (n = 8).
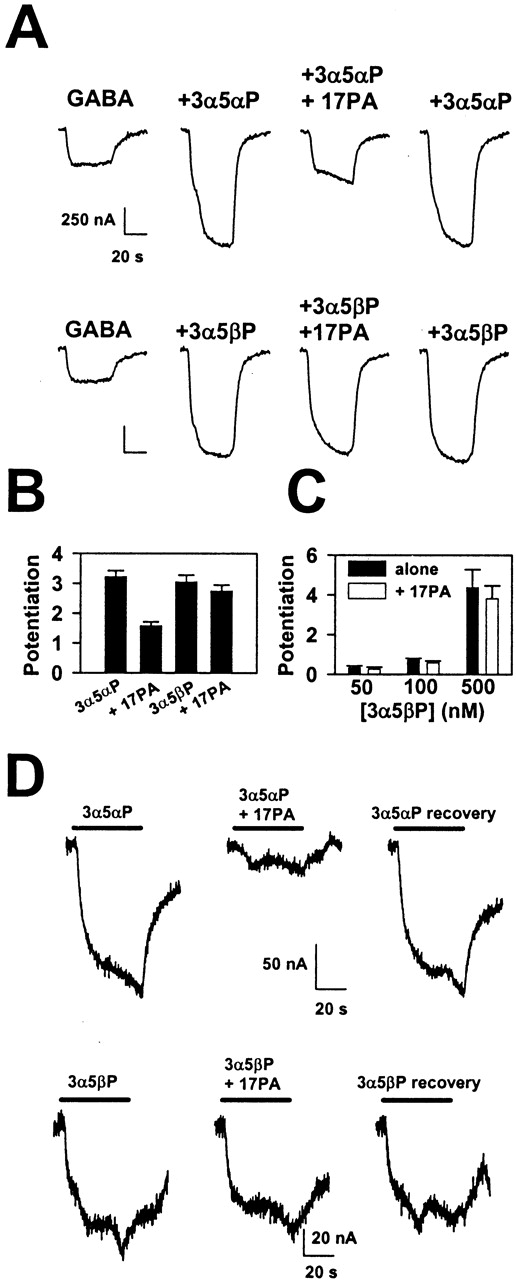
Specificity was further probed by examining the effect of 17PA on potentiation by 5β-reduced steroids. 3α5βP potentiated GABA responses nearly as well as 3α5αP, but 17PA had essentially no effect on responses enhanced by 3α5βP (Fig. 3, A and B). Even at the lowest concentrations of 3α5βP at which potentiation was observed, there was no detectable effect of 17PA (Fig. 3C). Thus, 17PA ineffectiveness did not result from saturation of potentiation by 3α5βP. We also examined the ability of 17PA to antagonize potentiation by another pair of 5α-reduced and 5β-reduced steroids, (3α,5α)-3,21-dihydroxypregnan-20-one and (3α,5β)-3,21-dihydroxypregnan-20-one (500 nM). Again, although the 5α-reduced steroid response was significantly reduced (38 ± 4%, n = 5), the response to co-application of GABA and the 5β-reduced steroid was nearly unaffected (13 ± 4%).
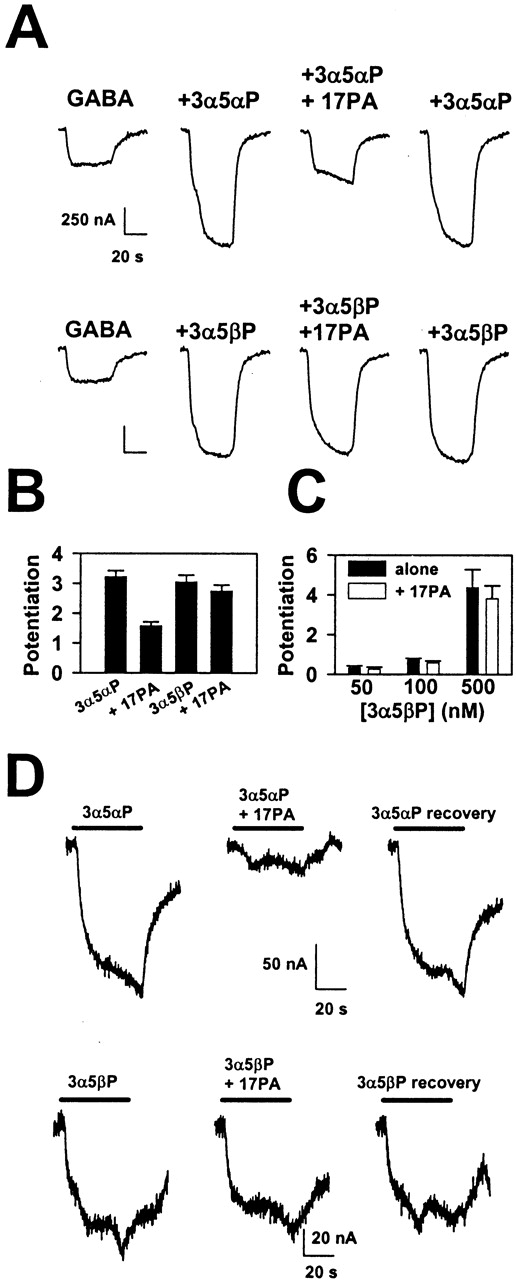
17PA only weakly affects 5β-reduced steroids. A, GABA responses under the indicated conditions. GABA was used at 2 μM, 3α5αP and 3α5βP at 0.5 μM, and 17PA at 10 μM. B, summary of effects like that shown in A from four oocytes. C, summary of potentiation by 3α5βP at very low concentrations and the effect of 10 μM 17PA (n = 3, 4, and 7, respectively, at the low, medium, and high concentrations of potentiator). D, 17 PA inhibited currents directly gated by a 5α-reduced steroid. Top, 3α5αP (5 μM) was applied and gated a slowly developing and decaying current in an oocyte expressing the α1β2γ2 subunit combination (left and right traces). The current was attenuated when 10 μM 17PA was co-applied (middle trace). Bottom, the same protocol was used, but 3α5βP was substituted for 3α5αP.
Pregnane steroids sulfated at carbon 3 represent another class of GABA-active steroids, but these steroids inhibit GABA receptor function (Majewska and Schwartz, 1987). We assessed whether 17PA interacted with effects of sulfated steroids. Using a sulfated pregnane steroid with 5α-reduced stereochemistry, (3α,5α)-3-hydroxypregnan-20-one sulfate, we found no change in the degree of inhibition. GABA responses were depressed by 36 ± 4% by 0.5 μM (3α,5α)-3-hydroxypregnan-20-one sulfate (data not shown). In the presence of 10 μM 17PA, responses were depressed similarly (38 ± 3%, n = 6). In sum, 17PA effects are selective to potentiating neurosteroids.
Effects of 17PA on Direct Gating of the GABAA Receptor by Steroids. Potentiating neuroactive steroids also are able to gate the GABAA receptor directly in the absence of GABA (Barker et al., 1987). 17PA effectively blocked currents directly gated by 3α5αP (Fig. 3D), but currents gated by 5 μM 3α5βP were unaffected (Fig. 3D), consistent with diastereoselective effects of 17PA on GABA potentiation. Currents gated by pentobarbital were not affected by 17PA (data not shown). On average, 17PA blocked currents gated by 5 μM 3α5αP by 74 ± 5% and currents gated by 5 μM 3α5βP by 7 ± 6% (n = 4). Notably, at a similar concentration of 3α5αP, 17PA was more effective at reversing that steroid's direct gating effects than it was at reversing its potentiation effects (Fig. 1, E and F).
Effects of 17PA in Hippocampal Neurons. We also examined the effects of 17PA in cultured hippocampal neurons and found that 10 μM failed to detectably alter the holding current of neurons when administered by itself and had no significant effect on responses to 0.5 μM GABA when co-applied with GABA (12 ± 8% potentiation, n = 6). As in oocytes, 17PA significantly inhibited the GABA potentiating actions of 3α5αP (Fig. 4, A and B). Thus, in native cells, where subunits, posttranslational processing, and other factors are likely to differ from the oocyte heterologous expression system, 17PA also antagonized potentiating steroid effects.
We used the more rapid solution exchanges attainable with hippocampal neurons, along with the characteristic slow kinetics of potentiating steroid dissociation, to probe the idea that 17PA acts competitively with respect to potentiating steroid. We reasoned that if 17PA competitively antagonizes potentiation, block of potentiation should not develop faster than the rate of dissociation of potentiator from receptors. If block develops faster than potentiator dissociation, then it is unlikely that 17PA acts in a competitive manner. Figure 4C shows an experiment designed to test the rate of 17PA block of prepotentiated receptors. Figure 4C, upper left, shows a hippocampal neuron's response to 0.5 μM GABA. At upper right is shown the same cell's response to GABA, potentiated by 100 nM 3α5αP. Midway through the co-application of GABA and 3α5αP, the solution was switched back to GABA alone. The current relaxed slowly back toward levels of GABA alone. Note that even after washing away potentiator for >20 s, the current still had not returned to the level of GABA alone, reflecting the extremely slow rate of potentiator dissociation. Washout of GABA at the end of the protocol was rapid on the time scale of the sweep, so it is unlikely that drug delivery was rate limiting in this experiment.
Figure 4C, lower left, shows the effect of again co-applying GABA plus 3α5αP for the first half of the drug application protocol. Midway through the protocol, the solution was switched to one containing a mixture of GABA, potentiator, and 10 μM 17PA. The rate of block in this phase of the experiment was similar to the rate of potentiator dissociation, suggesting that potentiator dissociation is probably rate limiting in the development of 17PA block. Comparable results were obtained in nine neurons, where the time constant of potentiator washout (determined from single exponential fits to the decay of the potentiated current) was indistinguishable from the time constant of block development (17 ± 3 s versus 13 ± 2 s; p > 0.05). As a positive control, we examined the kinetics of bicuculline block, which acts non-competitively with respect to 3α5αP (Ueno et al., 1996). The kinetics of inhibition were clearly faster than those of 17PA (0.5 ± 0.08 s, n = 4).
The slow kinetics of 17PA block in this experiment alone do not exclude the possibility that the antagonist acts noncompetitively with an extremely slow association or inhibition rate, although the results of co-application experiments like those in Fig. 4A suggest that 17PA can equilibrate with receptors rapidly when simultaneously applied with potentiator. These results suggest that competition between 17PA and 5α-reduced neurosteroids is the mechanism by which antagonism occurs. However, the data do not preclude models in which potentiator binding allosterically depresses binding of potentiating steroid at a different site or models in which bound potentiator allosterically alters the ability of bound antagonist to inhibit the potentiated response.
We also examined directly gated currents in hippocampal neurons. 3α5αP (100-200 nM) gated extremely slowly rising and decaying currents in hippocampal neurons, and these currents were more effectively blocked by 17PA than potentiated currents in the presence of GABA (Fig. 4D; compare Fig. 4A). Overall, steroid-gated currents were inhibited by 84 ± 6% by 10 μM 17PA (n = 5). It is interesting that when the steroid-gated current was challenged with bicuculline, bicuculline completely blocked the steroid-gated current along with a small tonic current, which 17PA did not affect (Fig. 4D, top). Bicuculline removal caused a tail current that mimicked the washout of 3α5αP alone (Fig. 4D, bottom). This tail current is consistent with a noncompetitive interaction between bicuculline and neurosteroid agonist (Ueno et al., 1996) and suggests that 3α5αP still interacts with receptors in the presence of bicuculline. This tail current was not observed when bicuculline was administered in the absence of 3α5αP (data not shown) and was never observed in the presence of 17PA (Fig. 4D, top).
Effects of 17PA on Anesthesia. Neuroactive steroids are potent anesthetics. Although it is believed that the relevant site of action for anesthetic steroids is the GABAA receptor, evidence for this assumption is largely correlative (Zorumski et al., 2000; Lambert et al., 2001). If steroids induce anesthesia through actions at GABAA receptors, we would expect 17PA to antagonize the anesthetic effect of steroids. We evaluated behavioral effects of 17PA in tadpoles, a well-validated anesthesia model (Wittmer et al., 1996). When administered alone, 17PA had no effect on LSR at any concentration tested (up to 10 μM). At 10 μM, LRR was observed in only 2 of 10 animals (data not shown). As hypothesized, when combined with steroid anesthetics, 17PA antagonized steroid anesthesia induced by 3α5αP but not anesthesia produced by 3α5βP (Fig. 5). At sufficiently high anesthetic concentrations, antagonism was not apparent (Fig. 5).

17PA effects on steroid anesthesia in tadpoles. A, concentration-response curve for LRR in tadpoles using 3α5αP as anesthetic in one group of animals (X) and combined with 10 μM 17PA (open circles) in another group of animals. B, concentration-response curve for LSR using 3α5αP. Each point represents the fraction of 10 animals that met the criteria for LRR and LSR. The fits estimated a shift from an EC50 of 1.2 to 3.2 μM (LRR) and 2.9 to 10.2 μM. C and D, similar plots using 3α5βP as anesthetic. Note that points in the presence and absence of antagonist superimpose.
The experiments in Fig. 5 examined co-administration of anesthetic and 17PA and demonstrate that 17PA prevents anesthesia in a manner that can be overcome by sufficiently high anesthetic concentration. We also assessed whether 17PA reverses anesthesia after induction. We evaluated concentrations of 3α5αP (2 μM) and 3α5βP (1 μM) that yielded equivalent and near-maximum LRR anesthesia (Fig. 5). An observer naive to the experimental conditions ranked tadpole behavioral responses and found that after a 2-h exposure, 18 of 20 animals exposed to 3α5αP, and 20 of 20 animals exposed to 3α5βP showed LRR. 17PA (10 μM) was then added to the anesthetic bath solution for half of the animals. After an additional 3 h., 8 of 10 animals exposed continuously (5 h) to 3α5αP alone and 10 of 10 animals exposed to 3α5βP alone showed LRR. In contrast, 0 of 10 animals exposed to the combination of 3α5αP plus 17PA showed LRR. 17PA failed to reverse anesthesia induced by 3α5βP; 10 of 10 animals exposed to 3α5βP plus 17PA exhibited LRR. Overall, these results are consistent with the cellular experiments and with the hypothesis that steroid actions at GABAA receptors participate critically in behavioral anesthesia.
Discussion
17PA represents a lead compound in the development of GABA-active neurosteroid antagonists and is the first selective antagonist of GABA-active neurosteroids. Our results suggest several novel conclusions regarding steroid interaction with GABA receptors. First, 17PA distinguishes steroid modulation from modulation by other potentiators, notably barbiturates and benzodiazepines. Second, 17PA selectively reduces the effects of 5α-reduced steroids compared with 5β-reduced steroids, suggesting differences in the mechanism and/or site of action for these classes. Finally, 17PA reduces both potentiation and direct gating by 5α-reduced steroids, suggesting a relationship between these two mechanisms not previously recognized. The development of selective steroid antagonists should be important for studies aimed at understanding the endogenous role of neurosteroids, which have been implicated in seizure disorders (Reddy et al., 2001), depression, sleep disorders, and ethanol effects (for review, see Gasior et al., 1999; Zorumski et al., 2000). In addition, neuroactive steroids may participate in modulation of GABAergic activity in the central nervous system.
The results with 17PA are consistent with at least a partially competitive mechanism. Although the pharmacological profile of 17PA suggests a competitive component with respect to potentiating steroids (Fig. 1, E and F), 17PA consistently depressed the maximum potentiated response. One possibility to explain this result and retain the hypothesis that 17PA acts through competition is that direct steroid gating and steroid potentiation both contribute to total response amplitude at high concentrations of potentiating steroid. Because 17PA is a stronger blocker of direct steroid gating (e.g., Fig. 4), apparent maximum potentiation values are depressed in the concentration-response relationship shown in Fig. 1F.
Another caveat of the concentration-response relationships is that the ability to overcome antagonism with high agonist concentrations does not necessarily imply competition for a binding site. For instance, inverse benzodiazepine agonists shift the concentration-response curve for GABA to the right without changing maximum responses to GABA (Jensen and Lambert, 1986; Sigel and Baur, 1988), but benzodiazepines apparently do not compete with GABA for binding. Similarly, at δ-containing GABA receptor subunit combinations, Zn2+ shifts the GABA concentration-response curve to the right without affecting maximum responses (Krishek et al., 1998). Again, it is unlikely that Zn2+ and GABA compete for binding. Block kinetics for prepotentiated receptors largely followed the time course of agonist dissociation. In addition, 17PA inhibition was not associated with large tail currents observed with bicuculline washout (Fig. 4D), consistent with the idea that 17PA, but not bicuculline, prevents potentiator association with receptors. Therefore, if 17PA and 3α5αP do not directly compete for a site, binding of one occludes the other's actions.
17PA was particularly effective at inhibiting directly gated 5α-reduced steroid responses. For instance, 10 μM 17PA was relatively weak against 100 nM 3α5αP as a potentiator in hippocampal neurons but nearly completely blocked currents directly gated by 100 to 200 nM 3α5αP. Oocytes yielded similar results (compare Figs. 1, E and F, and 3D). Therefore, gating is clearly more strongly blocked by 17PA than potentiation. The relationship between gating and potentiation has been unclear in the literature (Wittmer et al., 1996). One possible explanation for our results is that gating and potentiation may result from 3α5αP acting at a single binding site, whose accessibility to 3α5αP is altered by GABA presence on the receptor. Future work using 17PA and similar compounds can be directed at explicit tests of this model and others.
In summary, this is the first report, to our knowledge, of a selective antagonist of neurosteroid actions at GABAA receptors. 17PA exhibits selectivity for 5α-reduced steroid potentiation of GABA responses and 5α-reduced steroid direct receptor gating. Several aspects of the antagonist are consistent with a competitive mechanism. 17PA and future derivatives may be useful for probing the mechanism of neuroactive steroid actions at GABAA receptors and for probing the endogenous role of neurosteroids in nervous system excitability.
Acknowledgments
We thank Christopher Fields for help in collecting initial data, and we thank laboratory members Gustav Akk and Joe Henry Steinbach for discussion.
Footnotes
-
This study was supported by National Institutes of Health grant MH45493, by a gift from the Bantly Foundation (to C.F.Z.), by National Institutes of Health grants GM47969 (to D.F.C., C.F.Z., and A.S.E.), NS40488 and AA12952 (to S.M.), and a Klingenstein Foundation grant (to S.M.). M.W. was supported by a postdoctoral fellowship from Umeå University-Washington University Network and EU regional fund.
-
ABBREVIATIONS: 3α5αP, (3α,5α)-3-hydroxypregnan-20-one; 3α5βP, (3α,5β)-3-hydroxypregnan-20-one; 17PA, (3α,5α)-17-phenylandrost-16-en-3-ol; DMSO, dimethyl sulfoxide; LRR, loss of righting reflex; LSR, loss of swimming reflex.
-
↵1 Present address: Systems and Cognitive Neuroscience, National Institute of Neurological Disorders and Stroke, Neuroscience Center, Suite 2115, 6001 Executive Boulevard, Bethesda, MD 20892-9521.
-
↵2 Present address: Department of Clinical Science, Section of Obstetrics and Gynecology, Umeå, University, S-90187 Umeå, Sweden.
- Received November 5, 2003.
- Accepted January 23, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}